Anticancer Alkaloid Lamellarins Inhibit Protein Kinases

Abstract
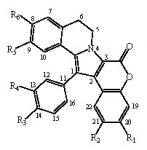
:
1. Introduction
2. Results and Discussion
2.1. Lamellarins inhibit protein kinases
- removal of the hydroxyl at R1 modestly modifies the activity (except GSK-3, DYRK1A and CK1), compare lamellarin D (1) and lamellarin 4 (10).
- removal of the hydroxyl at R2 results in minor changes in activity (except DYRK1A and CK1), compare lamellarin D (1) and lamellarin 3 (9).
- removal of the hydroxyl at R3 results in major reduction in activity, compare lamellarin D (1) and lamellarin 7 (13)
- removal of the hydroxyl at R4 results in enhanced inhibitory activity, compare lamellarin D (1) and lamellarin 6 (12).
- O-methylation at R6 results in massive loss of inhibitory activity, compare lamellarin D (1) and lamellarin α (2).
- replacing the hydroxyl groups of lamellarin D (1) at R1, R3, and R6 by O-isopropyl completely abolishes inhibitory activity, see lamellarin 31 (18) and 33 (19). This is also the case when substituting hydroxyl to isopropyl at positions R1, R4, and R6 of lamellarin N (6), lamellarin 32 (21) and 34 (20).
- transposition of the substitutions at R3 and R4 of lamellarin D (1), resulting in lamellarin N (6), lead to 10-fold enhanced kinase inhibitory activity.
- Reduction in activity due to saturation of D-ring double bond (C5=C6) has previously been reported to be due to loss of planarity and therefore steric hindrance in ATP pocket of targets [19]. Kinase inhibitory activities of lamellarins are generally reduced when the D-ring double bond is saturated, compare lamellarins D (1) with di-H-lamellarin (3), and lamellarins N (6) with lamellarin L (7).
2.2. Selectivity of lamellarins
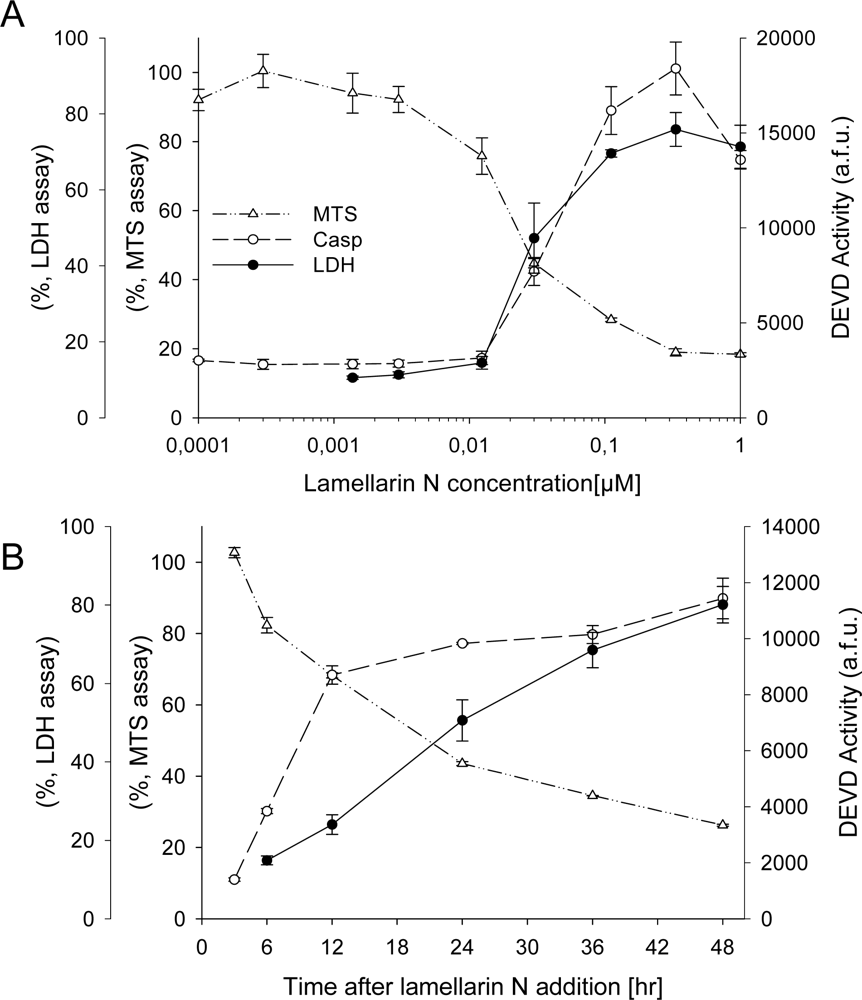
2.3. Cell death induction by lamellarins
3. Conclusions
5. Experimental Procedures
5.1. Chemistry
5.2. Kinase preparation and assays
5.3. Cell viability and cell death
5.4. Protein extraction, SDS-PAGE and Western Blotting
Acknowledgments
References and Notes
- Bourguet-Kondracki, ML; Kornprobst, JM. Marine pharmacology: potentialities in the treatment of infectious diseases, osteoporosis and Alzheimer's disease. Adv. Biochem. Eng. Biotechnol 2005, 97, 105–131. [Google Scholar]
- Simmons, TL; Andrianasolo, E; McPhail, K; Flatt, P; Gerwick, WH. Marine natural products as anticancer drugs. Mol. Cancer Ther 2005, 4, 333–342. [Google Scholar]
- Cironi, P; Albericio, F; Alvarez, M. Lamellarins: isolation, activity and synthesis. Progr. Heterocycl. Chem 2004, 16, 1–26. [Google Scholar]
