
Endocrine Disruptors Leading to Obesity and Related Diseases





Abstract
1. Introduction
2. Endocrine Disruptorss in Obesity Induction
3. Endocrine Disruptors—Role in Obesity Related Diseases
3.1. Endocrine Disruptors Involvement in Metabolic Syndrome
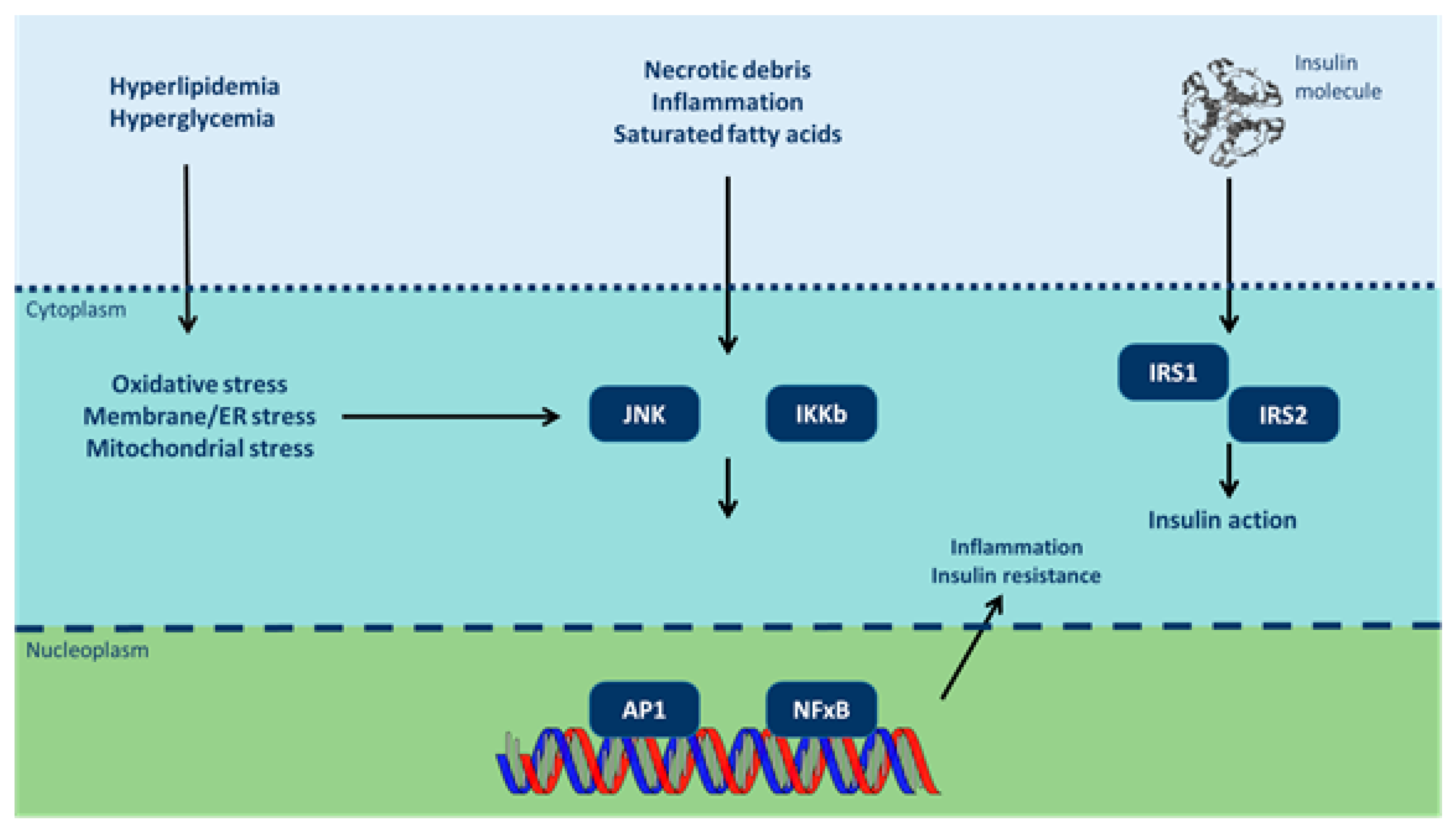
3.2. Endocrine Disruptors and Insulin Resistance
3.3. Endocrine Disruptors Involvement in the Manifestation of Cardiovascular Diseases
3.4. Endocrine Disruptors Involvement in Hepatic Steatosis
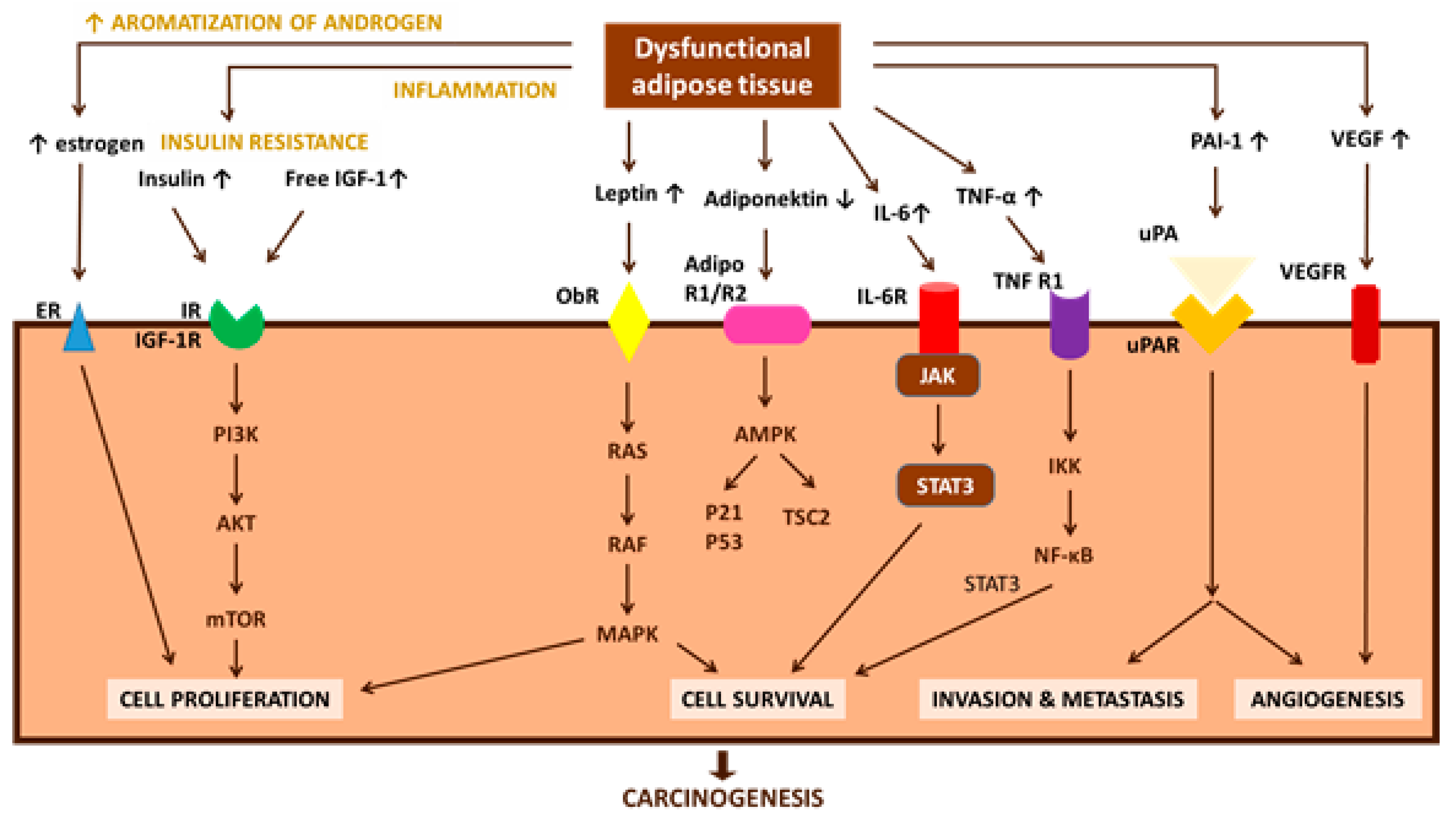
3.5. Endocrine Disruptors Involvement in Carcinogenesis
3.6. Endocrine Disruptors Effect in Female and Male Reproductive Function
4. Conclusions
Conflicts of Interest
References
- Vassilopoulou, L.; Psycharakis, C.; Petrakis, D.; Tsiaoussis, J.; Tsatsakis, A.M. Obesity, Persistent Organic Pollutants and Related Health Problems. Adv. Exp. Med. Biol. 2017, 960, 81–110. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization: Obesity and Overweight. Available online: http://www.who.int/mediacentre/factsheets/fs311/en/ (accessed on 3 August 2017).
- OECD Obesity Update. 2017. Available online: http://www.oecd.org/health/obesity-update.htm (accessed on 3 August 2017).
- Environment and Climate Research Programme of DG XII of the European Commission. EUR 17549, The European Workshop on the Impact of Endocrine Disrupters on Human Health and Wildlife, Report of Proceedings Weybridge, UK, 1996; Environment and Climate Research Programme of DG XII of the European Commission: Weybridge, UK, 1996. [Google Scholar]
- Saiyed, H.; Dewan, A.; Bhatnagar, V.; Shenoy, U.; Shenoy, R.; Rajmohan, H.; Patel, K.; Kashyap, R.; Kulkarni, P.; Rajan, B.; et al. Effect of endosulfan on male reproductive development. Environ. Health Perspect. 2003, 111, 1958–1962. [Google Scholar] [CrossRef] [PubMed]
- Sifakis, S.; Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Human exposure to endocrine disrupting chemicals: Effects on the male and female reproductive systems. Environ. Toxicol. Pharmacol. 2017, 51, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.M.; Chung, K.L.; Sonnenschein, C. The pesticides endosulfan, toxaphene, and dieldrin have estrogenic effects on human estrogen-sensitive cells. Environ. Health Perspect. 1994, 102, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Tong, W.; Branham, W.S.; Moland, C.L.; Dial, S.L.; Hong, H.; Xie, Q.; Perkins, R.; Owens, W.; Sheehan, D.M. Study of 202 natural, synthetic, and environmental chemicals for binding to the androgen receptor. Chem. Res. Toxicol. 2003, 16, 1338–1358. [Google Scholar] [CrossRef] [PubMed]
- Vinggaard, A.M.; Breinholt, V.; Larsen, J.C. Screening of selected pesticides for oestrogen receptor activation in vitro. Food Addit. Contam. 1999, 16, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, H.; Yanase, T.; Nomura, M.; Okabe, T.; Goto, K.; Harada, N.; Nawata, H. A benzimidazole fungicide, benomyl, and its metabolite, carbendazim, induce aromatase activity in a human ovarian granulose-like tumor cell line (KGN). Endocrinology 2004, 145, 1860–1869. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.R.; Vinggaard, A.M.; Rasmussen, T.H.; Gjermandsen, I.M.; Bonefeld-Jorgensen, E.C. Effects of currently used pesticides in assays for estrogenicity, androgenicity, and aromatase activity in vitro. Toxicol. Appl. Pharmacol. 2002, 179, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, J.T.; Seinen, W.; Giesy, J.P.; van den Berg, M. 2-Chloro-s-triazine herbicides induce aromatase (CYP19) activity in H295R human adrenocortical carcinoma cells: A novel mechanism for estrogenicity? Toxicol. Sci. 2000, 54, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Akingbemi, B.T.; Ge, R.S.; Klinefelter, G.R.; Gunsalus, G.L.; Hardy, M.P. A metabolite of methoxychlor, 2,2-bis(p-hydroxyphenyl)-1,1,1-trichloroethane, reduces testosterone biosynthesis in rat leydig cells through suppression of steady-state messenger ribonucleic acid levels of the cholesterol side-chain cleavage enzyme. Biol. Reprod. 2000, 62, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.F.; Parron, T.; Tsatsakis, A.M.; Requena, M.; Alarcon, R.; Lopez-Guarnido, O. Toxic effects of pesticide mixtures at a molecular level: Their relevance to human health. Toxicology 2013, 307, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, V.; Cardoso, R.C.; Puttabyatappa, M. Developmental Programming, a Pathway to Disease. Endocrinology 2016, 157, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Kirkley, A.G.; Sargis, R.M. Environmental endocrine disruption of energy metabolism and cardiovascular risk. Curr. Diab. Rep. 2014, 14, 494. [Google Scholar] [CrossRef] [PubMed]
- Trevino, L.S.; Wang, Q.; Walker, C.L. Hypothesis: Activation of rapid signaling by environmental estrogens and epigenetic reprogramming in breast cancer. Reprod. Toxicol. 2015, 54, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Grun, F.; Blumberg, B. Perturbed nuclear receptor signaling by environmental obesogens as emerging factors in the obesity crisis. Rev. Endocr. Metab. Disord. 2007, 8, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Snedeker, S.M.; Hay, A.G. Do interactions between gut ecology and environmental chemicals contribute to obesity and diabetes? Environ. Health Perspect. 2012, 120, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, B. Obesogens, stem cells and the maternal programming of obesity. J. Dev. Orig. Health Dis. 2011, 2, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J. The obesogen hypothesis of obesity: Overview and human evidence. Maternal and prenatal influences of the offspring. In Obesity before Birth; Lustig, R.H., Ed.; Springer: New York, NY, USA, 2011; pp. 355–366. ISBN 978-1-4419-7034-3. [Google Scholar]
