
Adrenal Tumors in Young Adults: Case Reports and Literature Review

 , ,
, ,
Abstract
1. Introduction
2. Materials
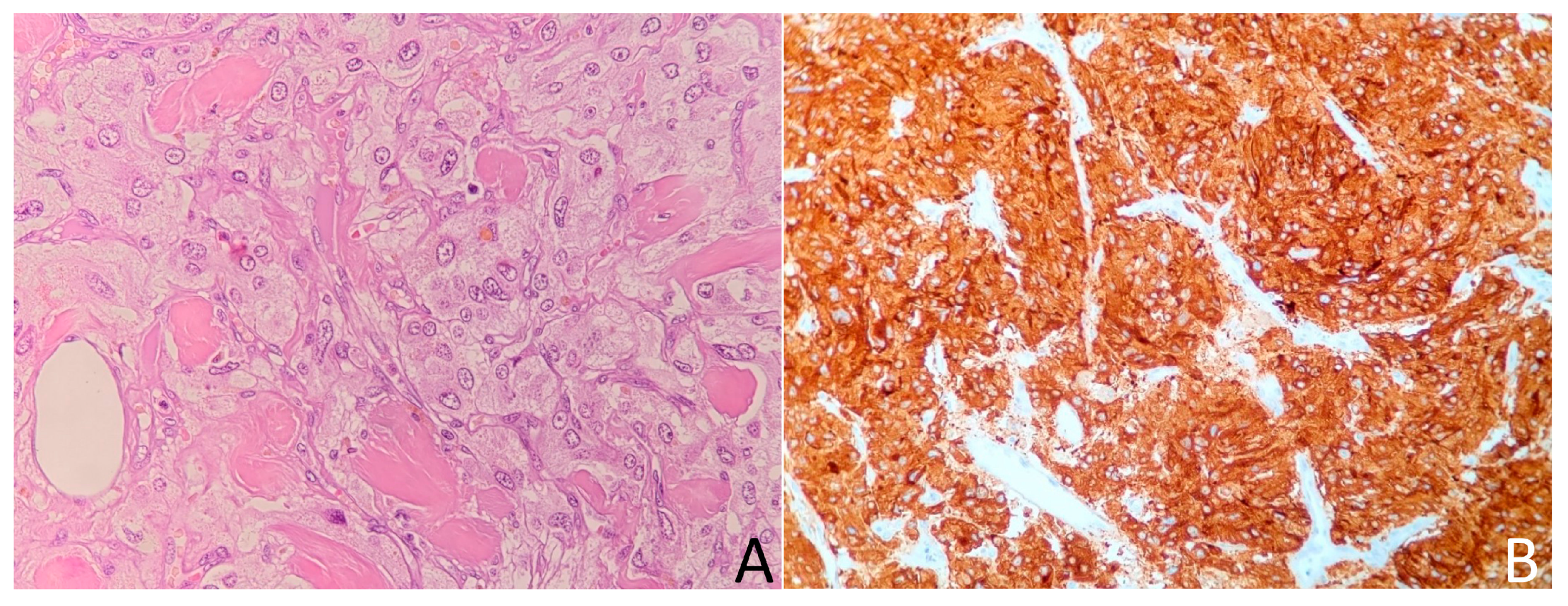
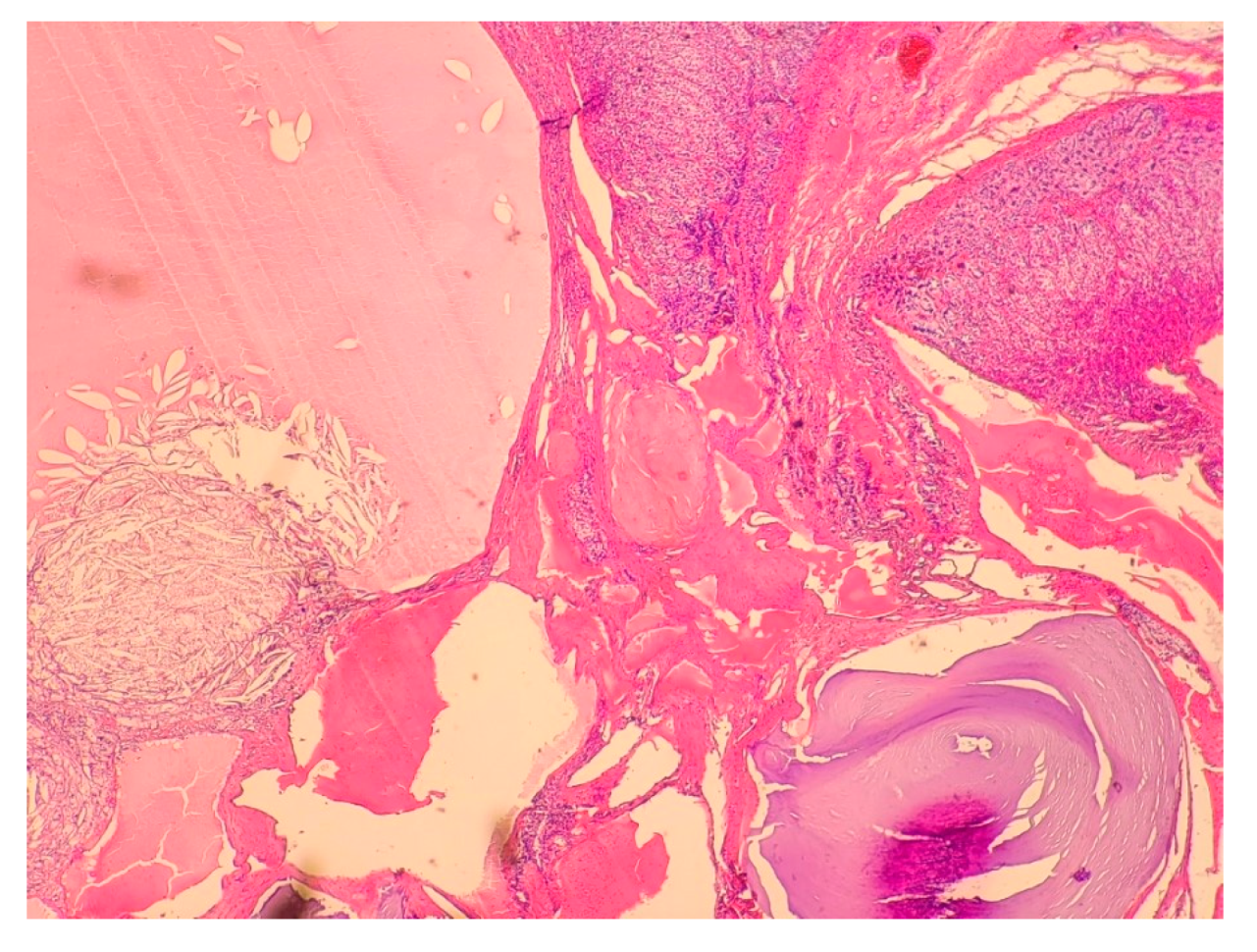
2.1. Case 1
2.2. Case 2
2.3. Case 3
2.4. Case 4
2.5. Case 5
2.6. Case 6
2.7. Case 7
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bednarczuk, T.; Bolanowski, M.; Sworczak, K.; Górnicka, B.; Cieszanowski, A.; Otto, M.; Ambroziak, U.; Pachucki, J.; Kubicka, E.; Babińska, A.; et al. Adrenal incidentaloma in adults—Management recommendations by the Polish Society of Endocrinology. Endokrynol. Pol. 2016, 67, 234–258. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Arlt, W.; Bancos, I.; Dralle, H.; Newell-Price, J.; Sahdev, A.; Tabarin, A.; Terzolo, M.; Tsagarakis, S.; Dekkers, O.M. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 2016, 175, G1–G34. [Google Scholar] [CrossRef] [PubMed]
- Kloos, R.T.; Gross, M.D.; Francis, I.R.; Korobkin, M.; Shapiro, B. Incidentally discovered adrenal masses. Endocr. Rev. 1995, 16, 460–484. [Google Scholar]
- Cyranska-Chyrek, E.; Szczepanek-Parulska, E.; Olejarz, M.; Ruchala, M. Malignancy Risk and Hormonal Activity of Adrenal Incidentalomas in a Large Cohort of Patients from a Single Tertiary Reference Center. Int. J. Environ. Res. Public Health 2019, 16, 1872. [Google Scholar] [CrossRef]
- di Dalmazi, G.; Vicennati, V.; Garelli, S.; Casadio, E.; Rinaldi, E.; Giampalma, E.; Mosconi, C.; Golfieri, R.; Paccapelo, A.; Pagotto, U.; et al. Cardiovascular events and mortality in patients with adrenal incidentalomas that are either non-secreting or associated with intermediate phenotype or subclinical Cushing’s syndrome: A 15-year retrospective study. Lancet Diabetes Endocrinol. 2014, 2, 396–405. [Google Scholar] [CrossRef]
- Lee, J.; Kim, M.K.; Ko, S.; Koh, J.; Kim, B.; Kim, S.W.; Kim, S.; Kim, H.J.; Ryu, O.; Park, J. Clinical Guidelines for the Management of Adrenal Incidentaloma. Endocrinol. Metab. 2017, 32, 200–218. [Google Scholar] [CrossRef]
