Abstract
The red seaweed Laurencia viridis is a rich source of oxygenated secondary metabolites that were derived from squalene. We report here the structures of three novel compounds, (+)-longilene peroxide (1), longilene (2), and (+)-prelongilene (3) that were isolated from this alga, in addition to other substances, 4 and 5, resulting from their acid-mediated degradation. The effect of compounds 1 and 3 against Ser-Thr protein phosphatase type 2A (PP2A) was evaluated, showing that (+)-longilene peroxide (1) inhibited PP2A (IC50 11.3 μM). In order to explain the interaction between PP2A and compounds 1 and 3, molecular docking simulations onto the PP2A enzyme-binding region were used.
1. Introduction
The phosphorylation and dephosphorylation processes that were controlled by protein kinases and phosphatases are essential for the appropriate functioning of all living cells [1]. However, while kinases were established as drug targets several years ago, their counterpart phosphatases have only been considered recently. In particular, Ser-Thr protein phosphatases type 1 (PP1) and 2A (PP2A) are involved in the control of the cell cycle, apoptosis, signal transduction, metabolism, protein synthesis, as well as tumour promoters and suppressors [2,3]. Therefore, inhibitors of these enzymes have been investigated as a potential novel therapeutic approach in diseases, such as cancer, osteoporosis, diabetes, and Alzheimer’s. disease [4,5]. A number of marine natural products have been identified as inhibitors of Ser-Thr protein phosphatases. The first example was okadaic acid—which was isolated in 1981 from the marine sponge Halicondria okadai—a natural toxin that is known to cause diarrhetic shellfish poisoning [6]. Other protein phosphatase inhibitors include cyclic peptides, such as microcystin-LR and motuporin, polyketides, such as spirastrellolide A, tautomycin, and calliculin A and C, and terpenes, such as thyrsiferyl-23-acetate and cantharidin [4,7,8].
Squalene-derived polyethers belonging to the thyrsiferol and venustatriol series have shown different biological activities, such as cytotoxicity, integrin antagonist activity, and protein phosphate type 2A inhibition [9,10,11]. As part of our continuing interest in new bioactive molecules from marine sources, we report herein the isolation and the structural elucidation of three new marine polyether natural products, (+)-longilene peroxide (1), longilene (2), and (+)-prelongilene (3), and another two compounds (4–5), which were derived from the rapid degradation of 2 and 3 (Figure 1). Additionally, the PP2A inhibitory effect of the less labile compounds (+)-longilene peroxide (1) and (+)-prelongilene (3) was tested, and their likely binding conformations proposed on the basis of molecular docking simulations.
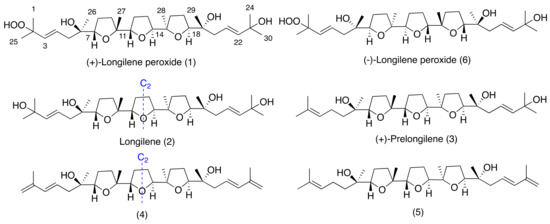
Figure 1.
Structures of longilene metabolites isolated from Laurencia viridis.
2. Results and Discussion
The molecular formula of (+)-longilene peroxide (1) C30H52O8Na, was established by HRESIMS analysis, where its sodiated molecular ion was observed at m/z 563.3571, calculated 563.3559, [M + Na]+. Analysis of the two-dimensional (2D) NMR spectra (COSY, HSQC, HMBC, and ROESY) of 1 indicated the presence of eight methyl, eight methylene and eight methine groups (four of the latter on sp2 carbons), in addition to six oxygenated quaternary carbons. Analysis of such spectroscopic and spectrometric data (Table 1) suggested that the structure of 1 was identical to that of (−)-longilene peroxide (6), as reported by Itokawa et al. in 1991 [12], isolated from the methanol extract of the Eurycoma longifolia wood (Simaroubaceae) (Figure 1 and Figure 2).

Table 1.
NMR data for (+)-longilene peroxide (1) and longilene (2) (500 MHz, 125 MHz, CDCl3).

Figure 2.
(a) Selected NMR-derived correlations HMBC and COSY and (b) perspective view with significant ROESY relationships (dashed line and distances in Ångstrom) observed for (+)-longilene peroxide (1).
