Pharmacological Therapy of Non-Alcoholic Fatty Liver Disease: What Drugs Are Available Now and Future Perspectives

 , ,
, ,  and
and
Abstract
1. Introduction
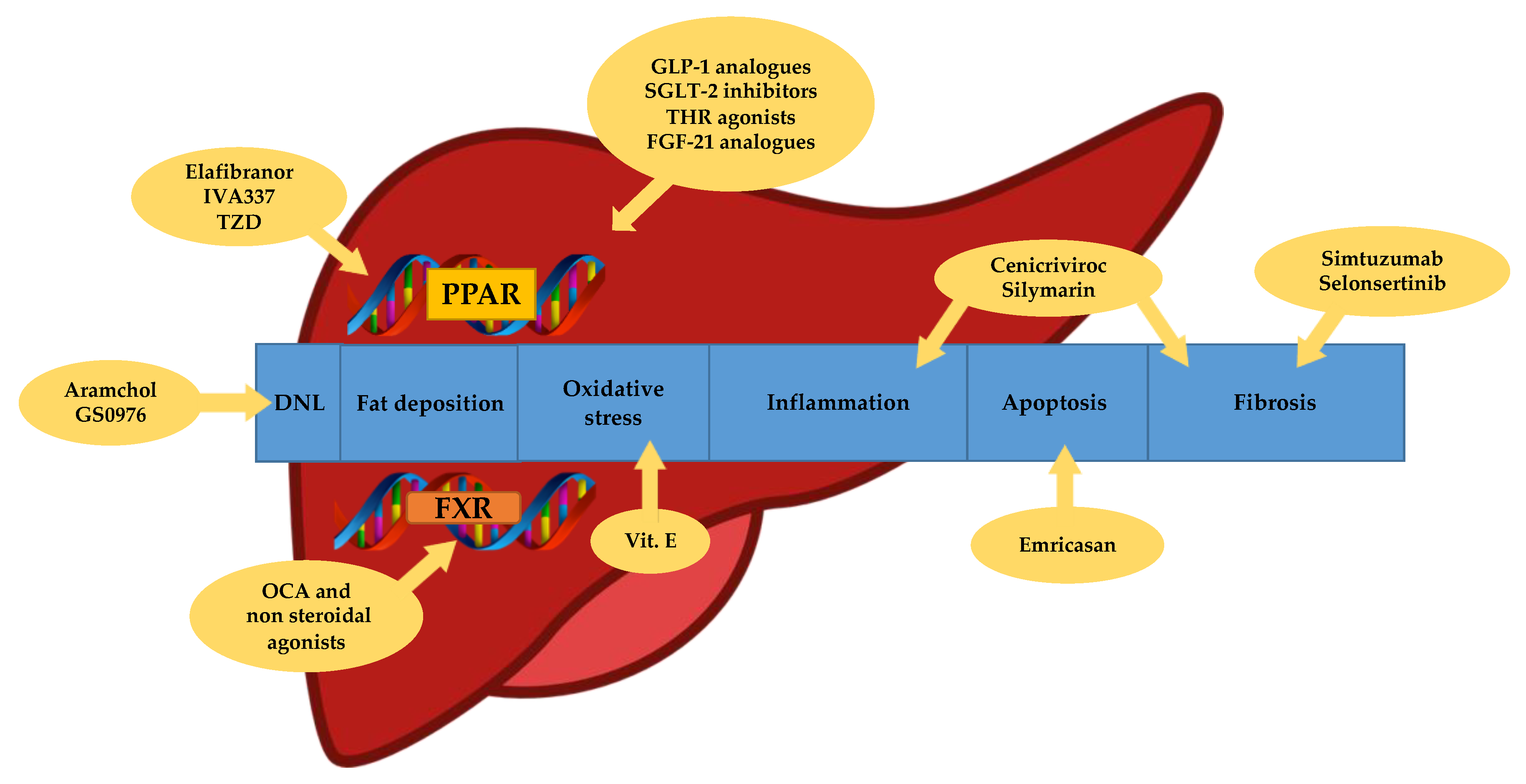
2. Pharmacological Treatment: Different Drugs for Different Therapeutic Targets
2.1. Insulin Sensitizers and Antioxidants
2.2. Vitamin E
2.3. Silymarin
3. Drugs in Development
3.1. Farnesoid X receptor (FXR) Agonists
3.2. Peroxisome proliferator-activated receptors (PPAR) Agonists
3.3. Inhibitors of De Novo Lipogenesis
3.4. Agonist of Thyroid Hormone Receptor
3.5. Antidiabetic Drugs
3.6. Emricasan
3.7. Antifibrotic Drugs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H., Jr.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Sinn, D.H.; Kang, D.; Chang, Y.; Ryu, S.; Gu, S.; Kim, H.; Seong, D.; Cho, S.J.; Yi, B.K.; Park, H.D.; et al. Non-alcoholic fatty liver disease and progression of coronary artery calcium score: A retrospective cohort study. Gut 2017, 66, 323–329. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e5. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef]
- Tacelli, M.; Celsa, C.; Magro, B.; Giannetti, A.; Pennisi, G.; Spatola, F.; Petta, S. Antidiabetic Drugs in NAFLD: The Accomplishment of Two Goals at Once? Pharmaceuticals 2018, 11, 121. [Google Scholar] [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef]
- Hardwick, R.N.; Fisher, C.D.; Canet, M.J.; Lake, A.D.; Cherrington, N.J. Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 2010, 38, 2293–2301. [Google Scholar] [CrossRef]
- Erhardt, A.; Stahl, W.; Sies, H.; Lirussi, F.; Donner, A.; Haussinger, D. Plasma levels of vitamin E and carotenoids are decreased in patients with Nonalcoholic Steatohepatitis (NASH). Eur. J. Med. Res. 2011, 16, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Daniel, K.; Tekeste, T.; Ditah, I.C. Low Vitamin E Levels in NAFLD Patients are Associated with Increased All-Cause Mortality: A 23-Year Prospective Population-Based Study. Gastroenterology 2017, 152, S1201. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Vuppalanchi, R.; Gawrieh, S.; Ghabril, M.; Saxena, R.; Cummings, O.W.; Chalasani, N. Vitamin E Improves Transplant-Free Survival and Hepatic Decompensation Among Patients with Nonalcoholic Steatohepatitis and Advanced Fibrosis. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R., 3rd; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef]
- Schurks, M.; Glynn, R.J.; Rist, P.M.; Tzourio, C.; Kurth, T. Effects of vitamin E on stroke subtypes: Meta-analysis of randomised controlled trials. BMJ 2010, 341, c5702. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Flora, K.; Hahn, M.; Rosen, H.; Benner, K. Milk thistle (Silybum marianum) for the therapy of liver disease. Am. J. Gastroenterol. 1998, 93, 139–143. [Google Scholar] [CrossRef]
