
Neuroprotective Effect of Cyclo-(L-Pro-L-Phe) Isolated from the Jellyfish-Derived Fungus Aspergillus flavus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
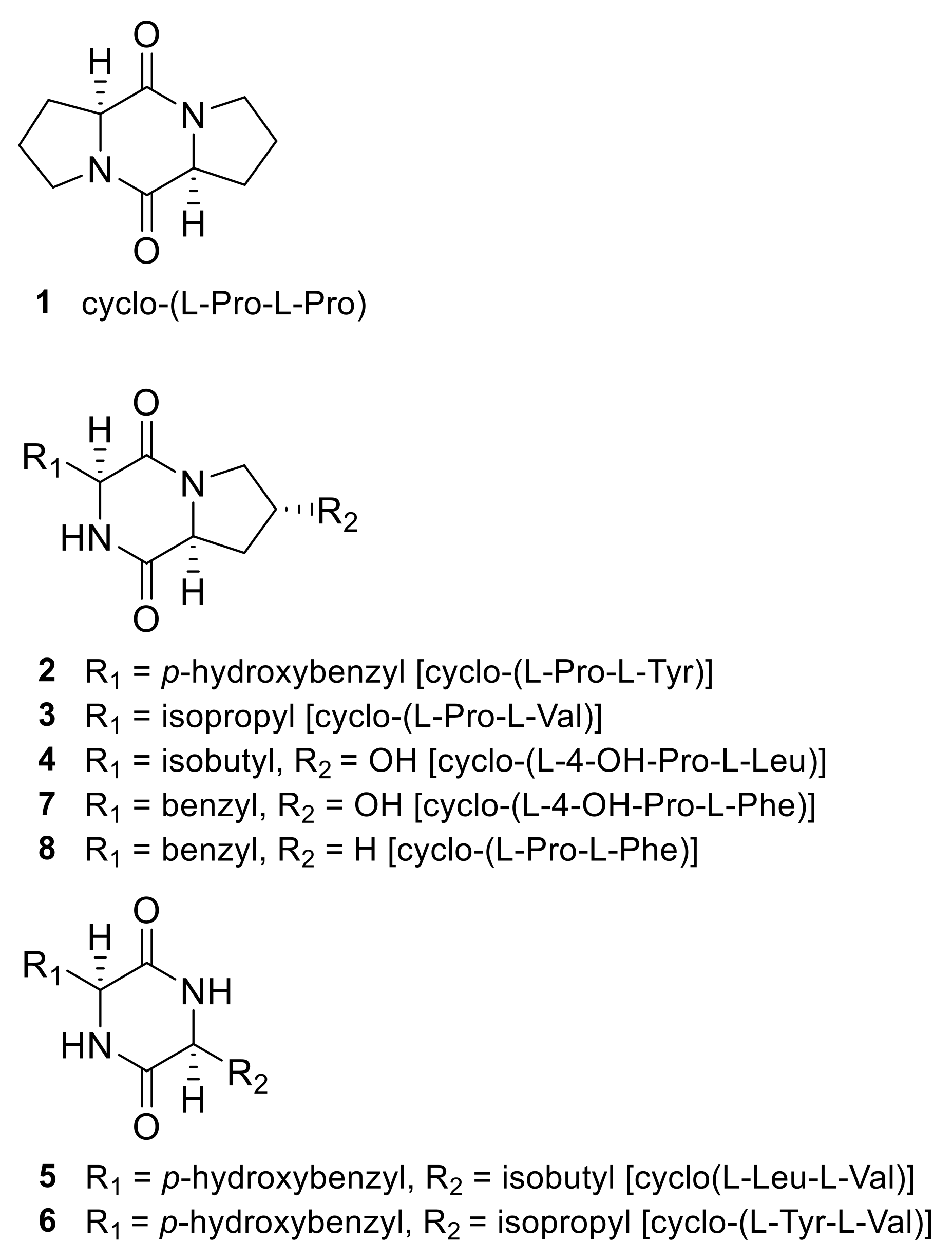
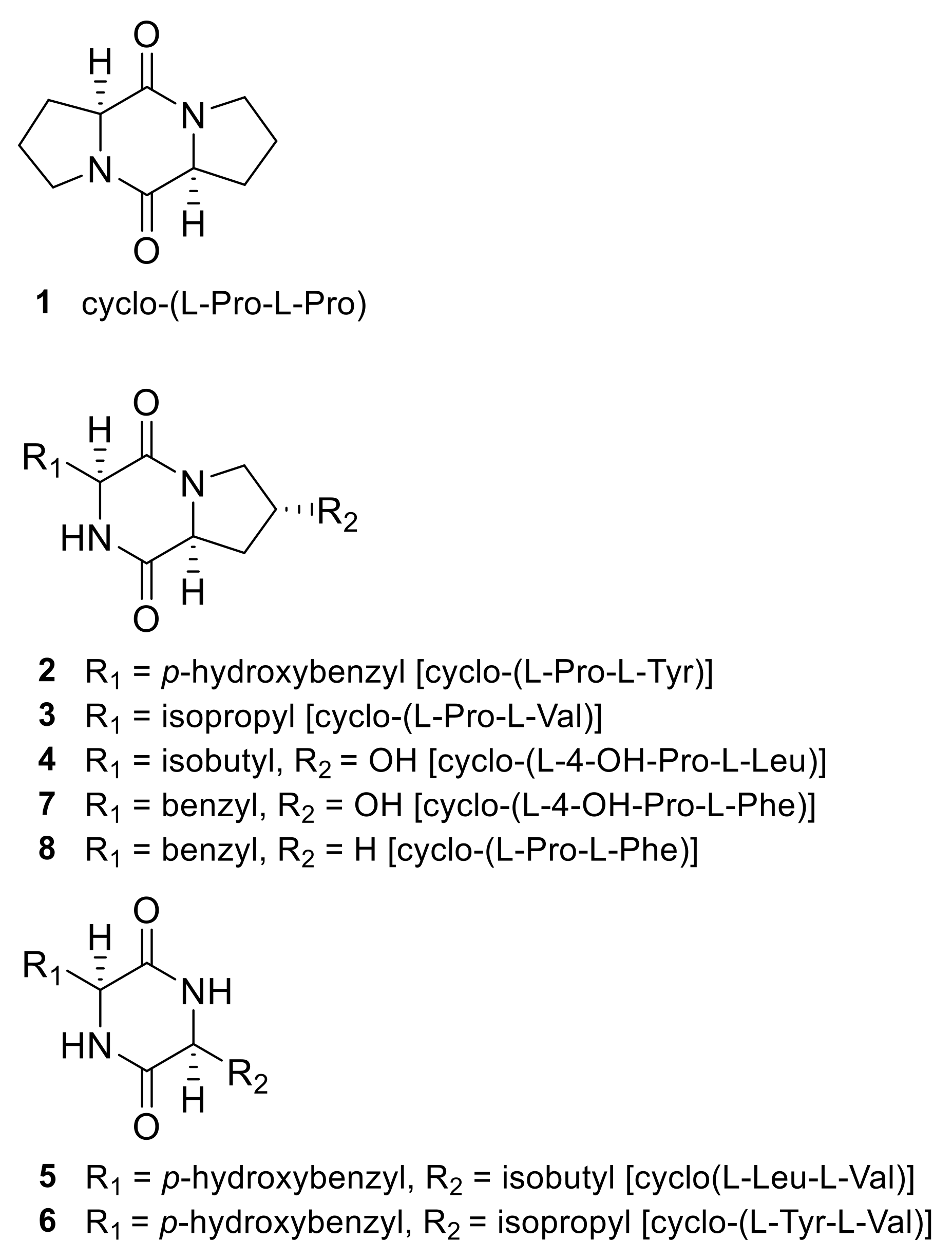
2.1. Identification of 2,5-DKPs
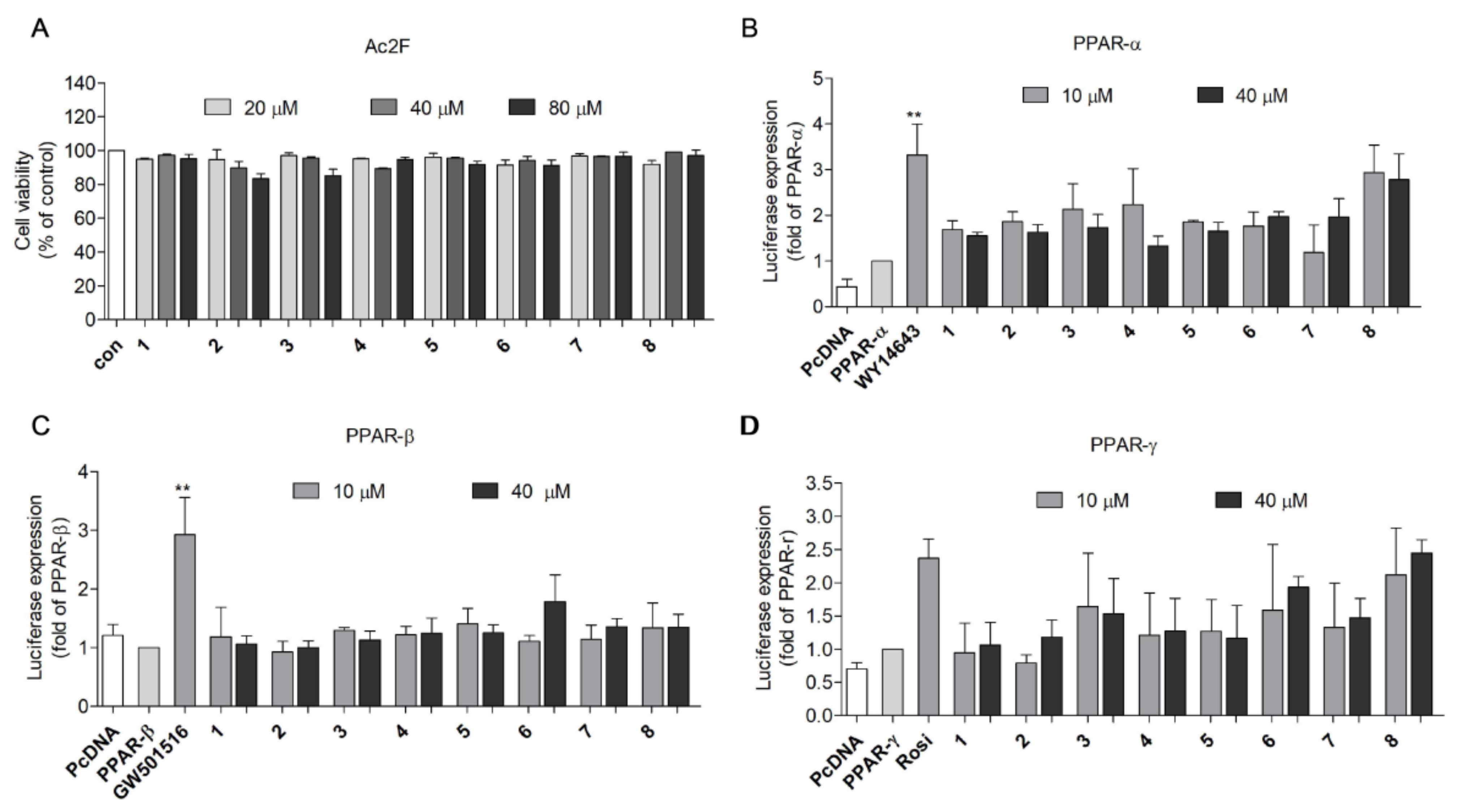
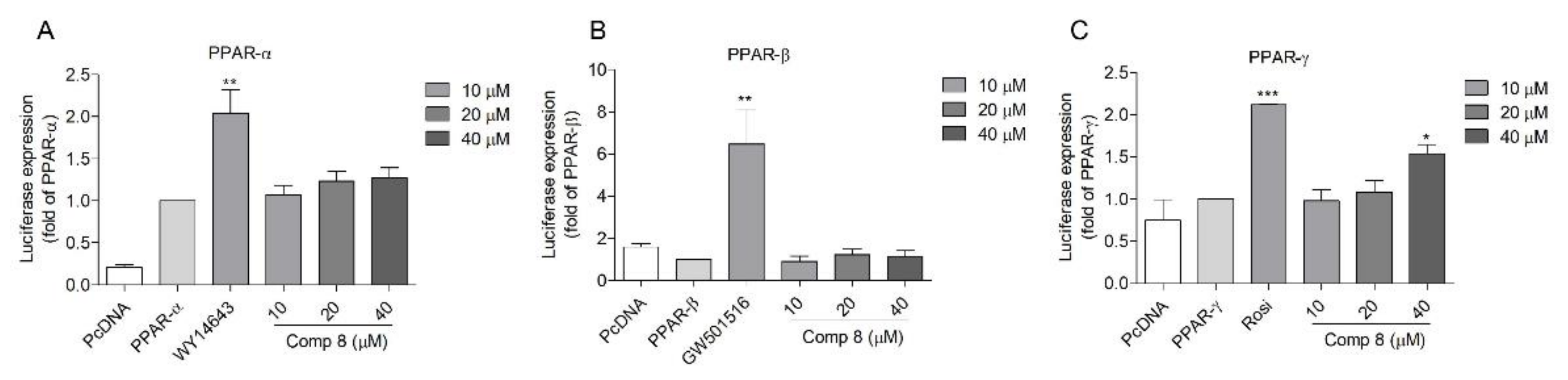
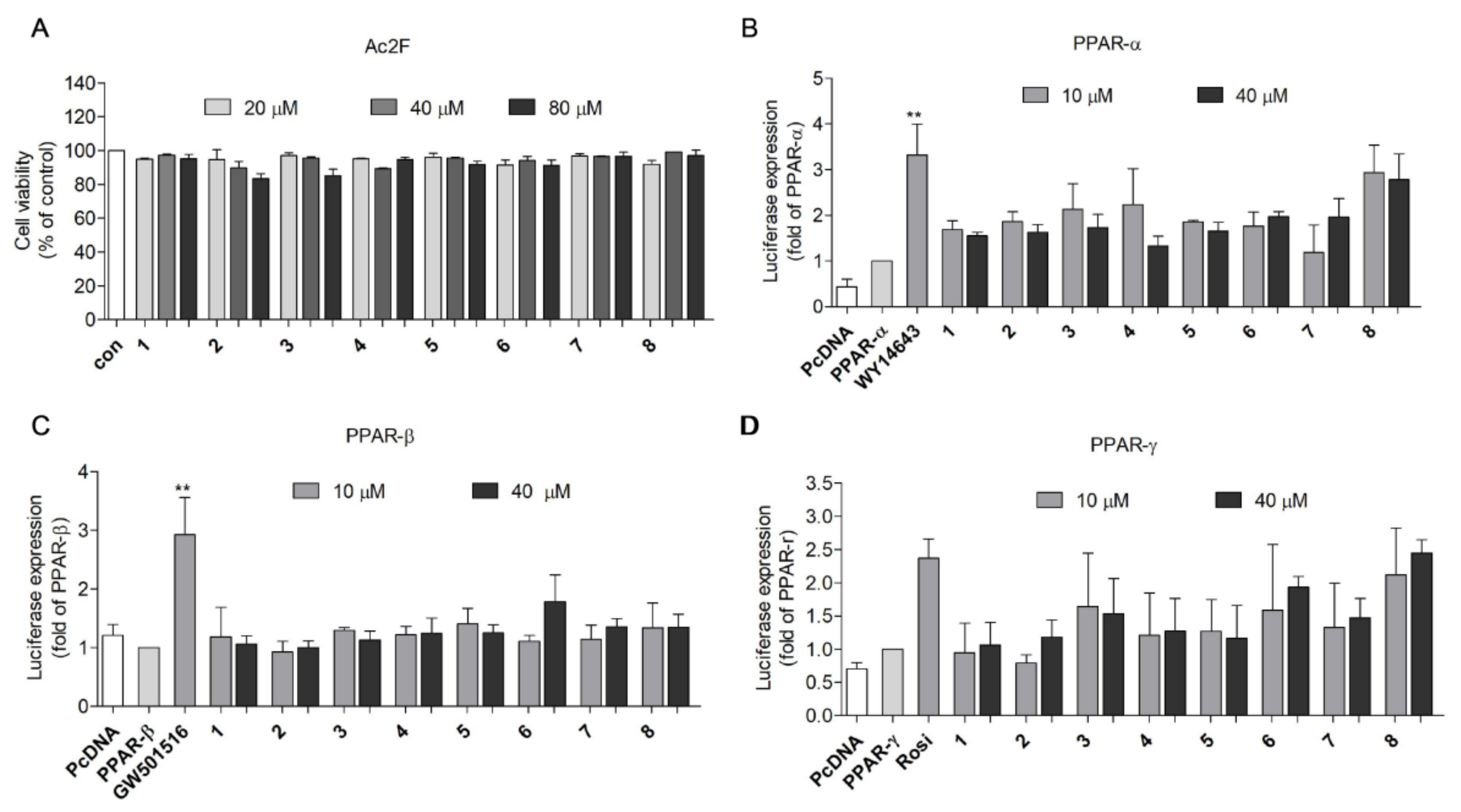
2.2. PPAR Transactivation by 2,5-DKPs
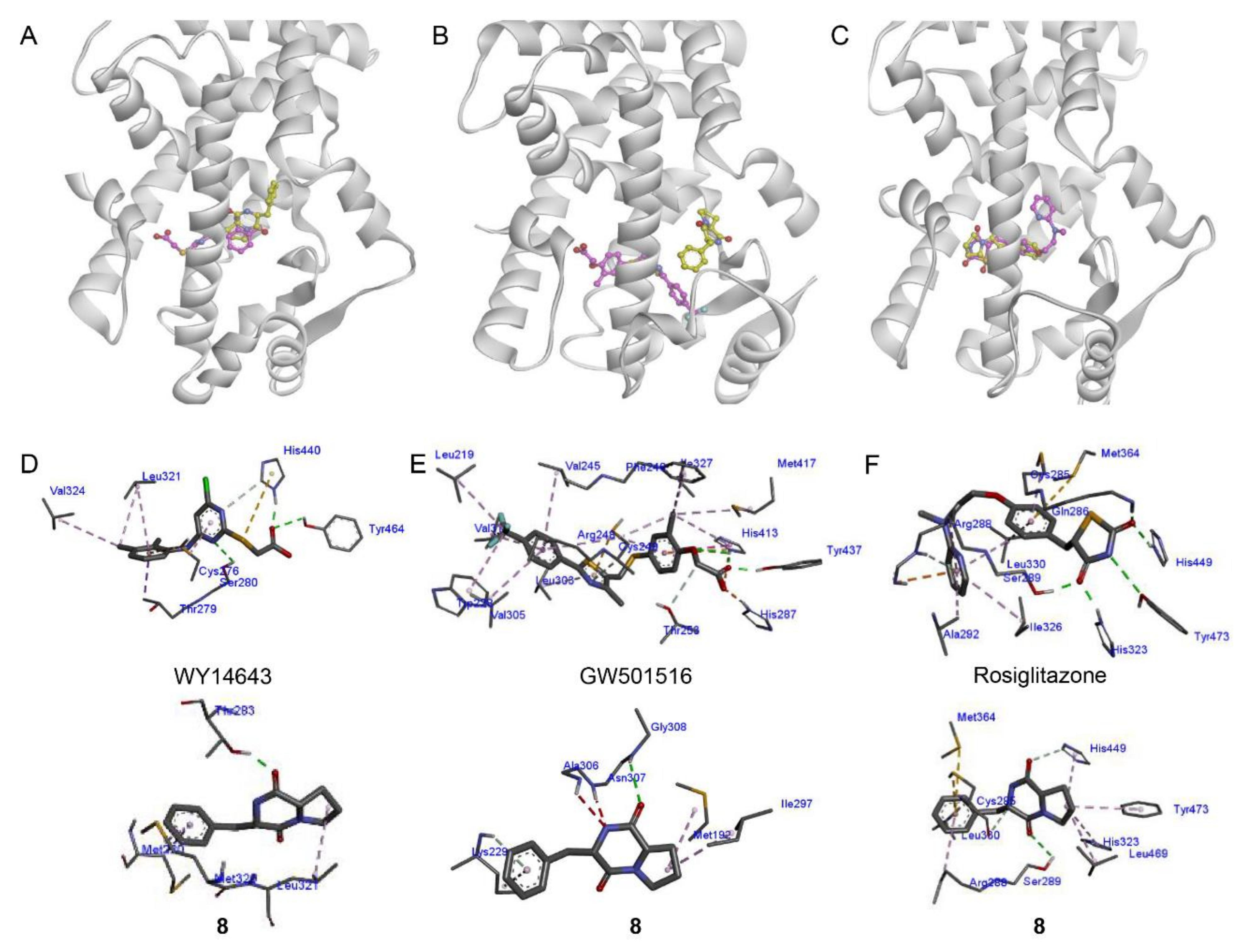
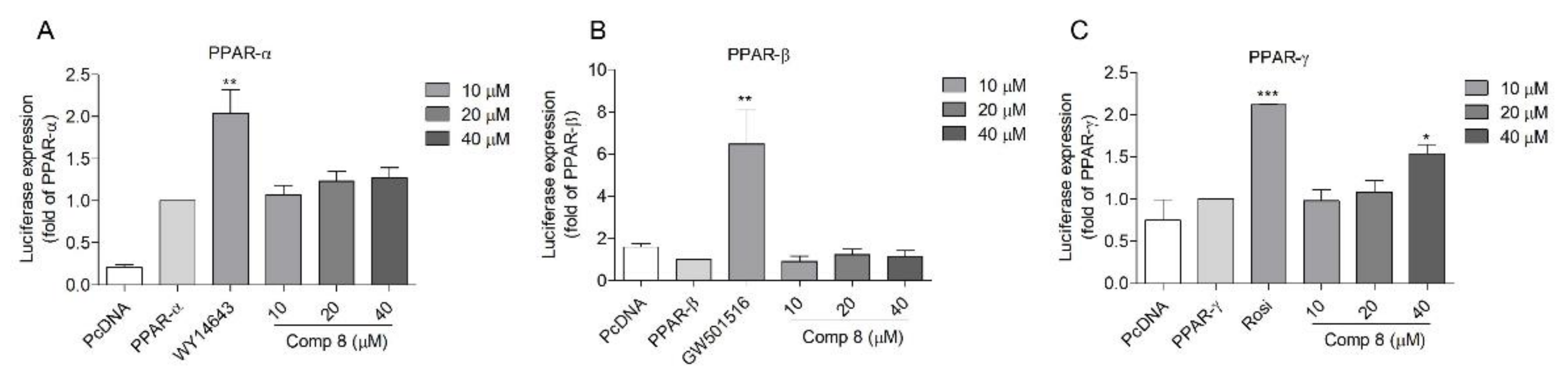
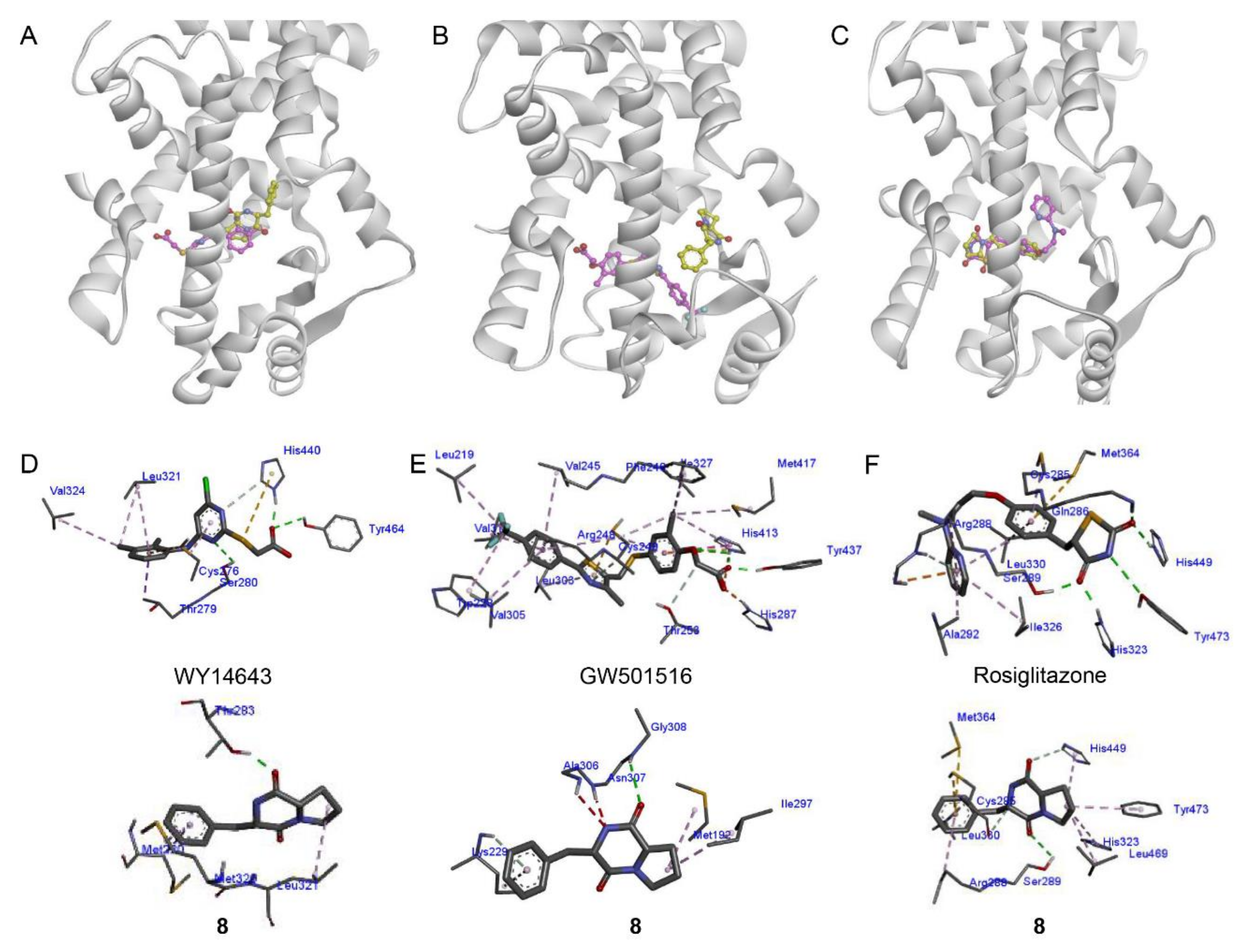
2.3. Docking Analysis of Compound 8 with PPAR-α, -β/δ, and -γ
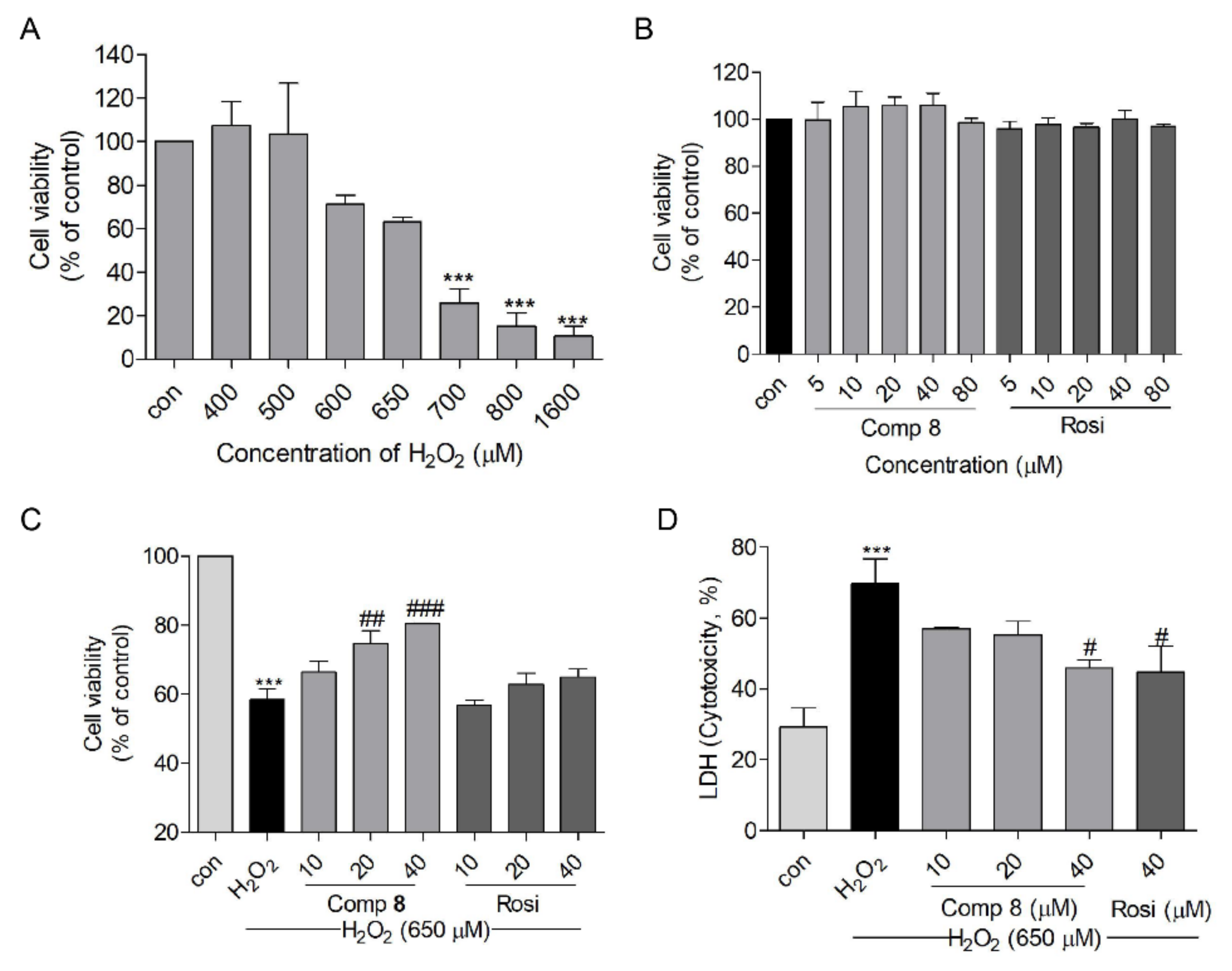
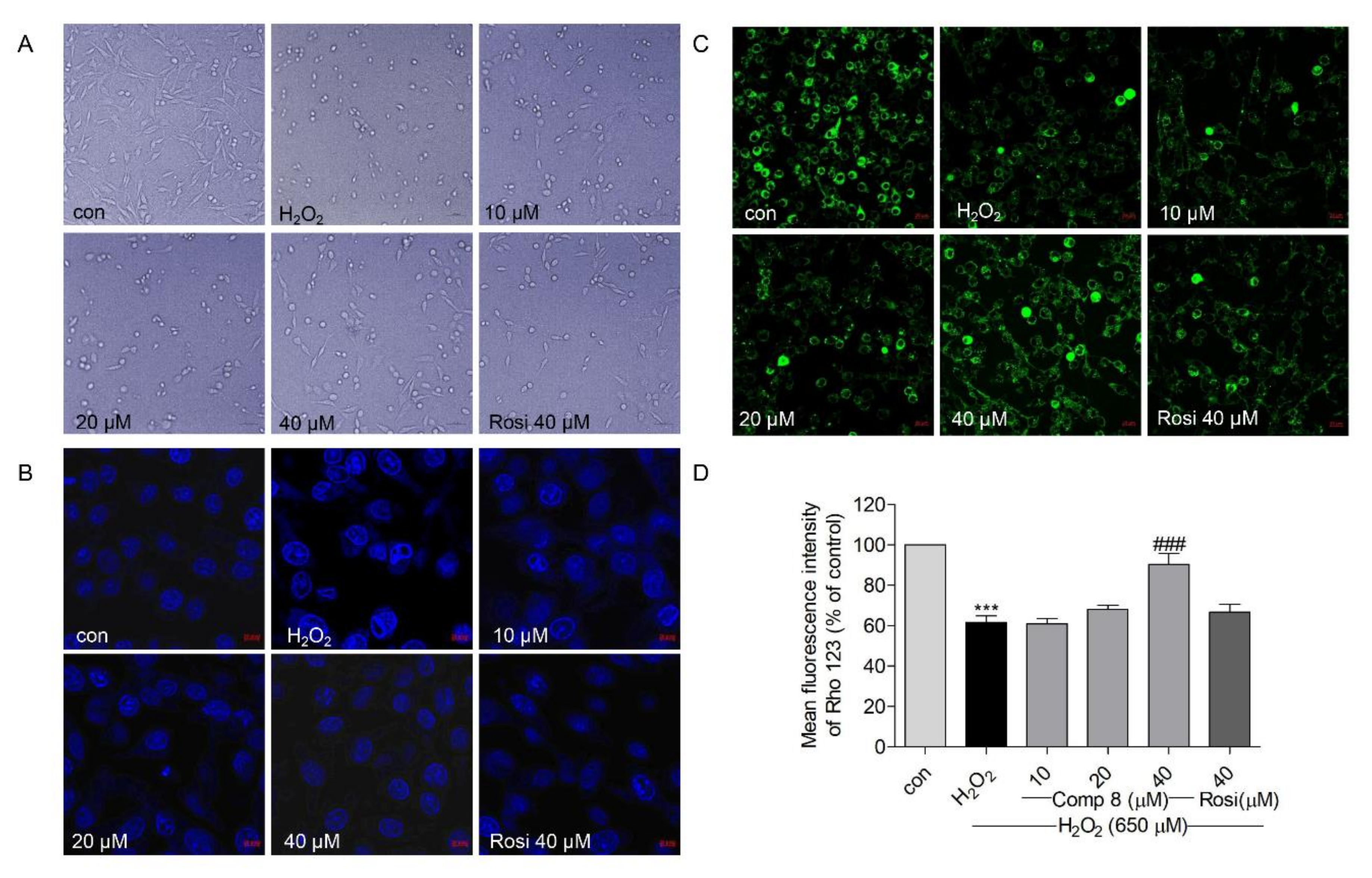
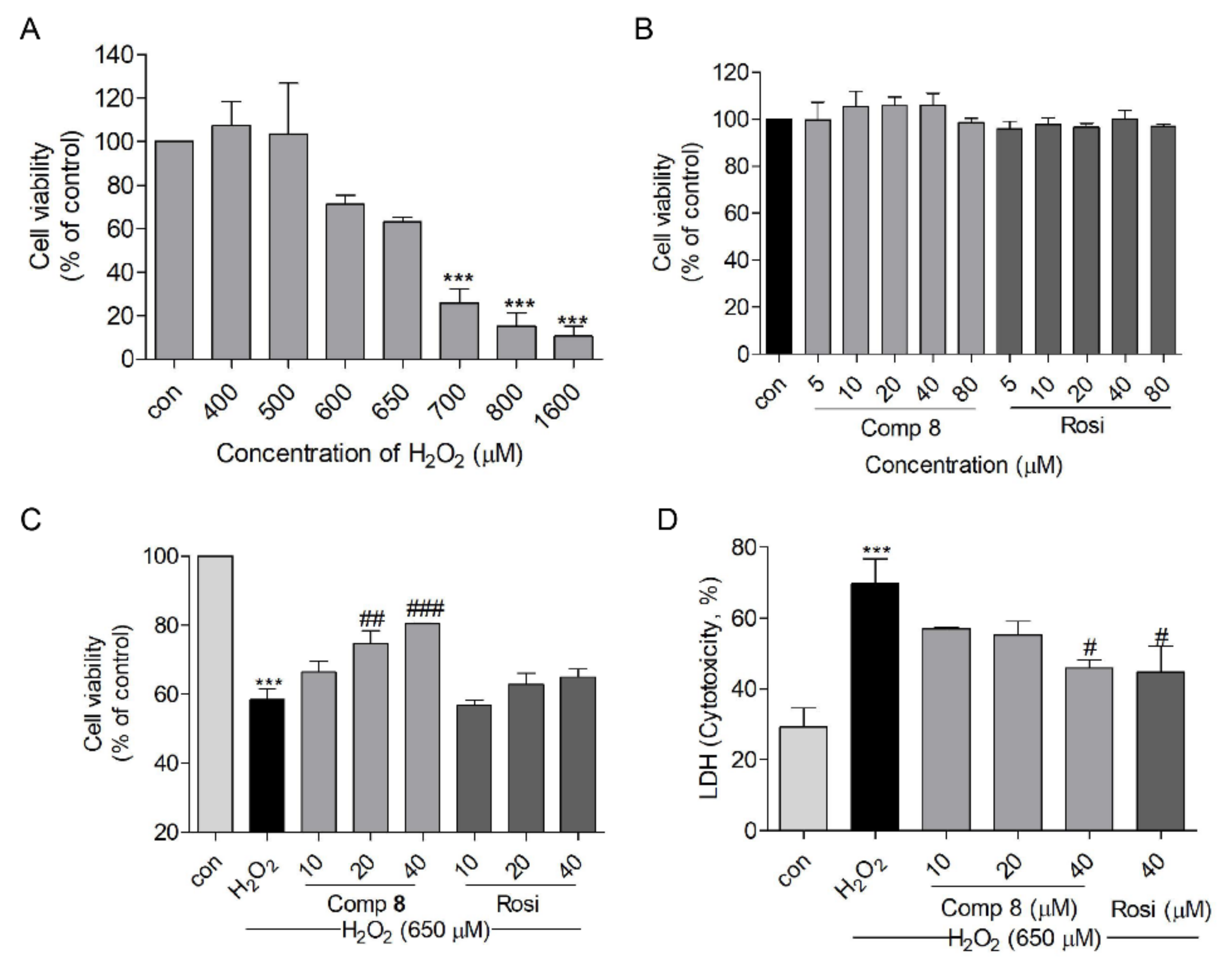
2.4. Protective Effects of Compound 8 against H2O2-Induced Cell Injury
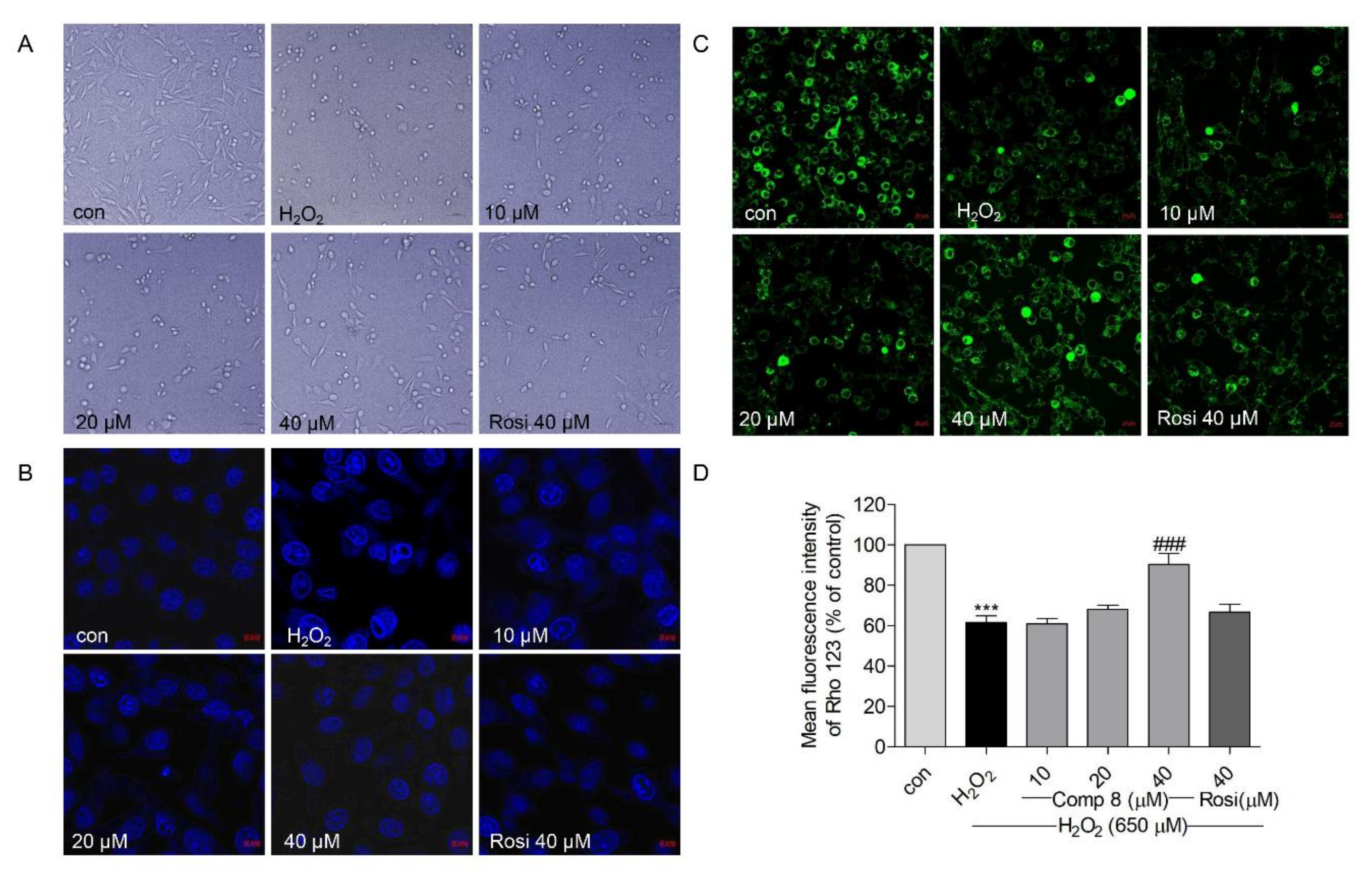
2.5. Effects of Compound 8 on H2O2-Induced Apoptosis
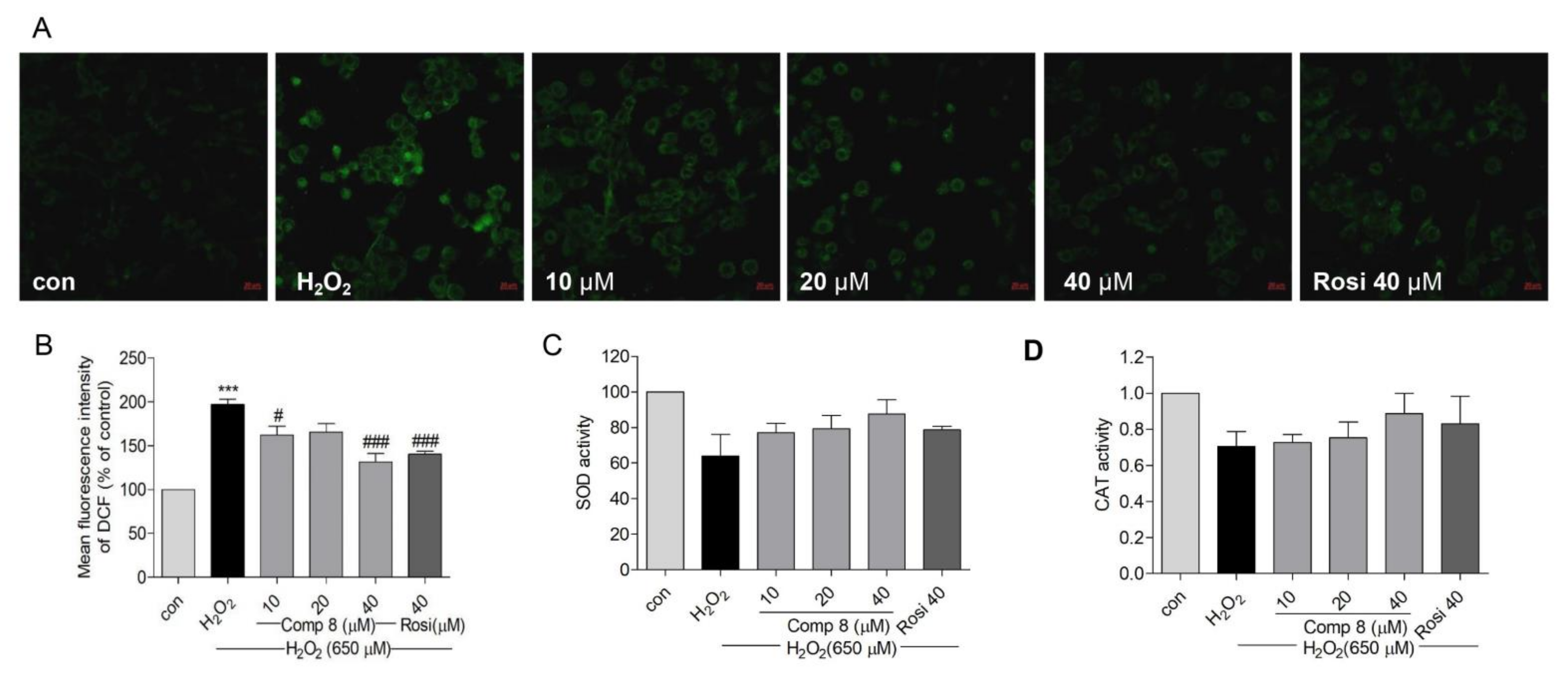
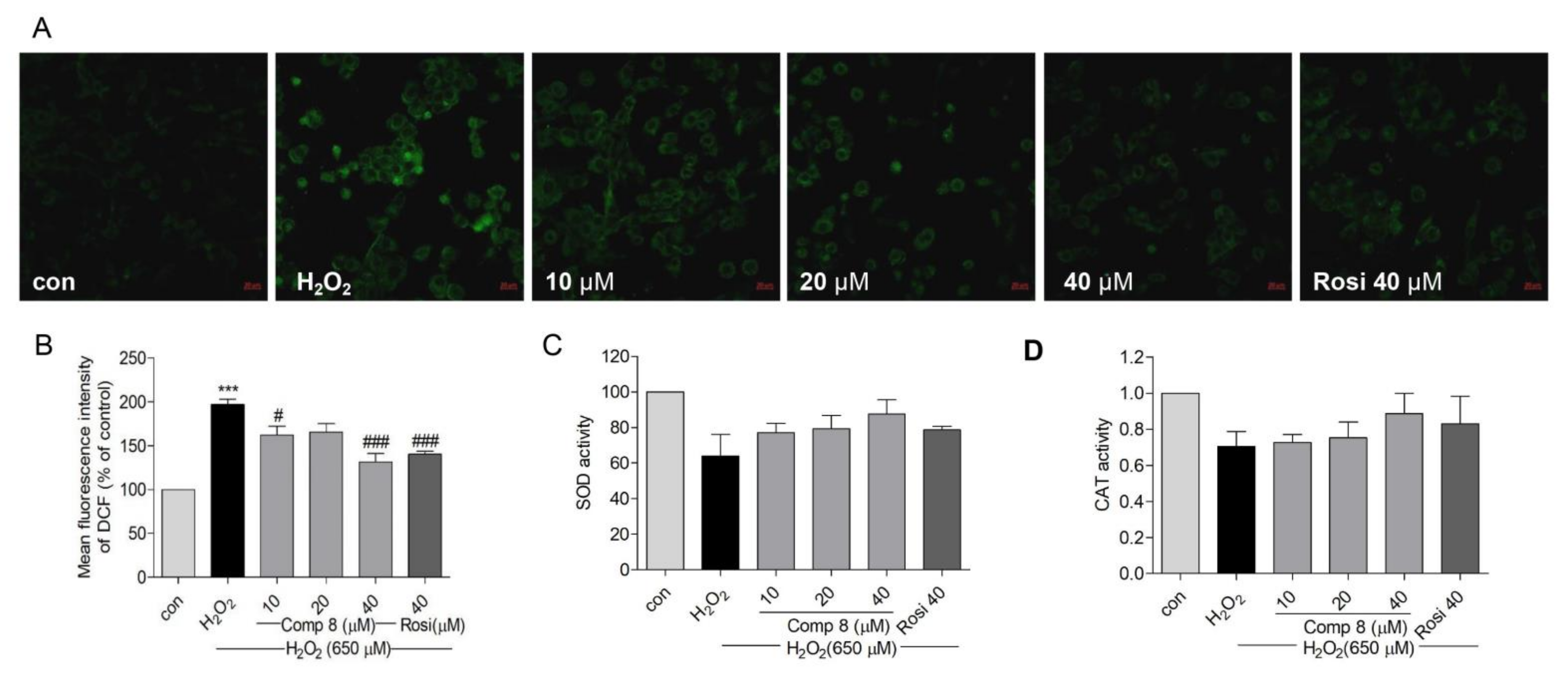
2.6. Effects of Compound 8 on H2O2-Induced Oxidative Stress
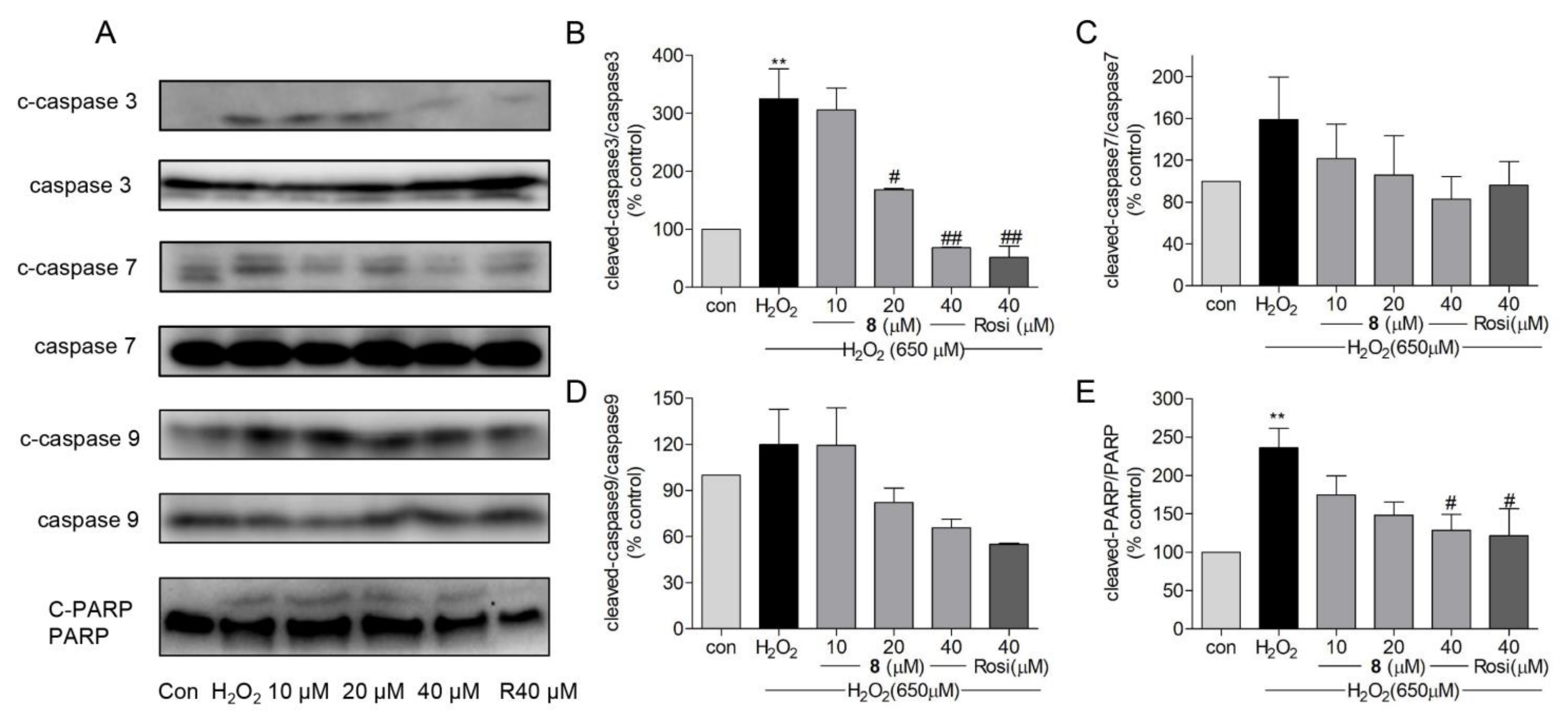
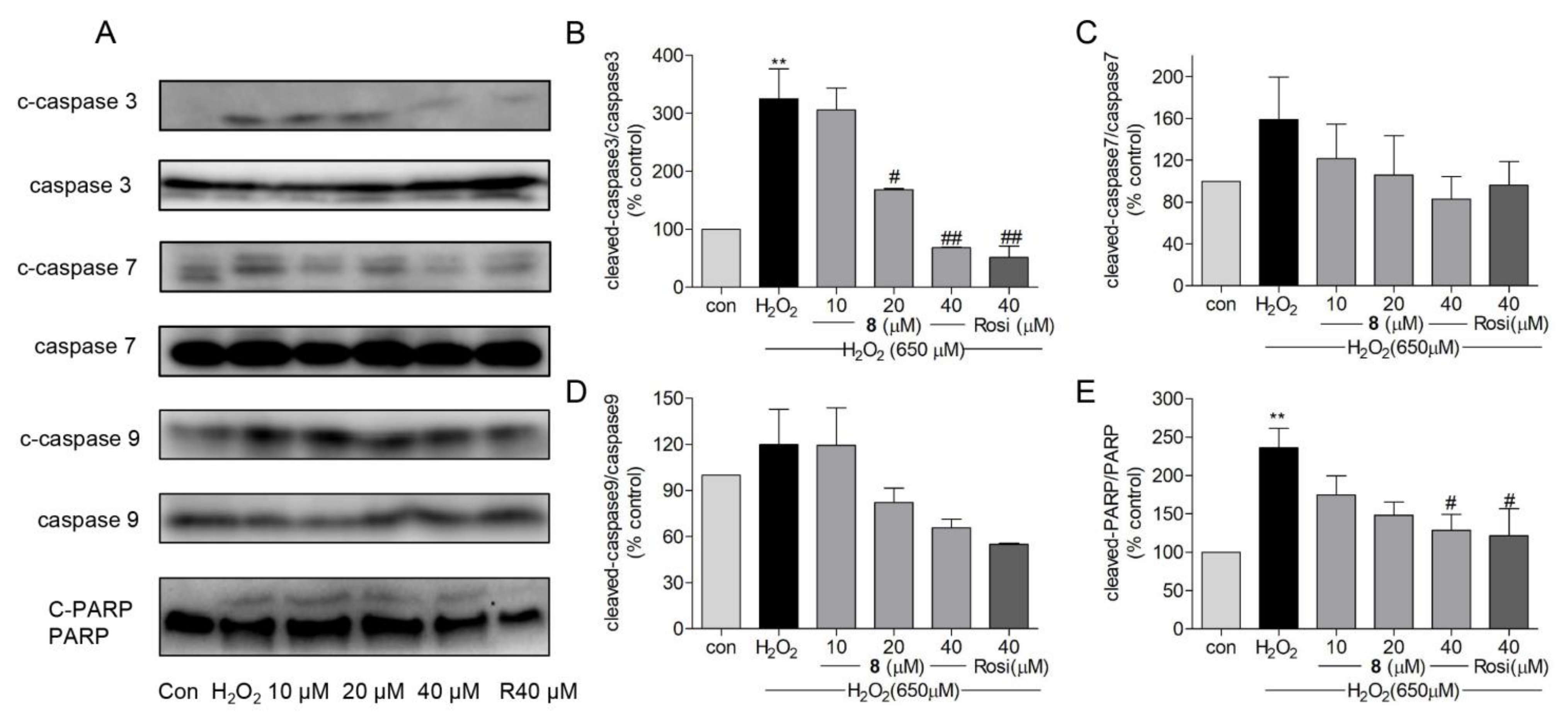
2.7. Effects of Compound 8 on H2O2-Induced Apoptosis-Related Proteins
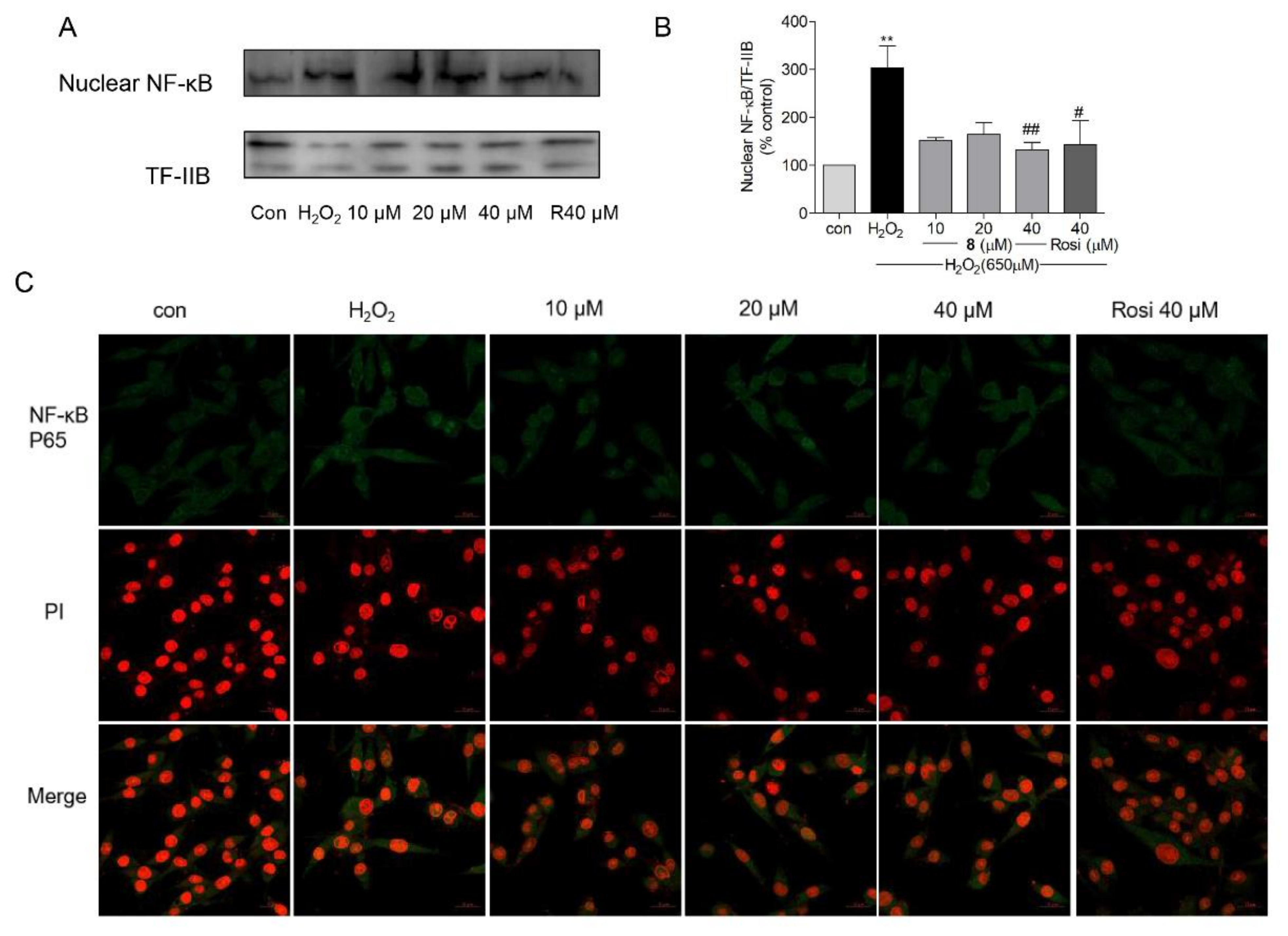
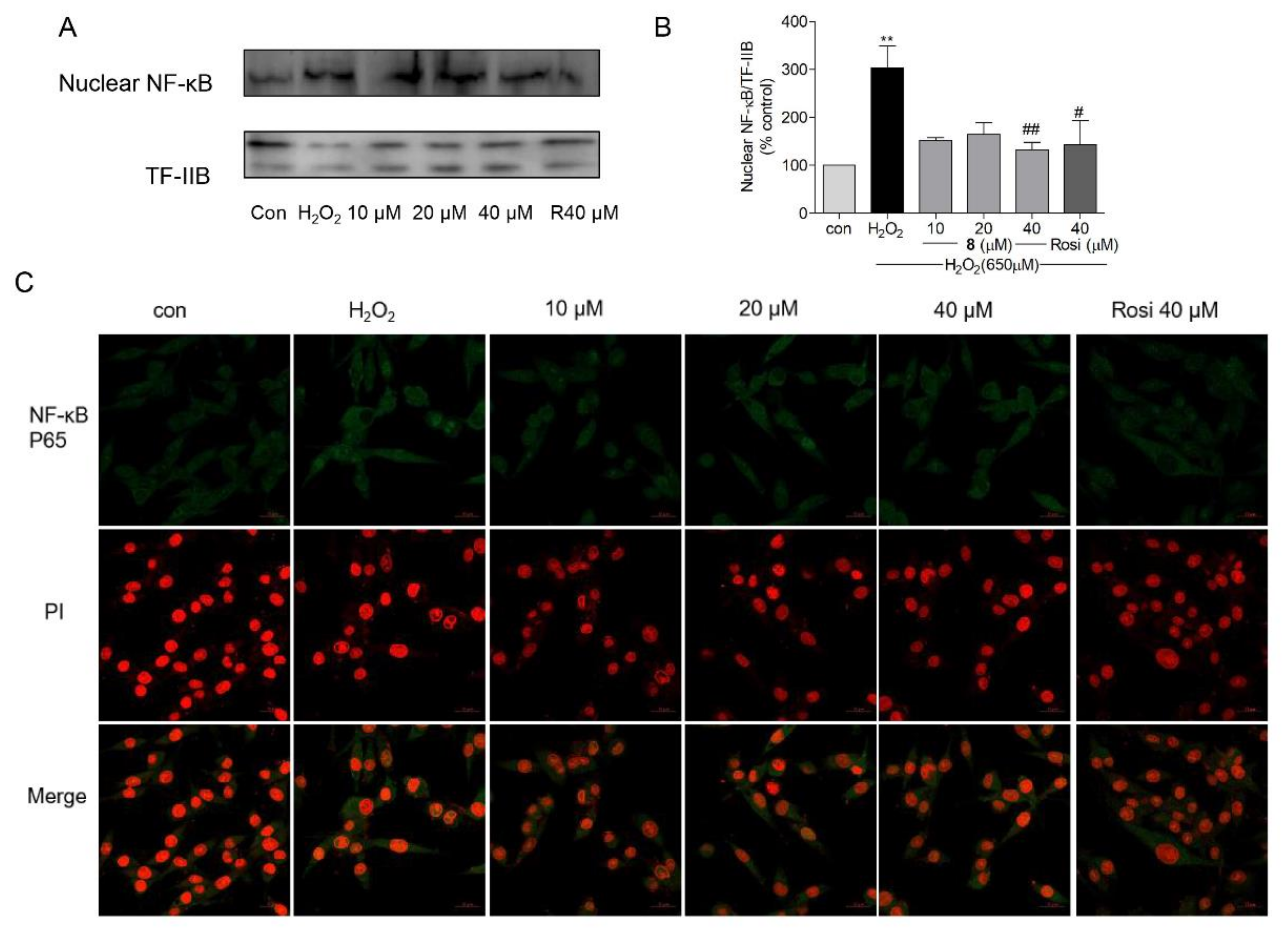
2.8. Effects of Compound 8 on NF-κB Activation and Nuclear Translocation
3. Materials and Methods
3.1. Materials
3.2. Isolation of 2,5-DKPs
3.3. Molecular Docking Study
3.4. Cell Culture and Cell Viability Assay
3.5. LDH Release
3.6. Luciferase Assay
3.7. Hoechst 33342 Staining
3.8. MMP Assay
3.9. ROS Generation
3.10. SOD and CAT Activities
3.11. Western Blotting
3.12. Immunofluorescence Assay
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Akizu, N.; Cantagrel, V.; Schroth, J.; Cai, N.; Vaux, K.; McCloskey, D.; Naviaux, R.K.; van Vleet, J.; Fenstermaker, A.G.; Silhavy, J.L.; et al. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell 2013, 154, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 2014, 42 (Suppl. 3), S125–S152. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.D. The Role of PPARs in Disease. Cells 2020, 9, 2367. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The novel role of PPAR alpha in the brain: Promising target in therapy of Alzheimer’s disease and other neurodegenerative disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.; Su, L.Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.X.; Zhang, D.F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 2020, 16, 52–69. [Google Scholar] [CrossRef]