The stereostructure and the conformation of the terrestrial molecule was assigned on the basis of X-ray crystallographic analysis, although the absolute stereochemistry was not defined. Afterward, Morimoto et al. solved this problem by means of an elegant total synthesis approach [13]. The specific rotation value that was measured for the marine triterpene 1 was +47, as opposed to −23, reported for the metabolite isolated from Eurycoma longifolia, and −45 for the synthetic substance 6. According to these value, the marine polyether that was isolated from Laurencia viridis must be the enantiomeric form of the terrestrial (−)-longilene peroxide (6) as shown in Figure 1. Therefore, the absolute configuration of 1 should be 6R, 7S, 10R, 11S, 14S, 15R, 18S, and 19R.
The HRESIMS analysis of the very unstable metabolite longilene (2) provided the molecular formula C30H52O7 that was based on the m/z 524.3716 (calculated for 524.3713 [M]+), as observed. However, its spectroscopic data revealed only half of the expected signals in both the 1H and 13C NMR spectra, suggesting the presence of a Cs symmetry element in this metabolite. Further study of the spectroscopic and spectrometric data of 2 permitted a full NMR assignment. In addition, the equivalence of the observed 13C chemical shifts at C-2 and C-23 confirmed the presence of a hydroxyl group at C-2 in 2, rather than the hydroperoxide functional group in compound 1. The resemblance of the NMR data in 1 and 2 suggested that the relative configurations of the stereocentres in 2 match those that were previously described for (+)-longilene peroxide (1) in accordance with a C2 symmetry (Table 1 and Figure 3).
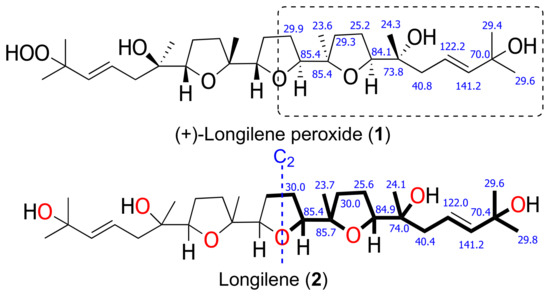
Figure 3.
Selected 13C NMR chemical shift comparison between similar fragment in (+)-longilene peroxide (1) and longilene (2) (bold line).
Compound 3, (+)-prelongilene, was obtained as an amorphous white solid. Its molecular formula was established by ESI-HRMS as C30H52O6 (m/z 531.3655; calculated for 531.3662, [M + Na]+). The structure of this metabolite was determined by comparison of its spectroscopic data with those of (+)-longilene peroxide (1) (Table 1 and Table 2). Thus, the main difference in their NMR spectra was in the C-1→C-5 fragment. A 1H-1H spin system was built for this fragment, starting from the proton signal H-3 (δH 5.08, t, J = 6.9 Hz), which was coupled with the methylene H2-4 (δH 1.97/2.04), and this sequentially connected with the diastereotopic protons H2-5 (δH 1.29/1.43). Long-range 1H-13C connectivities that were extracted from the HMBC experiment allowed us to connect this substructure within the molecule. Thus, the planar structure of 3 was established by correlations from the geminal methyls H3-1/H3-25 (δH 1.60/1.66) to C-2 (δC 131.1) and C-3 (δC 124.8), and those that were observed from the protons H2-5, H3-26 (δH 1.27) and H-7 (δH 3.73, dd, J = 6.9 and 7.6 Hz) to the quaternary oxygen-bearing carbon C-6 (δC 72.5). The relative stereochemistry we propose here derives from comparison of the spectroscopical data of 3 (chemical shift and ROESY correlations) with those of 1 and 2. Additionally it seems likely that this compound is a precursor of (+)-longilene peroxide (1), by action of peroxidase enzymes. This would follow a similar pathway to that observed in the biogenesis of datylomelanes in the Laurencia genus [14].
Table 2.
NMR data for (+)-prelongilene (3) (500 MHz, 125 MHz, CDCl3).