- Babinska, A.; Peksa, R.; Sworczak, K. Primary malignant lymphoma combined with clinically “silent” pheochromocytoma in the same adrenal gland. World J. Surg. Oncol. 2015, 30, 289. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, J.I.; Bae, J.M. Significant growth of adrenal lymphangioma: A case report and review of the literature. Int. J. Surg. Case Rep. 2015, 17, 48–50. [Google Scholar] [CrossRef]
- Sanal, H.T.; Kocaoglu, M.; Yildirim, D.; Bulakbasi, N.; Guvenc, I.; Tayfun, C.; Ucoz, T. Imaging features of benign adrenal cysts. Eur. J. Radiol. 2006, 60, 465–469. [Google Scholar] [CrossRef]
- Bibi, M.; Sellami, A.; Taktak, T.; Chelly, B.; Ghorbel, Z.; Zouari, H.; Boukriba, S.; Boussafa, H.; Chehida, M.a.B.; Rhouma, S.B.; et al. Giant cystic lymphangioma of adrenal gland: A case report and review of the literature. Urol. Case Rep. 2018, 22, 6–7. [Google Scholar] [CrossRef]
- Haines, I.; Macallister, A. Giant adrenal pseudocyst: A rare diagnosis. J. Med. Imaging Radiat. Oncol. 2018, 62, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Zografos, G.N.; Kothonidis, K.; Ageli, C.; Kopanakis, N.; Dimitriou, K.; Papaliodi, E.; Kaltsas, G.; Pagoni, M.; Papastratis, G. Laparoscopic resection of large adrenal ganglioneuroma. JSLS J. Soc. Laparoendosc. Surg. 2007, 11, 487–492. [Google Scholar]
- Rondeau, G.; Nolet, S.; Latour, M.; Braschi, S.; Gaboury, L.; Lacroix, A.; Panzini, B.; Arjane, P.; Cohade, C.; Bourdeau, I. Clinical and biochemical features of seven adult adrenal ganglioneuromas. J. Clin. Endocrinol. Metab. 2010, 95, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Sarwal, A.; Khullar, R.; Sharma, A.; Soni, V.; Baijal, M.; Chowbey, P. Laparoscopic adrenalectomy for ganglioneuroma presenting as an adrenal incidentaloma. J. Minim. Access Surg. 2019, 15, 226–259. [Google Scholar] [CrossRef] [PubMed]
- Babinska, A.; Peksa, R.; Swiątkowska-Stodulska, R.; Sworczak, K. The collection of five interesting cases of adrenal tumors from one medical center. World J. Surg. Oncol. 2014, 12, 377. [Google Scholar] [CrossRef] [PubMed]