- Loguercio, C.; Festi, D. Silybin and the liver: From basic research to clinical practice. World J. Gastroenterol. 2011, 17, 2288–2301. [Google Scholar] [CrossRef]
- Bosisio, E.; Benelli, C.; Pirola, O. Effect of the flavanolignans of Silybum marianum L. on lipid peroxidation in rat liver microsomes and freshly isolated hepatocytes. Pharmacol. Res. 1992, 25, 147–154. [Google Scholar] [CrossRef]
- Schumann, J.; Prockl, J.; Kiemer, A.K.; Vollmar, A.M.; Bang, R.; Tiegs, G. Silibinin protects mice from T cell-dependent liver injury. J. Hepatol. 2003, 39, 333–340. [Google Scholar] [CrossRef]
- Trappoliere, M.; Caligiuri, A.; Schmid, M.; Bertolani, C.; Failli, P.; Vizzutti, F.; Novo, E.; di Manzano, C.; Marra, F.; Loguercio, C.; et al. Silybin, a component of sylimarin, exerts anti-inflammatory and anti-fibrogenic effects on human hepatic stellate cells. J. Hepatol. 2009, 50, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hai, J.; Cao, M.; Zhang, Y.; Pei, S.; Wang, J.; Zhang, Q. Silibinin ameliorates steatosis and insulin resistance during non-alcoholic fatty liver disease development partly through targeting IRS-1/PI3K/Akt pathway. Int. Immunopharmacol. 2013, 17, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Salomone, F.; Barbagallo, I.; Godos, J.; Lembo, V.; Currenti, W.; Cina, D.; Avola, R.; D’Orazio, N.; Morisco, F.; Galvano, F.; et al. Silibinin Restores NAD(+) Levels and Induces the SIRT1/AMPK Pathway in Non-Alcoholic Fatty Liver. Nutrients 2017, 9, 1086. [Google Scholar] [CrossRef] [PubMed]
- Wah Kheong, C.; Nik Mustapha, N.R.; Mahadeva, S. A Randomized Trial of Silymarin for the Treatment of Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2017, 15, 1940–1949.e8. [Google Scholar] [CrossRef] [PubMed]
- Filozof, C.; Chow, S.C.; Dimick-Santos, L.; Chen, Y.F.; Williams, R.N.; Goldstein, B.J.; Sanyal, A. Clinical endpoints and adaptive clinical trials in precirrhotic nonalcoholic steatohepatitis: Facilitating development approaches for an emerging epidemic. Hepatol. Commun. 2017, 1, 577–585. [Google Scholar] [CrossRef]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Li, Y.; Jadhav, K.; Zhang, Y. Bile acid receptors in non-alcoholic fatty liver disease. Biochem. Pharmacol. 2013, 86, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; John, L.M.; Adams, S.H.; Yu, X.X.; Tomlinson, E.; Renz, M.; Williams, P.M.; Soriano, R.; Corpuz, R.; Moffat, B.; et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 2004, 145, 2594–2603. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Jiang, C.; Shi, J.; Gao, X.; Sun, D.; Sun, L.; Wang, T.; Takahashi, S.; Anitha, M.; Krausz, K.W.; et al. An Intestinal Farnesoid X Receptor-Ceramide Signaling Axis Modulates Hepatic Gluconeogenesis in Mice. Diabetes 2017, 66, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Fiorucci, S.; Camaioni, E.; Clerici, C.; Costantino, G.; Maloney, P.R.; Morelli, A.; Parks, D.J.; Willson, T.M. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem. 2002, 45, 3569–3572. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.; Flint, R. Response from Kelly and Flint. N. Z. Med. J. 2015, 128, 80–81. [Google Scholar] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Ratziu, V.; Sanyal, A.J.; Loomba, R.; Rinella, M.; Harrison, S.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; MacConell, L.; Shringarpure, R.; et al. REGENERATE: Design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemp. Clin. Trials 2019, 84, 105803. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig. Dis. 2015, 33, 327–331. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, X.; Phung, V.; Lindhout, D.A.; Mondal, K.; Hsu, J.Y.; Yang, H.; Humphrey, M.; Ding, X.; Arora, T.; et al. Separating Tumorigenicity from Bile Acid Regulatory Activity for Endocrine Hormone FGF19. Cancer Res. 2014, 74, 3306–3316. [Google Scholar] [CrossRef]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef]
- Harrison, S.A.; Rossi, S.J.; Paredes, A.H.; Trotter, J.F.; Bashir, M.R.; Guy, C.D.; Banerjee, R.; Jaros, M.J.; Owers, S.; Baxter, B.A.; et al. NGM282 Improves Liver Fibrosis and Histology in 12 Weeks in Patients with Nonalcoholic Steatohepatitis. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S. The nonsteroidal FXR agonist GS-9674 leads to significant reductions in hepatic steatosis, serum bile acids, and liver biochemistry in a phase 2, randomized, placebo-controlled trial of patients with NASH. Hepatology 2018, 68, 1460A–1461A. [Google Scholar]
- Margolis, R.N.; Evans, R.M.; O’Malley, B.W.; Consortium, N.A. The Nuclear Receptor Signaling Atlas: Development of a functional atlas of nuclear receptors. Mol. Endocrinol. 2005, 19, 2433–2436. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E. Proof of concept study of an apoptosis-signal regulating kinase (ASK-1) inhibitor (selonsertib)n in comnbination with an acetyl-Coa carboxylase inhibitor (GS-0976) or a farnesoid X receptor (FXR) agonist (GS-9674) in NASH. Gastroenterology 2018, 154, 2. [Google Scholar] [CrossRef]
- Sanyal, A. Tropifexor (TXR), an FXR Agonist for the Treatment of NASH–Interim Results from First Two Parts of Phase 2b Study FLIGHT-FXR. 2018. Available online: http://www.natap.org/2018/AASLD/AASLD_168.htm (accessed on 6 November 2019).
- Stringer, Safety, Tolerability, and Efficacy of a Combination Treatment of Tropifexor (LJN452) and Cenicriviroc (CVC) in Adult Patients with Nonalcoholic Steatohepatitis (NASH) and Liver Fibrosis. (TANDEM) In Clinicaltrials.Gov, Ed. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03517540 (accessed on 6 November 2019).