- Bailly, C. Lamellarins, from A to Z: a family of anticancer marine pyrrole alkaloids. Curr. Med. Chem. Anti-Cancer Agents 2004, 4, 363–378. [Google Scholar]
- Fan, RH; Peng, J; Hamann, MT; Hu, JF. Lamellarins and related pyrrole-derived alakaloids from marine organisms. Chem. Rev 2008, 108, 264–287. [Google Scholar]
- Andersen, RJ; Faulkner, DJ; He, CH; Van Duyne, GD; Clardy, J. Metabolites of the marine prosobranch mollusk Lamellaria sp. J. Am. Chem. Soc 1985, 107, 5492–5495. [Google Scholar]
- Lindquist, N; Fenical, W; Van Duyne, GD; Clardy, J. New alkaloids of the lamellarin class from the marine ascidian Didemnum chartaceum (Sluiter, 1909). J. Org. Chem 1988, 53, 4570–4574. [Google Scholar]
- Carroll, AR; Bowden, BF; Coll, JC. Studies of Australian ascidians. I. Six new lamellarin-class alkaloids from a colonial ascidian, Didemnum sp. Austr. J. Chem 1993, 46, 489–501. [Google Scholar]
- Urban, S; Capon, RJ; Lamellarin, S. A New Aromatic Metabolite from an Australian Tunicate Didemnum sp. Aust. J. Chem 1996, 49, 711–713. [Google Scholar]
- Reddy, MVR; Faulkner, DJ; Venkateswarlu, Y; Rao, MR. New lamellarin alkaloids from an unidentified ascidian from the Arabian Sea. Tetrahedron 1997, 53, 3457–3466. [Google Scholar]
- Davis, RA; Carroll, AR; Pierens, GK; Quinn, RJ. New lamellarin alkaloids from the australian ascidian, Didemnum chartaceum. J. Nat. Prod 1999, 62, 419–424. [Google Scholar]
- Ham, J; Kang, H. A novel cytotoxic alkaloid of lamellarin class from a marine ascidian Didemnum sp. Bull. Korean Chem. Soc 2002, 23, 163–166. [Google Scholar]
- Reddy, SM; Srinivasulu, M; Satyanarayana, N; Kondapi, AK; Venkateswarlu, Y. New potent cytotoxic lamellarin alkaloids from Indian ascidian Didemnum obscurum. Tetrahedron 2005, 61, 9242–9247. [Google Scholar]
- Reddy, MV; Rao, MR; Rhodes, D; Hansen, MS; Rubins, K; Bushman, FD; Venkateswarlu, Y; Faulkner, DJ. Lamellarin alpha 20-sulfate, an inhibitor of HIV-1 integrase active against HIV-1 virus in cell culture. J. Med. Chem 1999, 42, 1901–1907. [Google Scholar]
- Urban, S; Butler, MS; Capon, RJ. Lamellarins O and P: New Aromatic Metabolites from the Australian Marine Sponge Dendrilla cactos. Aust. J. Chem 1994, 47, 1919–1924. [Google Scholar]
- Urban, S; Hobbs, L; Hooper, JNA; Capon, RJ. Lamellarins Q and R: New Aromatic Metabolites from an Australian Marine Sponge Dendrilla cactos. Aus t. J. Chem 1995, 48, 1491–1494. [Google Scholar]
- Quesada, AR; Garcia Gravalos, MDJL. Fernandez Puentes Polyaromatic alkaloids from marine invertebrates as cytotoxic compounds and inhibitors of multidrug resistance caused by P-glycoprotein. Br. J. Cancer 1996, 74, 677–682. [Google Scholar]