- Khorsand, A.; Khatami, F.; Sefidbakht, S.; Saffar, H.; Sadeghipour, A.; Tavangar, S.M. Adrenal Collision Tumor Composed of Pheochromocytoma and Diffuse Large B-Cell Lymphoma: A Case Report. Int. J. Hematol. Oncol. Stem. Cell Res. 2018, 12, 249–252. [Google Scholar] [CrossRef]
- Benchetrit, S.; Bernheim, J.; Podjarny, E. Normokalemic hyperaldosteronism in patients with resistant hypertension. Isr. Med. Assoc. J. 2002, 4, 17–20. [Google Scholar]
- Walz, M.K.; Gwosdz, R.; Levin, S.L.; Alesina, P.F.; Suttorp, A.; Metz, K.A.; Wenger, F.A.; Petersenn, S.; Mann, K.; Schmid, K.W. Retroperitoneoscopic adrenalectomy in Conn’s syndrome caused by adrenal adenomas or nodular hyperplasia. World J. Surg. 2008, 32, 847–853. [Google Scholar] [CrossRef]
- McAlister, F.A.; Lewanczuk, R.Z. Primary hyperaldosteronism and adrenal incidentaloma: An argument for physiologic testing before adrenalectomy. Can. J. Surg. 1998, 41, 299–305. [Google Scholar]
- Papierska, L.; Cichocki, A.; Sankowski, A.J.; Ćwikła, J.B. Adrenal incidentaloma imaging—The first steps in therapeutic management. Pol. J. Radiol. 2013, 78, 47–55. [Google Scholar]
- Sahdev, A. Recommendations for the management of adrenal incidentalomas: What is pertinent for radiologists? Br. J. Radiol. 2017, 90, 20160627. [Google Scholar] [CrossRef] [PubMed]
- Amodru, V.; Taieb, D.; Guerin, C.; Paladino, N.C.I.; Brue, T.Y.; Sebag, F.; Castinetti, F. Large adrenal incidentalomas require a dedicated diagnostic procedure. Endocr. Pract. 2019, 25, 669–677. [Google Scholar] [CrossRef]
- Blake, M.A.; Kalra, M.K.; Maher, M.M.; Sahani, D.V.; Sweeney, A.T.; Mueller, P.R.; Hahn, P.F.; Boland, G.W. Pheochromocytoma: An imaging chameleon. Radiographics 2004, 24, S87–S99. [Google Scholar] [CrossRef] [PubMed]
- Sturgeon, C.; Shen, W.T.; Clark, O.H.; Duh, Q.Y.; Kebebew, E. Risk assessment in 457 adrenal cortical carcinomas: How much does tumor size predict the likelihood of malignancy? J. Am. Coll. Surg. 2006, 202, 423–430. [Google Scholar] [CrossRef]
