
Identification of Anhydrodebromoaplysiatoxin as a Dichotomic Autophagy Inhibitor


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
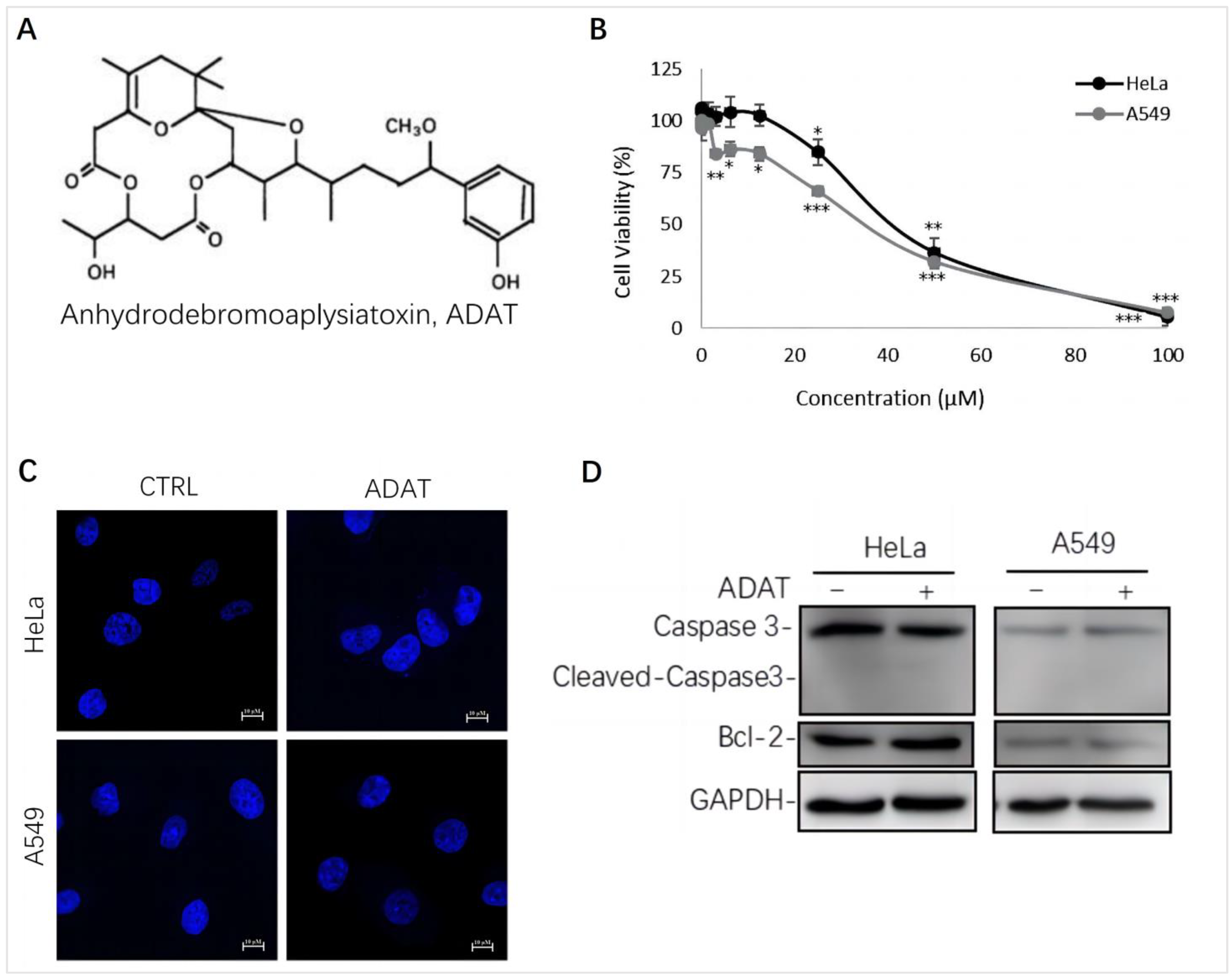
2.1. ADAT Inhibited Cell Proliferation of HeLa or A549 Cells without Inducing Apoptosis
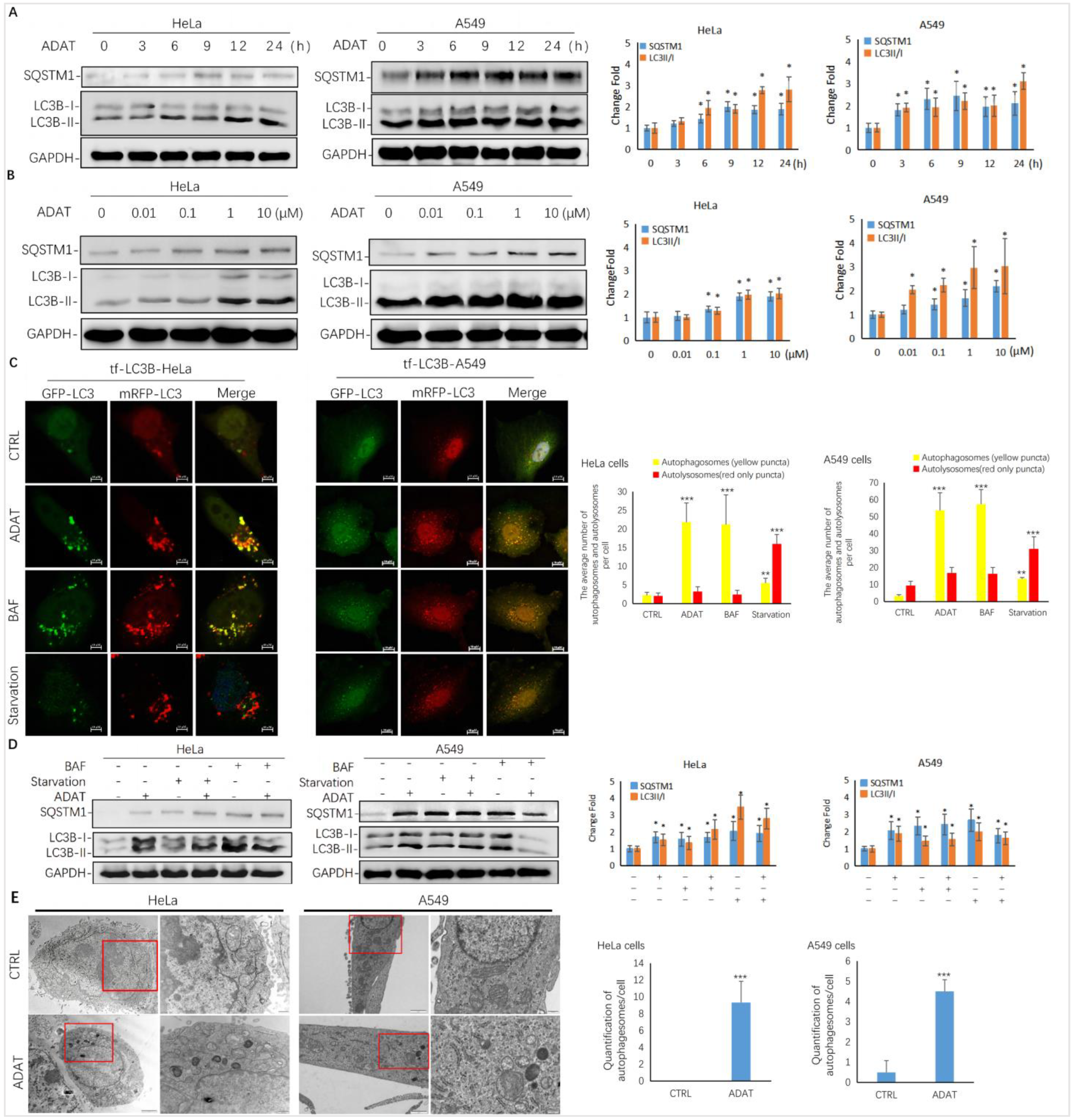
2.2. ADAT Is a Novel Autophagy Modulator
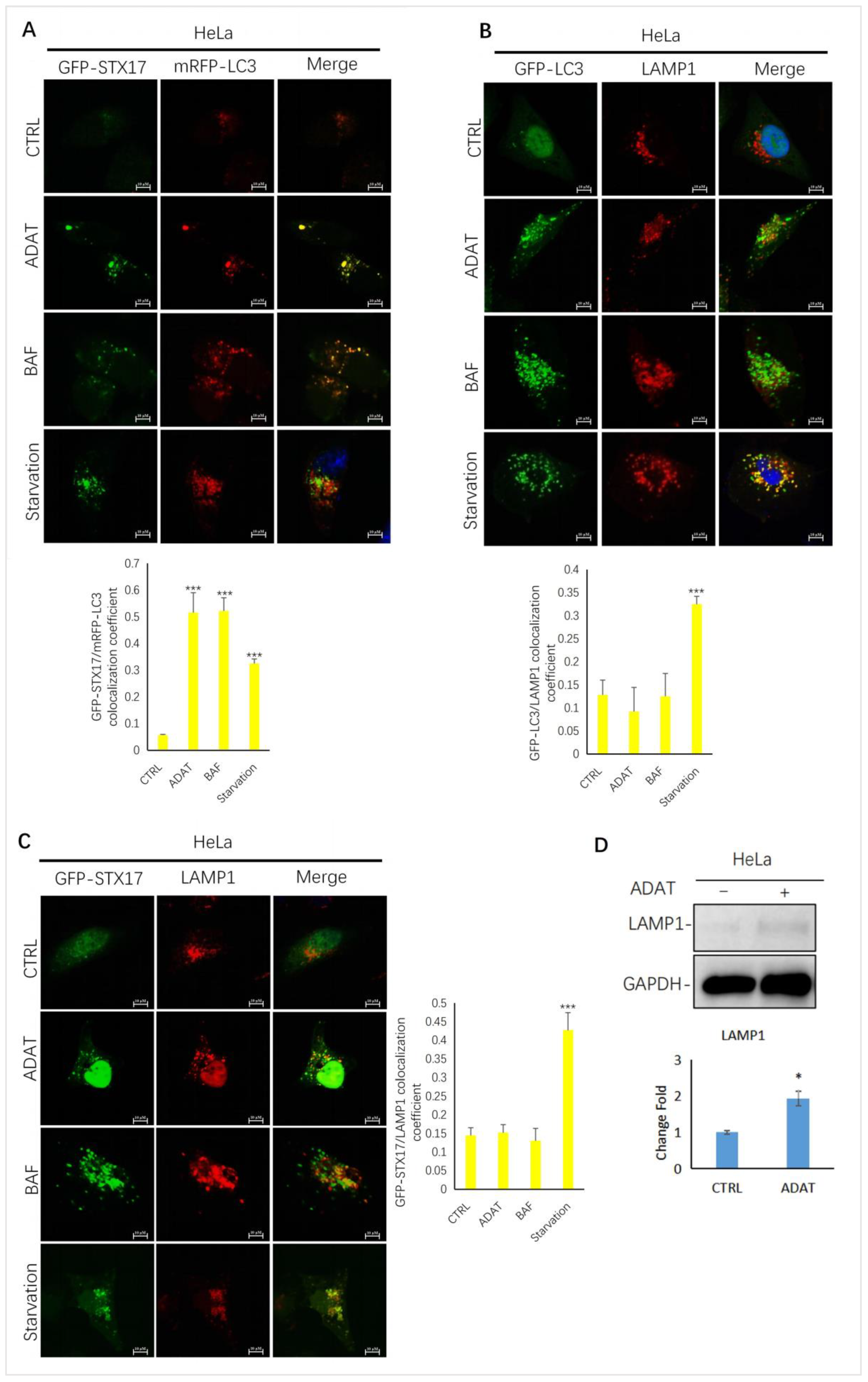
2.3. ADAT Inhibits Autophagic Flux by Blocking Autophagosome-Lysosome Fusion
2.4. ADAT Increasing Lysosomal pH
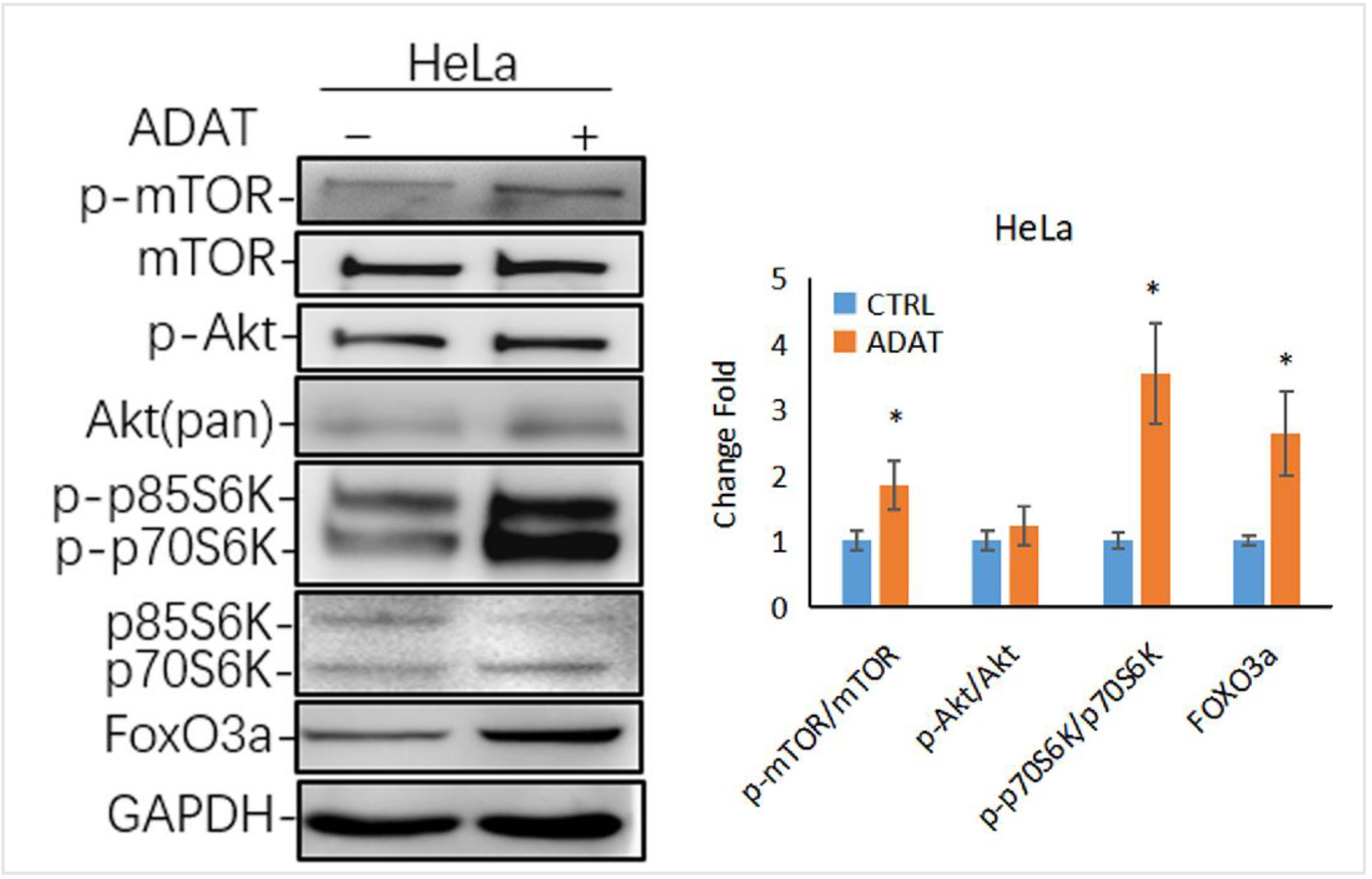
2.5. ADAT Might Inhibit Autophagy via Activating the mTOR/p70S6K/FOXO3a Pathway
3. Discussion
4. Materials and Methods
4.1. Materials and Antibodies
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. DAPI Staining
4.5. Confocal Microscopy Imaging
4.6. Transmission Electron Microscopy
4.7. Lysosomal pH Assessment
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Schneider, J.L.; Cuervo, A.M. Autophagy and human disease: Emerging themes. Curr. Opin. Genet. Dev. 2014, 26, 16–23. [Google Scholar] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Gavilán, E.; Sánchez-Aguayo, I.; Daza, P.; Ruano, D. GSK-3β signaling determines autophagy activation in the breast tumor cell line MCF7 and inclusion formation in the non-tumor cell line MCF10A in response to proteasome inhibition. Cell Death Dis. 2013, 4, e572. [Google Scholar]