- Ishibashi, F; Tanabe, S; Oda, T; Iwao, M. Synthesis and structure-activity relationship study of lamellarin derivatives. J. Nat. Prod 2002, 65, 500–504. [Google Scholar]
- Facompre, M; Tardy, C; Bal-Mahieu, C; Colson, P; Perez, C; Manzanares, I; Cuevas, C; Bailly, C. Lamellarin D: a novel potent inhibitor of topoisomerase I. Cancer Res 2003, 63, 7392–7399. [Google Scholar]
- Tardy, C; Facompre, M; Laine, W; Baldeyrou, B; Garcia-Gravalos, D; Francesch, A; Mateo, C; Pastor, A; Jimenez, JA; Manzanares, I; Cuevas, C; Bailly, C. Topoisomerase I-mediated DNA cleavage as a guide to the development of antitumor agents derived from the marine alkaloid lamellarin D: trimethylester derivatives incorporating amino acid residues. Bioorg. Med. Chem 2004, 12, 1697–1712. [Google Scholar]
- Marco, E; Laine, W; Tardy, C; Lansiaux, A; Iwao, M; Ishibashi, F; Bailly, C; Gago, F. Molecular determinants of topoisomerase I poisoning by lamellarins: comparison with camptothecin and structure-activity relationships. J. Med. Chem 2005, 48, 3796–3807. [Google Scholar]
- Vanhuyse, M; Kluza, J; Tardy, C; Otero, G; Cuevas, C; Bailly, C; Lansiaux, A. Lamellarin D: a novel pro-apoptotic agent from marine origin insensitive to P-glycoprotein-mediated drug efflux. Cancer Lett 2005, 22, 165–175. [Google Scholar]
- Kluza, J; Gallego, MA; Loyens, A; Beauvillain, JC; Sousa-Faro, JM; Cuevas, C; Marchetti, P; Bailly, C. Cancer cell mitochondria are direct proapoptotic targets for the marine antitumor drug lamellarin D. Cancer Res 2006, 66, 3177–3187. [Google Scholar]
- Gallego, MA; Ballot, C; Kluza, J; Hajji, N; Martoriati, A; Castéra, L; Cuevas, C; Formstecher, P; Joseph, B; Kroemer, G; Bailly, C; Marchetti, P. Overcoming chemoresistance of non-small cell lung carcinoma through restoration of an AIF-dependent apoptotic pathway. Oncogene 2008, 27, 1981–1992. [Google Scholar]
- Banwell, MG; Hamel, E; Hockless, DC; Verdier-Pinard, P; Willis, AC; Wong, DJ. 4,5-Diaryl-1H-pyrrole-2-carboxylates as combretastatin A-4/lamellarin T hybrids: Synthesis and evaluation as anti-mitotic and cytotoxic agents. Bioorg. Med. Chem 2006, 14, 4627–4638. [Google Scholar]
- Ridley, CP; Reddy, MV; Rocha, G; Bushman, FD; Faulkner, DJ. Total synthesis and evaluation of lamellarin alpha 20-Sulfate analogues. Bioorg. Med. Chem 2002, 10, 3285–3290. [Google Scholar]
- Yamaguchi, T; Fukuda, T; Ishibashi, F; Iwao, M. The first total synthesis of lamellarin α 20-sulfate, a selective inhibitor of HIV-1 integrase. Tetrahedron. in press..