- Thompson, G.B.; Young, W.F., Jr. Adrenal incidentaloma. Curr. Opin. Oncol. 2003, 15, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Terzolo, M.; Ali, A.; Osella, G.; Mazza, E. Prevalence of adrenal carcinoma among incidentally discovered adrenal masses. A retrospective study from 1989 to 1994. Arch. Surg. 1997, 132, 914–919. [Google Scholar] [CrossRef]
- Dworakowska, D.; Drabarek, A.; Wenzel, I.; Babińska, A.; Świątkowska-Stodulska, R.; Sworczak, K. Adrenocortical cancer (ACC)—Literature overview and own experience. Endokrynol. Pol. 2014, 65, 492–502. [Google Scholar]
- Stigliano, A.; Cerquetti, L.; Sampaoli, C.; Bucci, B.; Toscano, V. Current and emerging therapeutic options in adrenocortical cancer treatment. J. Oncol. 2012, 2012, 408131. [Google Scholar] [CrossRef]
- Patalano, A.; Brancato, V.; Mantero, F. Adrenocortical cancer treatment. Horm. Res. 2009, 71, 99–104. [Google Scholar] [CrossRef]
- Lu, Y.; Li, P.; Gan, W.; Zhao, X.; Shen, S.; Feng, W.; Xu, Q.; Bi, Y.; Guo, H.; Zhu, D. Clinical and Pathological Characteristics of Hypertensive and Normotensive Adrenal Pheochromocytomas. Exp. Clin. Endocrinol. Diabetes 2016, 124, 372–379. [Google Scholar] [CrossRef]
- Ohara, N.; Kaneko, M.; Yaguchi, Y.; Ishiguro, H.; Ishizaki, F.; Maruyama, R.; Suzuki, K.; Komeyama, T.; Usuda, H.; Yamazaki, Y. A case of normotensive incidentally discovered adrenal pheochromocytoma. Clin. Case Rep. 2018, 6, 2303–2308. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.D. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: A clinicopathologic and immunophenotypic study of 100 cases. Am. J. Surg. Pathol. 2002, 26, 551–556. [Google Scholar] [CrossRef] [PubMed]
- DeLellis, R.A.; Lloyd, R.V.; Heits, P.U.; Rosai, J. World Health Organization Classification of Tumours. Pathology and Genetics. Tumours of Endocrine Organs; IARC Press: Lyon, France, 2004. [Google Scholar]