- Gavilán, E.; Giráldez, S.; Sánchez-Aguayo, I.; Romero, F.; Ruano, D.; Daza, P. Breast cancer cell line MCF7 escapes from G1/S arrest induced by proteasome inhibition through a GSK-3β dependent mechanism. Sci. Rep. 2015, 5, 10027. [Google Scholar]
- Ruocco, N.; Costantini, S.; Costantini, M. Blue-Print Autophagy: Potential for Cancer Treatment. Mar. Drugs 2016, 14, 138. [Google Scholar] [CrossRef]
- Carr, G.; Williams, D.E.; Díaz-Marrero, A.R.; Patrick, B.O.; Bottriell, H.; Balgi, A.D.; Donohue, E.; Roberge, M.; Andersen, R.J. Bafilomycins produced in culture by Streptomyces spp. isolated from marine habitats are potent inhibitors of autophagy. J. Nat. Prod. 2010, 73, 422–427. [Google Scholar] [CrossRef]
- Kallifatidis, G.; Hoepfner, D.; Jaeg, T.; Guzmán, E.A.; Wright, A.E. The marine natural product manzamine A targets vacuolar ATPases and inhibits autophagy in pancreatic cancer cells. Mar. Drugs 2013, 11, 3500–3516. [Google Scholar] [CrossRef]
- Fujiki, H.; Suganuma, M.; Nakayasu, M.; Hoshino, H.; Moore, R.E.; Sugimura, T. The third class of new tumor promoters, polyacetates (debromoaplysiatoxin and aplysiatoxin), can differentiate biological actions relevant to tumor promoters. Gan 1982, 73, 495–497. [Google Scholar] [PubMed]