- Malumbres, M; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci 2005, 30, 630–641. [Google Scholar]
- Knockaert, M; Greengard, P; Meijer, L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol. Sci 2002, 23, 417–425. [Google Scholar]
- Meijer, L; Flajolet, M; Greengard, P. Pharmacological inhibitors of glycogen synthase kinase-3. Trends Pharmacol. Sci 2004, 25, 471–480. [Google Scholar]
- Bachmann, M; Moroy, T. The serine/threonine kinase Pim-1. Int. J. Biochem. Cell Biol 2005, 37, 726–730. [Google Scholar]
- Galceran, J; de Graaf, K; Tejedor, FJ; Becker, W. The MNB/DYRK1A protein kinase: genetic and biochemical properties. J. Neural. Transm. Suppl 2003, 67, 139–148. [Google Scholar]
- Hammerle, B; Elizalde, C; Galceran, J; Becker, W; Tejedor, FJ. The MNB/DYRK1A protein kinase: neurobiological functions and Down syndrome implications. J. Neural Transm. Suppl 2003, 67, 129–137. [Google Scholar]
- Ferrer, M Barrachina; Puig, B; Martinez de Lagran, M; Marti, E; Avila, J; Dierssen, M. Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, Down syndrome, Pick disease, and related transgenic models. Neurobiol. Dis 2005, 20, 392–400. [Google Scholar]
- Knippschild, U; Wolff, S; Giamas, G; Brockschmidt, C; Wittau, M; Wurl, PU; Eismann, T; Stoter, M. The role of the casein kinase 1 (CK1) family in different signaling pathways linked to cancer development. Onkologie 2005, 28, 508–514. [Google Scholar]
- Ishibashi, F; Miyazaki, Y; Iwao, M. Total syntheses of lamellarin D and H. The first synthesis of lamellarin-class marine alkaloids. Tetrahedron 1997, 53, 5951–5962. [Google Scholar]
- Fujikawa, N; Ohta, T; Yamaguchi, T; Fukuda, T; Ishibashi, F; Iwao, M. Total synthesis of lamellarin D, L, and N. Tetrahedron 2006, 62, 594–604. [Google Scholar]
- Meijer, L; Borgne, A; Mulner, O; Chong, JPJ; Blow, JJ; Inagaki, N; Inagaki, M; Delcros, JG; Moulinoux, JP. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem 1997, 24, 527–536. [Google Scholar]
- Primot; Baratte, B; Gompel, M; Borgne, A; Liabeuf, S; Romette, JL; Costantini, F; Meijer, L. Purification of GSK-3 by affinity chromatography on immobilised axin. Protein Expr. & Purif 2000, 20, 394–404. [Google Scholar]
- Reinhardt, J; Ferandin, Y; Meijer, L. Purification CK1 by affinity chromatography on immobilised axin. Protein Expr. & Purif 2007, 54, 101–109. [Google Scholar]
