Unexpected Enhancement of HDACs Inhibition by MeS Substitution at C-2 Position of Fluoro Largazole

Abstract
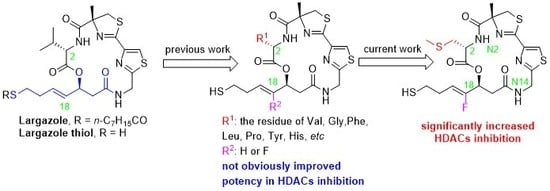
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biology
2.3. Molecular Modeling Study
3. Materials and Methods
3.1. Chemistry
3.1.1. 2-(Trimethylsilyl)ethyl (S,Z)-4-fluoro-3-hydroxy-7-(tritylthio)hept-4-enoate (9)
3.1.2. 2-(Trimethylsilyl)ethyl (S,Z)-3-((N-(((9H-fluoren-9-yl)methoxy)carbonyl)-S-methyl-l-cysteinyl)oxy)-4-fluoro-7-(tritylthio)hept-4-enoate (10c)
3.1.3. 2-(Trimethylsilyl)ethyl (S,Z)-3-((N-((R)-2′-(((tert-butoxycarbonyl)amino)methyl)-4-methyl-4,5-dihydro-[2,4′-bithiazole]-4-carbonyl)-S-methyl-L-cysteinyl)oxy)-4-fluoro-7-(tritylthio)hept-4-enoate (12c)
3.1.4. 2-(Trimethylsilyl)ethyl (S,Z)-3-((((R)-2′-(((tert-butoxycarbonyl)amino)methyl)-4-methyl-4,5-dihydro-[2,4′-bithiazole]-4-carbonyl)glycyl)oxy)-4-fluoro-7-(tritylthio)hept-4-enoate (12d)
3.1.5. (12Z,22Z,24R,5R,8S)-8-((Z)-1-fluoro-4-(tritylthio)but-1-en-1-yl)-24-methyl-5-((methylthio)methyl)-24,25-dihydro-7-oxa-4,11-diaza-1(4,2),2(2,4)-dithiazolacyclododecaphane-3,6,10-trione (14c)
3.1.6. (12Z,22Z,24R,8S)-8-((Z)-1-fluoro-4-(tritylthio)but-1-en-1-yl)-24-methyl-24,25-dihydro-7-oxa-4,11-diaza-1(4,2),2(2,4)-dithiazolacyclododecaphane-3,6,10-trione (14d)
3.1.7. (12Z,22Z,24R,8S)-8-((Z)-1-fluoro-4-mercaptobut-1-en-1-yl)-24-methyl-5-((methylthio)methyl)-24,25-dihydro-7-oxa-4,11-diaza-1(4,2),2(2,4)-dithiazolacyclododecaphane-3,6,10-trione (15c)
3.1.8. S-((Z)-4-fluoro-4-((12Z,22Z,24R,5R,8S)-24-methyl-5-((methylthio)methyl)-3,6,10-trioxo-24,25-dihydro-7-oxa-4,11-diaza-1(4,2),2(2,4)-dithiazolacyclododecaphane-8-yl)but-3-en-1-yl) octanethioate (16c)
3.1.9. S-((Z)-4-fluoro-4-((12Z,22Z,24R,8S)-24-methyl-3,6,10-trioxo-24,25-dihydro-7-oxa-4,11-diaza-1(4,2),2(2,4)-dithiazolacyclododecaphane-8-yl)but-3-en-1-yl) octanethioate (16d)
3.2. Biology
3.2.1. Recombinant Human HDAC1, HDAC2, HDAC3, HDAC6 and HDAC8 Enzymatic Assays
3.2.2. Cellular Assay
3.3. Molecular Modeling Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenetics 2012, 4, 5. [Google Scholar] [CrossRef]
- De Ruijter, A.J.M.; Van Gennip, A.H.; Caron, H.N.; Kemp, S.; Van Kuilenburg, A.B.P. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discovery 2002, 1, 287–299. [Google Scholar] [CrossRef]
- Zagni, C.; Floresta, G.; Monciino, G.; Rescifina, A. The search for potent, small-molecule HDACIs in cancer treatment: A decade after vorinostat. Med. Res. Rev. 2017, 37, 1373–1428. [Google Scholar]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Qin, H.-T.; Li, H.-Q.; Liu, F. Selective histone deacetylase small molecule inhibitors: Recent progress and perspectives. Expert Opin. Therap. Pat. 2017, 27, 621–636. [Google Scholar] [CrossRef]
- Wang, X.X.; Wan, R.Z.; Liu, Z.P. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem. 2018, 143, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Taori, K.; Paul, V.J.; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008, 130, 1806–1807. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Taori, K.; Kim, H.; Hong, J.; Hendrik Luesch, H. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130, 8455–8459. [Google Scholar] [CrossRef] [PubMed]