Together with the previously described metabolites, compounds 4 and 5 (Tables S4 and S5) were characterized as degradation by products of 2 and 3. Thus, the origin of 4 and 5 was explained by dehydration of the terminal alcohols present in 2 and 3, respectively. Briefly, when compound 2 was dissolved in CDCl3, we observed its total conversion into 4 after 48 h (see Supplementary Materials). Its NMR data confirmed the presence of conjugated double bonds in the C-1→C-4 and C-21→C-24 portions of 4. To confirm this hypothesis, the conjugated double bonds in the C-21→C-24 fragment were obtained in acid media by treatment of 3 with Dowex-50 in CDCl3, to obtain compound 5 (see Supplementary Materials).
Some marine polyethers derived from squalene show selective inhibition of protein phosphate type 2A. Taking this into account, in vitro inhibition assays for the stable compounds (1 and 3) were undertaken. These indicated that while (+)-longilene peroxide (1) showed inhibition on the Ser-Thr protein phosphatase type 2A (PP2A) (IC50 11.3 μM ±1.4), (+)-prelongilene (3) was inactive at a concentration of 100 μM.
In an attempt to understand the structural features that are underpinning the observed in vitro inhibitory activities, molecular docking simulations were performed to obtain the most favourable binding positions of the two ligands, (+)-longilene peroxide (1) and (+)-prelongilene (3), which are inside the Ser-Thr protein phosphatase type 2A (PP2A) binding-pocket enzyme. It has to be noted that PP2A is a metal-dependent enzyme that includes two Mn2+ ions within its active site. Metalloproteins are still challenging systems that can be tackled through molecular simulations, mainly because the atomic charges of those atoms that were included within the metal coordination sphere are difficult to parametrize. For that reason, electrostatic charges were established for those residues involved in a coordination sphere within 3 Å from the metal ion (Asp-57, His-59, Asp-85, Asn-117, His-167, His-241 and H2O-503), together with those of the two Mn2+ ions by ab initio calculations at the B3LYP/6-31G** theoretical level. Next, each inhibitor was docked into the active site of PP2A using the AutoDock software [15,16]. The structures that were resulting from the docking simulations were clustered on the basis of their binding energies and geometrical similarity. Nevertheless, all of the docking solutions where the ligand was located less than 4.5 Å from the Mn2+ ions were discarded. The criterion for this was that examination of different crystallographic structures showed that the distance between the ligand and the manganese ions was larger than 4.5 Å in all of them. Finally, in order to identify major contributions to binding, selected docking scenarios were evaluated using DrugScore [17]. As a control for the docking simulations, the structure of the complexes formed by okadaic acid—PP2A (pdb 2ie4) and calyculin—PP2A (pdb 1IT6) was predicted using the previous protocol, obtaining excellent results through comparing the corresponding crystallographic coordinates (RMSD 0.47 and 0.5 Å, respectively) (see Supplementary Materials) [18,19].
According to our simulation results (Figure 4), the binding modes of 1 and 3 to PP2A are very similar, as expected from their minor structural differences. However, the resulting (+)-longilene peroxide (1)—PP2A complex showed several favourable contacts that were not observed in the (+)-prelongilene (3)—PP2A complex. Among these, were the key hydrogen bonds that were formed between the hydroperoxide group with the Arg-214 and His-241 residues, as well as that between the hydroxyl group on C-23 and the Ile-123 residue. On the other hand, from the analysis of the structure of the (+)-prelongilene—PP2A complex, no important favourable contributions were identified, but small unfavourable contacts with Trp-200, Hys-118 and Leu-243 were observed. The overall orientation of the two ligands within the active site of PP2A was therefore very similar and the structural difference between these two molecules is the additional allylic hydroperoxide group at C-2 in 1. For these reasons, it seems clear that the two predicted hydrogen bonds in the (+)-longilene peroxide (1)—PP2A complex are the factor leading to their differences in bioactivity.
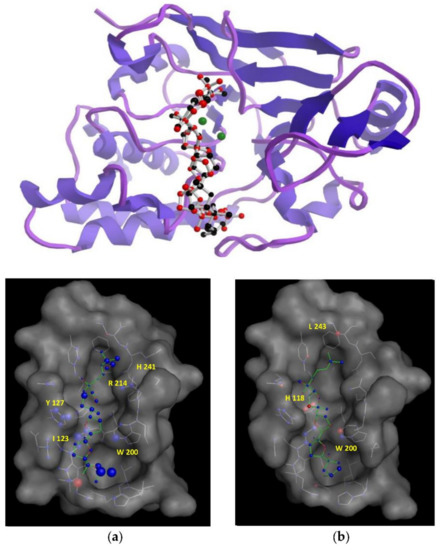
Figure 4.