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef]
- Carulli, L.; Maurantonio, M.; Hebbard, L.; Baldelli, E.; Loria, P.; George, J. Classical and innovative insulin sensitizing drugs for the prevention and treatment of NAFLD. Curr. Pharm. Des. 2013, 19, 5280–5296. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef]
- Pfutzner, A.; Hohberg, C.; Lubben, G.; Pahler, S.; Pfutzner, A.H.; Kann, P.; Forst, T. Pioneer study: PPARgamma activation results in overall improvement of clinical and metabolic markers associated with insulin resistance independent of long-term glucose control. Horm. Metab. Res. 2005, 37, 510–515. [Google Scholar] [CrossRef]
- Semple, R.K.; Chatterjee, V.K.; O’Rahilly, S. PPAR gamma and human metabolic disease. J. Clin. Investig. 2006, 116, 581–589. [Google Scholar] [CrossRef]
- Hashimoto, T.; Cook, W.S.; Qi, C.; Yeldandi, A.V.; Reddy, J.K.; Rao, M.S. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J. Biol. Chem. 2000, 275, 28918–28928. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Gervois, P.; Kleemann, R.; Pilon, A.; Percevault, F.; Koenig, W.; Staels, B.; Kooistra, T. Global suppression of IL-6-induced acute phase response gene expression after chronic in vivo treatment with the peroxisome proliferator-activated receptor-alpha activator fenofibrate. J. Biol. Chem. 2004, 279, 16154–16160. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Miranda, C.; Perez-Carreras, M.; Colina, F.; Lopez-Alonso, G.; Vargas, C.; Solis-Herruzo, J.A. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig. Liver Dis. 2008, 40, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Acbay, O.; Sonsuz, A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J. Hepatol. 1999, 31, 384. [Google Scholar] [CrossRef]
- Laurin, J.; Lindor, K.D.; Crippin, J.S.; Gossard, A.; Gores, G.J.; Ludwig, J.; Rakela, J.; McGill, D.B. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: A pilot study. Hepatology 1996, 23, 1464–1467. [Google Scholar] [CrossRef]
- Palmer, C.N.; Hsu, M.H.; Griffin, K.J.; Raucy, J.L.; Johnson, E.F. Peroxisome proliferator activated receptor-alpha expression in human liver. Mol. Pharmacol. 1998, 53, 14–22. [Google Scholar] [CrossRef]
- Bojic, L.A.; Huff, M.W. Peroxisome proliferator-activated receptor delta: A multifaceted metabolic player. Curr. Opin. Lipidol. 2013, 24, 171–177. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef]
- Riserus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zair, Y.; Sauvinet, V.; Noel, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Zair, Y.; Staels, B.; Bruckert, E. Effects of the new dual PPAR alpha/delta agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Bedossa, P. Liver histology and clinical trials for nonalcoholic steatohepatitis-perspectives from 2 pathologists. Gastroenterology 2015, 149, 1305–1308. [Google Scholar] [CrossRef]
- Goedeke, L.; Bates, J.; Vatner, D.F.; Perry, R.J.; Wang, T.; Ramirez, R.; Li, L.; Ellis, M.W.; Zhang, D.; Wong, K.E.; et al. Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatology 2018, 68, 2197–2211. [Google Scholar] [CrossRef]
- Lally, J.S.V.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182.e5. [Google Scholar] [CrossRef]
- Stiede, K.; Miao, W.; Blanchette, H.S.; Beysen, C.; Harriman, G.; Harwood, H.J., Jr.; Kelley, H.; Kapeller, R.; Schmalbach, T.; Westlin, W.F. Acetyl-coenzyme A carboxylase inhibition reduces de novo lipogenesis in overweight male subjects: A randomized, double-blind, crossover study. Hepatology 2017, 66, 324–334. [Google Scholar] [CrossRef]
- Lawitz, E.J.; Coste, A.; Poordad, F.; Alkhouri, N.; Loo, N.; McColgan, B.J.; Tarrant, J.M.; Nguyen, T.; Han, L.; Chung, C.; et al. Acetyl-CoA Carboxylase Inhibitor GS-0976 for 12 Weeks Reduces Hepatic De Novo Lipogenesis and Steatosis in Patients With Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2018, 16, 1983–1991.e3. [Google Scholar] [CrossRef]
