Abstract
The type strains of 10 small-spored species of the ascomycetous yeast genus Metschnikowia usually form a compact group on the phylogenetic trees inferred from barcode sequences. Based on the name of the species, which was described first (Metschnikowia pulcherrima), the group is frequently referred to as the pulcherrima clade. All strains produce the iron-chelate pigment pulcherrimin and have antagonistic effects on many microorganisms. Recent results of molecular phylogenetic, genetic, and genomic research raised doubts about the taxonomic division of the clade. Those data—combined with results obtained in this study by comprehensive analysis of primary and secondary barcode sequences, physiological tests, and hybridisation experiments—demonstrate that the species cannot be distinguished from each other by the criteria of any of the phenotypic, phylogenetic, and biological species concepts. Therefore, I propose that the species of the pulcherrima clade be merged into one species under the oldest species name, M. pulcherrima.
Keywords:
yeast; taxonomy; barcode; rDNA; reticulation; chimeric genome; hybridisation; pulcherrimin 1. Introduction
Metschnikowia (Saccharomycetales) is a large genus of budding yeasts currently comprising 71 species and a large number of related strains (Mycobank and NCBI Taxonomy Database, 31 October 2020). A group of species, the so-called M. pulcherrima clade (or pulcherrima clade), produces pulcherrimin, a chelate complex of pulcherriminic acid and ferric ions [1]. The complex turns the colony faintly violet to maroon-red depending on the concentration of ferric ions in the medium. As pulcherriminic acid can be secreted into the environment, pigmented halos can frequently be seen around the pigmented colonies. In contrast to its secreted precursor, pulcherrimin is not soluble in water, so its production immobilises the iron in the medium [2]. The process makes the iron inaccessible to microorganisms. Numerous pulcherrima-clade strains have been found to have antagonistic activity against bacteria and fungi (e.g., [2,3,4,5,6,7]). The strength of their antagonistic effect correlates with the intensity of the extracellular production of pulcherrimin, and supplementation of the medium with ferric salts reduces the size of both the pigmented halo and the inhibition zone around the Metschnikowia colony. From these results, it was inferred that the pulcherrimin-producing strains antagonise fungal and bacterial growth by iron depletion [2]. Although iron sequestration seems to be the major element in the antimicrobial antagonism, other factors—such as the secretion of cell-wall degrading enzymes (e.g., [8]) and the release of volatile compounds [9]—may also contribute to the antagonistic effect. Due to their antimicrobial activities, these strains can be used as biological agents to protect agricultural commodities and food products against pathogenic and destructive microorganisms (for a review, see [10]).
Pulcherrima-clade yeasts occur in a broad spectrum of natural substrates, fermenting beverages, and also in processed food products. However, their taxonomic position within the clade cannot be easily determined because of the frequent occurrence of ambiguous (di- or polymorphic) nucleotides in the D1/D2 and ITS barcode sequences routinely used for taxonomic identification of yeast strains. The situation is further complicated by the presence of such nucleotides in the database sequences of the type strains of certain species. The clade consists of 10 species (M. andauensis, M. citriensis, M. fructicola, M. leonuri, M. persimmonesis, M. pulcherrima, M. rubicola, M. shanxiensis, M. sinensis, and M. ziziphicola) [3,5,6,11,12,13,14]. However, the taxon name M. citriensis is listed in MycoBank as invalid. All but one species were delimited on the basis of differences between the above-mentioned barcode sequences of their ex-type strains (living strains of types permanently preserved in a metabolically inactive state). Cloning and sequencing of the D1/D2 and ITS segments of individual rDNA repeats revealed that the nucleotide ambiguities are caused by intragenomic rDNA repeat heterogeneity [15,16]. A recent study found that secondary barcode sequences are also heterogeneous in the ex-type strains for which such sequences are available [17]. Thus, the type strains of the species of the clade are not separated by clear barcode gaps and therefore they cannot represent distinct species in terms of the phylogenetic species concept. This concept is based on the premise that a species represents a distinct entity containing its own synapomorphic characteristics (e.g., specific barcode sequences) not shared with neighbouring taxa [18]. From a recent analysis of a larger group of orthologous genes of seven whole-genome sequences, including those of three ex-type strains, it was inferred that the failure to differentiate the species by clear barcode gaps might be attributed to their admixed chimeric genomes consisting of sequences of diverse phylogenetic histories [17].
Delimitation of fungal species by the application of the phylogenetic species concept is based on the analysis of DNA barcodes. However, at least 30 other species concepts have been described in the literature [19]. Those preferably applied to fungi have been critically reviewed in numerous recent works (e.g., [20,21,22]). One of these concepts is the biological concept which defines a species as a group of interbreeding natural populations that are reproductively isolated from other such groups [23]. In yeasts, it was successfully applied, e.g., to the delimitation of Saccharomyces species separated by postzygotic sterility barriers [24]. All Saccharomyces strains can form viable hybrids, but only the hybrids of conspecific strains can produce functional (mating-competent) gametes. Interspecies hybrids either do not form viable gametes (ascospores) or their gametes (spores and spore clones) are sterile due to the second sterility barrier underlain by MAT heterozygosity (for a review, see [25]). Formation of viable hybrids was also reported for certain combinations of auxotrophic mutants of Metschnikowia ex-type strains, and mating-competent segregants were detected in a M. andauensis × M. fructicola hybrid [16,17] which implies that the reproductive (biological) isolation of the species of the clade may be incomplete.
Since both the barcoding results and the hybridisation tests raised doubts as to whether the taxonomic division of the clade is sufficiently justified, I set out to perform barcode analyses with more sequences and hybridisation tests with more strains. Certain phenotypic traits not tested or found variable in previous studies were also re-examined. The results show that the species of the clade cannot be distinguished from each other by any of the phenotypic, phylogenetic, and biological species concepts. Besides, their non-homogenised rDNA repeats seem to evolve by reticulation, a complex process that involves horizontal exchange and recombination of genetic elements, e.g., [15,17]. Therefore, I propose their unification into one species.
2. Materials and Methods
2.1. Strains and Culture Conditions
The Metschnikowia strains used in this study are listed in Table S1. The composition of the growth media YEA (yeast-extract agar), YEL (yeast-extract liquid), and SMA (synthetic minimal agar) were described previously [26]. Assimilation tests were performed on YNB plates supplemented with the test carbon sources. Sporulation was examined in cultures grown on vegetable juice medium prepared from diluted BIO Gemüsesaft (Josef Pölz, Bio-Produkte, 84,518 Garching an der Alz, Germany) and supplemented with 2% agar [16].
