Abstract
Marine-derived fungi are a promising and untapped reservoir for discovering structurally interesting and pharmacologically active natural products. In our efforts to identify novel bioactive compounds from marine-derived fungi, four breviane spiroditerpenoids, including a new compound, brevione O (1), and three known compounds breviones I (2), J (3), and H (4), together with a known diketopiperazine alkaloid brevicompanine G (5), were isolated and identified from an ethyl acetate extract of the fermented rice substrate of the coral-derived fungus Penicillium sp. TJ403-1. The absolute structure of 1 was elucidated by HRESIMS, one- and two-dimensional NMR spectroscopic data, and a comparison of its electronic circular dichroism (ECD) spectrum with the literature. Moreover, we confirmed the absolute configuration of 5 by single-crystal X-ray crystallography. All the isolated compounds were evaluated for isocitrate dehydrogenase 1 (IDH1) inhibitory activity and cytotoxicity, and compound 2 showed significant inhibitory activities against HL-60, A-549, and HEP3B tumor cell lines with IC50 values of 4.92 ± 0.65, 8.60 ± 1.36, and 5.50 ± 0.67 µM, respectively.
1. Introduction
Since the ocean covers over 70% of the Earth’s surface, marine organisms are regarded as a prolific and under-explored resource of bioactive natural products [1,2]. Over the past few decades, with the discovery of plenty of new chemicals, marine-derived fungi have increasingly attracted the attention of natural product chemists and biologists, largely due to their surprising potentials for drug discovery [3,4,5].
Breviane spiroditerpenoids, which are biosynthesized from geranylgeranyl and pyrone derived from three molecules of acetyl CoA and one methyl from methionine, are an important group of architecturally complex and bioactive meroterpenoids [6]. Since the first breviane spiroditerpenoid, namely brevione A, was found from Penicillium brevicompactum Dierckx in 2000 [7], a total of 14 naturally occurring compounds with identical skeletons, showing intriguing allelopathic, anti-HIV, cytotoxic, and Aβ aggregate-induced neurotoxic inhibitory effects have been reported to date [7,8,9,10,11]. Notably, their architecturally complex frameworks with multiple chiral centers and attractive biological profiles made these natural products target molecules for total synthesis [12,13,14].
Previously, we performed a chemical investigation on a mangrove-derived fungus, Daldinia eschscholzii, resulting in the isolation and identification of three novel polyketide glycosides, daldinisides A–C, and two new alkaloids, 1-(3-indolyl)-2R,3-dihydroxypropan-1-one and 3-ethyl-2,5-pyrazinedipropanoic acid [15]. Interestingly, daldinisides A–C represented a rare class of d-ribose-containing natural products. In our continuous exploration for structurally unique and biologically active natural products from the marine-derived fungi, our attention was focused on the coral-derived fungi, a neglected and insufficiently explored natural resource. A chemical investigation of the fermented rice substrate of the coral-derived fungus Penicillium sp. TJ403-1 afforded four breviane spiroditerpenoids, including a new compound, brevione O (1), and three known compounds breviones I (2), J (3), and H (4), together with a known diketopiperazine alkaloid brevicompanine G (5). Herein, the details of the isolation, structural elucidation, and bioactivity evaluations of these compounds (Figure 1) are reported.
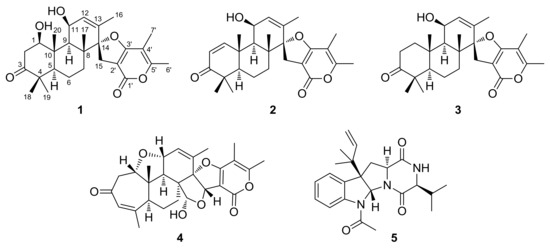
Figure 1.
Structures of compounds 1–5.