- Loomba, R.; Kayali, Z.; Noureddin, M.; Ruane, P.; Lawitz, E.J.; Bennett, M.; Wang, L.; Harting, E.; Tarrant, J.M.; McColgan, B.J.; et al. GS-0976 Reduces Hepatic Steatosis and Fibrosis Markers in Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1463–1473.e6. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed]
- Leikin-Frenkel, A. Treatment of preestablished diet-induced fatty liver by oral fatty acid-bile acid conjugates in rodents. Gastroenterology 2008, 20, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Allen, B. Aramchol Downregulates SCD1 and Induces PPARγ in Hepatic Stellate Cells to Attenuate Cellular Activation and Fibrogenesis; AASLD: Boston, MA, USA, 2018. [Google Scholar]
- Safadi, R.; Konikoff, F.M.; Mahamid, M.; Zelber-Sagi, S.; Halpern, M.; Gilat, T.; Oren, R.; Group, F. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2014, 12, 2085–2091.e1. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V. One-Year Results of the Global Phase 2b Randomized Placebo-Controlled ARREST Trial of Aramchol, a Stearoyl CoA Desaturasemodulator in NASH Patients; AASLD: Boston, MA, USA, 2018. [Google Scholar]
- Michalaki, M.A.; Vagenakis, A.G.; Leonardou, A.S.; Argentou, M.N.; Habeos, I.G.; Makri, M.G.; Psyrogiannis, A.I.; Kalfarentzos, F.E.; Kyriazopoulou, V.E. Thyroid function in humans with morbid obesity. Thyroid 2006, 16, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Boutati, E.; Maratou, E.; Panagiotakos, D.B.; Koukkou, E.; Tzanela, M.; Thalassinos, N.; Raptis, S.A. Insulin action in adipose tissue and muscle in hypothyroidism. J. Clin. Endocrinol. Metab. 2006, 91, 4930–4937. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.; Dinneen, S.F.; O’Brien, P.C.; Palumbo, P.J. Hyperlipidemia in patients with primary and secondary hypothyroidism. Mayo Clin. Proc. 1993, 68, 860–866. [Google Scholar] [CrossRef]
- Rodondi, N.; den Elzen, W.P.; Bauer, D.C.; Cappola, A.R.; Razvi, S.; Walsh, J.P.; Asvold, B.O.; Iervasi, G.; Imaizumi, M.; Collet, T.H.; et al. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA 2010, 304, 1365–1374. [Google Scholar] [CrossRef]
- Eshraghian, A.; Hamidian Jahromi, A. Non-alcoholic fatty liver disease and thyroid dysfunction: A systematic review. World J. Gastroenterol. 2014, 20, 8102–8109. [Google Scholar] [CrossRef]
- Pagadala, M.R.; Zein, C.O.; Dasarathy, S.; Yerian, L.M.; Lopez, R.; McCullough, A.J. Prevalence of hypothyroidism in nonalcoholic fatty liver disease. Dig. Dis. Sci. 2012, 57, 528–534. [Google Scholar] [CrossRef]
- Kim, D.; Kim, W.; Joo, S.K.; Bae, J.M.; Kim, J.H.; Ahmed, A. Subclinical Hypothyroidism and Low-Normal Thyroid Function Are Associated With Nonalcoholic Steatohepatitis and Fibrosis. Clin. Gastroenterol. Hepatol. 2018, 16, 123–131.e1. [Google Scholar] [CrossRef]
- Harrison, S. MGL-3196, a selective thyroid hormone receptor-beta agonist significantly decreases hepatic fat in NASH patients at 12 weeks, the primary endpoint in a 36 week serial liver biopsy study. Hepatology 2018, 68, 38. [Google Scholar] [CrossRef]
- Loomba, R. VK2809, a novel liver-directed thyroid receptor beta agonist, significantly reduces liver fat in patients with non-alcoholic fatty liver disease: A phase 2 randomized, placebo-controlled trial. Hepatology 2018, 68, e150–e151. [Google Scholar] [CrossRef]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Charlotte, F.; Fedchuk, L.; Bedossa, P.; Lebray, P.; Poynard, T.; Ratziu, V.; Group, L.S. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J. Hepatol. 2013, 59, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic reviewand meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Marchesini, G. Time for Glucagon like peptide-1 receptor agonists treatment for patients with NAFLD? J. Hepatol. 2016, 64, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Houlihan, D.D.; Rowe, I.A.; Clausen, W.H.; Elbrond, B.; Gough, S.C.; Tomlinson, J.W.; Newsome, P.N. Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: Individual patient data meta-analysis of the LEAD program. Aliment. Pharmacol. Ther. 2013, 37, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; LEAN Trial Team; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N.; et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef]