2.2. Phenotypic Examination
Pulcherrimin production was examined on YEA plates supplemented with various amounts of FeCl3. Colonies producing pulcherrimin turn red on this medium. The higher the concentration of FeCl3, the more intense the pigmentation. If pulcherrimin is also produced extracellularly, a coloured halo can be seen around the pigmented colony. Segregants producing no or less pulcherrimin form white or pale red sectors in the colonies. To test the segregants for stability, samples were taken from the sectors by sterile loops and inoculated onto a fresh medium supplemented with 0.02 mg/mL FeCl3. Sectors differing in pigmentation intensity in the cultures of the segregants indicated instability.
To test the strain for the ability to penetrate the agar medium, 10 μL of dense cell suspension (107 cells/mL) was dropped onto YEA. After two weeks of incubation at room temperature, the colony was washed off from the plate with water to visualise the intrusions formed beneath the colony. The cell morphology in the intrusions was examined microscopically.
For the examination of assimilation of carbon sources, 5 μL of cell suspension (106 cells/mL) was dropped onto the YNB plate supplemented with a carbon source. The plates were incubated at room temperature for two weeks.
Formation of asci and chlamydospores was tested on vegetable juice medium. A 10 μL measure of dense suspension of cells (107 cells/mL) was dropped onto the medium and incubated at 14 °C. After 4 weeks of incubation, samples of the culture were examined microscopically. For germination tests, ascospores were released from the asci by Zymolyase 20T treatment, (1.0 mg/mL, 25 °C, 3 h), spread on thin YEA films on glass slides, covered with cover slips and examined microscopically at regular intervals for 7 days.
2.3. Barcode Amplification and Sequencing
DNA was isolated from overnight cultures grown in YEL broth as described previously [27]. The D1/D2 domains and the ITS1-5.8S-ITS2 segments of the rDNA repeats were amplified with the primer pairs NL1-NL4 [28] and ITS1-ITS4 [29], respectively. The ACT1 sequences were amplified as described in [13]. The primer pairs used for the amplification of barcode segments of the RPB2 were RPB2F5 (aggagcctgacgacaga), RPB2R5 (gcgtgaaggtgtgtgtgga, PBR2FreF (gaggaggcccagctgaaggt), and PBR2FreR (ggtaagctttttgaataagag). The amplified fragments were sequenced in both directions using the same primer pairs. The sequences were deposited in GenBank under accession numbers listed in Tables S2 and S3.
2.4. Sequence Analysis
The accession numbers of the rDNA barcode sequences (‘primary barcodes’) used in this study are listed in Table S2. The accession numbers of the ACT1, TEF1, and RPB2 barcode sequences (‘secondary barcodes’) are listed in Table S3.
Multiple alignments were produced with the Clustal W 1.7 [30] or the MUSCLE 3.8.31 [31] algorithms. Maximum-likelihood phylogenetic trees were generated from the alignments with the PhyML 3.0 algorithm [32] using the best model suggested by the Akaike information criterion (AIC) in jMedelTest version 1.0.2 [33].
2.5. Hybridisation and Segregation
Mutants of different (complementary) auxotrophic markers were hybridised by the replica-plate method described previously [16]. Briefly, the mutants were streaked on YEA plates, incubated for 3 days, then replica-plated onto fresh YEA plates perpendicularly to each other to produce grids of prints in which the mutants intersected each other. After 7 days of incubation, the grids were replica-plated onto SMA plates. After two weeks of incubation, individual prototrophic (hybrid) colonies were isolated from the intersections and maintained on SMA plates.
The hybrids were tested for segregation by cultivating their cultures on YEA plates where both the protrophic hybrid cells and the auxotrophic segregants could propagate. After three days, samples were taken from the cultures and spread on YEA plates to obtain at least 1000 colonies. The colonies were replica-plated on SMA plates to identify auxotrophic segregants. The segregants were tested for auxotrophies by inoculating them onto SMA plates supplemented with compounds for which the parental strains were auxotrophic.
3. Results
3.1. Phenotypic Examination: Lack of Taxonomically Relevant Differences
In the taxonomic descriptions, the type strains of the clade had highly similar phenotypic properties [5,6,11,34]. Since different laboratories used different test methods and/or certain strains were not tested for the utilisation of complete sets of carbon sources, the assimilation tests were repeated in this study. All ex-type strains assimilated the same subset of carbon sources, albeit there were slight differences in the efficiency of the assimilation of glucitol, D-gluconate, mannitol, methyl-alpha-D-glucoside, salicin, L-sorbose, trehalose, and D-xylose. The differences were too small for reliable and reproducible differentiation of strains. When grown on YEA plates, all ex-type cultures formed pseudohyphae capable of invasive growth in the medium associated with the formation of blastoconidia along the pseudohyphae (Figure 1A–C). All cultures grown on the vegetable medium contained chlamydospores (5–60%) (Figure 1D) and asci (1–14%) (Figure 1F). The asci contained two, not always clearly discernible ascopores. Ascospores were released from asci of the ex-type strains of M. andauensis, M, fructicola, M. pulcherrima, M. shanxiensis, M sinensis, and M. ziziphicola by Zymolyase treatment and tested for germination. No germination was observed among the 24–84 ascospores examined for each strain. On the vegetable medium, cell pairs connected with protuberances reminiscent of conjugation tubes formed by cells of opposite mating types in other yeast species were also observed at low frequency in all ex-type cultures (Figure 1E).
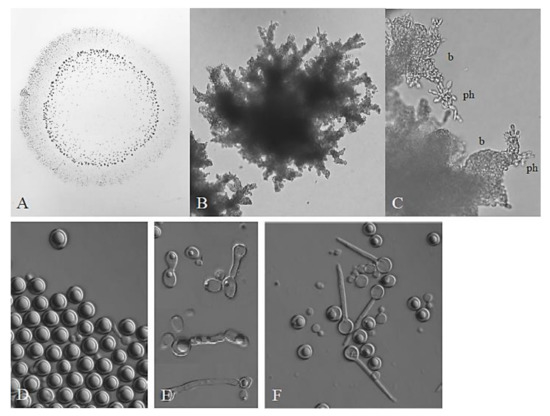
Figure 1.
Morphology. (A) Invasive growth (penetration the medium) made visible by washing off the colony from the surface of the medium. Penetrating structures are visible as dark spots. (B) Penetrating pseudomycelium. (C) Pseudohyphae growing in the medium. Ph: pseudohypha; b: blastoconidia. (D) Chlamidospores formed on vegetable agar. (E) Conjugation-like connections between cells. (F) Asci.