- Chamorro-Garcia, R.; Blumberg, B. Transgenerational effects of obesogens and the obesity epidemic. Curr. Opin. Pharmacol. 2014, 19, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulou, L.; Psycharakis, C.; Petrakis, D.; Tsiaoussis, J.; Tsatsakis, A.M. Persistent organic pollutants and related health problems. In Obesity and Lipotoxicity. Advances in Experimental Medicine and Biology; Engin, A.B., Engin, A., Eds.; Springer: New York, NY, USA, 2017; pp. 81–110. ISBN 978-3-319-48382-5. [Google Scholar]
- Cioboata, R.; Gaman, A.; Trasca, D.; Ungureanu, A.; Docea, A.O.; Tomescu, P.; Gherghina, F.; Arsene, A.L.; Badiu, C.; Tsatsakis, A.M.; et al. Pharmacological management of non-alcoholic fatty liver disease: Atorvastatin versus pentoxifylline. Exp. Ther. Med. 2017, 13, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Mangum, L.H.; Crow, J.A.; Stokes, J.V.; Howell, G.E., 3rd; Ross, M.K.; Pruett, S.B.; Chambers, J.E. Exposure to p,p′-DDE alters macrophage reactivity and increases macrophage numbers in adipose stromal vascular fraction. Toxicol. Sci. 2016, 150, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Koppaka, S.; Kehlenbrink, S.; Carey, M.; Li, W.; Sanchez, E.; Lee, D.E.; Lee, H.; Chen, J.; Carrasco, E.; Kishore, P.; et al. Reduced adipose tissue macrophage content is associated with improved insulin sensitivity in thiazolidinedione-treated diabetic humans. Diabetes 2013, 62, 1843–1854. [Google Scholar] [CrossRef] [PubMed]
- Boudalia, S.; Belloir, C.; Miller, M.L.; Canivenc-Lavier, M.C. Early endocrine disruptors exposure acts on 3T3-L1 differentiation and endocrine activity. Bioimpacts 2017, 7, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arevalo, M.; Alonso-Magdalena, P.; Servitja, J.M.; Boronat-Belda, T.; Merino, B.; Villar-Pazos, S.; Medina-Gomez, G.; Novials, A.; Quesada, I.; Nadal, A. Maternal Exposure to Bisphenol-A During Pregnancy Increases Pancreatic beta-Cell Growth During Early Life in Male Mice Offspring. Endocrinology 2016, 157, 4158–4171. [Google Scholar] [CrossRef] [PubMed]
- Howell, G., 3rd; Mangum, L. Exposure to bioaccumulative organochlorine compounds alters adipogenesis, fatty acid uptake, and adipokine production in NIH3T3-L1 cells. Toxicol. In Vitro 2011, 25, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.W.; Tang, Y.; Li, X.; Liu, Y.; Zhang, Y.Y.; Huang, H.Y.; Xue, R.D.; Yu, H.Y.; Guo, L.; Gao, H.D.; et al. BMP4-mediated brown fat-like changes in white adipose tissue alter glucose and energy homeostasis. Proc. Natl. Acad. Sci. USA 2013, 110, E798–E807. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, T.; Yokoyama, S.; Matsuo, S.; Akira, S.; Ozawa, K. Myeloid differentiation factor 88 (MyD88)-deficiency increases risk of diabetes in mice. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Tuncman, G.; Hirosumi, J.; Solinas, G.; Chang, L.; Karin, M.; Hotamisligil, G.S. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2006, 103, 10741–10746. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Li, W.; Sciullo, E.; Newman, J.; Hammock, B.; Reader, J.R.; Tuscano, J.; Matsumura, F. Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression. Am. J. Pathol. 2007, 171, 1538–1548. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Filho, M.A.; Ueno, M.; Hirabara, S.M.; Seabra, A.B.; Carvalheira, J.B.; de Oliveira, M.G.; Velloso, L.A.; Curi, R.; Saad, M.J. S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: A novel mechanism of insulin resistance. Diabetes 2005, 54, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Magdalena, P.; Laribi, O.; Ropero, A.B.; Fuentes, E.; Ripoll, C.; Soria, B.; Nadal, A. Low doses of bisphenol A and diethylstilbestrol impair Ca2+ signals in pancreatic alpha-cells through a nonclassical membrane estrogen receptor within intact islets of Langerhans. Environ. Health Perspect. 2005, 113, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.Z.; Speck, M.; Yoganathan, P.; Nackiewicz, D.; Hansen, A.M.; Ladefoged, M.; Rabe, B.; Rose-John, S.; Voshol, P.J.; Lynn, F.C.; et al. Glycoprotein 130 receptor signaling mediates alpha-cell dysfunction in a rodent model of type 2 diabetes. Diabetes 2014, 63, 2984–2995. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, Q.; Xu, C.; Shao, W.; Zhang, C.; Liu, H.; Jiang, Z.; Gu, A. Organochloride pesticides impaired mitochondrial function in hepatocytes and aggravated disorders of fatty acid metabolism. Sci. Rep. 2017, 7, 46339. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.; Lensu, S.; Pohjanvirta, R. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on hormones of energy balance in a TCDD-sensitive and a TCDD-resistant rat strain. Int. J. Mol. Sci. 2014, 15, 13938–13966. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Matsui, H. Triphenyltin impairs a protein kinase A (PKA)-dependent increase of cytosolic Na+ and Ca2+ and PKA-independent increase of cytosolic Ca2+ associated with insulin secretion in hamster pancreatic beta-cells. Toxicol. Appl. Pharmacol. 2006, 216, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.J.; Zhou, H.R.; Wagner, M.A. Polychlorinated biphenyls release insulin from RINm5F cells. Life Sci. 1996, 59, 2041–2049. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, H.; Geng, N.; Zhang, B.; Ren, X.; Chen, J. New Insights into the Cytotoxic Mechanism of Hexabromocyclododecane from a Metabolomic Approach. Environ. Sci. Technol. 2016, 50, 3145–3153. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, A.; Toyoshima, M.; Fujimiya, M.; Harada, K.; Ataka, K.; Inoue, K.; Koizumi, A. Perfluorooctane sulfonate influences feeding behavior and gut motility via the hypothalamus. Int. J. Mol. Med. 2007, 19, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Betts, K.S. Perfluoroalkyl acids: What is the evidence telling us? Environ. Health Perspect. 2007, 115, A250–A256. [Google Scholar] [CrossRef] [PubMed]
- Birks, L.; Casas, M.; Garcia, A.M.; Alexander, J.; Barros, H.; Bergstrom, A.; Bonde, J.P.; Burdorf, A.; Costet, N.; Danileviciute, A.; et al. Occupational exposure to endocrine-disrupting chemicals and birth weight and length of gestation: A European meta-analysis. Environ. Health Perspect. 2016, 124, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Janesick, A.S.; Blumberg, B. Obesogens: An emerging threat to public health. Am. J. Obstet. Gynecol. 2016, 214, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Tang-Peronard, J.L.; Heitmann, B.L.; Andersen, H.R.; Steuerwald, U.; Grandjean, P.; Weihe, P.; Jensen, T.K. Association between prenatal polychlorinated biphenyl exposure and obesity development at ages 5 and 7 y: A prospective cohort study of 656 children from the Faroe Islands. Am. J. Clin. Nutr. 2014, 99, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Vafeiadi, M.; Roumeliotaki, T.; Myridakis, A.; Chalkiadaki, G.; Fthenou, E.; Dermitzaki, E.; Karachaliou, M.; Sarri, K.; Vassilaki, M.; Stephanou, E.G.; et al. Association of early life exposure to bisphenol A with obesity and cardiometabolic traits in childhood. Environ. Res. 2016, 146, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.J.; Cheng, X.J.; Xia, H.F.; Ma, X. The endocrine disruptor 4-nonylphenol promotes adipocyte differentiation and induces obesity in mice. Cell. Physiol. Biochem. 2012, 30, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Kamstra, J.H.; Hruba, E.; Blumberg, B.; Janesick, A.; Mandrup, S.; Hamers, T.; Legler, J. Transcriptional and epigenetic mechanisms underlying enhanced in vitro adipocyte differentiation by the brominated flame retardant BDE-47. Environ. Sci. Technol. 2014, 48, 4110–4119. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Webster, T.F.; Ferguson, P.L.; Stapleton, H.M. Characterizing the peroxisome proliferator-activated receptor (PPARgamma) ligand binding potential of several major flame retardants, their metabolites, and chemical mixtures in house dust. Environ. Health Perspect. 2015, 123, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Watt, J.; Schlezinger, J.J. Structurally-diverse, PPARgamma-activating environmental toxicants induce adipogenesis and suppress osteogenesis in bone marrow mesenchymal stromal cells. Toxicology 2015, 331, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Braga-Basaria, M.; Dobs, A.S.; Muller, D.C.; Carducci, M.A.; John, M.; Egan, J.; Basaria, S. Metabolic syndrome in men with prostate cancer undergoing long-term androgen-deprivation therapy. J. Clin. Oncol. 2006, 24, 3979–3983. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Gelman, L.; Rossi, D.; Zoete, V.; Metivier, R.; Tudor, C.; Anghel, S.I.; Grosdidier, A.; Lathion, C.; Engelborghs, Y.; et al. The endocrine disruptor monoethyl-hexyl-phthalate is a selective peroxisome proliferator-activated receptor gamma modulator that promotes adipogenesis. J. Biol. Chem. 2007, 282, 19152–19166. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.A.; Leffers, H. Emerging endocrine disrupters: Perfluoroalkylated substances. Int. J. Androl. 2008, 31, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.E. Metabolic imprinting: critical impact of the perinatal environment on the regulation of energy homeostasis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1107–1121. [Google Scholar] [CrossRef] [PubMed]