- Ribas, J; Boix, J. Cell differentiation, caspase inhibition, and macromolecular synthesis blockage, but not BCL-2 or BCL-XL proteins, protect SH-SY5Y cells from apoptosis triggered by two CDK inhibitory drugs. Exp. Cell Res 2004, 295, 9–24. [Google Scholar]
- Bettayeb, K; Oumata, N; Echalier, A; Ferandin, Y; Endicott, J; Galons, H; Meijer, L. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene in press.. 2008. [Google Scholar]
- Nguyen, M; Marcellus, RC; Roulston, A; Watson, M; Serfass, L; Murthy Madiraju, SR; Goulet, D; Viallet, J; Bélec, L; Billot, X; Acoca, S; Purisima, E; Wiegmans, A; Cluse, L; Johnstone, RW; Beauparlant, P; Shore, GC. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. U.S.A 2007, 104, 19512–19517. [Google Scholar]
- Bach, S; Knockaert, M; Lozach, O; Reinhardt, J; Baratte, B; Schmitt, S; Coburn, SP; Tang, L; Jiang, T; Liang, DC; Galons, H; Dierick, JF; Totzke, F; Schächtele, C; Lerman, AS; Carnero, A; Wan, Y; Gray, N; Meijer, L. Roscovitine targets: protein kinases and pyridoxal kinase. J. Biol. Chem 2005, 280, 31208–31219. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
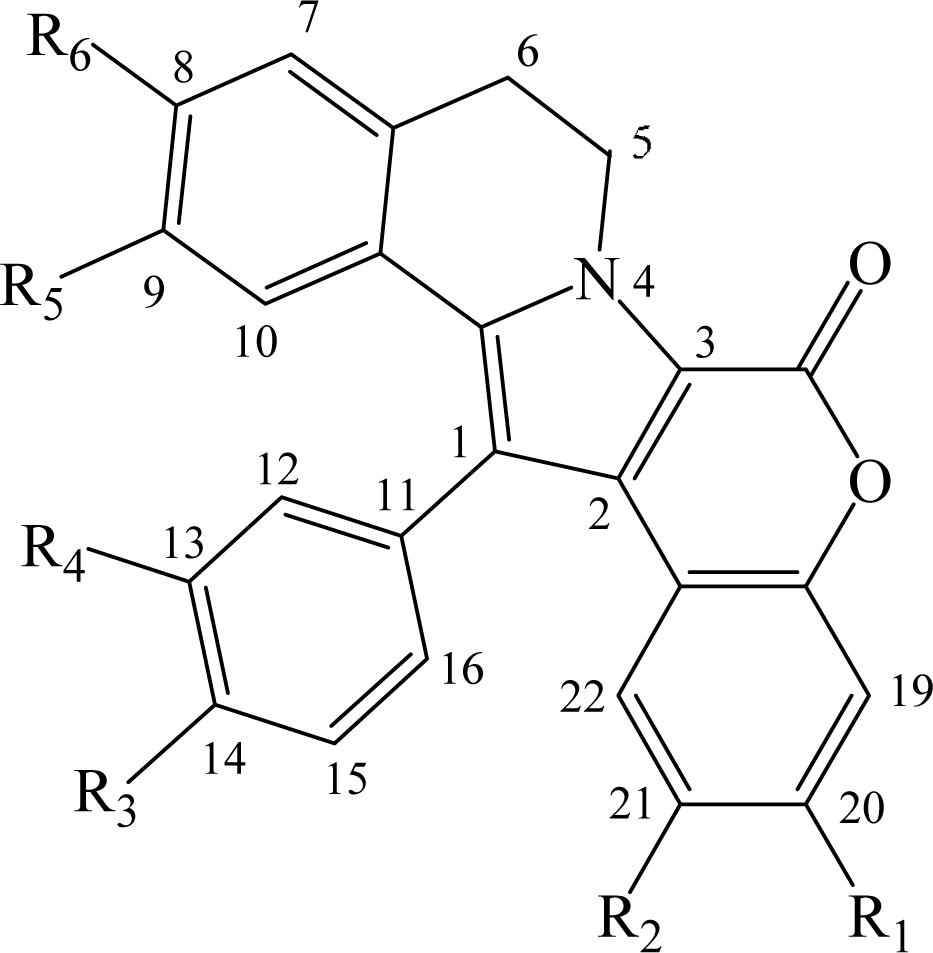
| # | Lamellarin | R1 | R2 | R3 | R4 | R5 | R6 | 5–6 |
|---|---|---|---|---|---|---|---|---|
| 1 | lamellarin D | OH | OMe | OH | OMe | OMe | OH | = |
| 2 | lamellarin α | OH | OMe | OH | OMe | OMe | OMe | = |
| 3 | di-H-lamellarin D | OH | OMe | OH | OMe | OMe | OH | = |
| 4 | lamellarin H | OH | OH | OH | OH | OH | OH | = |
| 5 | di-H-lamellarin H | OH | OH | OH | OH | OH | OH | = |
| 6 | lamellarin N | OH | OMe | OMe | OH | OMe | OH | = |
| 7 | lamellarin L | OH | OMe | OMe | OH | OMe | OH | = |
| 8 | lamellarin G tri-OMe | OMe | OMe | OMe | OMe | OMe | OMe | = |
| 9 | lamellarin 3 | OH | H | OH | OMe | OMe | OH | = |
| 10 | lamellarin 4 | H | OMe | OH | OMe | OMe | OH | = |
| 11 | lamellarin 5 | OH | OMe | OMe | OMe | OMe | OMe | = |
| 12 | lamellarin 6 | OH | OMe | OH | H | OMe | OH | = |
| 13 | lamellarin 7 | OH | OMe | H | OMe | OMe | OH | = |
| 14 | lamellarin 8 | H | H | OH | OMe | OMe | OH | = |
| 15 | lamellarin 9 | H | H | OH | OH | OH | OH | = |
| 16 | lamellarin 11 | H | H | OMe | OMe | OMe | OMe | = |
| 17 | lamellarin 12 | O-CH2-O | OMe | OMe | OMe | OMe | = | |
| 18 | lamellarin 33 | Oi-Pr | OMe | Oi- Pr | OMe | OMe | Oi-Pr | = |
| 19 | lamellarin 31 | Oi-Pr | OMe | Oi-Pr | OMe | OMe | Oi-Pr | = |
| 20 | lamellarin 34 | Oi-Pr | OMe | OMe | Oi-Pr | OMe | Oi-Pr | = |
| 21 | lamellarin 32 | Oi-Pr | OMe | OMe | Oi-Pr | OMe | Oi-Pr | = |
| 22 | lamellarin K | OH | OMe | OH | OMe | OMe | OMe | = |