3.2. Pulcherrimin Production: A Variable and Unstable Trait
The formation of the maroon-red complex pulcherrimin is a characteristic feature of the clade [10,34]. Pulcherrimin accumulation in the cells pigments the colonies on most laboratory media. To find out whether the intensity of pulcherrimin synthesis can be used in the differentiation of the species, in this study the ex-type cultures of M. andauensis, M. fructicola, M. pulcherrima, M. shanxiensis, M. sinensis, and M. ziziphicola were inoculated on the yeast extract medium YEA supplemented with various amounts (0, 0.005, 0.01, 0.02, 0.04 mg/mL) of FeCl3. As expected, all formed pigmented colonies after 3–4 days of incubation on the media supplemented with iron, and the intensity of pigmentation increased with the concentration of the salt (examples are shown in Figure 2).
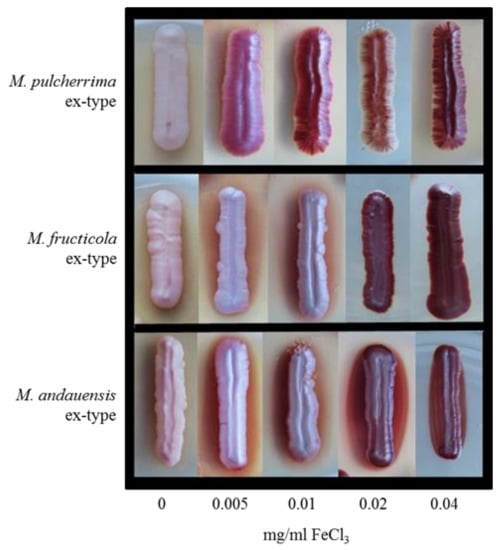
Figure 2.
Effect of FeClš concentration on pigmentation. The cultures shown are ex-types of M. pulcherrima CBS5833T, M. fructicola CBS 8853T, and M. andauensis CBS 10809T, respectively.
M. pulcherrima and M. sinensis formed darker colonies than the other strains at the lowest FeCl3 concentration, but at 0.04 Mg/mL all cultures were equally pigmented. However, the weaker colony pigmentation does not necessarily imply less efficient pulcherrimin synthesis. It can also be due to the formation of some of the pigment in the medium around the colonies (generating a pigmented halo). Upon prolonged incubation, less pigmented sectors appeared at the edges of all cultures (Figure 3). When isolated and inoculated onto fresh medium, these sectors (segregants) formed pale or pink cultures. As the pigment is the chelate of ferric irons and pulcherriminic acid [1], the reduced pigmentation implies that pulcherriminic acid was synthesised at a reduced rate in these segregants. The dark sectors formed dark colonies when inoculated onto fresh medium. However, neither of the segregants was stable; their colonies occasionally contained segments or spots differing from the rest of the colony in pigmentation intensity (Figure 3). Because of the instability of colony pigmentation and the pigmented halos around the colonies, the intensity of pulcherrimin production cannot be used as a distinctive taxonomic trait either.

Figure 3.
Segregation of pulcherrimin production (pigmentation intensity).
3.3. Analysis of Primary Barcodes
3.3.1. Analysis of D1/D2 Sequences: A Clade-Specific Consensus Sequence Can Be Defined
The sequences containing D1/D2 LSU domains of the type strains of the pulcherrima clade were downloaded from the databases and compared with each-other by generating multiple alignments. For the M. andauensis, M. fructicola, and M. pulcherrima type strains, multiple slightly different sequences were deposited in the databases by different laboratories and certain sequences contained ambiguous nucleotides. The presence of ambiguous, mostly dimorphic (SND), rarely polymorphic (SNP) sites, and the intra-strain differences hinted at rDNA heterogeneity (rDNA repeat diversity) in the genomes of the strains. For the rest of the type strains, single unambiguous sequences were deposited. Since the database entries do not provide information about the ‘pre-submission processing’ of the nascent Sanger sequences, the D1/D2 domains of six type strains were resequenced in this study. The accession numbers of the new sequences of M. pulcherrima CBS 5833T, M. fructicola CBS 8853T, M. sinensis CBS 10357T, M. ziziphicola CBS 10357T, M. shanxiensis CBS 10359T, and M. andauensis CBS 10809T are listed in Table S2. All newly generated sequences contained ambiguous nucleotides and differed from all amplicon sequences previously deposited in databases for these strains. As a control, the D1/D2 domain of the ex-type strain (CBS 9803T) of the closely related species M. chrysoperlae was also sequenced; its sequence had no ambiguous positions and was identical with that available in the GenBank database.
In previous studies, individual D1/D2 domain sequences were cloned from six ex-type strains and the clones representing individual rDNA repeats showed high intrastrain diversity and formed a continuous cloud in the phylogenetic analyses [10,15,17]. In this study, all cloned sequences were aligned with all non-cloned amplicon sequences (including those produced here) (Figure 4A). The cloned sequences differed from one another in positions corresponding to the positions of ambiguous nucleotides in the sequences obtained by direct amplicon sequencing. From the multiple alignment of all sequences, a consensus D1/D2 sequence could be generated (Figure 4B) in which the variable positions were denoted with IUPAC codes of alternating nucleotides. A Blast search with this sequence in GenBank identified all type strains of the clade among the most similar entries with 91.78–94.39% identity (100% coverage, E value: 0.0). The most similar related type-strain sequence was that of M. pimensisCBS:9805T (87.28% identity). Thus, despite its high number of variable (mostly dimorphic) sites, the deduced consensus sequence is suitable for the assignment of isolates to the pulcherrima clade.

Figure 4.
Comparison of the D1/D2 sequences of the pulcherrima clade. (A) Multiple alignment of variable segments. Only examples of the database type-strain sequences are shown (GenBank accession numbers are given in parentheses). The cloned sequences are consensus sequences of clones. Numbering of the positions corresponds to that of the M. pulcherrima sequence NG_075437. Ma: M. andauensis; Mc: M. citriensis; Mf: M. fructicola; Ml: M. leonuri; Mpe: M. persimmonesis; Mpu: M. pulcherrima; Mr: M. rubicola; Msh: M. shanxiensis; Msi: M. sinensis; Mz: M. zizyphicola; M: A or C; R: A or G; Y: C or T; V: A or C or G; W: A or T. (B) Consensus sequence of the entire D1/D1 domain. Variable positions are highlighted with red colour.