- Lewandowska-Graban, K.; Zdrojewska, M.; Jendrzejewski, J.; Śledziński, M.; Pęksa, R.; Sworczak, K. A case report of melanoma metastasis to adrenal gland. Pol. Arch. Intern. Med. 2019, 129, 636–637. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Sex/Age | Cortisol Urinary 24-h Excretion (nmol/24 h) | ACTH Serum (pg/mL) | Cortisol Serum 1 mg DXM Suppression Test (nmol/L) | DHEA-S Serum (µg/dL) | Androstenedione Serum (ng/mL) | Metanephrines/Normetanephrines/3-Metoksythyramine Urinary 24 h Excretion (µg/24 h) |
|---|---|---|---|---|---|---|
| P1 M/31 | - | 7.8 | <28 | 242 | 3.6 | 236/220/133 |
| P2 M/34 | 144.0 | 31.3 | - | 306 | 2.8 | 89961/229/19 |
| P3 M/37 | 798.0 | 6 | 169 | 77.1 | <0.30 | 254/327/234 |
| P4 F/27 | 212.0 | 8.5 | <28 | 248 | 2.2 | 154/300/345 |
| P5 F/19 | 100 | 32.2 | <28 | 366 | 3.8 | 98/220/- |
| P5 F/27 | 87.4 | 22.3 | <28 | 233 | 3.5 | -/-/- |
| P6 F/37 | 141.0 | 19.4 | <28 | 165 | 2.31 | 129/188/461 |
| P7 F/33 | 288.0 | 15.5 | <28 | 120 | 2.6 | 113/212/298 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zdrojewska, M.; Mech-Siebieszuk, E.; Świątkowska-Stodulska, R.; Regent, B.; Kunc, M.; Zdrojewski, Ł.; Sworczak, K. Adrenal Tumors in Young Adults: Case Reports and Literature Review. Medicina 2022, 58, 746. https://doi.org/10.3390/medicina58060746
Zdrojewska M, Mech-Siebieszuk E, Świątkowska-Stodulska R, Regent B, Kunc M, Zdrojewski Ł, Sworczak K. Adrenal Tumors in Young Adults: Case Reports and Literature Review. Medicina. 2022; 58(6):746. https://doi.org/10.3390/medicina58060746
Chicago/Turabian StyleZdrojewska, Małgorzata, Emilia Mech-Siebieszuk, Renata Świątkowska-Stodulska, Bartosz Regent, Michał Kunc, Łukasz Zdrojewski, and Krzysztof Sworczak. 2022. "Adrenal Tumors in Young Adults: Case Reports and Literature Review" Medicina 58, no. 6: 746. https://doi.org/10.3390/medicina58060746
APA StyleZdrojewska, M., Mech-Siebieszuk, E., Świątkowska-Stodulska, R., Regent, B., Kunc, M., Zdrojewski, Ł., & Sworczak, K. (2022). Adrenal Tumors in Young Adults: Case Reports and Literature Review. Medicina, 58(6), 746. https://doi.org/10.3390/medicina58060746