| # | Lamellarin | CDK1/cyclin B | CDK5/p25 | GSK-3α/ß | PIM 1 | DYRK1A | CK1 | SH-SY5Y | HeLa [18] |
|---|---|---|---|---|---|---|---|---|---|
| 1 | lamellarin D | 0.50 | 0.55 | 0.3 | 0.10 | 0.45 | 13.0 | 0.019 | 0.011 |
| 2 | lamellarin α | 8.0 | > 10 | 1.4 | 0.59 | 5.0 | 7.9 | − (10) | |
| 3 | di-H-lamellarin D | 1.85 | 0.11 | 0.9 | 0.20 | 0.50 | 5.9 | 0.41 | nt |
| 4 | lamellarin H | − (10) | − (10) | 9.5 | − (10) | − (10) | 5.3 | 0.45 | > 100 |
| 5 | di-H-lamellarin H | − (10) | − (10) | 0.67 | − (10) | − (10) | 5.2 | 2.55 | nt |
| 6 | lamellarin N | 0.070 | 0.025 | 0.005 | 0.055 | 0.035 | − (10) | 0.025 | nt |
| 7 | lamellarin L | 0.38 | 0.1 | 0.041 | 0.25 | 0.14 | − (10) | 0.7 | nt |
| 8 | lamellarin G tri- | ||||||||
| OMe | − (10) | − (10) | − (10) | − (10) | > 10 | − (10) | − (100) | nt | |
| 9 | lamellarin 3 | 0.53 | 0.60 | 0.58 | 0.15 | 0.06 | 0.41 | 0.056 | 0.04 |
| 10 | lamellarin 4 | 2.0 | 0.6 | 0.05 | 0.05 | 0.08 | 1.3 | 0.79 | 0.85 |
| 11 | lamellarin 5 | − (10) | − (10) | − (10) | 2.0 | − (10) | − (10) | 8.0 | 2.5 |
| 12 | lamellarin 6 | 0.10 | 0.03 | 0.13 | 0.33 | 0.09 | 0.8 | 0.11 | 0.04 |
| 13 | lamellarin 7 | 4.3 | 2.1 | 2.1 | − (10) | − (10) | − (10) | 0.14 | 0.07 |
| 14 | lamellarin 8 | 5 | 0.9 | 2.2 | 0.7 | 1.0 | − (10) | 2.65 | 4.0 |
| 15 | lamellarin 9 | − (10) | − (10) | − (10) | − (10) | − (10) | − (10) | − (10) | 1.1 |
| 16 | lamellarin 11 | − (10) | − (10) | − (10) | − (10) | − (10) | − (10) | − (10) | 5.7 |
| 17 | lamellarin 12 | − (10) | − (10) | − (10) | − (10) | − (10) | − (10) | − (10) | > 100 |
| 18 | lamellarin 33 | − (10) | − (10) | − (10) | − (10) | − (10) | − (10) | − (100) | nt |
| 19 | lamellarin 31 | − (10) | − (10) | − (10) | − (10) | − (10) | − (10) | − (100) | nt |
| 20 | lamellarin 34 | − (10) | − (10) | − (10) | − (10) | − (10) | − (10) | − (100) | nt |
| 21 | lamellarin 32 | − (10) | − (10) | − (10) | − (10) | − (10) | − (10) | − (100) | nt |
| 22 | lamellarin K | − (10) | − (10) | − (10) | 0.6 | − (10) | 6.0 | − (30) | nt |
| Protein Kinase | Activity (% of control) | SEM (%) |
|---|---|---|
| Abl kinase (h) | 61.4 | 0.8 |
| Akt1/PKB α (h) | 104.6 | 2.6 |
| AMPKα | 42.4 | 0.9 |
| BMX kinase (h) (Etk) | 60.8 | 5.4 |
| Brk (h) | 72.8 | 4.7 |
| CaMK2α (h) | 38.2 | 2.2 |
| CaMK4 (h) | 89.9 | 3.1 |
| CDC2/CDK1 (h) (cycB) | 20.4 | 0.6 |
| CDK2 (h) (cycE) | 57.1 | 0.3 |
| CHK1 (h) | 93.3 | 1.9 |
| CHK2 (h) | 47.6 | 0.6 |
| c-Met kinase (h) | 92.1 | 1.6 |