3.3.2. Analysis of ITS Sequences: No Clade-Specific Consensus Sequence Can Be Defined
At the beginning of this study, ITS1-5.8S-ITS2 (Internal Transcribed Sequence 1, 5.8 S rRNA coding region and Internal Transcribed Sequence 2) sequences were available in databases for all but two species of the clade. These sequences were diverse in the ITS1 segments but almost identical in the ITS2 segments. No ITS sequences were deposited for the M. andauensis and M. fructicola type strains. The attempts to sequence their ITS regions in a previous work [16] and in the current work were only partially successful. The reactions primed with the ITS4 primer (complementary to the end of ITS2) produced ITS2 sequences containing a few ambiguous nucleotides, but the ITS1 segments were practically unreadable. When the ITS1 primer was used for sequencing, both ITS1 and ITS2 were full of ambiguous positions. The high number of ambiguous nucleotides in these sequences was puzzling because the sequences of individual repeats cloned in a previous study from these strains differed by much lower numbers of substitutions [16]. Obviously, these substitutions cannot account for all the ambiguous nucleotides of the amplicon sequences. Rather, they could be attributed to the presence of indels in the ITS segments of certain rDNA repeats. When the cloned sequences were realigned in this study, single-nucleotide indels were detected in some of them. Obviously, the shifts caused by these indels during the sequencing process resulted in mass ambiguities in the final amplicon sequences.
Figure 5A shows the alignment of the database ITS1 and ITS2 sequences of the type strains, the consensus sequences of the individual repeats cloned from six ex-type strains in a previous study, and the ITS2 segments of the M. andauensis and M. fructicola ex-type strains sequenced in this study. From the alignment, a consensus ITS1 and a consensus ITS2 sequence were generated for the entire clade (Figure 5B). The former contained a very high number of di- and polymorphic sites. Both turned out to be unsuitable for taxonomic identification by Blast searches in sequence databases. ITS1 is not recognised as nucleotide sequence by the NCBI Blast algorithm (due to the high proportion of symbols different from A, G, T, and C, Blast recognises it as an amino-acid sequence) and ITS2 cannot distinguish the pulcherrima-clade type strains from the type strains of closely related species.
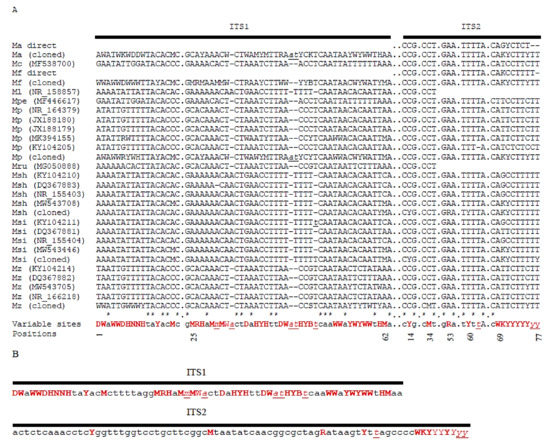
Figure 5.
Comparison of the ITS sequences of the pulcherrima clade. (A) Multiple alignment of variable segments. Only examples of the database type-strain sequences are shown (GenBank accession numbers are given in parenthesis). Direct: sequence obtained in this study by direct sequencing of the PCR amplicon. The cloned sequences are consensus sequences of clones. Numbering of the positions corresponds to that of the M. pulcherrima sequence NR_164379. Ma: M. andauensis; Mc: M. citriensis; Mf: M. fructicola; Ml: M. leonuri; Mpe: M. persimmonesis; Mpu: M. pulcherrima; Mr: M. rubicola; Msh: M. shanxiensis; Msi: M. sinensis; Mz: M. zizyphicola; M: A or C; R: A or G; Y: C or T; V: A or C or G; W: A or T. Nucleotide symbols are lower-case and underlined at positions in which indels or insertions occur. (B) Consensus sequences of the entire ITS1 and ITS2 segments. Variable positions are highlighted with red colour.
3.4. Analysis of Secondary Barcodes
3.4.1. Analysis of ACT1 (Actin) Sequences: Insufficient Variability
To explore the diversity of the ACT1 barcodes in the clade, all ACT1 sequences available in databases (Table S3) were downloaded and examined for the presence of ambiguous nucleotides and for interstrain differences. For the M. andauensis type strain, two different sequences are available in GenBank; they have ambiguous nucleotides in 8 and 10 positions, respectively. The database sequences of a group of related strains isolated in the same Austrian wine-growing region [13] and the M. shanxiensis, M. sinensis, and M. ziziphicola type strains also had a few ambiguous nucleotides, mostly in corresponding positions. However, when re-sequenced in this study, the latter three strains had clean sequences which only differed from each other by one nucleotide. The ACT1 genes had identical sequences in all sequenced genomes, including those of the type strains and only differing from those of all but one type (M. andauensis) strains by 1 to 2 nucleotides. The very low interstrain diversity precludes the application of ACT1 to taxonomic differentiation of the type strains of the pulcherrima clade.
When all type-strain and non-type-strain sequences were aligned, nucleotide differences were detected at 15 sites, but these differences were mainly due to the high number of ambiguous positions of the M. andauensis sequences. All these sites were dimorphic; no transversion was detected and the C/T transitions were more frequent than the A/G transitions. All but two dimorphic sites were third nucleotides of triplets encoding amino acids. It can be assumed that the strains having ambiguous positions have two (or more) different ACT1 genes. This assumption seems to be corroborated by the genome sequences of the M. fructicola type strain and the strain AF47 which both have two ACT1 genes, although one is incomplete in both cases. The consensus sequence derived from the multiple alignment identified all type strains of the clade in the GenBank Blast search (96.61% to 98.57% sequence identity), but did not distinguish them from M. picachoensis (97.32% sequence identity) and M. rancensis (96.96% sequence identity).
3.4.2. Analysis of TEF1 (Translation Elongation Factor 1-α) Sequences: Intragenomic Diversity Obscures Species Boundaries
The TEF1 barcode sequences turned out to be more diverse. Although listed in Table S3, the TEF1 sequences of certain sequenced genomes could not be used in the analysis because they were incomplete and did not overlap all regions regularly used as barcode sequences. Large parts of the TEF1 ORFs were missing in unitig 43 of the strain M. fructicola CBS8853T, unitig 43 of AP47, the NODES 547 and 1051 of UCD127, and scaffold 137 of Bath1 (the deposited M. fructicola CBS8853T genome sequence is organised in 93 unitigs, high-confidence contigs; the deposited Bath1 genome sequence is organised in 524 scaffolds, ordered sets of contigs). In the alignment of 890 nt-long segments of the rest of the TEF1 sequences, C/T, A/G, and G/C dimorphism were detected at 22, 2, and 2 sites, respectively. All dimorphic sites were third nucleotides of triplets, and none of the substitutions caused amino-acid replacements in the proteins. Hence, these differences in the gene sequences are unlikely to affect the fitness of the organisms. Remarkably, the M. fructicola CBS 8853T, M. citriensis FL01T, and AP47 genomes had 3 to 6 TEF1 genes which differed in sequence (intrastrain diversity). The phylogenetic analysis (Figure 6) grouped the sequences into clusters that did not correspond to their origin. The genes showing higher interstrain than intrastrain (intragenomic) similarity are most probably not paralogs of the other genes of the genome, but their orthologs of different evolutionary histories.