2. Results
Brevione O (1), purified as a white powder, was assigned the molecular formula C27H36O6, based on the HRESIMS m/z 479.2382 [M + Na]+ (calcd. for C27H36O6Na, 479.2410) and 13C-NMR analysis (see Supplementary Materials), corresponding to 10 degrees of unsaturation. Its 1H-NMR data (Table 1) showed signals of seven methyl groups (δH 1.76 (d, J = 1.6 Hz, H3-16), 1.21 (s, H3-17), 1.09 (s, H3-18), 1.03 (s, H3-19), 1.48 (s, H3-20), 1.92 (s, H3-6′), and 2.25 (s, H3-7′)), one olefinic proton (δH 5.74 (dd, J = 1.6, 4.8 Hz, H-12)), and two oxygenated methine protons (δH 3.95 (dd, J = 6.2, 9.2 Hz, H-1) and 4.68 (m, H-11)). Detailed analysis of the 13C- and DEPT NMR spectroscopic data (Table 1) of 1 indicated the presence of seven sp3 methyls, four sp3 methylenes, five methines (including two oxygenated and one olefinic), eleven non-protonated carbons (including one oxygenated, five olefinic, one ester carbonyl, and one ketone). Comparing the HRESIMS and NMR data with previously reported C27 meroterpenoids from the Penicillium species [7,8,9,10,11], these data were indicative of a breviane spiroditerpenoid, and interpretations of the 1H–1H COSY and HMBC spectra (Figure 2) of 1 established its planar structure as the C-1 hydroxylated analogue of the known compound brevione J (3), a co-isolated known metabolite that was initially characterized from a marine-derived Penicillium sp. [10].

Table 1.
1H- and 13C-NMR data for brevione O (1) in methanol-d4 (δ in ppm, J in Hz).
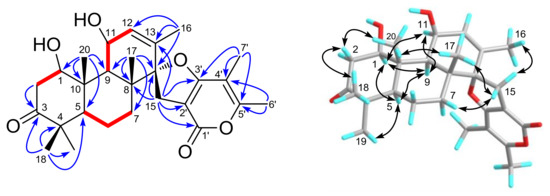
Figure 2.
Selected 1H–1H COSY (red lines), HMBC (blue arrows), and NOESY (black arrows) correlations of compound 1.
In the NOESY experiment (Figure 2), the cross-peaks of H3-18/H-2β (δH 2.81), H-2β/H3-20, H3-20/H3-17, and H3-19/H-5, H-5/H-9, H-9/H-11, and correlations of H-1 with H-5, H-9, and H-11 indicated that H3-17, H3-18, and H3-20 were all β-oriented, while H-1, H-5, H-9, H-11, and H3-19 were all on the opposite side with α-orientations. Moreover, the NOE correlations of H2-15 with H-7β (δH 1.62), H3-16, and H3-17 indicated that two planes of C- and D-rings were vertically arranged and that C-15 was on the upside of C-ring. Thus, the relative configuration of compound 1 was established.
To determine its absolute stereochemistry, the experiment electronic circular dichroism (ECD) spectrum (Figure 3) of 1, which was measured in MeOH, was compared to that of brevione J (3). Their nearly identical ECD spectra indicated that the absolute stereochemistry of 1 should be identified as 1R,5R,8R,9R,10S,11S,14S.
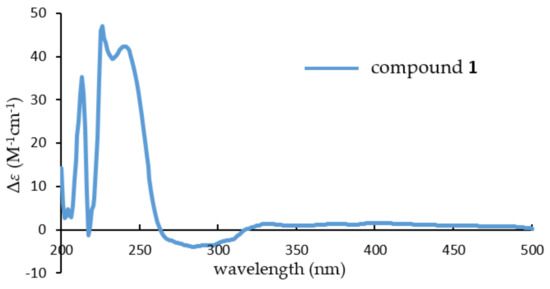
Figure 3.
Experimental electronic circular dichroism (ECD) spectrum of compound 1.