- Fujiki, H.; Tanaka, Y.; Miyake, R.; Kikkawa, U.; Nishizuka, Y.; Sugimura, T. Activation of calcium-activated, phospholipid-dependent protein kinase (protein kinase C) by new classes of tumor promoters: Teleocidin and debromoaplysiatoxin. Biochem. Biophys. Res. Commun. 1984, 120, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, M.; Fujiki, H.; Tahira, T.; Cheuk, C.; Moore, R.E.; Sugimura, T. Estimation of tumor promoting activity and structure-function relationships of aplysiatoxins. Carcinogenesis 1984, 5, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Arcoleo, J.P.; Weinstein, I.B. Activation of protein kinase C by tumor promoting phorbol esters, teleocidin and aplysiatoxin in the absence of added calcium. Carcinogenesis 1985, 6, 213–217. [Google Scholar] [CrossRef]
- Nakamura, H.; Kishi, Y.; Pajares, M.A.; Rando, R.R. Structural basis of protein kinase C activation by tumor promoters. Proc. Natl. Acad. Sci. USA 1989, 86, 9672–9676. [Google Scholar] [CrossRef]
- Mynderse, J.S.; Moore, R.E.; Kashiwagi, M.; Norton, T.R. Antileukemia activity in the Osillatoriaceae: Isolation of Debromoaplysiatoxin from Lyngbya. Science 1977, 196, 538–540. [Google Scholar] [CrossRef]
- Nagai, H.; Sato, S.; Iida, K.; Hayashi, K.; Kawaguchi, M.; Uchida, H.; Satake, M. Oscillatoxin I: A New Aplysiatoxin Derivative, from a Marine Cyanobacterium. Toxins 2019, 11, 366. [Google Scholar] [CrossRef]