| CSK (h) | 47.4 | 2.5 |
| EphB4 kinase (h) | 89.7 | 0.9 |
| ERK1 (h) | 94.1 | 0.1 |
| ERK2 (h) (P42mapk) | 79.9 | 1.4 |
| FGFR2 kinase (h) | 19.8 | 0.6 |
| FGFR4 kinase (h) | 62.5 | 0.3 |
| FLT-1 kinase (h) (VEGFR1) | 4.0 | 0.2 |
| FLT-3 kinase (h) | 0.4 | 0.4 |
| Fyn kinase (h) | 20.1 | 1.2 |
| IGF1R kinase (h) | 87.2 | 1.6 |
| IRK (h) (InsR) | 46.7 | 1.6 |
| JNK 2 (h) | 15.7 | 1.8 |
| KDR kinase (h) (VEGFR2) | 7.4 | 1.1 |
| Lck kinase (h) | 7.1 | 0.7 |
| Lyn kinase (h) | 7.2 | 0.5 |
| MAPKAPK2 (h) | 96.6 | 2.1 |
| MEK1/MAP2K1 (h) | 81.5 | 1.0 |
| p38α kinase (h) | 97.4 | 1.4 |
| p38δ kinase (h) | 96.0 | 0.9 |
| p38γ kinase (h) | 79.6 | 0.6 |
| PDGFRβ kinase (h) | 1.8 | 0.4 |
| PDK1 (h) | 89.8 | 0.6 |
| PKA (h) | 80.0 | 4.6 |
| PKCα (h) | 91.7 | 7.6 |
| PKCβ 1 (h) | 102.0 | 1.8 |
| PKCγ (h) | 94.1 | 1.9 |
| Ret kinase (h) | 1.8 | 0.4 |
| ROCK2 (h) | 103.3 | 0.9 |
| RSK2 (h) | 44.5 | 0.5 |
| Src kinase (h) | 56.2 | 0.4 |
| Syk (h) | 97.1 | 0.7 |
| TRKA (h) | 0.8 | 0.3 |
Abbreviations:
| CDK | cyclin-dependent kinase |
| CK1 | casein kinase-1 |
| DYRK1A | dual-specificity tyrosine-phosphorylated and regulated kinase 1A |
| FCS | fetal calf serum |
| GSK-3 | glycogen synthase kinase-3 |
| GST | Glutathione-S-transferase |
| MBP | myelin basic protein |
| MTS | 3- (4,5-dimethylthiazol-2-yl)-5- (3-carboxymethoxyphenyl)-2- (4-sulfophenyl)-2H–tetrazolium |
| PARP | poly (ADP-ribose) polymerase |
Share and Cite
Baunbæk, D.; Trinkler, N.; Ferandin, Y.; Lozach, O.; Ploypradith, P.; Rucirawat, S.; Ishibashi, F.; Iwao, M.; Meijer, L. Anticancer Alkaloid Lamellarins Inhibit Protein Kinases. Mar. Drugs 2008, 6, 514-527. https://doi.org/10.3390/md20080026
Baunbæk D, Trinkler N, Ferandin Y, Lozach O, Ploypradith P, Rucirawat S, Ishibashi F, Iwao M, Meijer L. Anticancer Alkaloid Lamellarins Inhibit Protein Kinases. Marine Drugs. 2008; 6(4):514-527. https://doi.org/10.3390/md20080026
Chicago/Turabian StyleBaunbæk, Dianne, Nolwenn Trinkler, Yoan Ferandin, Olivier Lozach, Poonsakdi Ploypradith, Somsak Rucirawat, Fumito Ishibashi, Masatomo Iwao, and Laurent Meijer. 2008. "Anticancer Alkaloid Lamellarins Inhibit Protein Kinases" Marine Drugs 6, no. 4: 514-527. https://doi.org/10.3390/md20080026
APA StyleBaunbæk, D., Trinkler, N., Ferandin, Y., Lozach, O., Ploypradith, P., Rucirawat, S., Ishibashi, F., Iwao, M., & Meijer, L. (2008). Anticancer Alkaloid Lamellarins Inhibit Protein Kinases. Marine Drugs, 6(4), 514-527. https://doi.org/10.3390/md20080026