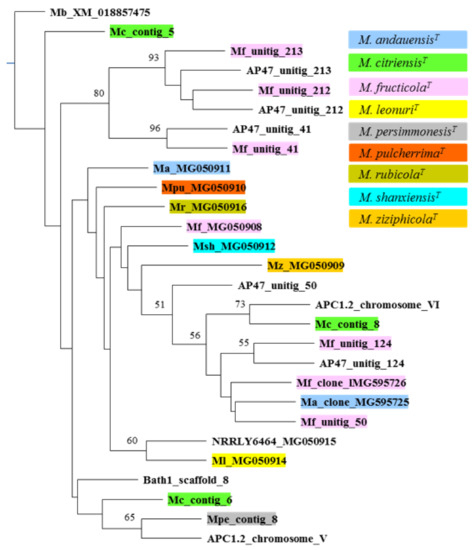
Figure 6.
The neighbour joining tree of TEF1 barcode sequences. Sequences from type materials are marked with colours (see colour key at the top right). Boostrap values higher than 50 are shown at nodes.
3.4.3. Analysis of RPB2 (Second Largest Subunit of RNA-Polymerase II) Sequences: Genomes Contain Genes of Different Phylogenetic Relationships
The RPB2 barcode sequences of the pulcherrima clade available in databases are diverse in length and their locations within the genes. The sequences analysed in this work are listed in Table S3. Their multiple alignment identified a 398-long segment (segment I: 1183–1580 in Figure 7A) which was present in most of them and also in most genes extracted from the whole-genome sequences. The genes of contig 43 of the genome Bath1 and NODE 247 of the genome UCD 127 lacked it, most probably due to incompleteness of the genome sequences. As no M. andauensis RPB2 sequences were available at the beginning of this study, this region was amplified and sequenced in this study from an ex-type culture and from its two auxotrophic mutants generated for hybridisation purposes (see below).

Figure 7.
Neighbour joining trees of RPB2 segments. (A) Segments used for analysis. (B) Tree inferred from the sequences of segment I. Outgroup: M. bicuspidata Mb_KC859681. (C) Tree inferred from the sequences of segment II. Outgroup: M. bicuspidata Mb_XM_018855993. On both trees: sequences from type materials are marked with colours (for the colour key, see Figure 6) and boostrap values higher than 50 are shown at nodes.
All but one of these sequences contained ambiguous nucleotides in positions corresponding to third nucleotides of triplets. The exception was the M. rubicola type strain. All these sites were dimorphic (SND), in which either C/T (12) or A/G (6) transitions occurred. No transversion and no polymorphic sites (SNP) were detected. The intrastrain variability hints at the presence of two or more gene copies in the genomes. To test this assumption, individual fragments were cloned from the ex-type cultures of M. andauensis and M. fructicola (accession numbers are listed in Table S3). The cloned sequences contained no ambiguous nucleotides. On the phylogenetic tree (Figure 7B), both the clones and the mutants grouped together with the corresponding type strains, but the M. fructicola sequences shared a branch with their M. leonuri counterpart and with one of the genes of the M. citriensis genome.
Because of the close relationship of its sequences to sequences of other type strains, a longer upstream region (segment II: 460-1264 in Figure 7A) was also sequenced for the M. fructicola ex-type strain, both directly and after cloning. As expected, the direct amplicon sequencing resulted in a sequence containing numerous ambiguous nucleotides, but the clones were clean (for accession numbers, see Table S3). When compared with each other by aligning all new sequences, five types of cloned sequences could be distinguished that differed from each other at positions corresponding to the ambiguous sites of the amplicon sequence. Again, all variability was due to SNDs (7 C/T, 7 A/G, 3 A/C, 2 G/C, and 2 A/T) in third positions of triplets, causing no amino-acid substitutions. On the PhyML tree (Figure 7C), they formed two well-separated clusters. One of the clusters shared a branch with the sequence located in unitig 185 of the genome sequence of the ex-type strain.
3.5. Hybridisation and Segregation
As shown above, the tested ex-type strains sporulated poorly and no spore germination was observed. There can be several reasons for the low sporulation efficiency and the inability of the spores to germinate. One is the complexity of genome structures. The results of the analysis of the secondary barcode genes in this study (see above) and 35 non-barcode genes described in a recent comparative analysis [17] pointed to segmental mosaic genome structures in the strains of the clade. Segmental genomes are usually poor at meiosis because of poor chromosome paring in Meiosis I, and the abruption of meiosis results in low gamete viability (e.g., [35,36]).
Mosaic (chimeric) genomes can evolve from hybrids by gradual loss of segments of the subgenomes during propagation of the hybrid cells (postzygotic genome evolution). Similar processes can be assumed to take place in the pulcherrima clade. Recently, the ex-type strains of M. andauensis and M. fructicola were found to be able to form hybrids with the M. pulcherrima ex-type strain (e.g., [16,17]). In this study, prototrophic hybrids were also obtained between auxotrophic mutants of M. pulcherrima CBS 5833T and auxotrophic mutants of the rest of the ex-type strains (all mutants are listed in Table S1) but not in all possible mutant combinations. The M. sinensis and M. ziziphicola mutants only formed hybrids with the M. pulcherrima mutant of a mating type. Apparently, the mutants of these species had alpha mating type. Consistently, no M. sinensis × M. ziziphicola hybrids were obtained. The microscopic examination of six M. pulcherrima × M. andauensis, five M. pulcherrima × M. fructicola, two M. pulcherrima × M. shanxiensis, two M. pulcherrima × M. sinensis, and six M. pulcherrima × M. ziziphicola hybrids, numerous ‘intrastrain’ M. pulcherrima and M. andauensis hybrids and the parental mutants cultured on vegetable agar revealed no increase in sporulation efficiency in the hybrids.