- Nagai, H.; Kan, Y.; Fujita, T.; Sakamoto, B.; Hokama, Y. Manauealide C and Anhydrodebromoaplysiatoxin, Toxic Constituents of the Hawaiian Red Alga, Gracilaria coronopifolia. Biosci. Biotechnol. Biochem. 1998, 62, 1011–1013. [Google Scholar] [CrossRef]
- Gupta, D.K.; Kaur, P.; Leong, S.T.; Tan, L.T.; Prinsep, M.R.; Chu, J.J.H. Anti-Chikungunya viral activities of aplysiatoxin-related compounds from the marine cyanobacterium Trichodesmium erythraeum. Mar. Drugs 2014, 12, 115–127. [Google Scholar] [CrossRef]
- Tang, Y.; Wu, J.; Fan, T.; Zhang, H.; Han, B. Chemical and biological study of aplysiatoxin derivatives showing inhibition of potassium channel Kv 1.5. RSC Adv. 2019, 14, 7594–7600. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, K.; Takats, S.; Kovacs, A.L.; Juhasz, G. Evolutionarily conserved role and physiological relevance of a STX17/Syx17 (syntaxin 17)-containing SNARE complex in autophagosome fusion with endosomes and lysosomes. Autophagy 2013, 9, 1642–1646. [Google Scholar] [CrossRef]
- Davis-Kaplan, S.R.; Ward, D.M.; Shiflett, S.L.; Kaplan, J. Genome-wide analysis of iron-dependent growth reveals a novel yeast gene required for vacuolar acidification. J. Biol. Chem. 2004, 279, 4322–4329. [Google Scholar] [CrossRef]
- Agnieszka, P.M.; Paweł, A.J.; Jurek, W.D. Evaluation of acridine orange, LysoTracker Red, and quinacrine as fluorescent probes for long-term tracking of acidic vesicles. Cytometry A 2014, 85, 729–737. [Google Scholar]