- Khoo, J.; Hsiang, J.; Taneja, R.; Law, N.M.; Ang, T.L. Comparative effects of liraglutide 3 mg vs structured lifestyle modification on body weight, liver fat and liver function in obese patients with non-alcoholic fatty liver disease: A pilot randomized trial. Diabetes Obes. Metab. 2017, 19, 1814–1817. [Google Scholar] [CrossRef] [PubMed]
- Hayashizaki-Someya, Y.; Kurosaki, E.; Takasu, T.; Mitori, H.; Yamazaki, S.; Koide, K.; Takakura, S. Ipragliflozin, an SGLT2 inhibitor, exhibits a prophylactic effect on hepatic steatosis and fibrosis induced by choline-deficient l-amino acid-defined diet in rats. Eur. J. Pharmacol. 2015, 754, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Imajo, K.; Kato, T.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Kato, S.; Mawatari, H.; Fujita, K.; Yoneda, M.; et al. The Selective SGLT2 Inhibitor Ipragliflozin Has a Therapeutic Effect on Nonalcoholic Steatohepatitis in Mice. PLoS ONE 2016, 11, e0146337. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Shirakami, Y.; Maruta, A.; Ideta, T.; Miyazaki, T.; Kochi, T.; Sakai, H.; Tanaka, T.; Seishima, M.; Shimizu, M. Preventive effects of the sodium glucose cotransporter 2 inhibitor tofogliflozin on diethylnitrosamine-induced liver tumorigenesis in obese and diabetic mice. Oncotarget 2017, 8, 58353–58363. [Google Scholar] [CrossRef]
- Ito, D. Comparison of Ipragliflozin and Pioglitazone Effects on Nonalcoholic Fatty Liver Disease in Patients with Type 2 Diabetes: A Randomized, 24-Week, Open-Label, Active-Controlled Trial. Diabetes Care 2017, 40, 1364–1372. [Google Scholar] [CrossRef]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Farooqui, K.J.; Singh, M.K.; Wasir, J.S.; Bansal, B.; Kaur, P.; Jevalikar, G.; Gill, H.K.; et al. Effect of Empagliflozin on Liver Fat in Patients With Type 2 Diabetes and Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial (E-LIFT Trial). Diabetes Care 2018, 41, 1801–1808. [Google Scholar] [CrossRef]
- Vasilakou, D.; Karagiannis, T.; Athanasiadou, E.; Mainou, M.; Liakos, A.; Bekiari, E.; Sarigianni, M.; Matthews, D.R.; Tsapas, A. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 262–274. [Google Scholar] [CrossRef]
- Faillie, J.L. Pharmacological aspects of the safety of gliflozins. Pharmacol. Res. 2017, 118, 71–81. [Google Scholar] [CrossRef]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef]
- Nies, V.J.; Sancar, G.; Liu, W.; van Zutphen, T.; Struik, D.; Yu, R.T.; Atkins, A.R.; Evans, R.M.; Jonker, J.W.; Downes, M.R. Fibroblast Growth Factor Signaling in Metabolic Regulation. Front. Endocrinol. (Lausanne) 2015, 6, 193. [Google Scholar] [CrossRef]
- Mai, K.; Andres, J.; Biedasek, K.; Weicht, J.; Bobbert, T.; Sabath, M.; Meinus, S.; Reinecke, F.; Mohlig, M.; Weickert, M.O.; et al. Free fatty acids link metabolism and regulation of the insulin-sensitizing fibroblast growth factor-21. Diabetes 2009, 58, 1532–1538. [Google Scholar] [CrossRef]
- Hale, C.; Chen, M.M.; Stanislaus, S.; Chinookoswong, N.; Hager, T.; Wang, M.; Veniant, M.M.; Xu, J. Lack of overt FGF21 resistance in two mouse models of obesity and insulin resistance. Endocrinology 2012, 153, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Coskun, T.; Bina, H.A.; Schneider, M.A.; Dunbar, J.D.; Hu, C.C.; Chen, Y.; Moller, D.E.; Kharitonenkov, A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 2008, 149, 6018–6027. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Adams, A.C.; Brozinick, J.T.; Bui, H.H.; Miyauchi, Y.; Kusminski, C.M.; Bauer, S.M.; Wade, M.; Singhal, E.; Cheng, C.C.; et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 2013, 17, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.S.; Lindberg, R.A.; et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef]
- Luo, Y. BMS-986036, a PEGylated fibroblast growth factor 21 analogue, reduces fibrosis and PRO-C3 in a mouse model of non-alcoholic steatohepatitis. Hepatology 2018, 68, S396–S397. [Google Scholar] [CrossRef]