- Hall, M.G.; Quignodon, L.; Desvergne, B. Peroxisome Proliferator-Activated Receptor beta/delta in the Brain: Facts and Hypothesis. PPAR Res. 2008, 2008, 780452. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Yan, K.; Wang, Y.; Wang, Y.; Chen, H.; Xu, J.; Lu, Y.; Wang, X.; Liang, J.; Zhang, X. Activation of PPAR-beta/delta Attenuates Brain Injury by Suppressing Inflammation and Apoptosis in a Collagenase-Induced Intracerebral Hemorrhage Mouse Model. Neurochem. Res. 2020, 45, 837–850. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, N.S.; Kumar, N.; Duseja, A. Peroxisome Proliferator-Activated Receptors and Their Agonists in Nonalcoholic Fatty Liver Disease. J. Clin. Exp. Hepatol. 2019, 9, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla, R.A.; Utreras, E.; Cabezas-Opazo, F.A. Role of PPAR gamma in the Differentiation and Function of Neurons. PPAR Res. 2014, 2014, 768594. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Wu, J.S.; Tsai, H.D.; Huang, C.Y.; Chen, J.J.; Sun, G.Y.; Lin, T.N. Peroxisome proliferator-activated receptor gamma (PPAR-gamma) and neurodegenerative disorders. Mol. Neurobiol. 2012, 46, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Villapol, S. Roles of Peroxisome Proliferator-Activated Receptor Gamma on Brain and Peripheral Inflammation. Cell Mol. Neurobiol. 2018, 38, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef]
- Seok, H.; Lee, M.; Shin, E.; Yun, M.R.; Lee, Y.H.; Moon, J.H.; Kim, E.; Lee, P.H.; Lee, B.W.; Kang, E.S.; et al. Low-dose pioglitazone can ameliorate learning and memory impairment in a mouse model of dementia by increasing LRP1 expression in the hippocampus. Sci. Rep. 2019, 9, 4414. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.C.; Nicol, C.J.; Cheng, Y.C.; Lin, K.H.; Yen, C.H.; Lin, C.H. Rosiglitazone activation of PPARgamma-dependent pathways is neuroprotective in human neural stem cells against amyloid-beta-induced mitochondrial dysfunction and oxidative stress. Neurobiol. Aging 2016, 40, 181–190. [Google Scholar] [CrossRef]
- Yi, J.H.; Park, S.W.; Brooks, N.; Lang, B.T.; Vemuganti, R. PPARgamma agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Res. 2008, 1244, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Cho, J.-H.; Lee, S.; Lee, W.; Chang, S.-C.; Chung, H.Y.; Moon, H.R.; Lee, J. Neuroprotective effects of MHY908, a PPAR α/γ dual agonist, in a MPTP-induced Parkinson’s disease model. Brain Res. 2019, 1704, 47–58. [Google Scholar] [CrossRef]
- Qin, Z.H.; Tao, L.Y.; Chen, X. Dual roles of NF-κB in cell survival and implications of NF-κB inhibitors in neuroprotective therapy. Acta Pharm. Sin. 2007, 28, 1859–1872. [Google Scholar] [CrossRef] [Green Version]