- Zhang, L.; Wang, H.; Zhu, J.; Xu, J.; Ding, K. Mollugin induces tumor cell apoptosis and autophagy via the PI3K/AKT/mTOR/p70S6K and ERK signaling pathways. Biochem. Biophys. Res. Commun. 2014, 450, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Zhang, C.; Yu, B.; Chen, B.; Liu, Z.; Hou, C.; Wang, F.; Shen, H.; Chen, Z. Autophagic degradation of FOXO3a represses the expression of PUMA to block cell apoptosis in cisplatin-resistant osteosarcoma cells. Am. J. Cancer Res. 2017, 7, 1407–1422. [Google Scholar]
- Fitzwalter, B.E.; Towers, C.G.; Sullivan, K.D.; Andrysik, Z.; Hoh, M.; Ludwig, M.; O′Prey, J.; Ryan, K.M.; Espinosa, J.M.; Morgan, M.J.; et al. Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev. Cell 2018, 44, 555–565. [Google Scholar] [CrossRef]
- Blankson, H.; Holen, I.; Seglen, P.O. Disruption of the cytokeratin cytoskeleton and inhibition of hepatocytic autophagy by okadaic acid. Exp. Cell Res. 1995, 218, 522–530. [Google Scholar] [CrossRef]
- Morgan, M.J.; Gamez, G.; Menke, C.; Hernandez, A.; Thorburn, J.; Gidan, F.; Staskiewicz, L.; Morgan, S.; Cummings, C.; Maycotte, P.; et al. Regulation of autophagy and chloroquine sensitivity by oncogenic RAS in vitro is context-dependent. Autophagy 2014, 10, 1814–1826. [Google Scholar] [CrossRef]