Two 1591 M. pulcherrima his− × 1622 M. ziziphicola ade− and two 1591 M. pulcherrima his− × 1605 M. shanxiensis asp− hybrids were tested for segregation. After cultivating their cultures for two days under non-selective conditions, cells were spread on a complete medium to obtain detached colonies. Colonies with auxotrophic markers of the parental mutants (4 and 6 among 989 and 1201 colonies, respectively) segregated from both M. pulcherrima × M. ziziphicola hybrids. Two his− and two ade− segregants were then tested for hybridisation with parental mutants of complementary auxotrophies. Prototrophs were formed in all backcrosses. The M. pulcherrima × M. shanxiensis hybrids did not segregate.
4. Discussion
The experimental results and the analysis of barcode sequences presented in this study reinforce doubts about the validity of the taxonomic division of the pulcherrima clade of Metschnikowia.
When the utilisation of carbon sources was examined, all tested ex-type cultures assimilated the same set of compounds and only slightly differed in growth intensity. The morphological examinations also found the ex-type strains very similar. All formed chlamydospores and asci and could penetrate the solid medium by means of pseudohyphal growth. All produced the pigment pulcherrimin, albeit with somewhat different intensities. However, the differences in the production of the pigment turned out to be unstable. The cultures segregated into clones of different pigmentation intensities and the segregants were not stable either. Because of the absence of characteristic differences, phenotypic species boundaries cannot be defined in the clade.
A similarity search in databases identified multiple D1/D2 and ITS sequences for most type strains that differed at certain positions or contained ambiguous nucleotides. In this study, the rDNA segments of the ex-type strains of six species were re-sequenced, including those originally delimited on the basis of single cloned sequences. All newly generated amplicon sequences contained ambiguous positions. In other cases, the sequences used for species delimitation differed from those found in the whole-genome sequences of the ex-type strains of the species. Thus, all known species of the clade have non-homogenised heterogeneous rDNA repeats. This conclusion is consistent with previous studies that revealed enormous internal diversity among individual D1/D2 and ITS segments cloned from the genomes of these strains (e.g., [17]). On the phylogenetic trees and networks, the D1/D2 and ITS sequences of the strains had intermixed positions (forming no distinct clusters separated by clear barcode gaps), indicating conspecificity [17]. Additionally, the secondary barcode analysis did not identify clear barcode gaps between the type strains. Most strains harboured more than one copy of the barcode genes that usually differed in sequence. Like the rDNA repeats, these copies frequently did not group with each other on the trees but with their counterparts from other strains. A recent analysis of the genome sequences of three ex-type strains and three strains of uncertain position in the clade found that other protein-encoding genes also have copies that differ more from each other than from their counterparts in other genomes [17]. Thus, neither phylogenetic boundaries can be clearly defined in the pulcherrima clade.
The intragenomic (intrastrain) barcode diversity implies that segmental chimeric genomes are common in the clade. Chimeric genomes consisting of sequences of different phylogenetic histories can evolve from hybrids of organisms (strains) of different genomes by postzygotic evolution of the hybrid genomes. For example, in the hybrids of the Saccharomyces species, the hybrid genomes are usually unstable and convert to chimeric genomes during meiotic and mitotic divisions of the cells by gradually losing large parts of the subgenomes (for a review, see [25]). The chimeric composition of the genomes of the pulcherrima-clade strains can be attributed to a similar process. Like the cultures of the Saccharomyces species, the ex-type cultures of the tested pulcherrima-clade species also produced viable hybrids and the spores (gametes) of the hybrids also were unable to germinate. However, the analogy between the two groups of strains is only partial because, unlike the non-chimeric (“pure”) genomes of the natural Saccharomyces species, the pulcherrima-clade type strains seem to have composite genomes. This is probably the reason why their ascospores do not germinate, whereas the ascospores of the natural Saccharomyces species are viable. However, despite the fact that the spores appear unviable, the hybrids investigated in this and a previous study [16] segregated, and the segregants were mating-competent. As the culturing conditions were not favourable for sporulation, these segregants must have arisen by mitotic segregation. This assumption is consistent with the observations of Pitt and Miller [37] who found that the life cycle of the M. pulcherrima type strain is parasexual. Due to the hybridisation–segregation cycle, the pulcherrima-clade strains can evolve in interactions including horizontal exchange of genetic material and reticulation of repeat sequences. Thus, the species of the clade do not meet the criteria of the biological species concept either.
In conclusion, the type strains of the species of the pulcherrima clade cannot be distinguished from each other by the criteria of any of the phenotypic, phylogenetic, and biological species concepts. The lack of reliable phenotypic differences and barcode gaps hampers the taxonomic identification of new isolates. The high numbers of misidentified records and entries labelled as “Metschnikowia sp.” or “Metschnikowia aff.” among the pulcherrima-clade strains in sequence databases can be attributed to this problem. Therefore, I propose that the species of the pulcherrima clade be merged into one species.
As the unified taxon the name M. pulcherrima has to be chosen because according to the International Code of Nomenclature for Algae, Fungi, and Plants [38] the oldest epithet has nomenclatural priority when one of the competing legitimate names is to be chosen. In this case, the oldest name is M. pulcherrima. The other species names have to be relegated to synonyms of M. pulcherrima. However, in contrast to other mergers of fungal species which were usually based on high barcode similarity or sequence identity, here diverse sequences coexist even within strains. Which barcode sequences can then be assigned to the merged species? The pragmatic solution is to retain all amplicon sequences deposited in the databases for the ex-type strains of CBS 5833. These sequences can be considered various (slightly different) consensus sequences of all repeats and copies of the barcode genes in the CBS 5833T genome. The problem with this option is that even closely related strains can differ significantly in the D1/2 and ITS sequences if they differ in the proportion of the different rDNA repeats (that determine the ‘consensus’ Sanger sequence) in their genomes. The unorthodox alternative could be the use of consensus barcode sequences inferred from all known gene and repeat sequences. I compiled one consensus sequence for each of the D1/D2 domain, the ITS1, and the ITS2 segments. The D1/D2 sequence, despite its numerous dimorphic positions, is suitable for the taxonomic identification of pulcherrima-clade strains. However, the ITS1 consensus sequence contains too many ambiguous positions to be recognisable as DNA sequence for the GenBank blast algorithm. The ITS2 consensus sequence is not specific enough for the differentiation of the clade from other species of the genus. The usage of such consensus sequences in taxonomic identification is not only impossible because of these problems, but also because databases do not accept consensus sequences. Thus, the typological, type-centric approach to species delimitation and strain identification has limits in this group of yeasts. In their case, Mayr’s population approach (“population thinking”) is more appropriate [39,40]. Population thinking rejects the use of a central representative type, and emphasizes the variation among individuals. According to Mayr, a species is a genetic unit consisting of a large intercommunicating gene pool.