- Bowers, A.; West, N.; Taunton, J.; Schreiber, S.L.; Bradner, J.E.; Williams, R.M. Total synthesis and biological mode of action of largazole: A potent class I histone deacetylase inhibitorc. J. Am. Chem. Soc. 2008, 130, 11219–11222. [Google Scholar] [CrossRef]
- Maolanon, A.R.; Kristensen, H.M.E.; Leman, L.J.; Ghadiri, M.R.; Olsen, C.A. Natural and synthetic macrocyclic inhibitors of the histone deacetylase enzymes. ChemBioChem 2017, 18, 5–49. [Google Scholar] [CrossRef]
- Reddy, D.N.; Ballante, F.; Chuang, T.; Pirolli, A.; Marrocco, B.; Marshall, G.R. Design and synthesis of simplified largazole analogues as isoform-selective human lysine deacetylase inhibitors. J. Med. Chem. 2016, 59, 1613–1633. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Tu, Z.; Liao, C.; Wang, Z.; Li, S.; Yao, H.; Li, Z.; Jiang, S. Discovery of novel class I histone deacetylase inhibitors with promising in vitro and in vivo antitumor activities. J. Med. Chem. 2015, 58, 7672–7680. [Google Scholar] [CrossRef]
- Poli, G.; Fabio, R.D.; Ferrante, L.; Summa, V.; Maurizio Botta, M. Largazole Analogues as Histone Deacetylase Inhibitors and Anticancer Agents: An Overview of Structure–Activity Relationships. ChemMedChem 2017, 12, 1917–1926. [Google Scholar] [CrossRef]
- Kim, B.; Ratnayake, R.; Lee, H.; Shi, G.; Zeller, S.L.; Li, C.; Luesch, H.; Hong, J. Synthesis and biological evaluation of largazole zinc-binding group analogs. Bioorg. Med. Chem. 2017, 25, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Liu, Y.X.; Byeon, S.R.; Kim, H.; Luesch, H.; Hong, J. Synthesis and activity of largazole analogues with linker and macrocycle modification. Org. Lett. 2008, 10, 4021–4024. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.X.; Salvador, L.A.; Byeon, S.; Ying, Y.; Kwan, J.C.; Law, B.K.; Hong, J.; Luesch, H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yin, B.L.; Hu, Z.; Liao, C.Z.; Liu, J.L.; Li, S.; Li, Z.; Nicklaus, M.C.; Zhou, G.B.; Jiang, S. Total synthesis and biological evaluation of largazole and derivatives with promising selectivity for cancers cells. Org. Lett. 2010, 12, 1368–1371. [Google Scholar] [CrossRef] [PubMed]
- Benelkebir, H.; Marie, S.; Hayden, A.L.; Lyle, J.; Loadman, P.M.; Crabb, S.J.; Packham, G.; Ganesan, A. Total synthesis of largazole and analogues: HDAC inhibition, antiproliferative activity and metabolic stability. Bioorg. Med. Chem. 2011, 19, 3650–3658. [Google Scholar] [CrossRef]
- Bowers, A.A.; West, N.; Newkirk, T.L.; Troutman-Youngman, A.E.; Schreiber, S.L.; Wiest, O.; Bradner, J.E.; Williams, R.M. Synthesis and histone deacetylase inhibitory activity of largazole analogs: Alteration of the zinc-binding domain and macrocyclic scaffold. Org. Lett. 2009, 11, 1301–1304. [Google Scholar] [CrossRef]