By comparing their specific rotation and NMR data with the literature, compounds 2–5 were identified as breviones I (2) [10], J (3) [10], H (4) [9], and brevicompanine G (5) [16], respectively. Remarkably, the absolute configuration of 5 was first confirmed via single-crystal X-ray crystallography with a Flack parameter = −0.03(4) (Figure 4).
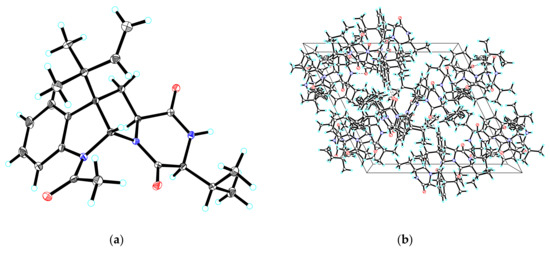
Figure 4.
(a) ORTEP drawing of compound 5; (b) View of the pack drawing of 5 and hydrogen-bonds are shown as dashed lines.
Isocitrate dehydrogenase 1 (IDH1) catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG). Neomorphic mutations in IDH1, mostly occurring at arginine 132, are frequently found in several human cancer types, including glioma, acute myeloid leukemia (AML), and myeloproliferative neoplasm [17]. When our research group screened for the IDH1 inhibitors from our natural product libraries, all the isolates 1–5 were evaluated for IDH1 inhibitory activity; unfortunately, none of them was active at a concentration of 20 μM. Additionally, compounds 1–5 were investigated for cytotoxic activities against several human tumor cell lines, including HL-60 (acute leukemia), MM231 (breast cancer), A-549 (lung cancer), HEP3B (hepatic cancer), SW-480 (colon cancer), and one normal colonic epithelial cell, NCM460. As shown in Table 2, compound 2, with no cytotoxic activity against the NCM460 cell, showed significant inhibitory potency against the HL-60, A-549, and HEP3B cell lines, with IC50 values of 4.92 ± 0.65, 8.60 ± 1.36, and 5.50 ± 0.67 µM, respectively. However, other compounds showed no obvious cytotoxicity against all these cell lines. Comparison of the IC50 values of 2 and 3 implied that the α,β-unsaturated carbonyl group plays an essential role in the cytotoxic activity. This group might interact with biological molecules by forming covalent bonds with free thiol of cysteine [18]; thus, the potential mechanism deserves further investigation in a following study.
Table 2.
Cytotoxic activities of compounds 1–5 against human tumor cell lines.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations, ultraviolet (UV), and ECD data were recorded on a PerkinElmer 341 polarimeter (Waltham, MA, USA), a Varian Cary 50 (Santa Clara, CA, USA), and a JASCO-810 CD (Oklahoma City, OK, USA) spectrometer instrument, respectively. FT-IR spectra were measured by using a Bruker Vertex 70 instrument (Billerica, MA, USA). One- and two-dimensional NMR data were collected from a Bruker AM-400 instrument (Bruker, Karlsruhe, Germany). The 1H- and 13C-NMR chemical shifts of the solvent peaks for methanol-d4 (δH 3.31 and δC 49.0) were referenced. The positive ion mode on a Thermo Fisher LC-LTQ-Orbitrap XL instrument (Waltham, MA, USA) was used for the measurement of high-resolution electrospray ionization mass spectra (HRESIMS). Semi-preparative HPLC separations were performed on an Agilent 1200 instrument with a reversed-phased C18 column (5 μm, 10 × 250 mm), using a UV detector or a Dionex HPLC system (Sunnyvale, CA, USA) which was equipped with an Ultimate 3000 autosampler injector, an Ultimate 3000 pump, and an Ultimate 3000 DAD controlled by Chromeleon software (version 6.80). Column chromatography (CC) was performed using silica gel (200−300 mesh, Qingdao Marine Chemical, Inc., Qingdao, China), Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden), and Lichroprep RP-C18 gel (40–63 μm, Merck, Darmstadt, Germany). Thin-layer chromatography was performed with the silica gel 60 F254 and RP-C18 F254 plates (Merck, Darmstadt, Germany), and spots were visualized by spraying heated silica gel plates with 10% H2SO4 in EtOH.