- Lanzillotta, A.; Porrini, V.; Bellucci, A.; Benarese, M.; Branca, C.; Parrella, E.; Spano, P.F.; Pizzi, M. NF-κB in innate neuroprotection and age-related neurodegenerative diseases. Front. Neurol. 2015, 6, 98. [Google Scholar] [CrossRef] [Green Version]
- Bright, J.J.; Kanakasabai, S.; Chearwae, W.; Chakraborty, S. PPAR regulation of inflammatory signaling in CNS diseases. PPAR Res. 2008, 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, M.M.F.; Bassani, T.B.; Cóppola-Segovia, V.; Moura, E.L.R.; Zanata, S.M.; Andreatini, R.; Vital, M.A.B.F. PPAR-γ agonist pioglitazone reduces microglial proliferation and NF-κB activation in the substantia nigra in the 6-hydroxydopamine model of Parkinson’s disease. Pharmacol. Rep. 2019, 71, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, A.D.; da Costa, N.C. 2,5-diketopiperazines in food and beverages: Taste and bioactivity. Crit. Rev. Food Sci. Nutr. 2017, 57, 718–742. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.M.; Yi, X.X.; Zhou, Y.; Su, X.; Peng, Y.; Gao, C.H. An update on 2,5-diketopiperazines from marine organisms. Mar. Drugs 2014, 12, 6213–6235. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, P.; Ma, H.; Zhu, W. Developments around the bioactive diketopiperazines: A patent review. Expert Opin. Ther. Pat. 2013, 23, 1415–1433. [Google Scholar] [CrossRef] [PubMed]
- McCleland, K.; Milne, P.; Lucieto, F.; Frost, C.; Brauns, S.; van de Venter, M.; Plessis, J.D.; Dyason, K. An investigation into the biological activity of the selected histidine-containing diketopiperazines cyclo (His-Phe) and cyclo (His-Tyr). J. Pharm. Pharmacol. 2004, 56, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Costante, R.; Fiorito, S.; Genovese, S.; Stefanucci, A.; Mathieu, V.; Kiss, R.; Epifano, F. Synthesis and anti-cancer activity of naturally occurring 2, 5-diketopiperazines. Fitoterapia 2014, 98, 91–97. [Google Scholar] [CrossRef]
- Huang, D.Y.; Nong, X.H.; Zhang, Y.Q.; Xu, W.; Sun, L.Y.; Zhang, T.; Chen, G.Y.; Han, C.R. Two new 2,5-diketopiperazine derivatives from mangrove-derived endophytic fungus Nigrospora camelliae-sinensis S30. Nat. Prod. Res. 2021, 35. Online ahead of print. [Google Scholar]
- Cornacchia, C.; Cacciatore, I.; Baldassarre, L.; Mollica, A.; Feliciani, F.; Pinnen, F. 2, 5-Diketopiperazines as neuroprotective agents. Mini Rev. Med. Chem. 2012, 12, 2–12. [Google Scholar] [CrossRef]