The proposed unification of the clade in M. pulcherrima requires emendation of its original taxonomic description. I propose the following new description.
Metschnikowia pulcherrima J.I. Pitt & M.W. Miller (1968) emend M. Sipiczki
The description is as before (Pitt and Miller, 1968) with the following addition:
Pseudohyphae capable of invasive growth are formed on solid media. During invasive growth, blastoconidia are formed on the pseudohyphae. The pigment pulcherrimin can be produced both intracellularly (pigmented colonies) and extracellularly (pigmented zones around colonies), but certain strains only produce it intracellularly. The intensity of pigmentation is variable and unstable. Colonies can segregate into more and less pigmented sectors. Reduction in pigment formation by segregation is reversible. Strains can have antimicrobial activity which correlates with pigmentation intensity. Hybridisation can take place between sexually competent strains and the hybrids can segregate to form mating-competent segregants. Genomes are variable in size and can have chimeric structures associated with intragenomic barcode diversity.
MycoBank: MB#334124
New synonyms:
Metschnikowia andauensis O. Molnar & H. Prillinger, Systematic and Applied Microbiology 28 (8): 723 (2005) [MB#356956]
Metschnikowia fructicola C.P. Kurtzman & S. Droby, Systematic and Applied Microbiology 24 (3): 396 (2002) [MB#375792]
Metschnikowia leonuri C.P. Kurtzman, C.J. Robnett & E. Basehoar, Antonie van Leeuwenhoek 111 (11): 2026 (2018) [MB#823166]
Metschnikowia persimmonesis Y.M. Kang, J.E. Choi, R. Komakech, J.H. Park, D.W. Kim, K.M. Cho, S.M. Kang, S.H. Choi, K.C. Song, C.M. Ryu, K.C. Lee & J.S. Lee, AMB Express Journal 7 (199): 1 (2017) [MB#823579]
Metschnikowia rubicola C.P. Kurtzman, C.J. Robnett & E. Basehoar, Antonie van Leeuwenhoek 111 (11): 2014 (2018) [MB#823165]
Metschnikowia shanxiensis M.L. Xue & L.Q. Zhang, International Journal of Systematic and Evolutionary Microbiology 56: 2249 (2006) [MB#501113]
Metschnikowia sinensis M.L. Xue & L.Q. Zhang, International Journal of Systematic and Evolutionary Microbiology 56: 2248 (2006) [MB#529862]
Metschnikowia ziziphicola M.L. Xue & L.Q. Zhang, International Journal of Systematic and Evolutionary Microbiology 56:2248 (2006) [MB#529862]
Metschnikowia citriensis Y. Liu, S.X. Yao, L.L. Deng, J. Ming & K.F. Zeng, Biological Control 125: 17 (2018) [MB#838084] is not listed among the new synonyms because this name is invalid
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/taxonomy2010009/s1, Table S1: List of strains; Table S2: Accession numbers of rDNA (primary) barcode sequences; Table S3: Accession numbers of secondary barcode sequences.
Funding
This research was funded by the National Research, Development, and Innovation Office of Hungary (grant nos. 2020-1.1.2-PIACI-KFI-2020-00130 and 2019-2.1.11-TÉT-2019-00001).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All sequences produced in this study are available in GenBank under accession numbers listed in Tables S2 and S3.
Acknowledgments
The author thanks Anita Kovacs and Anita Olah for excellent technical assistance.
Conflicts of Interest
The author declares no conflict of interest.
References
- Cook, A.H.; Slater, C.A. The structure of pulcherrimin. J. Chem. Soc. 1956, 4133–4135. [Google Scholar] [CrossRef]
- Sipiczki, M. Metschnikowia strains isolated from botrytized grapes antagonize fungal and bacterial growth by iron depletion. Appl. Environ. Microbiol. 2006, 72, 6716–6724. [Google Scholar] [CrossRef] [PubMed]
- Kurtzman, C.P.; Droby, S. Metschnikowia fructicola, a new ascosporic yeast with potential for biocontrol of postharvest fruit rots. Syst. Appl. Microbiol. 2001, 24, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Türkel, S.; Korukluoglu, M.; Yavuz, M. Biocontrol activity of the local strain of Metschnikowia pulcherrima on different postharvest pathogens. Biotechnol. Res. Int. 2014, 2014, 1–6. [Google Scholar] [CrossRef]
- Kang, Y.M.; Choi, J.E.; Komakech, R.; Park, J.H.; Kim, D.W.; Cho, K.M.; Kang, S.M.; Choi, S.H.; Song, K.C.; Ryu, C.M.; et al. Characterization of a novel yeast species Metschnikowia persimmonesis KCTC 12991BP (KIOM G15050 type strain) isolated from a medicinal plant, Korean persimmon calyx (Diospyros kaki Thumb). AMB Express 2017, 7, 199. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, S.; Deng, L.; Ming, J.; Zeng, K. Metschnikowia citriensis sp. nov., a novel yeast species isolated from leaves with potential for biocontrol of postharvest fruit rot. Biol. Control 2018, 125, 15–19. [Google Scholar] [CrossRef]
- Gore-Lloyd, D.; Sumann, I.; Brachmann, A.O.; Schneeberger, K.; Ortiz-Merino, R.A.; Moreno-Beltrán, M.; Schläfli, M.; Kirner, P.; Santos Kron, A.; Rueda-Mejia, M.P.; et al. Snf2 controls pulcherriminic acid biosynthesis and antifungal activity of the biocontrol yeast Metschnikowia pulcherrima. Mol. Microbiol. 2019, 112, 317–332. [Google Scholar] [CrossRef]
- Saravanakumar, D.; Spadaro, D.; Garibaldi, A.; Gullino, M.L. Detection of enzymatic activity and partial sequence of a chitinase gene in Metschnikowia pulcherrima strain MACH1 used as postharvest biocontrol agent. Eur. J. Plant Pathol. 2009, 123, 183–193. [Google Scholar] [CrossRef]
- Oro, L.; Feliziani, E.; Ciani, M.; Romanazzi, G.; Comitini, F. Volatile organic compounds from Wickerhamomyces anomalus, Metschnikowia pulcherrima and Saccharomyces cerevisiae inhibit growth of decay causing fungi and control postharvest diseases of strawberries. Int. J. Food Microbiol. 2018, 265, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Sipiczki, M. Metschnikowia pulcherrima and related pulcherrimin-producing yeasts: Fuzzy species boundaries and complex antimicrobial antagonism. Microorganisms 2020, 8, 1029. [Google Scholar] [CrossRef]