- Charles, E.D. Pegbelfermin (BMS-986036), PEGylated FGF21, in patients with obesity and type 2 diabetes: Results from a randomized phase 2 study. Obesity 2019, 27, 41–49. [Google Scholar] [CrossRef]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 2019, 392, 2705–2717. [Google Scholar] [CrossRef]
- Syn, W.K.; Choi, S.S.; Diehl, A.M. Apoptosis and cytokines in non-alcoholic steatohepatitis. Clin. Liver Dis. 2009, 13, 565–580. [Google Scholar] [CrossRef][Green Version]
- Shiffman, M.L.; Pockros, P.; McHutchison, J.G.; Schiff, E.R.; Morris, M.; Burgess, G. Clinical trial: The efficacy and safety of oral PF-03491390, a pancaspase inhibitor-a randomized placebo-controlled study in patients with chronic hepatitis C. Aliment. Pharmacol. Ther. 2010, 31, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Shiffman, M.; Freilich, B.; Vuppalanchi, R.; Watt, K.; Chan, J.L.; Spada, A.; Hagerty, D.T.; Schiff, E. Randomised clinical trial: Emricasan versus placebo significantly decreases ALT and caspase 3/7 activation in subjects with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2019, 49, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tsao, G.; Fuchs, M.; Shiffman, M.; Borg, B.B.; Pyrsopoulos, N.; Shetty, K.; Gallegos-Orozco, J.F.; Reddy, K.R.; Feyssa, E.; Chan, J.L.; et al. Emricasan (IDN-6556) Lowers Portal Pressure in Patients with Compensated Cirrhosis and Severe Portal Hypertension. Hepatology 2019, 69, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tsao, G. LBO-01-Multicenter, double-blind, placebo-controlled, randomized trial of emricasan in subjects with NASH cirrhosis and severe portal hypertension. Hepatology 2019, 70, e127. [Google Scholar] [CrossRef]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell. Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef]
- Raeman, R.; Anania, F.A. Therapy for steatohepatitis: Do macrophages hold the clue? Hepatology 2018, 67, 1204–1206. [Google Scholar] [CrossRef]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef]
- Mossanen, J.C.; Krenkel, O.; Ergen, C.; Govaere, O.; Liepelt, A.; Puengel, T.; Heymann, F.; Kalthoff, S.; Lefebvre, E.; Eulberg, D.; et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 2016, 64, 1667–1682. [Google Scholar] [CrossRef]
- Friedman, S.; Sanyal, A.; Goodman, Z.; Lefebvre, E.; Gottwald, M.; Fischer, L.; Ratziu, V. Efficacy and safety study of cenicriviroc for the treatment of non-alcoholic steatohepatitis in adult subjects with liver fibrosis: CENTAUR Phase 2b study design. Contemp. Clin. Trials 2016, 47, 356–365. [Google Scholar] [CrossRef]
- Lefebvre, E.; Moyle, G.; Reshef, R.; Richman, L.P.; Thompson, M.; Hong, F.; Chou, H.L.; Hashiguchi, T.; Plato, C.; Poulin, D.; et al. Antifibrotic Effects of the Dual CCR2/CCR5 Antagonist Cenicriviroc in Animal Models of Liver and Kidney Fibrosis. PLoS ONE 2016, 11, e0158156. [Google Scholar] [CrossRef] [PubMed]
- Puengel, T.; Krenkel, O.; Kohlhepp, M.; Lefebvre, E.; Luedde, T.; Trautwein, C.; Tacke, F. Differential impact of the dual CCR2/CCR5 inhibitor cenicriviroc on migration of monocyte and lymphocyte subsets in acute liver injury. PLoS ONE 2017, 12, e0184694. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, E.; Gottwald, M.; Lasseter, K.; Chang, W.; Willett, M.; Smith, P.F.; Somasunderam, A.; Utay, N.S. Pharmacokinetics, Safety, and CCR2/CCR5 Antagonist Activity of Cenicriviroc in Participants with Mild or Moderate Hepatic Impairment. Clin. Transl. Sci. 2016, 9, 139–148. [Google Scholar] [CrossRef]
- Thompson, M.; Saag, M.; DeJesus, E.; Gathe, J.; Lalezari, J.; Landay, A.L.; Cade, J.; Enejosa, J.; Lefebvre, E.; Feinberg, J. A 48-week randomized phase 2b study evaluating cenicriviroc versus efavirenz in treatment-naive HIV-infected adults with C-C chemokine receptor type 5-tropic virus. AIDS 2016, 30, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Hong, F. Significant anti-fibrotic activity of cenicriviroc, a dual CCR2/CCR5 antagonist, in a rat model of thioacetamide-induced liver fibrosis and cirrhosis. Hepatology 2013, 58, 1381A–1382A. [Google Scholar]