- Stahlhut, R.W.; van Wijngaarden, E.; Dye, T.D.; Cook, S.; Swan, S.H. Concentrations of urinary phthalate metabolites are associated with increased waist circumference and insulin resistance in adult U.S. males. Environ. Health Perspect. 2007, 115, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Ciais, D.; Bailly, S. BMPs go for apelin to regulate angiogenesis. Focus on “inhibition of apelin expression by BMP signaling in endothelial cells”. Am. J. Physiol. Cell Physiol. 2012, 303, C1127–C1128. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Casals-Casas, C.; Desvergne, B. Endocrine disruptors: From endocrine to metabolic disruption. Annu. Rev. Physiol. 2011, 73, 135–162. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.T.; Lee, H.K. Metabolic syndrome and the environmental pollutants from mitochondrial perspectives. Rev. Endocr. Metab. Disord. 2014, 15, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Jellyman, J.K.; Ross, M.G. Epigenomics, gestational programming and risk of metabolic syndrome. Int. J. Obes. (Lond.) 2015, 39, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Giulivo, M.; Lopez de Alda, M.; Capri, E.; Barcelo, D. Human exposure to endocrine disrupting compounds: Their role in reproductive systems, metabolic syndrome and breast cancer. A review. Environ. Res. 2016, 151, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Kern, P.A.; Dicker-Brown, A.; Said, S.T.; Kennedy, R.; Fonseca, V.A. The stimulation of tumor necrosis factor and inhibition of glucose transport and lipoprotein lipase in adipose cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Metabolism 2002, 51, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Chawla, A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 2013, 339, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Hauner, H.; Petruschke, T.; Russ, M.; Rohrig, K.; Eckel, J. Effects of tumour necrosis factor alpha (TNF alpha) on glucose transport and lipid metabolism of newly-differentiated human fat cells in cell culture. Diabetologia 1995, 38, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Paz-Filho, G.; Mastronardi, C.; Wong, M.L.; Licinio, J. Leptin therapy, insulin sensitivity, and glucose homeostasis. Indian J. Endocrinol. Metab. 2012, 16, S549–S555. [Google Scholar] [CrossRef] [PubMed]
- Hivert, M.F.; Sullivan, L.M.; Fox, C.S.; Nathan, D.M.; D'Agostino, R.B., Sr.; Wilson, P.W.; Meigs, J.B. Associations of adiponectin, resistin, and tumor necrosis factor-alpha with insulin resistance. J. Clin. Endocrinol. Metab. 2008, 93, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Olszanecka-Glinianowicz, M.; Kocelak, P.; Nylec, M.; Chudek, J.; Zahorska-Markiewicz, B. Circulating visfatin level and visfatin/insulin ratio in obese women with metabolic syndrome. Arch. Med. Sci. 2012, 8, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Song, N.J.; Kim, S.; Jang, B.H.; Chang, S.H.; Yun, U.J.; Park, K.M.; Waki, H.; Li, D.Y.; Tontonoz, P.; Park, K.W. Small Molecule-Induced Complement Factor D (Adipsin) Promotes Lipid Accumulation and Adipocyte Differentiation. PLoS ONE 2016, 11, e0162228. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Biddinger, S.B. Dissecting the role of insulin resistance in the metabolic syndrome. Curr. Opin. Lipidol. 2009, 20, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wei, J.; Li, Y.; Chen, J.; Zhou, Z.; Song, L.; Wei, Z.; Lv, Z.; Chen, X.; Xia, W.; et al. Developmental exposure to di(2-ethylhexyl) phthalate impairs endocrine pancreas and leads to long-term adverse effects on glucose homeostasis in the rat. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E527–E538. [Google Scholar] [CrossRef] [PubMed]
- Bodin, J.; Bolling, A.K.; Becher, R.; Kuper, F.; Lovik, M.; Nygaard, U.C. Transmaternal bisphenol A exposure accelerates diabetes type 1 development in NOD mice. Toxicol. Sci. 2014, 137, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Bohacek, J.; Mansuy, I.M. Epigenetic inheritance of disease and disease risk. Neuropsychopharmacology 2013, 38, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Bodin, J.; Stene, L.C.; Nygaard, U.C. Can exposure to environmental chemicals increase the risk of diabetes type 1 development? BioMed Res. Int. 2015, 2015, 208947. [Google Scholar] [CrossRef] [PubMed]
- Katsikantami, I.; Sifakis, S.; Tzatzarakis, M.N.; Vakonaki, E.; Kalantzi, O.I.; Tsatsakis, A.M.; Rizos, A.K. A global assessment of phthalates burden and related links to health effects. Environ. Int. 2016, 97, 212–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jin, T. The role of insulin signaling in the development of beta-cell dysfunction and diabetes. Islets 2009, 1, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Timme-Laragy, A.R.; Sant, K.E.; Rousseau, M.E.; Diiorio, P.J. Deviant development of pancreatic beta cells from embryonic exposure to PCB-126 in zebrafish. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2015, 178, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Lind, L.; Zethelius, B.; Salihovic, S.; van Bavel, B.; Lind, P.M. Circulating levels of perfluoroalkyl substances and prevalent diabetes in the elderly. Diabetologia 2014, 57, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Hectors, T.L.; Vanparys, C.; van der Ven, K.; Martens, G.A.; Jorens, P.G.; Van Gaal, L.F.; Covaci, A.; De Coen, W.; Blust, R. Environmental pollutants and type 2 diabetes: A review of mechanisms that can disrupt beta cell function. Diabetologia 2011, 54, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Steffes, M.W.; Sjodin, A.; Jones, R.S.; Needham, L.L.; Jacobs, D.R., Jr. Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS ONE 2011, 6, e15977. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.L.; Shaw, A.C.; Gagne, A.X.; Chan, H.M. Chronic exposure to PCBs (Aroclor 1254) exacerbates obesity-induced insulin resistance and hyperinsulinemia in mice. J. Toxicol. Environ. Health A 2013, 76, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Heppner, K.M.; Kirigiti, M.; Secher, A.; Paulsen, S.J.; Buckingham, R.; Pyke, C.; Knudsen, L.B.; Vrang, N.; Grove, K.L. Expression and distribution of glucagon-like peptide-1 receptor mRNA, protein and binding in the male nonhuman primate (Macaca mulatta) brain. Endocrinology 2015, 156, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Matsuoka, T.A. Role of pancreatic transcription factors in maintenance of mature beta-cell function. Int. J. Mol. Sci. 2015, 16, 6281–6297. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.S.; Kang, L.; Wasserman, D.H. The extracellular matrix and insulin resistance. Trends Endocrinol. Metab. 2015, 26, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, M.C.; Chiche, J.; Pouyssegur, J. Hypoxia and cancer. J. Mol. Med. 2007, 85, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.P.; Wirth, J.J.; Saed, G.M. PCBs enhance collagen I expression from human peritoneal fibroblasts. Fertil. Steril. 2008, 90, 1372–1375. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Brown, M. Advances in estrogen receptor biology: Prospects for improvements in targeted breast cancer therapy. Breast Cancer Res. 2004, 6, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.A.; Bhat-Nakshatri, P.; Patel, N.M.; Constantinidou, D.; Ali, S.; Nakshatri, H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: A new model for anti-estrogen resistance. J. Biol. Chem. 2001, 276, 9817–9824. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Dong, X.; Zou, D.; Yu, Y.; Fang, Q.; Zhang, Q.; Zhao, M. Enantioselective Effects of o,p′-DDT on Cell Invasion and Adhesion of Breast Cancer Cells: Chirality in Cancer Development. Environ. Sci. Technol. 2015, 49, 10028–10037. [Google Scholar] [CrossRef] [PubMed]
- Marchand, A.; Tomkiewicz, C.; Marchandeau, J.P.; Boitier, E.; Barouki, R.; Garlatti, M. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces insulin-like growth factor binding protein-1 gene expression and counteracts the negative effect of insulin. Mol. Pharmacol. 2005, 67, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.W.; Jong, C.J.; Mozaffari, M. Role of oxidative stress in diabetes-mediated vascular dysfunction: unifying hypothesis of diabetes revisited. Vascul. Pharmacol. 2012, 57, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- D'Souza, A.; Hussain, M.; Howarth, F.C.; Woods, N.M.; Bidasee, K.; Singh, J. Pathogenesis and pathophysiology of accelerated atherosclerosis in the diabetic heart. Mol. Cell. Biochem. 2009, 331, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity: Implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes. Res. Clin. Pract. 2013, 7, e330–e341. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Wu, C.W.; Chang, A.Y.; Hsu, K.S.; Chan, J.Y. Transcriptional upregulation of brain-derived neurotrophic factor in rostral ventrolateral medulla by angiotensin II: Significance in superoxide homeostasis and neural regulation of arterial pressure. Circ. Res. 2010, 107, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.C.; Goubern, M.; Surwit, R.; et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000, 26, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.L.; Liu, J.C. Role of Notch signaling in the mammalian heart. Braz. J. Med. Biol. Res. 2014, 47, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Walker, N.J.; Wang, D. Dioxin (2,3,7,8-tetrachlorodibenzo-p-dioxin) enhances triggered afterdepolarizations in rat ventricular myocytes. Cardiovasc. Toxicol. 2006, 6, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.R.; Burton, F.; Salama, G. Cytosolic Ca2+ triggers early afterdepolarizations and Torsade de Pointes in rabbit hearts with type 2 long QT syndrome. J. Physiol. 2002, 543, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Cranefield, P.F. Action potentials, afterpotentials, and arrhythmias. Circ. Res. 1977, 41, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Jo, S. Differential Effects of environmental toxicants, PCB 126 and PCB 77, on cardiac electrophysiology. In Proceedings of the 37th Congress of IUPS Poster Communications, Birmingham, UK, 21–26 July 2013. [Google Scholar]
- Moon, J.M.; Chun, B.J. Acute endosulfan poisoning: A retrospective study. Hum. Exp. Toxicol. 2009, 28, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J.P.; Lipscomb, J.C.; Wesselkamper, S.C. Putative mechanisms of environmental chemical-induced steatosis. Int. J. Toxicol. 2012, 31, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome—An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Kit, B.K.; Kuklina, E.; Carroll, M.D.; Ostchega, Y.; Freedman, D.S.; Ogden, C.L. Prevalence of and trends in dyslipidemia and blood pressure among US children and adolescents, 1999–2012. JAMA Pediatr. 2015, 169, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.L.; Howe, L.D.; Jones, H.E.; Higgins, J.P.; Lawlor, D.A.; Fraser, A. The Prevalence of Non-Alcoholic Fatty Liver Disease in Children and Adolescents: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0140908. [Google Scholar] [CrossRef] [PubMed]