(Top) Structures minimum energy obtained of the molecular docking of the PP2A with the (+)-longilene peroxide (1, red) and the (+)-prelongilene (3, black). (Bottom) Analysis in DrugScore of the structures minimum energy obtained of the molecular Docking of the PP2A with the (+)-longilene peroxide (1) (a), and the (+)-prelongilene (3) (b).
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were determined on a Perkin-Elmer 241 polarimeter (Waltham, MA, USA), using a sodium lamp operating at 589 nm. The IR spectrum was measured on a Bruker IFS55 spectrometer (Billerica, MA, USA), using a chloroform solution to place a film of the compounds on the NaCl disk. UV spectra were determined on a spectrophotometer V-560, Jasco Inc. (Easton, MD, USA) NMR spectra were obtained on Bruker AVANCE 500 MHz instruments at 300 K, and coupling constants are given in Hz. NMR experiments, COSY, HSQC, HMBC, and ROESY, were performed using standard pulse sequences. Phase-sensitive ROESY spectra were measured using a mixing time of 500 ms. 3JH,H values were measured from one-dimensional (1D) 1H-NMR. NMR data were processed using Topspin or MestReNova software (v.10., Santiago de Compostela, Spain). Mass spectra were recorded on a VG AutoSpec FISON spectrometer (Danvers, MA, USA). HPLC (High performance liquid chromatography) separations were carried out with a LKB 2248 system (LKB-Producter AB, Bromma, Sweden) that was equipped with a photodiode array detector. All of the solvents used were HPLC-grade. HPLC chromatography was monitored by TLC, performed on AL Si gel Merck 60 F254 (Kenilworth, NJ, USA). TLC plates were visualized by UV light (365 nm) and phosphomolybdic acid solution 10 wt % in methanol.
3.2. Biological Material
Specimens of Laurencia viridis were collected in April 2016 in the intertidal zone at Paraiso Floral, Tenerife, Canary Islands (28°07′12″ N, 16°46′45″ W). Dried material from the sterile plants, sporophytes, and gametophytes was filed at TFC Phyc 7180. (Herbario de la Universidad de La Laguna, Departamento de Biología Vegetal, Botánica, Tenerife, Spain).
3.3. Extraction and Isolation
The dried red algae was extracted with CHCl3:MeOH (1:1) at room temperature (rt), and a dark-green, viscous oil was obtained (78.0 g) after concentration under reduced pressure. The extract was first chromatographed using Sephadex LH20 (7 Ø × 50 cm) using DCM/MeOH (1:1) as the mobile phase. This procedure was repeated until all of the extract was processed. The enriched polyether fractions (52.1 g) were collected between 225 and 360 mL was subsequently processed on a silica gel 0.2–0.5 mm (Sigma-Aldrich, St. Louis, MO, USA) column (7 Ø × 50 cm) using a linear gradient of n-Hex/EtOAc (8:2–2:8), and the fraction collected between 350 and 500 mL was dried (1.49 g). Next, medium-pressure chromatography was done using Lobar LiChroprep-RP18 MeOH/H2O (9:1) at 5 mL/min flow. Fractions collected between 138 and 146 mL, and 147 and 165 mL were pooled together (5D and 5E 100 and 1220 mg, respectively). Final purification was performed on an HPLC with a μ-Porasil column, n-Hex/Acetone (8:2) and DCM/Acetone (8:2), to afford (+)-prelongilene (3) (2.1 mg) and the compound 5 (0.91 mg), (in the case of the fraction 5D); while for the fraction 5E, n-Hex/EtOAc (7:3) and DCM/Acetone (8:2) were used to afford the polyethers: (+)-longilene peroxide (1) (3.1 mg), longilene (2) (1.5 mg), and the compound 4 (0.83 mg) (see Supplementary Materials).