- Kang, H.; Ku, S.-K.; Choi, H.; Bae, J.-S. Three diketopiperazines from marine-derived bacteria inhibit LPS-induced endothelial inflammatory responses. Bioorg. Med. Chem. Lett. 2016, 26, 1873–1876. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.-x.; Huang, J.-f.; Li, X.-m.; Kang, Q.-j.; Pan, Y.-t. Three new 2, 5-diketopiperazines from the fish intestinal Streptomyces sp. MNU FJ-36. Nat. Prod. Res. 2016, 30, 1771–1775. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, X.; Xu, T.; Yang, X.; Liu, Y. Diketopiperazines from marine organisms. Chem. Biodivers. 2010, 7, 2809–2829. [Google Scholar] [CrossRef]
- Martins, M.B.; Carvalho, I. Diketopiperazines: Biological activity and synthesis. Tetrahedron 2007, 63, 9923–9932. [Google Scholar] [CrossRef]
- Gos, F.M.; Savi, D.C.; Shaaban, K.A.; Thorson, J.S.; Aluizio, R.; Possiede, Y.M.; Rohr, J.; Glienke, C. Antibacterial activity of endophytic actinomycetes isolated from the medicinal plant Vochysia divergens (Pantanal, Brazil). Front. Microbiol. 2017, 8, 1642. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, R.; Borriello, G.; Piccirillo, G.; Lahoz, E.; Sorrentino, R.; Cermola, M.; Censi, S.B.; Grauso, L.; Mangoni, A.; Vinale, F. Antibiotic Activity of a Paraphaeosphaeria sporulosa-Produced Diketopiperazine against Salmonella enterica. J. Fungi 2020, 6, 83. [Google Scholar] [CrossRef]
- Ström, K.; Sjögren, J.; Broberg, A.; Schnürer, J. Lactobacillus plantarum MiLAB 393 produces the antifungal cyclic dipeptides cyclo (L-Phe-L-Pro) and cyclo (L-Phe-trans-4-OH-L-Pro) and 3-phenyllactic acid. Appl. Environ. Microbiol. 2002, 68, 4322–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.-X.; Xie, C.-L.; Zhou, M.; Xia, M.-L.; Zhou, T.-T.; Chen, H.-F.; Yang, X.-W.; Yang, Q. Chemical constituents from the deep sea-derived Streptomyces xiamenensis MCCC 1A01570 and their effects on RXRα transcriptional regulation. Nat. Prod. Res. 2020, 34, 1461–1464. [Google Scholar] [CrossRef]
- Vázquez-Rivera, D.; González, O.; Guzmán-Rodríguez, J.; Díaz-Pérez, A.L.; Ochoa-Zarzosa, A.; López-Bucio, J.; Meza-Carmen, V.; Campos-García, J. Cytotoxicity of cyclodipeptides from Pseudomonas aeruginosa PAO1 leads to apoptosis in human cancer cell lines. BioMed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, S.-Q.; Tang, H.-Y.; Li, X.-J.; Zhang, L.; Xiao, J.; Gao, Y.-Q.; Zhang, A.-L.; Gao, J.-M. Potential allelopathic indole diketopiperazines produced by the plant endophytic Aspergillus fumigatus using the one strain–many compounds method. J. Agric. Food Chem. 2013, 61, 11447–11452. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Castro, R.; Díaz-Pérez, C.; Martínez-Trujillo, M.; Rosa, E.; Campos-García, J.; López-Bucio, J. Transkingdom signaling based on bacterial cyclodipeptides with auxin activity in plants. Proc. Natl. Acad. Sci. USA 2011, 108, 7253–7258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furtado, N.A.; Pupo, M.T.; Carvalho, I.; Campo, V.L.; Duarte, M.C.T.; Bastos, J.K. Diketopiperazines produced by an Aspergillus fumigatus Brazilian strain. J. Braz. Chem. Soc. 2005, 16, 1448–1453. [Google Scholar] [CrossRef]
- Jayatilake, G.S.; Thornton, M.P.; Leonard, A.C.; Grimwade, J.E.; Baker, B.J. Metabolites from an Antarctic sponge-associated bacterium, Pseudomonas aeruginosa. J. Nat. Prod. 1996, 59, 293–296. [Google Scholar] [CrossRef]
- Tullberg, M.; Grøtli, M.; Luthman, K. Efficient synthesis of 2, 5-diketopiperazines using microwave assisted heating. Tetrahedron 2006, 62, 7484–7491. [Google Scholar] [CrossRef]
- Stark, T.; Hofmann, T. Structures, sensory activity, and dose/response functions of 2, 5-diketopiperazines in roasted cocoa nibs (Theobroma cacao). J. Agric. Food Chem. 2005, 53, 7222–7231. [Google Scholar] [CrossRef]
- Wang, G.; Dai, S.; Chen, M.; Wu, H.; Xie, L.; Luo, X.; Li, X. Two diketopiperazine cyclo (pro-phe) isomers from marine bacteria Bacillus subtilis sp. 13-2. Chem. Nat. Compd. 2010, 46, 583–585. [Google Scholar] [CrossRef]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-γ. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Waku, T.; Shiraki, T.; Oyama, T.; Morikawa, K. Atomic structure of mutant PPARγ LBD complexed with 15d-PGJ2: Novel modulation mechanism of PPARγ/RXRα function by covalently bound ligands. FEBS Lett. 2009, 583, 320–324. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Li, D.-d.; Wang, Y.; Kim, E.; Zhao, N.; Jin, S.-W.; Bai, D.-H.; Sun, L.-D.; Jung, J.H. Cyclooxygenase-2 Inhibitor Parecoxib Was Disclosed as a PPAR-γ Agonist by In Silico and In Vitro Assay. Biomol. Ther. 2021. [Google Scholar] [CrossRef]
- Mandrekar-Colucci, S.; Sauerbeck, A.; Popovich, P.G.; McTigue, D.M. PPAR agonists as therapeutics for CNS trauma and neurological diseases. ASN Neuro. 2013, 5, AN20130030. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Luo, J.Z.Q.; Jackson, I. Tripeptide Amide L-pyroglutamyl-Histidyl-L-Prolineamide (L-PHPThyrotropin-Releasing Hormone, TRH) Promotes Insulin-Producing Cell Proliferation. Curr. Aging Sci. 2013, 6, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Knoblach, S.M.; Movsesyan, V.A.; Lea, P.M., IV; Cernak, I. Novel neuroprotective tripeptides and dipeptides. Ann. N.Y. Acad. Sci. 2005, 1053, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Fox, G.B.; Fan, L.; Araldi, G.L.; Qiao, L.; Wang, S.; Kozikowski, A.P. Novel TRH analog improves motor and cognitive recovery after traumatic brain injury in rodents. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1999, 277, R1196–R1204. [Google Scholar] [CrossRef] [PubMed]