- Maranghi, F.; Rescia, M.; Macri, C.; Di Consiglio, E.; De Angelis, G.; Testai, E.; Farini, D.; De Felici, M.; Lorenzetti, S.; Mantovani, A. Lindane may modulate the female reproductive development through the interaction with ER-beta: An in vivo-in vitro approach. Chem. Biol. Interact. 2007, 169, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Attia, A.M.; El-Banna, S.G.; Nomeir, F.R.; Abd El-Basser, M.I. Lindane-induced biochemical perturbations in rat serum and attenuation by omega-3 and Nigella sativa seed oil. Indian J. Biochem. Biophys. 2011, 48, 184–190. [Google Scholar] [PubMed]
- Angrish, M.M.; Dominici, C.Y.; Zacharewski, T.R. TCDD-elicited effects on liver, serum, and adipose lipid composition in C57BL/6 mice. Toxicol. Sci. 2013, 131, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Fausther, M.; Pritchard, M.T.; Popov, Y.V.; Bridle, K. Contribution of Liver Nonparenchymal Cells to Hepatic Fibrosis: Interactions with the Local Microenvironment. BioMed Res. Int. 2017, 2017, 6824762. [Google Scholar] [CrossRef] [PubMed]
- Anway, M.D.; Cupp, A.S.; Uzumcu, M.; Skinner, M.K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005, 308, 1466–1469. [Google Scholar] [CrossRef] [PubMed]
- Dolinoy, D.C.; Huang, D.; Jirtle, R.L. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef] [PubMed]
- Cabia, B.; Andrade, S.; Carreira, M.C.; Casanueva, F.F.; Crujeiras, A.B. A role for novel adipose tissue-secreted factors in obesity-related carcinogenesis. Obes. Rev. 2016, 17, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.S. Obesity and carcinogenesis. J. Cancer Res. Pract. 2011, 27, 242–256. [Google Scholar]
- LaMarca, H.L.; Rosen, J.M. Estrogen regulation of mammary gland development and breast cancer: Amphiregulin takes center stage. Breast Cancer Res. 2007, 9, 304. [Google Scholar] [CrossRef] [PubMed]
- Kos, M.; Reid, G.; Denger, S.; Gannon, F. Minireview: Genomic organization of the human ERalpha gene promoter region. Mol. Endocrinol. 2001, 15, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- VoPham, T.; Brooks, M.M.; Yuan, J.M.; Talbott, E.O.; Ruddell, D.; Hart, J.E.; Chang, C.C.; Weissfeld, J.L. Pesticide exposure and hepatocellular carcinoma risk: A case-control study using a geographic information system (GIS) to link SEER-Medicare and California pesticide data. Environ. Res. 2015, 143, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Clary, T.; Ritz, B. Pancreatic cancer mortality and organochlorine pesticide exposure in California, 1989–1996. Am. J. Ind. Med. 2003, 43, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, J.; Tsuji, H.; Iida, T.; Nakagawa, R.; Matsueda, T.; Hirakawa, H.; Yanagawa, T.; Fukushige, J.; Watanabe, T. Immunologic effects of perinatal exposure to dioxins, PCBs and organochlorine pesticides in Japanese infants. Chemosphere 2007, 67, S393–S398. [Google Scholar] [CrossRef] [PubMed]
- Klymkowsky, M.W.; Savagner, P. Epithelial-mesenchymal transition: A cancer researcher’s conceptual friend and foe. Am. J. Pathol. 2009, 174, 1588–1593. [Google Scholar] [CrossRef] [PubMed]
- Stanculescu, D.; Margaritescu, C.; Stepan, A.; Mitrut, A.O. E-cadherin in gastric carcinomas related to histological prognostic parameters. Romanian J. Morphol. Embryol. 2011, 52, 1107–1112. [Google Scholar] [PubMed]
- Kawata, M.; Koinuma, D.; Ogami, T.; Umezawa, K.; Iwata, C.; Watabe, T.; Miyazono, K. TGF-beta-induced epithelial-mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by pro-inflammatory cytokines derived from RAW 264.7 macrophage cells. J. Biochem. 2012, 151, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Lim, J.W.; Kim, H.; Kim, K.H. Helicobacter pylori in a Korean isolate activates mitogen-activated protein kinases, AP-1, and NF-kappaB and induces chemokine expression in gastric epithelial AGS cells. Lab. Investig. 2004, 84, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Ochieng, J.; Nangami, G.N.; Ogunkua, O.; Miousse, I.R.; Koturbash, I.; Odero-Marah, V.; McCawley, L.J.; Nangia-Makker, P.; Ahmed, N.; Luqmani, Y.; et al. The impact of low-dose carcinogens and environmental disruptors on tissue invasion and metastasis. Carcinogenesis 2015, 36 (Suppl. 1), S128–S159. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, G.; Mnif, W.; Mauvais, P.; Balaguer, P.; Rahmani, R. Activation of alpha- and beta-estrogen receptors by persistent pesticides in reporter cell lines. Life Sci. 2006, 79, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Frigo, D.E.; Burow, M.E.; Mitchell, K.A.; Chiang, T.C.; McLachlan, J.A. DDT and its metabolites alter gene expression in human uterine cell lines through estrogen receptor-independent mechanisms. Environ. Health Perspect. 2002, 110, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.C.; Young, M.R.; Cmarik, J.; Colburn, N.H. Activator protein 1 (AP-1)- and nuclear factor kappaB (NF-kappaB)-dependent transcriptional events in carcinogenesis. Free Radic. Biol. Med. 2000, 28, 1338–1348. [Google Scholar] [CrossRef]