- Kurtzman, C.P.; Robnett, C.J.; Basehoar, E.; Ward, T.J. Four new species of Metschnikowia and the transfer of seven Candida species to Metschnikowia and Clavispora as new combinations. Antonie Van Leeuwenhoek 2018, 111, 2017–2035. [Google Scholar] [CrossRef]
- Pitt, J.I.; Miller, M.W. Sporulation in Candida pulcherrima, Candida reukaufii and Chlamydozyma species; their relationship with Metschnikowia. Mycologia 1968, 60, 663–685. [Google Scholar] [CrossRef]
- Molnar, O.; Prillinger, H. Analysis of yeast isolates related to Metschnikowia pulcherrima using the partialsequences of the large subunit rDNA and the actin gene; description of Metschnikowia andauensis sp. nov. Syst. Appl. Microbiol. 2005, 28, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.L.; Zhang, L.Q.; Wang, Q.M.; Zhang, J.S.; Bai, F.Y. Metschnikowia sinensis sp. nov., Metschnikowia zizyphicola sp. nov. and Metschnikowia shanxiensis sp. nov., novel yeast species from jujube fruit. Int. J. Syst. Evol. Microbiol. 2006, 56, 2245–2250. [Google Scholar] [CrossRef][Green Version]
- Sipiczki, M.; Pfliegler, W.P.; Holb, I.J. Metschnikowia species share a pool of diverse rRNA genes differing in regions that determine hairpin-loop structures and evolve by reticulation. PLoS ONE 2013, 8, e67384. [Google Scholar] [CrossRef]
- Sipiczki, M.; Horvath, E.; Pfliegler, W.P. Birth-and-death evolution and reticulation of ITS segments of Metschnikowia andauensis and Metschnikowia fructicola rDNA repeats. Front. Microbiol. 2018, 9, 1193. [Google Scholar] [CrossRef]
- Sipiczki, M. When barcoding fails: Genome chimerisation (admixing) and reticulation obscure phylogenetic and taxonomic relationships. Mol. Ecol. Resour. 2022. [Google Scholar] [CrossRef]
- Wheeler, Q.D.; Platnick, N.I. The Phylogenetic species concept (sensu Wheeler and Platnick). In Species Concepts and Phylogenetic Theory: A Debate; Wheeler, Q.D., Meier, R., Eds.; Columbia University Press: New York, NY, USA, 2000; pp. 55–69. [Google Scholar]
- Zachos, F.E. Species Concepts in Biology: Historical Development, Theoretical Foundations and Practical Relevance; Springer Nature: Cham, Switzerland, 2016. [Google Scholar]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M.; et al. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Boekhout, T.; Aime, M.C.; Begerow, D.; Gabaldón, T.; Heitman, J.; Kemler, M.; Khayhan, K.; Lachance, M.-A.; Louis, E.J.; Sun, S.; et al. The evolving species concepts used for yeasts: From phenotypes and genomes to speciation networks. Fungal Divers. 2021, 109, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Yurkov, A.; Alves, A.; Bai, F.Y.; Boundy-Mills, K.; Buzzini, P.; Čadež, N.; Cardinali, G.; Casaregola, S.; Chaturvedi, V.; Collin, V.; et al. Nomenclatural issues concerning cultured yeasts and other fungi: Why it is important to avoid unneeded name changes. IMA Fungus 2021, 12, 18. [Google Scholar] [CrossRef]
- Mayr, E. Speciation phenomena in birds. Am. Nat. 1940, 74, 249–278. [Google Scholar] [CrossRef]
- Naumov, G. Genetic identification of biological species in the Saccharomyces sensu stricto complex. J. Ind. Microbiol. 1996, 17, 295–302. [Google Scholar] [CrossRef]
- Sipiczki, M. Interspecies hybridisation and genome chimerisation in Saccharomyces: Combining of gene pools of species and its biotechnological perspectives. Front. Microbiol. 2018, 9, 3071. [Google Scholar] [CrossRef]
- Sipiczki, M. Pichia bruneiensis sp. nov., a biofilm-producing dimorphic yeast species isolated from flowers in Borneo. Int. J. Syst. Evol. Microbiol. 2012, 62, 3099–3104. [Google Scholar] [CrossRef]
- Sipiczki, M. Candida zemplinina sp. nov., an osmotolerant and psychrotolerant yeast that ferments sweet botrytized wines. Int. J. Syst. Evol. Microbiol. 2003, 53, 2079–2083. [Google Scholar] [CrossRef]
- O’Donnell, K. Fusarium and its near relatives. In The Fungal Holomorph: Mitotic, Meiotic and Pleomorphic Speciation in Fungal Systematics; Reynolds, D.R., Taylor, J.W., Eds.; CAB International: Wallingford, UK, 1993; pp. 225–233. [Google Scholar]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols. A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Thompson, J.D.; Higgions, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package), Version 3.67; Distributed by the Author; Department of Genome Sciences, University of Washington: Seattle, DC, USA, 2007. [Google Scholar]
- Lachance, M.A. Metschnikowia Kamienski (1899). In The Yeasts. A Taxonomic Study; Kurtzman, C.P., Fell, J.W., Boekhout, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; pp. 575–620. [Google Scholar]
- Kaur, D.; Singhal, V.K. Meiotic abnormalities affect genetic constitution and pollen viability in dicots from Indian cold deserts. BMC Plant Biol. 2019, 19, 10. [Google Scholar] [CrossRef]
- Mason, A.S.; Wendel, J.F. Homoeologous exchanges, segmental allopolyploidy, and polyploid genome evolution. Front. Genet. 2020, 11, 1014. [Google Scholar] [CrossRef]
- Pitt, J.I.; Miller, M.W. The parasexual cycle in yeasts from the genus Metschnikowia. Mycologia 1970, 62, 462–473. [Google Scholar] [CrossRef]
- Turland, N.J.; Wiersema, J.H.; Barrie, F.R.; Greuter, W.; Hawksworth, D.L.; Herendeen, P.S.; Knapp, S.; Kusber, W.-H.; Li, D.-Z.; Marhold, K.; et al. International Code of Nomenclature for algae, fungi, and plants (Shenzhen Code) adopted by the Nineteenth International Botanical Congress Shenzhen, China, July 2017; Regnum Vegetabile; Koeltz Botanical Books: Glashütten, Germany, 2018; Volume 159. [Google Scholar]
- Mayr, E. Animal Species and Evolution; Harvard University Press: Cambridge, MA, USA, 1963. [Google Scholar]
- Mayr, E. Populations, Species, and Evolution; Harvard University Press: Cambridge, MA, USA, 1970. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).