3.2. Fungal Material
Strain TJ403-1 was isolated from a piece of the inner tissues of a fresh soft coral of the genus Alcyonium (an unidentified sp.), which was collected from the Sanya Bay, Hainan Island, China. According to its morphology and sequence analysis of the ITS (Internal Transcribed Spacer) region of the rDNA, the strain was identified as Penicillium sp. and its sequence data have been submitted to the GenBank with the accession no. MG839539. This strain has been reserved in the culture collection of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China).
3.3. Cultivation, Extraction, and Isolation
The TJ403-1 strain was incubated on potato dextrose agar (PDA) at 28 °C for 6 days in a stationary phase to prepare the seed cultures, which were then cut into small pieces (about 0.4 × 0.4 × 0.4 cm) and inoculated into 350 × 500 mL sterilized Erlenmeyer flasks (each comprised of 200 g rice and 200 mL distilled water). After an incubation at 28 °C for 40 days, 300 mL EtOAc was added to all flasks to stop the growth of cells. Then, the filtrates were collected, and the fermented rice substrate was extracted with EtOAc (8 × 30 L) six times. Lastly, these two parts of the extracts were mixed together. Under reduced pressure, the organic solvent was evaporated to dryness to obtain a dark brown crude extract (700 g).
The EtOAc extract (700 g) was subjected to silica gel CC eluted with a gradient of petroleum ether/EtOAc/MeOH (stepwise 20:1:0 to 1:1:1, v/v/v) to afford eight fractions (Fr.1–Fr.8). Fr.5 was separated through RP-C18 CC (MeOH/H2O, 20% to 100%, v/v) to give five subfractions (Fr.5.1–Fr.5.5). Then, Fr.5.3 was purified via silica gel CC, eluted with CH2Cl2/MeOH (stepwise 50:1 to 30:1, v/v) to afford two additional subfractions (Fr.5.3.1−5.3.2). Further separations of Fr.5.3.1 through Sephadex LH-20 eluted with a mixture of CH2Cl2/MeOH (1:1, v/v) and repeated semi-preparative HPLC (MeOH/H2O, 70:30, v/v; 2 mL/min) yielded compounds 2 (16 mg), 3 (8 mg), and 5 (9 mg). Fr.6 was loaded onto RP-C18 CC (MeOH/H2O, 20% to 100%, v/v) to give five subfractions (Fr.6.1–Fr.6.5). By using the silica gel CC (CH2Cl2/MeOH, 40:1, v/v) and repeated semi-preparative HPLC (MeOH/H2O, 60:40, v/v; 2 mL/min) methods, Fr.6.3 was finally separated to yield compounds 1 (5 mg) and 4 (60 mg).
Brevione O (1): white powder; [α: +94.1 (c 0.14, MeOH); UV (MeOH) λmax (log ε): 218 (4.33), 253 (3.73), 307 (3.46) nm; CD (MeOH) λmax (Δε): 213 (+35.24), 225 (+45.98), 238 (+41.91), 283 (–3.94); IR (KBr) νmax: 3423, 2925, 2855, 1703, 1573, 1446, 1388, 1363, 1275, 1116, 1062, 983, 924, 856, 746 cm−1; HRESIMS m/z 479.2382 [M + Na]+ (calcd. for C27H36O6Na, 479.2410). For 1H- and 13C-NMR data, see Table 1.