- Shi, H.; Teng, C. Promoter-specific activation of mouse lactoferrin gene by epidermal growth factor involves two adjacent regulatory elements. Mol. Endocrinol. 1996, 10, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Daly, R.J.; King, R.J.; Darbre, P.D. Interaction of growth factors during progression towards steroid independence in T-47-D human breast cancer cells. J. Cell. Biochem. 1990, 43, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Angel, P.; Szabowski, A.; Schorpp-Kistner, M. Function and regulation of AP-1 subunits in skin physiology and pathology. Oncogene 2001, 20, 2413–2423. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.W.; Li, J.; Brost, B.C.; Xia, X.Y.; Chen, H.B.; Wang, C.X.; Jiang, S.W. Decreased expression and altered methylation of syncytin-1 gene in human placentas associated with preeclampsia. Curr. Pharm. Des. 2014, 20, 1796–1802. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Janesick, A.; Blumberg, B.; Heindel, J.J. Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol. 2011, 127, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Younglai, E.V.; Holloway, A.C.; Foster, W.G. Environmental and occupational factors affecting fertility and IVF success. Hum. Reprod. Update 2005, 11, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Bretveld, R.; Zielhuis, G.A.; Roeleveld, N. Time to pregnancy among female greenhouse workers. Scand. J. Work Environ. Health 2006, 32, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Ventura, S.J.; Curtin, S.C.; Abma, J.C.; Henshaw, S.K. Estimated pregnancy rates and rates of pregnancy outcomes for the United States, 1990–2008. Natl. Vital. Stat. Rep. 2012, 60, 1–21. [Google Scholar] [PubMed]
- Van der Stege, J.G.; Groen, H.; van Zadelhoff, S.J.; Lambalk, C.B.; Braat, D.D.; van Kasteren, Y.M.; van Santbrink, E.J.; Apperloo, M.J.; Weijmar Schultz, W.C.; Hoek, A. Decreased androgen concentrations and diminished general and sexual well-being in women with premature ovarian failure. Menopause 2008, 15, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Zama, A.M.; Uzumcu, M. Fetal and neonatal exposure to the endocrine disruptor methoxychlor causes epigenetic alterations in adult ovarian genes. Endocrinology 2009, 150, 4681–4691. [Google Scholar] [CrossRef] [PubMed]
- Zama, A.M.; Uzumcu, M. Targeted genome-wide methylation and gene expression analyses reveal signaling pathways involved in ovarian dysfunction after developmental EDC exposure in rats. Biol. Reprod. 2013, 88, 52. [Google Scholar] [CrossRef] [PubMed]
- Bernatchez, R.; Belkacemi, L.; Rassart, E.; Daoud, G.; Simoneau, L.; Lafond, J. Differential expression of membrane and soluble adenylyl cyclase isoforms in cytotrophoblast cells and syncytiotrophoblasts of human placenta. Placenta 2003, 24, 648–657. [Google Scholar] [CrossRef]
- Covaci, A.; Jorens, P.; Jacquemyn, Y.; Schepens, P. Distribution of PCBs and organochlorine pesticides in umbilical cord and maternal serum. Sci. Total Environ. 2002, 298, 45–53. [Google Scholar] [CrossRef]
- Backlin, B.M.; Persson, E.; Jones, C.J.; Dantzer, V. Polychlorinated biphenyl (PCB) exposure produces placental vascular and trophoblastic lesions in the mink (Mustela vison): A light and electron microscopic study. APMIS 1998, 106, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, R.M.; Skakkebaek, N.E. Are oestrogens involved in falling sperm counts and disorders of the male reproductive tract? Lancet 1993, 341, 1392–1395. [Google Scholar] [CrossRef]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Main, K.M. Testicular dysgenesis syndrome: An increasingly common developmental disorder with environmental aspects. Hum. Reprod. 2001, 16, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, H.E.; Rajpert-De Meyts, E.; Main, K.M.; Skakkebaek, N.E.; Toppari, J. Testicular dysgenesis syndrome and the development and occurrence of male reproductive disorders. Toxicol. Appl. Pharmacol. 2005, 207, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Giwercman, A.; Giwercman, Y.L. Epidemiology of male reproductive disorders. In Endotext [Internet]; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Kelce, W.R.; Stone, C.R.; Laws, S.C.; Gray, L.E.; Kemppainen, J.A.; Wilson, E.M. Persistent DDT metabolite p,p′-DDE is a potent androgen receptor antagonist. Nature 1995, 375, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Toppari, J.; Larsen, J.C.; Christiansen, P.; Giwercman, A.; Grandjean, P.; Guillette, L.J., Jr.; Jegou, B.; Jensen, T.K.; Jouannet, P.; Keiding, N.; et al. Male reproductive health and environmental xenoestrogens. Environ. Health Perspect. 1996, 104 (Suppl. 4), 741–803. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, S.; Baumann, K.; Lehnert, G. Occupational exposure to hexachlorocyclohexane. IV. Sex hormone alterations in HCH-exposed workers. Int. Arch. Occup. Environ. Health 1981, 48, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Multigner, L.; Ndong, J.R.; Giusti, A.; Romana, M.; Delacroix-Maillard, H.; Cordier, S.; Jegou, B.; Thome, J.P.; Blanchet, P. Chlordecone exposure and risk of prostate cancer. J. Clin. Oncol. 2010, 28, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Toppari, J.; Virtanen, H.; Skakkebaek, N.E.; Main, K.M. Environmental effects on hormonal regulation of testicular descent. J. Steroid Biochem. Mol. Biol. 2006, 102, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Waste Electrical and Electronic Equipment (WEEE) Directive. Available online: http://www.conformance.co.uk/directives/full_text/l_19720120724en00380071_bookmarked.pdf (accessed on 21 October 2017).