(+)-Longilene peroxide (1): Amorphous white solid, +47 (c 0.31, CHCl3); IR vmax (CHCl3) 3360, 2969, 1732, 1455, 1373 and 1077 cm−1; HR-ESI-MS m/z 563.3571 [M + Na]+ (calcd. 563.3559 for C30H52O8Na); NMR data 1H (500 MHz, CDCl3) and 13C (125 MHz, CDCl3) see Table 1.
Longilene (2): Amorphous white solid; +21 (c 0.15, CHCl3); IR vmax (CHCl3) 3365, 2971, 2929, 2872, 1455, 1373 and 1085 cm−1; HR-ESI-MS m/z 524.3716 [M]+ (calcd. 524.3713 for C30H52O7); NMR data 1H (500 MHz, CDCl3) and 13C (125 MHz, CDCl3) see Table 1.
(+)-Prelongilene (3): Amorphous white solid, +19 (c 0.21, CHCl3); IR vmax (CHCl3) 3363, 2971, 2931, 2872, 1453, 1372 and 1082 cm−1; HR-ESI-MS m/z 531.3655 [M + Na]+ (calcd. 531.3662 for C30H52O6Na); NMR data 1H (500 MHz, CDCl3) and 13C (125 MHz, CDCl3) see Table 2.
3.4. Transformation of (+)-Prelongilene (3) into Compound 5
Dowex-50 was added to a solution of 3 (1.1 mg, 2.2 μmol) in CDCl3 (0.4 mL), and the contents were mixed for 48 h at room temperature, while monitoring the reaction by NMR. Of the resulting solution was 0.91 mg (1.9 μmol) of pure compound 5 (see Supplementary Materials).
3.5. Protein Phosphatase 2A Inhibition Assay
The enzymatic assays were performed in 96-well plates. Each well contained 10 μL of MnCl2 (1 mM), 5 μL of BSA (5 mg/mL), 10 μL of buffer (50 mM Tris-HCl, 0.1 mM EDTA, 5 mM DTT, 0.025% Tween, pH 7.0), 15 μL of distilled water, and 5 μL of PP2A (Sigma-Aldrich®; 50 mM of Tris-HCl pH 7.0, 14 mM of 2-mercaptoethanol, 1 mM of benzamidine, 0.1 mM of PMSF, 1 mM of EDTA y 50% of glycerol). The blank was prepared in the same way as the sample, but with no enzyme (30 μL of distilled water); there were at least eight blanks per plate. The reaction mixture was incubated for 10 min at 37 °C, sample (10 μL) was added, except to the control and blank wells. This mixture was again incubated for 10 min at 37 °C and then the fluorescent substrate (PNPP) was added in 50 μL as an assay buffer at a final concentration of 1.5 mg/mL. Each determination was carried out in triplicate and each sample was tested in four different dilutions (100, 10, 1, and 0.1 µM). The reaction was incubated for 60 min at 37 °C and fluorescence was measured using a microplate reader fluorometer set at 492 nm (exc) and 514 nm (em).
3.6. Docking Studies
Docking simulations were performed with Autodock software (v.3.0.5; Molecular Graphics Laboratory, The Scripps Research Institute, La Jolla, CA, USA) using the GUI AutoDockTools (v.1.5.2). The receptor file was prepared using the coordinates of the crystallographic structure OA-PP2A (pdb 2ie4) that was obtained from the Protein Data Bank in which the ligand was removed. The atomic charges of the Mn2+ ions as well as that of those residues in their coordination spheres were calculated by ab initio calculations at the B3LYP/6-31G** theoretical level.
4. Conclusions
Three new squalene derived metabolites have been isolated from the red algae Laurencia. viridis and their structures were determined on the basis of a spectroscopic analysis. Subsequently, the protein phosphatase type 2A inhibitory activity of the less labile compounds (1 and 3) was tested, resulting in 1 as a moderately active compound (IC50 11.3 μM), while the very similar compound 3 was completely inactive up to a concentration of 100 μM. Due to the minor structural changes between them, it is clear that the allylic hydroperoxide functional group of 1 is responsible for the observed differences in activity.