- Lefebvre, E.; Gottwald, M.; Lasseter, K.; Chang, W.; Willett, M.; Smith, P.F.; Somasunderam, A.; Utay, N.S. Pharmacokinetics, Safety, and CCR2/CCR5 Antagonist Activity of Cenicriviroc in Participants With Mild or Moderate Hepatic Impairment. Clin Transl Sci. 2016, 9, 139–148. [Google Scholar] [CrossRef]
- Friedman, S. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef]
- Ratziu, V. Cenicriviroc Treatment for Adults with Non-Alcoholic Steatohepatitis: Year 2 Analysis of the Phase 2b CENTAUR Study; Congress, I.L.: The international liver congress: Paris, France, 2018. [Google Scholar]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J.; American Association for the Study of Liver Diseases; American College of Gastroenterologyh; et al. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Am. J. Gastroenterol. 2012, 107, 811–826. [Google Scholar] [CrossRef]
- Hayakawa, R.; Hayakawa, T.; Takeda, K.; Ichijo, H. Therapeutic targets in the ASK1-dependent stress signaling pathways. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 434–453. [Google Scholar] [CrossRef]
- Yamamoto, E.; Dong, Y.F.; Kataoka, K.; Yamashita, T.; Tokutomi, Y.; Matsuba, S.; Ichijo, H.; Ogawa, H.; Kim-Mitsuyama, S. Olmesartan prevents cardiovascular injury and hepatic steatosis in obesity and diabetes, accompanied by apoptosis signal regulating kinase-1 inhibition. Hypertension 2008, 52, 573–580. [Google Scholar] [CrossRef]
- Diehl, A.M. Efficacy and safety of simtuzumab for the treatment of NASH with bridging fibrosis or cirrhosis: Results of two phase 2b, dose-ranging, randomized, placebo-controlled trials. Hepatology 2017, 66, S54. [Google Scholar]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Talal, A.H. Simtuzumab, an antifibrotic monoclonal antibody against lysyl oxidase-like 2 (LOXL2) enzyme, appears safe and well tolerated in patients with liver disease of diverse etiology [Abstract]; Congress I.L.: The International liver congress: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Ikenaga, N. Dual combination therapy directed against lysyl oxidase-like 2 (LOXL2) and apoptosis signal–regulating kinase 1 (ASK1) potently inhibits fibrosis and portal hypertension in a new mouse model of PSC-like liver disease [Abstract]. Hepatology 2015, 62, 881A. [Google Scholar]


{kind=link}
| Drugs | Mechanisms of Action | Scientific Evidence |
|---|---|---|
| Thiazolidinediones |
|
|
| Vitamin E |
|
|
| Silymarin |
|
|
| Obeticholic acid |
|
|
| NGM282 |
|
|
| Cilofexor |
|
|
| Tropifexor |
|
|
| Elafibranor |
|
|
| Lanifibranor |
|
|
| GS-0976 |
|
|
| Aramchol |
|
|
| Resmetirom |
|
|
| VK-2809 |
|
|
| Liraglutide |
|
|
| Ipraglifozin |
|
|
| Pegbelfermin |
|
|
| Emricasan |
|
|
| Cenicriviroc |
|
|
| Selonsertinib |
|
|
| Simtuzumab |
|
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pennisi, G.; Celsa, C.; Spatola, F.; Dallio, M.; Federico, A.; Petta, S. Pharmacological Therapy of Non-Alcoholic Fatty Liver Disease: What Drugs Are Available Now and Future Perspectives. Int. J. Environ. Res. Public Health 2019, 16, 4334. https://doi.org/10.3390/ijerph16224334
Pennisi G, Celsa C, Spatola F, Dallio M, Federico A, Petta S. Pharmacological Therapy of Non-Alcoholic Fatty Liver Disease: What Drugs Are Available Now and Future Perspectives. International Journal of Environmental Research and Public Health. 2019; 16(22):4334. https://doi.org/10.3390/ijerph16224334
Chicago/Turabian StylePennisi, Grazia, Ciro Celsa, Federica Spatola, Marcello Dallio, Alessandro Federico, and Salvatore Petta. 2019. "Pharmacological Therapy of Non-Alcoholic Fatty Liver Disease: What Drugs Are Available Now and Future Perspectives" International Journal of Environmental Research and Public Health 16, no. 22: 4334. https://doi.org/10.3390/ijerph16224334
APA StylePennisi, G., Celsa, C., Spatola, F., Dallio, M., Federico, A., & Petta, S. (2019). Pharmacological Therapy of Non-Alcoholic Fatty Liver Disease: What Drugs Are Available Now and Future Perspectives. International Journal of Environmental Research and Public Health, 16(22), 4334. https://doi.org/10.3390/ijerph16224334