- Minelli, A.; Bellezza, I.; Grottelli, S.; Galli, F. Focus on cyclo (His-Pro): History and perspectives as antioxidant peptide. Amino Acids 2008, 35, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Prakash, K.; Tang, Y.; Kozikowski, A.P.; Flippen-Anderson, J.L.; Knoblach, S.M.; Faden, A.I. Synthesis and biological activity of novel neuroprotective diketopiperazines. Bioorg. Med. Chem. 2002, 10, 3043–3048. [Google Scholar] [CrossRef]
- Faden, A.I.; Movsesyan, V.A.; Fang, X.; Wang, S. Identification of novel neuroprotective agents using pharmacophore modeling. Chem. Biodivers. 2005, 2, 1564–1570. [Google Scholar] [CrossRef]
- Luo, Y.; Yin, W.; Signore, A.P.; Zhang, F.; Hong, Z.; Wang, S.; Graham, S.H.; Chen, J. Neuroprotection against focal ischemic brain injury by the peroxisome proliferator-activated receptor-γ agonist rosiglitazone. J. Neurochem. 2006, 97, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.K.; Lee, I.H.; Kim, H.J.; Chang, G.S.; Chung, J.E.; No, K.T. The PreADME Approach: Web-Based Program for Rapid Prediction of Physico-Chemical, Drug Absorption and Drug-Like Properties. In EuroQSAR 2002 Designing Drugs and Crop Protectants: Processes, Problems and Solutions; Wiley-Blackwell Publishing: Hoboken, NJ, USA, 2003; pp. 418–420. [Google Scholar]
- Banks, W.A.; Kastin, A.J.; Akerstrom, V.; Jaspan, J.B. Radioactively iodinated cyclo (His-Pro) crosses the blood-brain barrier and reverses ethanol-induced narcosis. Am. J. Physiol. Endocrinol. Metab. 1993, 264, E723–E729. [Google Scholar] [CrossRef]
- Xiao, H.; Li, P.; Hu, X.; Shi, X.; Zhang, W.; Tang, B. Simultaneous fluorescence imaging of hydrogen peroxide in mitochondria and endoplasmic reticulum during apoptosis. Chem. Sci. 2016, 7, 6153–6159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Liu, J.; Fu, S.; Zhang, Y.; Li, Y.; He, D.; Ran, X.; Yan, X.; Du, J.; Meng, T. α-Cyperone attenuates H2O2-induced oxidative stress and apoptosis in SH-SY5Y cells via activation of Nrf2. Front. Pharmacol. 2020, 11, 281. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E.; Armour, S.; Harris, M.; Thompson, C. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003, 10, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, S.A.; Masud, A.; Kuida, K.; Porter, G.A.; Booth, C.J.; Mehal, W.Z.; Inayat, I.; Flavell, R.A. Caspases 3 and 7: Key mediators of mitochondrial events of apoptosis. Science 2006, 311, 847–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, H.; Zhou, Z.; Qin, J.; Liu, W.; Wang, B.; Gu, Y. Erastin disrupts mitochondrial permeability transition pore (mPTP) and induces apoptotic death of colorectal cancer cells. PLoS ONE 2016, 11, e0154605. [Google Scholar] [CrossRef] [Green Version]
- Baracca, A.; Sgarbi, G.; Solaini, G.; Lenaz, G. Rhodamine 123 as a probe of mitochondrial membrane potential: Evaluation of proton flux through F0 during ATP synthesis. BBA Bioenergetics 2003, 1606, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural. Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [PubMed]
- Takahashi, A.; Masuda, A.; Sun, M.; Centonze, V.E.; Herman, B. Oxidative stress-induced apoptosis is associated with alterations in mitochondrial caspase activity and Bcl-2-dependent alterations in mitochondrial pH (pHm). Brain Res. Bull. 2004, 62, 497–504. [Google Scholar] [CrossRef]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.; Gibson, S. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Hadwan, M.H. Simple spectrophotometric assay for measuring catalase activity in biological tissues. BMC Biochem. 2018, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vendemiale, G.; Grattagliano, I.; Altomare, E. An update on the role of free radicals and antioxidant defense in human disease. Int. J. Clin. Lab. Res. 1999, 29, 49–55. [Google Scholar] [CrossRef]
- Ighodaro, O.; Akinloye, O. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Brentnall, M.; Rodriguez-Menocal, L.; de Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- D’Amelio, M.; Sheng, M.; Cecconi, F. Caspase-3 in the central nervous system: Beyond apoptosis. Trends Neurosci. 2012, 35, 700–709. [Google Scholar] [CrossRef]
- Wang, X.; Ohnishi, K.; Takahashi, A.; Ohnishi, T. Poly (ADP-ribosyl) ation is required for p53-dependent signal transduction induced by radiation. Oncogene 1998, 17, 2819–2825. [Google Scholar] [CrossRef] [Green Version]
- Srinivasula, S.M.; Ahmad, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol. Cell 1998, 1, 949–957. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. BBA Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.-P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-κB is increased in dopaminergic neurons of patients with Parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-H.; Deng, Y.-Y.; Li, F.; Shi, J.-S.; Gong, Q.