3.4. X-ray Crystallographic Analysis
At room temperature, after attempts with various organic solvents, compound 5 was obtained as colorless crystals from MeOH/H2O (15:1, v/v) by slow evaporation. The intensity data for compound 5 was collected with a Bruker APEX DUO diffractometer, which was outfitted with an APEX II CCD by using graphite-monochromated Cu Kα radiation (100 K). The Bruker SAINT was used for the cell refinement and data reduction of compound 5, whose structure was then solved and refined through direct means by using SHELXS-97 program (Göttingen, Germany) [19]. The crystallographic data for compound 5 have been deposited in the Cambridge Crystallographic Data Center (CCDC 1827348 for 5). Copies of the data can be obtained free of charge from the CCDC, 12 Union Road, Cambridge CB 1EZ, UK (fax: Int. +44(0) (1223) 336 033); e-mail: deposit@ccdc.cam.ac.uk).
Crystallographic data for brevicompanine G (5): C23H29N3O3, M = 395.49, a = 18.2192(5) Å, b = 13.1497(3) Å, c = 19.6255(5) Å, α = 90°, β = 116.2910(10)°, γ = 90°, V = 4215.44(19) Å3, T = 100(2) K, space group P21, Z = 8, μ(CuKα) = 0.668 mm−1, 38,513 reflections measured, 14,878 independent reflections (Rint = 0.0360). The final R1 values were 0.0364 (I > 2σ(I)). The final wR(F2) values were 0.0949 (I > 2σ(I)). The final R1 values were 0.0366 (all data). The final wR(F2) values were 0.0952 (all data). The goodness of fit on F2 was 1.045. Flack parameter = −0.03(4).
3.5. In Vitro IDH1(R132H) Inhibition Assay
The activity and inhibition of IDH1 (R132H) was determined by measuring the initial linear consumption of NADPH of the reaction. The enzyme activity assay was carried out in a 96-well microplate, using the purified IDH1 (R132H) protein in buffer solution containing 50 mM HEPES pH = 7.5, 4 mM MgCl2, 100 mM NaCl, and 0.1 mg/mL bovine serum albumin. For inhibition assay, triplicate samples of compounds (10 µL) were incubated with the protein (2 µg/mL, 20 µL) and 60 µL buffer for 5 min. The reaction was initiated by adding 2 mM α-KG, 100 μM NADPH (10 µL) into the 96-well microplate. The consumption of NADPH was measured by monitoring the optical absorbance of each well every 30 s at 340 nm, which was the maximum absorption wavelength of NADPH, using a Biotek Synergy HT microplate reader.
3.6. Cytotoxicity Assay
Five human tumor cell lines (HL-60, MM231, A-549, HEP3B, and SW480), together with one non-cancerous cell line, the human normal colonic epithelial cell NCM460, were used in the cytotoxic activity assay. All cells were cultured in DMEM or RPMI-1640 medium (HyClone, Logan, UT, USA), supplemented with 10% fetal bovine serum (HyClone) at 37 °C in a humidified atmosphere with 5% CO2. The cell survival assay was performed using the MTT method. Briefly, 100 μL suspended cells at an initial density of 1 × 105 cells/mL were seeded into each well of the 96-well culture plates and allowed to adhere for 12 h before addition of the test compounds. Each tumor cell line was exposed for 48 h to the test compounds at concentrations ranging from 0.0625 to 40 μM, with DDP (cis-platin, Sigma, St. Louis, MO, USA) and paclitaxel as positive controls. After incubation, culture supernatants were removed and exchanged with medium containing 0.5 mg/mL MTT. Then, after 4 h incubation in darkness at 37 °C, the medium was removed, and cells were added with 100 μL dimethyl sulfoxide. The absorbance at 570 nm was measured and data are expressed as averages of three replicates. The value of inhibition was calculated by using the following formula: % inhibition = (1 − ODtreated/ODcontrol) × 100. The IC50 values were calculated by using a standard dose-response curve fitting with Prism (version 5.0, GraphPad Software, La Jolla, CA, USA).