- RC Annual Report 2006. Available online: https://ec.europa.eu/jrc/sites/jrcsh/files/jrc_ar_2006_0.pdf, Archived at http://www.webcitation.org/6uNpUhilW (accessed on 21 October 2017).
- KEMI Swedish Chemicals Agency. Available online: https://www.kemi.se/global/rapporter/2015/report-7–15-occurrence-and-use-of-highly-fluorinated-substances-and-alternatives.pdf (accessed on 21 October 2017).




{kind=link}
{kind=link}
{kind=link}
| Test Compound | Cell Proliferation | Estrogen Receptor Transactivation | Androgen Receptor Transactivation | Aromatase Activity |
|---|---|---|---|---|
| Dieldrin | ↑ | ↑ | ↓ | – |
| Endosulfan | ↑ | ↑ | ↓ | ↓ |
| Methiocarb | ↑ | ↑ | ↓ | – |
| Pirimicarb | – | ↑ | – | ↑ |
| Propamocarb | – | ↑ | – | ↑ |
| Fenarimol | ↑ | ↑ | ↓ | ↓ |
| Prochloraz | ↓ | ↓ | ↓ | ↓ |
| Chemical | Obesity | Diabetes Mellitus Type 2 | Lipid Disorders/Fatty Liver |
|---|---|---|---|
| Bisphenol A | xxx | xxx | xxx |
| Di(2-ethylhexyl)phthalate | xxx | xxx | xxx |
| Dichlorodiphenyltrichloroethane/ Dichlorodiphenyldichloroethylene | xxx | xx | x |
| Polybrominated diphenyl ether | x | ||
| Perfluorooctanoic acid | xx | xxx | |
| Perfluorooctanesulfonic acid | x | xxx | |
| Tributyltin | xxx | xxx | xxx |
| Air Pollution | xx | xxx | xxx |
| Polycyclic aromatic hydrocarbons | |||
| Polychlorinated biphenyls | x | xxx | xxx |
| 2,3,7,8-tetrachlorodibenzo-p-dioxin | xx | xxx | |
| Atrazine | x | xx | |
| Benzo(a)pyrene | x | xx |
| Biomarkers in Oxidative Stress | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EDs | DNA | MDA | LPO | EROD | GST | GSH | SOD | ROS | Vit. C | Vit. E | CAT |
| Organochlorines | + | + | + | + | + | – | – | + | – | – | – |
| Polychlorinated biphenyls | + | + | + | + | – | – | – | + | – | – | + |
| 2,3,7,8-tetrachlorodibenzo-p-dioxin | + | + | + | + | – | – | – | + | – | – | – |
| Polycyclic aromatic hydrocarbons | + | + | + | + | + | – | – | + | – | – | + |
| Perfluorinated compound | + | + | + | + | – | – | + | + | – | – | + |
| Di(2-ethylhexyl) phthalate | + | + | + | non | – | – | + | + | – | – | + |
| Diethylstilbestrol | + | + | + | + | – | – | – | + | – | – | – |
| Bisphenol A | + | + | + | non | – | – | – | + | – | –1 | – |
| Tributyltin | + | + | + | non | – | – | + | + | – | – | + |
| Short-chain chlorinated paraffins | non | ? | + | non | + | ? | ? | + | ? | ? | ? |
| Polychlorinated naphthalene | ? | + | + | + | ? | – | ? | + | ? | non | ? |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrakis, D.; Vassilopoulou, L.; Mamoulakis, C.; Psycharakis, C.; Anifantaki, A.; Sifakis, S.; Docea, A.O.; Tsiaoussis, J.; Makrigiannakis, A.; Tsatsakis, A.M. Endocrine Disruptors Leading to Obesity and Related Diseases. Int. J. Environ. Res. Public Health 2017, 14, 1282. https://doi.org/10.3390/ijerph14101282
Petrakis D, Vassilopoulou L, Mamoulakis C, Psycharakis C, Anifantaki A, Sifakis S, Docea AO, Tsiaoussis J, Makrigiannakis A, Tsatsakis AM. Endocrine Disruptors Leading to Obesity and Related Diseases. International Journal of Environmental Research and Public Health. 2017; 14(10):1282. https://doi.org/10.3390/ijerph14101282
Chicago/Turabian StylePetrakis, Demetrios, Loukia Vassilopoulou, Charalampos Mamoulakis, Christos Psycharakis, Aliki Anifantaki, Stavros Sifakis, Anca Oana Docea, John Tsiaoussis, Antonios Makrigiannakis, and Aristides M. Tsatsakis. 2017. "Endocrine Disruptors Leading to Obesity and Related Diseases" International Journal of Environmental Research and Public Health 14, no. 10: 1282. https://doi.org/10.3390/ijerph14101282
APA StylePetrakis, D., Vassilopoulou, L., Mamoulakis, C., Psycharakis, C., Anifantaki, A., Sifakis, S., Docea, A. O., Tsiaoussis, J., Makrigiannakis, A., & Tsatsakis, A. M. (2017). Endocrine Disruptors Leading to Obesity and Related Diseases. International Journal of Environmental Research and Public Health, 14(10), 1282. https://doi.org/10.3390/ijerph14101282