Supplementary Materials
The following are available online at http://www.mdpi.com/1660-3397/16/4/131/s1, Table S1: NMR data of (+)-longilene peroxide (1) in CDCl3 at 300 K, 500 MHz; Table S2: NMR data of longilene (2) in CDCl3 at 300 K, 500 MHz; Table S3: NMR data of (+)-prelongilene (3) in CDCl3 at 300 K, 500 MHz; Table S4: NMR data of compound 5 in CDCl3 at 300 K, 500 MHz; Table S5: NMR data of compound 4 in CDCl3 at 300 K, 500 MHz; Figure S1: 1H-NMR spectrum of (+)-longilene peroxide (1) in CDCl3 at 300 K, 500 MHz; Figure S2: COSY spectrum of (+)-longilene peroxide (1) in CDCl3 at 300 K, 500 MHz; Figure S3: HSQC spectrum of (+)-longilene peroxide (1) in CDCl3 at 300 K, 500 MHz; Figure S4: HMBC spectrum of (+)-longilene peroxide (1) in CDCl3 at 300 K, 500 MHz; Figure S5: ROESY spectrum of (+)-longilene peroxide (1) in CDCl3 at 300 K, 500 MHz; Figure S6: MS spectrum of (+)-longilene peroxide (1); Figure S7: 1H-NMR spectrum of longilene (2) in CDCl3 at 300 K, 500 MHz; Figure S8: COSY spectrum of longilene (2) in CDCl3 at 300 K, 500 MHz; Figure S9: HSQC spectrum of longilene (2) in CDCl3 at 300 K, 500 MHz; Figure S10: HMBC spectrum of longilene (2) in CDCl3 at 300 K, 500 MHz; Figure S11: MS spectrum of longilene (2); Figure S12: 1H-NMR spectrum of (+)-prelongilene (3) in CDCl3 at 300 K, 500 MHz; Figure S13: COSY spectrum of (+)-prelongilene (3) in CDCl3 at 300 K, 500 MHz; Figure S14: HSQC spectrum of (+)-prelongilene (3) in CDCl3 at 300 K, 500 MHz; Figure S15: HMBC spectrum of (+)-prelongilene (3) in CDCl3 at 300 K, 500 MHz; Figure S16: ROESY spectrum of (+)-prelongilene (3) in CDCl3 at 300 K, 500 MHz; Figure S17: MS spectrum of (+)-prelongilene (3); Figure S18: Conversion of (+)-prelongilene (3) to compound 5 for 1H-NMR; 0 h; Figure S19: Conversion of (+)-prelongilene (3) to compound 5 for 1H-NMR; 24 h; Figure S20: Conversion of (+)-prelongilene (3) to compound 5 for 1H-NMR; 48 h; Figure S21: Conversion of (+)-prelongilene (3) to compound 5 for 1H-NMR; 72 h; Figure S22: 1H-NMR spectrum of compound 5 in CDCl3 at 300 K, 500 MHz; Figure S23: COSY spectrum of compound 5 in CDCl3 at 300 K, 500 MHz; Figure S24: HSQC spectrum of compound 5 in CDCl3 at 300 K, 500 MHz; Figure S25: HMBC spectrum of compound 5 in CDCl3 at 300 K, 500 MHz; Figure S26: NOESY spectrum of compound 5 in CDCl3 at 300 K, 500 MHz; Figure S27: MS spectrum of compound 5; Figure S28: 1H-NMR spectrum of compound 4 in CDCl3 at 300 K, 500 MHz; Figure S29: HSQC spectrum of compound 4 in CDCl3 at 300 K, 500 MHz; Figure S30: HMBC spectrum of compound 4 in CDCl3 at 300 K, 500 MHz; Figure S31: NOESY spectrum of compound 4 in CDCl3 at 300 K, 500 MHz; Figure S32: In vitro inhibitory effect of (+)-longilene peroxide (1) on PP2A.
Acknowledgments
This work was supported by grant CTQ2014-55888-C03-01-R (Ministry of Economy and Competitiveness of Spain, MINECO). Authors are grateful to M. C. Gil-Rodríguez (Departamento de Biología Vegetal, ULL) for the taxonomic classification of the alga.