- Kim, B.; Park, H.; Salvador, L.A.; Serrano, P.E.; Kwan, J.C.; Zeller, S.L.; Chen, Q.Y.; Ryu, S.; Liu, Y.; Byeon, S.; et al. Evaluation of class I HDAC isoform selectivity of largazole analogues. Bioorg. Med. Chem. Lett. 2014, 24, 3728–3731. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, B.; Shan, G.; Wu, Y.; Yang, F.-L.; Lei, X. Synthesis of the molecular hybrid inspired by Largazole and Psammaplin A. Tetrahedron 2018, 74, 549–555. [Google Scholar] [CrossRef]
- Zhang, B.; Shan, G.; Zheng, Y.; Yu, X.; Ruan, Z.-W.; Li, Y.; Lei, X. Synthesis and preliminary biological evaluation of two fluoroolefin analogs of Largazole inspired by the structural similarity of the side chain unit in Psammaplin A. Mar. Drugs 2019, 17, 333. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.; Gao, D.; Yu, X.; Wang, J.; Lei, X. A fluorine scan on the Zn2+-binding thiolate side chain of HDAC inhibitor largazole: Synthesis, biological evaluation, and molecular modeling. Eur. J. Med. Chem. 2019, 182. [Google Scholar] [CrossRef]
- Cole, K.E.; Dowling, D.P.; Boone, M.A.; Phillips, A.J.; Christianson, D.W. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 a (µM) | ||||
|---|---|---|---|---|
| A549 | HCT116 | MDA-MB-231 | SK-OV-3 | |
| largazole | 0.52 ± 0.17 | 0.11 ± 0.01 | 4.75 ± 1.95 | 0.25 ± 0.17 |
| 16c | 2.92 ± 0.53 | 1.41 ± 0.54 | >10 | 3.13 ± 0.50. |
| 16d | >10 | 6.62 ± 1.86 | >10 | >10 |
| IC50 a (µM) | ||||
|---|---|---|---|---|
| Hela | Eca-109 | Bel 7402 | U937 | |
| largazole | 0.17 ± 0.01 | 0.10 ± 0.00 | 0.17 ± 0.02 | 0.02 ± 0.00 |
| 16c | 0.10 ± 0.01 | 0.09 ± 0.02 | 0.25 ± 0.07 | 0.08 ± 0.02 |

| IC50 (nM) a | Selectivity (HDAC1/6) | |||||
|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC6 | ||
| 15ab | 4.39 ± 0.17 | 21.0 ± 0.3 | 39.2 ± 2.3 | 8.38 ± 2.46 | 300 ± 5 | 68 |
| 15bb | 4.24 ± 0.38 | 16.3 ± 0.4 | 37.1 ± 0.8 | 17.5 ± 4.2 | 338 ± 17 | 80 |
| 15c | 0.27 ± 0.02 | 1.33 ± 0.02 | 2.33 ± 0.15 | 0.44 ± 0.16 | 23.9 ± 0.2 | 89 |
| largazole thiol | 2.01 ± 0.01 | 9.49 ± 0.19 | 14.4 ± 0.2 | 3.75 ± 0.20 | 121 ± 5 | 60 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Ruan, Z.-W.; Luo, D.; Zhu, Y.; Ding, T.; Sui, Q.; Lei, X. Unexpected Enhancement of HDACs Inhibition by MeS Substitution at C-2 Position of Fluoro Largazole. Mar. Drugs 2020, 18, 344. https://doi.org/10.3390/md18070344
Zhang B, Ruan Z-W, Luo D, Zhu Y, Ding T, Sui Q, Lei X. Unexpected Enhancement of HDACs Inhibition by MeS Substitution at C-2 Position of Fluoro Largazole. Marine Drugs. 2020; 18(7):344. https://doi.org/10.3390/md18070344
Chicago/Turabian StyleZhang, Bingbing, Zhu-Wei Ruan, Dongdong Luo, Yueyue Zhu, Tingbo Ding, Qiang Sui, and Xinsheng Lei. 2020. "Unexpected Enhancement of HDACs Inhibition by MeS Substitution at C-2 Position of Fluoro Largazole" Marine Drugs 18, no. 7: 344. https://doi.org/10.3390/md18070344
APA StyleZhang, B., Ruan, Z.-W., Luo, D., Zhu, Y., Ding, T., Sui, Q., & Lei, X. (2020). Unexpected Enhancement of HDACs Inhibition by MeS Substitution at C-2 Position of Fluoro Largazole. Marine Drugs, 18(7), 344. https://doi.org/10.3390/md18070344