- Maes, H.; Kuchnio, A.; Peric, A.; Moens, S.; Nys, K.; De Bock, K.; Quaegebeur, A.; Schoors, S.; Georgiadou, M.; Wouters, J.; et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 2014, 26, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Antal, C.E.; Hudson, A.M.; Kang, E.; Zanca, C.; Wirth, C.; Stephenson, N.L.; Trotter, E.W.; Gallegos, L.L.; Miller, C.J.; Furnari, F.B.; et al. Cancer-associated protein kinase C mutations reveal kinase′s role as tumor suppressor. Cell 2015, 160, 489–502. [Google Scholar] [CrossRef]
- Long, Z.; Chen, B.; Liu, Q.; Zhao, J.; Yang, Z.; Dong, X.; Xia, L.; Huang, S.; Hu, X.; Song, B.; et al. The reverse-mode NCX1 activity inhibitor KB-R7943 promotes prostate cancer cell death by activating the JNK pathway and blocking autophagic flux. Oncotarget 2016, 7, 42059–42070. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Fang, Y.; Yang, Y.; Qin, Y.; Wu, P.; Wang, T.; Lai, H.; Meng, L.; Wang, D.; Zheng, Z.; et al. Elaiophylin, a novel autophagy inhibitor, exerts antitumor activity as a single agent in ovarian cancer cells. Autophagy 2015, 11, 1849–1863. [Google Scholar] [CrossRef] [PubMed]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Park, Y.J.; Park, J.Y.; Jeong, H.O.; Kim, D.H.; Ha, Y.M.; Kim, J.M.; Song, Y.M.; Heo, H.-S.; Yu, B.P.; et al. Inhibitory effect of mTOR activator MHY1485 on autophagy: Suppression of lysosomal fusion. PLoS ONE 2012, 7, e43418. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhu, H.; Xu, X.; Li, L.; Tan, H.; Cai, X. Inactivated Sendai virus induces apoptosis and autophagy via the PI3K/Akt/mTOR/p70S6K pathway in human non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2015, 465, 64–70. [Google Scholar] [CrossRef]
- Fitzwalter, B.E.; Thorburn, A. FOXO3 links autophagy to apoptosis. Autophagy 2018, 14, 1467–1468. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J. Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim. Et Biophys. Acta 2011, 1813, 1961–1964. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, L.; Lu, C.-K.; Wu, J.; Chan, L.L.; Yue, J. Identification of Anhydrodebromoaplysiatoxin as a Dichotomic Autophagy Inhibitor. Mar. Drugs 2023, 21, 46. https://doi.org/10.3390/md21010046
Feng L, Lu C-K, Wu J, Chan LL, Yue J. Identification of Anhydrodebromoaplysiatoxin as a Dichotomic Autophagy Inhibitor. Marine Drugs. 2023; 21(1):46. https://doi.org/10.3390/md21010046
Chicago/Turabian StyleFeng, Limin, Chung-Kuang Lu, Jiajun Wu, Leo Lai Chan, and Jianbo Yue. 2023. "Identification of Anhydrodebromoaplysiatoxin as a Dichotomic Autophagy Inhibitor" Marine Drugs 21, no. 1: 46. https://doi.org/10.3390/md21010046
APA StyleFeng, L., Lu, C.-K., Wu, J., Chan, L. L., & Yue, J. (2023). Identification of Anhydrodebromoaplysiatoxin as a Dichotomic Autophagy Inhibitor. Marine Drugs, 21(1), 46. https://doi.org/10.3390/md21010046