Author Contributions
Antonio Hernández Daranas, María Luisa Souto and José Javier Fernández designed and analyzed the experiments. Francisco Cen-Pacheco, Claudia Pérez Manríquez and Manuel Norte performed the experiments. All authors contributed to the final version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zolnierowicz, S. Type 2A protein phosphatase, the complex regulator of numerous signaling pathways. Biochem. Pharmacol. 2000, 60, 1225–1235. [Google Scholar] [CrossRef]
- Wera, S.; Hemmings, B.A. Serine/threonine protein phosphatases. Biochem. J. 1995, 311, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T. Protein kinases and phosphatases: The Yin and Yang of protein phosphorylation and signaling. Cell 1995, 80, 225–236. [Google Scholar] [CrossRef]
- McCluskey, A.; Sim, A.T.R.; Sakoff, J.A. Serine-Threonine Protein Phosphatase Inhibitors: Development of Potential Therapeutic Strategies. J. Med. Chem. 2002, 45, 1151–1175. [Google Scholar] [CrossRef] [PubMed]
- Honkanen, R.E.; Golden, T. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr. Med. Chem. 2002, 9, 2055–2075. [Google Scholar] [CrossRef] [PubMed]
- Bialojan, C.; Takai, A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J. 1988, 256, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Dilip de Silva, E.; Williams, D.E.; Andersen, R.J.; Klix, H.; Holmes, C.F.B.; Allen, T.M. Motuporin, a Potent Protein Phosphatase Inhibitor Isolated from the Papua New Guinea Sponge Theonella swinhoei Gray. Tetrahedron Lett. 1992, 33, 1561–1564. [Google Scholar] [CrossRef]
- Williams, D.E.; Lapawa, M.; Feng, X.D.; Tarling, T.; Roberge, M.; Andersen, R.J. Spirastrellolide A: Revised structure, progress toward the relative configuration, and inhibition of protein phosphatase 2A. Org. Lett. 2004, 6, 2607–2610. [Google Scholar] [CrossRef] [PubMed]
- Cen-Pacheco, F.; Nordström, L.; Souto, M.L.; Martín, M.N.; Fernández, J.J.; Daranas, A.H. Studies on Polyethers Produced by Red Algae. Mar. Drugs 2010, 8, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Cen-Pacheco, F.; Villa-Pulgarin, J.A.; Mollinedo, F.; Norte, M.; Daranas, A.H.; Fernández, J.J. Cytotoxic oxasqualenoids from the red alga Laurencia viridis. Eur. J. Med. Chem. 2011, 46, 3302–3308. [Google Scholar] [CrossRef] [PubMed]
- Cen-Pacheco, F.; Villa-Pulgarin, J.A.; Mollinedo, F.; Norte, M.; Fernández, J.J.; Daranas, A.H. New Polyether Triterpenoids from Laurencia viridis and Their Biological Evaluation. Mar. Drugs 2011, 9, 2220–2235. [Google Scholar] [CrossRef] [PubMed]
- Itokawa, H.; Kishi, E.; Morita, H.; Takeya, K.; Iitaka, Y. A New Squalene-type Triterpene from the Woods of Eurycoma longifolia. Chem. Lett. 1991, 2221–2222. [Google Scholar] [CrossRef]
- Morimoto, Y.; Iwai, T.; Kinoshita, T. Total synthesis and determination of the absolute configuration of (−)-longilene peroxide. Tetrahedron Lett. 2001, 42, 6307–6309. [Google Scholar] [CrossRef]
- Fernández, J.J.; Souto, M.L.; Gil, L.V.; Norte, M. Isolation of naturally occurring dactylomelane metabolites as Laurencia constituents. Tetrahedron 2005, 61, 8910–8915. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olsen, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Velec, H.F.; Gohlke, H.; Klebe, G. DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J. Med. Chem. 2005, 48, 6296–6303. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Xu, Y.; Chen, Y.; Jeffrey, P.D.; Chao, Y.; Lin, Z.; Strack, Z.L.J.; Stock, J.B.; Shi, Y. Structure of Protein Phosphatase 2A Core Enzyme Bound to Tumor-Inducing Toxins. Cell 2006, 127, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Kita, A.; Matsunaga, S.; Takai, A.; Kataiwa, H.; Wakimoto, T.; Fusetani, N.; Isobe, M.; Miki, K. Crystal structure of the complex between calyculin A and the catalytic subunit of protein phosphatase 1. Structure 2002, 10, 715–724. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).