4. Conclusions
To sum up, four breviane spiroditerpenoids (1–4), including a new compound, brevione O (1), and three known compounds breviones I (2), J (3), and H (4), together with a known diketopiperazine alkaloid (5), were isolated and identified from an ethyl acetate extract of the fermented rice substrate of the coral-derived fungus Penicillium sp. TJ403-1. The absolute structure of 1 was elucidated on the basis of HRESIMS, one- and two-dimensional NMR spectroscopic data, and a comparison of its ECD spectrum with data gathered from the literature. Remarkably, despite the fact that the structure of brevicompanine G (5) was once established via a combination of NMR spectroscopic data and biosynthetic logic-based consideration, our current work is the first to confirm the absolute configuration of 5 by single-crystal X-ray crystallography, which will be of great significance for the structure elucidation of complex diketopiperazine alkaloids. Remarkably, compound 2 showed significant inhibitory activities against HL-60, A-549, and HEP3B tumor cell lines, with IC50 values of 4.92 ± 0.65, 8.60 ± 1.36, and 5.50 ± 0.67 µM, respectively.
Supplementary Materials
The following are available online at http://www.mdpi.com/1660-3397/16/4/110/s1, 1D (1H- and 13C-) and 2D (HMBC, HSQC, COSY, NOESY) NMR, HRESIMS, IR, and UV spectra of 1.
Acknowledgments
We thank the Analytical and Testing Center at HUST for the ECD and IR analyses. The support from the Program for Changjiang Scholars of Ministry of Education of the People’s Republic of China (No. T2016088), the Innovative Research Groups of the National Natural Science Foundation of China (No. 81721005), the National Science Fund for Distinguished Young Scholars (No. 8172500151), the China Postdoctoral Science Foundation Funded Project (No. 2017M610479), the National Natural Science Foundation of China (Nos. 81573316, 21702067), the Academic Frontier Youth Team of HUST, and the Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College (HUST) are greatly acknowledged.
Author Contributions
Beiye Yang contributed to the extraction, isolation, identification, and manuscript preparation. Weiguang Sun contributed to the bioactivity tests. Shuang Lin contributed to the fungal isolation and fermentation. Jianping Wang advised and assisted Yang’s experiments. Xiao-Nian Li contributed to the X-ray diffraction experiment. Hucheng Zhu and Zengwei Luo contributed to the structure elucidation of isolated compounds and shared in the tasks of the manuscript preparation. Yongbo Xue contributed to the NMR experiments. Zhengxi Hu guided the experiments and wrote the manuscript. Yonghui Zhang designed the experiments and revised the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sueyoshi, K.; Yamano, A.; Ozaki, K.; Sumimoto, S.; Iwasaki, A.; Suenaga, K.; Teruya, T. Three new malyngamides from the marine cyanobacterium Moorea producens. Mar. Drugs 2017, 15, 367. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F. Natural products from marine fungi—Still an underrepresented resource. Mar. Drugs 2016, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.S.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Current Status of marine-derived compounds as warheads in anti-tumor drug candidates. Mar. Drugs 2017, 15, 99. [Google Scholar] [CrossRef] [PubMed]
- Cherigo, L.; Lopez, D.; Martinez-Luis, S. Marine natural products as breast cancer resistance protein inhibitors. Mar. Drugs 2015, 13, 2010–2029. [Google Scholar] [CrossRef] [PubMed]