-H. Neuroprotective effects of sodium hydrosulfide against β-amyloid-induced neurotoxicity. Int. J. Mol. Med. 2016, 38, 1152–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, K.N.; Amstad, P.; Cerutti, P.; Baeuerle, P.A. The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-κB. Chem. Biol. 1995, 2, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Berghe, W.V.; Vermeulen, L.; Delerive, P.; de Bosscher, K.; Staels, B.; Haegeman, G. A paradigm for gene regulation: Inflammation, NF-κB and PPAR. Peroxisomal Disord. Regul. Genes 2003, 544, 181–196. [Google Scholar]
- Cai, W.; Yang, T.; Liu, H.; Han, L.; Zhang, K.; Hu, X.; Zhang, X.; Yin, K.-J.; Gao, Y.; Bennett, M.V. Peroxisome proliferator-activated receptor γ (PPARγ): A master gatekeeper in CNS injury and repair. Prog. Neurobiol. 2018, 163, 27–58. [Google Scholar] [CrossRef]
- Satyanarayanan, S.K.; Shih, Y.-H.; Chien, Y.-C.; Huang, S.-Y.; Gałecki, P.; Kasper, S.; Chang, J.P.-C.; Su, K.-P. Anti-oxidative effects of melatonin receptor agonist and omega-3 polyunsaturated fatty acids in neuronal SH-SY5Y cells: Deciphering synergic effects on anti-depressant mechanisms. Mol. Neurobiol. 2018, 55, 7271–7284. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Vina, O.A.A. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Biovia, D.S. Discovery Studio Modeling Environment. Release 4.5; Dassault Systemes: San Diego, CA, USA, 2015. [Google Scholar]
- Eom, S.H.; Liu, S.; Su, M.; Noh, T.H.; Hong, J.; Kim, N.D.; Chung, H.Y.; Yang, M.H.; Jung, J.H. Synthesis of phthalimide derivatives as potential PPAR-γ ligands. Mar. Drugs 2016, 14, 112. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.-d.; Wang, Y.; Kim, E.L.; Hong, J.; Jung, J.H. Neuroprotective Effect of Cyclo-(L-Pro-L-Phe) Isolated from the Jellyfish-Derived Fungus Aspergillus flavus. Mar. Drugs 2021, 19, 417. https://doi.org/10.3390/md19080417
Li D-d, Wang Y, Kim EL, Hong J, Jung JH. Neuroprotective Effect of Cyclo-(L-Pro-L-Phe) Isolated from the Jellyfish-Derived Fungus Aspergillus flavus. Marine Drugs. 2021; 19(8):417. https://doi.org/10.3390/md19080417
Chicago/Turabian StyleLi, Dan-dan, Ying Wang, Eun La Kim, Jongki Hong, and Jee H. Jung. 2021. "Neuroprotective Effect of Cyclo-(L-Pro-L-Phe) Isolated from the Jellyfish-Derived Fungus Aspergillus flavus" Marine Drugs 19, no. 8: 417. https://doi.org/10.3390/md19080417
APA StyleLi, D.-d., Wang, Y., Kim, E. L., Hong, J., & Jung, J. H. (2021). Neuroprotective Effect of Cyclo-(L-Pro-L-Phe) Isolated from the Jellyfish-Derived Fungus Aspergillus flavus. Marine Drugs, 19(8), 417. https://doi.org/10.3390/md19080417