- Geris, R.; Simpson, T.J. Meroterpenoids produced by fungi. Nat. Prod. Rep. 2009, 26, 1063–1094. [Google Scholar] [CrossRef] [PubMed]
- Macías, F.A.; Varela, R.M.; Simonet, A.M.; Cutler, H.G.; Cutler, S.J.; Ross, S.A.; Dunbar, D.C.; Dugan, F.M.; Hill, R.A. (+)-Brevione A. The first member of a novel family of bioactive spiroditerpenoids isolated from Penicillium brevicompactum Dierckx. Tetrahedron Lett. 2000, 41, 2683–2686. [Google Scholar] [CrossRef]
- Macías, F.A.; Varela, R.M.; Simonet, A.M.; Cutler, H.G.; Cutler, S.J.; Dugan, F.M.; Hill, R.A. Novel bioactive breviane spiroditerpenoids from Penicillium brevicompactum Dierckx. J. Org. Chem. 2000, 65, 9039–9046. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, D.; Chen, X.; Lu, X.; Shao, Z.; Zhang, H.; Che, Y. Breviane spiroditerpenoids from an extreme-tolerant Penicillium sp. isolated from a deep sea sediment sample. J. Nat. Prod. 2009, 72, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, D.; Shao, Z.; Cui, C.; Che, Y. A sterol and spiroditerpenoids from a Penicillium sp. isolated from a deep sea sediment sample. Mar. Drugs 2012, 10, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Seo, Y.H.; Lee, J.E.; Seo, E.K.; Li, S.; Guo, Y.; Hong, S.B.; Park, S.Y.; Lee, D. Spiroindole alkaloids and spiroditerpenoids from Aspergillus duricaulis and their potential neuroprotective effects. J. Nat. Prod. 2015, 78, 2572–2579. [Google Scholar] [CrossRef] [PubMed]
- Yokoe, H.; Mitsuhashi, C.; Matsuoka, Y.; Yoshimura, T.; Yoshida, M.; Shishido, K. Enantiocontrolled total syntheses of breviones A, B, and C. J. Am. Chem. Soc. 2011, 133, 8854–8857. [Google Scholar] [CrossRef] [PubMed]
- Macías, F.A.; Carrera, C.; Chinchilla, N.; Fronczek, F.R.; Galindo, J.C.G. Synthesis of the western half of breviones C, D, F and G. Tetrahedron 2010, 66, 4125–4132. [Google Scholar] [CrossRef]
- Takikawa, H.; Hirooka, M.; Sasaki, M. The first synthesis of (±)-brevione B, an allelopathic agent isolated from Penicillium sp. Tetrahedron Lett. 2003, 44, 5235–5238. [Google Scholar] [CrossRef]
- Hu, Z.X.; Xue, Y.B.; Bi, X.B.; Zhang, J.W.; Luo, Z.W.; Li, X.N.; Yao, G.M.; Wang, J.P.; Zhang, Y.H. Five new secondary metabolites produced by a marine-associated fungus, Daldinia eschscholzii. Mar. Drugs 2014, 12, 5563–5575. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Yang, X.; Zhu, T.; Wang, F.; Xiao, X.; Park, H.; Gu, Q. Diketopiperazine alkaloids from a deep ocean sediment derived fungus Penicillium sp. Chem. Pharm. Bull. 2009, 57, 873–876. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zheng, M.; Li, Q.; Hu, Z.; Zhu, H.; Liu, J.; Wang, J.; Xue, Y.; Li, H.; Zhang, Y. Asperspiropene A, a novel fungal metabolite as an inhibitor of cancer-associated mutant isocitrate dehydrogenase 1. Org. Chem. Front. 2017, 4, 1137–1144. [Google Scholar] [CrossRef]
- Sun, X.; Wang, W.; Chen, J.; Cai, X.; Yang, J.; Yang, Y.; Yan, H.; Cheng, X.; Ye, J.; Lu, W.; et al. The natural diterpenoid isoforretin A inhibits thioredoxin-1 and triggers potent ROS-mediated antitumor effects. Cancer Res. 2017, 77, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.X.; Liu, M.; Wang, W.G.; Li, X.N.; Hu, K.; Li, X.R.; Du, X.; Zhang, Y.H.; Puno, P.T.; Sun, H.D. 7α,20-Epoxy-ent-kaurane diterpenoids from the aerial parts of Isodon pharicus. J. Nat. Prod. 2018, 81, 106–116. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).