Abstract
In recent years, amino-gem-bisphosphonic acids and their esters have been considered a family of compounds of great chemical and pharmacological interest due to their important biological properties and their value as key synthons in the synthesis of more complex molecules with biological interest. This explains why several research groups are interested in developing new methods for the preparation of these compounds. Therefore, we would like to report here a summary of the synthetic strategies published in the last fifteen years for the synthesis of acyclic and heterocyclic α-, β- and γ-amino-gem-bisphosphonates, as well as their application in the preparation of selected compounds of chemical and pharmacological interest. This information can be of general knowledge to researchers working in this area, as it provides the starting point for new methods and applications of these compounds.
1. Introduction
In recent decades, organophosphorus compounds have been the focus of attention of several research groups, due to the chemical and pharmacological importance of this type of compound. For example, α-aminophosphonic, α-amino-H-phosphonic and α-amino-C-phosphonic are possibly the most important analogs of α-amino acids, in which the planar carboxylic acid group (-CO2H) is replaced by a sterically more demanding tetrahedral phosphonic acid group [-P(O)(OH)2] or phosphinic acid [-P(O)(OH)R]. These analogs represent an important class of compounds with multiple applications in medicinal and organic chemistry. This is due to the ability of phosphonic and phosphinic groups to mimic the high-energy transition state of peptide bond hydrolysis, enabling them to act as enzyme inhibitors or receptor ligands in pathological conditions associated with amino acid metabolism [1,2,3,4,5]. Due to the importance of these compounds, excellent methods for their preparation have been published in the last decades [6,7,8,9,10,11,12,13,14,15,16,17,18]. Additionally, gem-bisphosphonic acids and gem-bisphosphonates are analogs of pyrophosphoric acid and pyrophosphate, respectively, in which the hydrolytically labile P-O-P oxygen bridge is replaced by the hydrolysis-resistant P-C-P linkage, making these compounds metabolically more stable. Furthermore, replacing the methylene hydrogen P-CH2-P with other substituents such as the amino group gives rise to compounds of high chemical and biological interest such as the α-amino gem-bisphosphonic acids and their corresponding phosphonic esters (Figure 1).
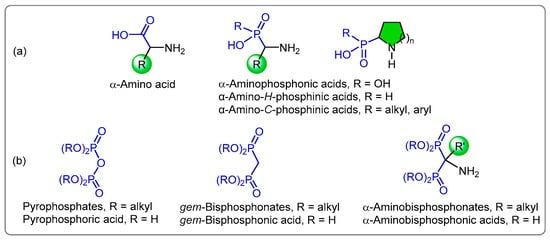
Figure 1.
General structure for (a) α-amino acids and their phosphonic and phosphinic acid analogs and (b) pyrophosphoric acids and their α-gem-bisphosphonic acid analogs.
The amino-gem-bisphosphonates are an important class of compounds and have received great attention in recent years due to their relevant pharmacological applications. For example, Incadronate 1 is an anticancer agent [19,20,21]; 2 is herbicidal [22]; 3 acts as an antiparasitic agent [23]; the α-amino-gem-bisphosphonic acid (BP) linked to Bortezomib (BP-Btz)-4 is an FDA-approved drug for the treatment of patients with multiple myeloma, more effective and with less side effects than Btz [24]; the α-amino-gem-bisphosphonic acid 5 (BPAMD) and α-amino-gem-bisphosphonic acid 6 (BPAPD) bearing 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid have been used in imaging (111In and 68Ga) for single-photon emission computed tomography (SPECT) and positron emission tomography (PET) [25,26]; HBED-CC linked to α-amino-gem-bisphosphonic acid (68Ga) 7 is a potential PET/CT bone imaging agent, and biodistribution, autoradiography and imaging studies clearly demonstrate that the tracer is taken up almost exclusively by the skeletal bone system, with minimal activity accumulation in other organs [27] (Figure 2).
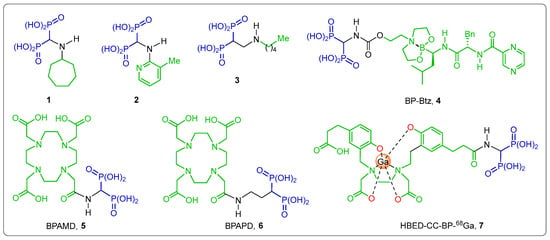
Figure 2.
Representative compounds containing α-, β- and γ-amino-gem-bisphosphonic acids with biological activity.
The biological activity of the selected amino-gem-bisphosphonates and amino-gem-bisphosphonic acids described in this review are summarized in Table 1.

Table 1.
Biological activity of selected amino-gem-bisphosphonate derivatives.
Despite the wide biological application of the amino-gem-bisphosphonic acids, its administration is complicated by poor bioavailability and poor gastrointestinal tolerability. Therefore, the synthesis of amino-gem-bisphosphonic acids and their esters with better bioavailability, greater biological activity and lower secondary toxicity is a challenge for chemists [64,65,66,67,68]. In this context, this review aims to summarize the strategies reported in the literature for its synthesis in the last 15 years, as well as their biological properties.
2. Synthesis of α-Amino-gem-Bisphosphonate Derivatives
The synthesis of α-amino-gem-bisphosphonate derivatives can be obtained by four routes: (1) three-component reaction involving orthoformates, amines and dialkyl phosphites; (2) phosphonylation of imidates; (3) phosphonylation of amides; (4) phosphonylation of nitriles; (5) phosphonylation of isonitriles; and (6) electrophilic amination of gem-bisphosphonates (Scheme 1).
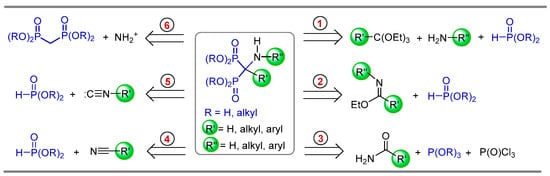
Scheme 1.
Retrosynthetic analysis for the preparation of α-amino-gem-bisphosphonate derivatives.
2.1. Three-Component Reaction of Orthoformates, Amines and Dialkyl Phosphites
One of the most convenient and common methods for the synthesis of α-amino-gem-bisphosphonate derivatives is the three-component reaction involving orthoformates, amines and dialkyl phosphites. For example, Rodriguez et al. [29] carried out the preparation of a series of N-alkyl α-amino-gem-bisphosphonates 8a–i by reacting triethyl orthoformate with alkyl or cycloalkyl amines and diethyl phosphite at 135 °C, obtaining the corresponding N-alkyl α-amino-gem-bisphosphonates 8a–i in moderate yields, which, by hydrolysis with HCl at 100 °C, gave the N-alkyl α-amino-gem-bisphosphonic acids 9a–i in good yields, which were tested against Trypanosoma cruzi (amastigotes) and Toxoplasma gondii (tachyzoites) (Scheme 2).
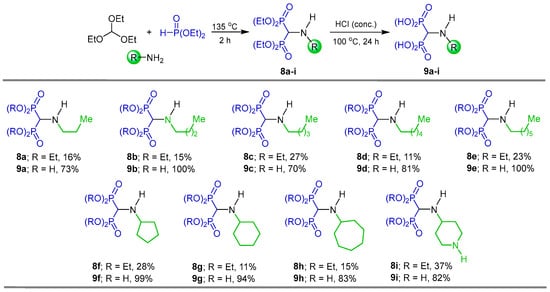
Scheme 2.
Synthesis of N-alkyl α-amino-gem-bisphosphonates 8a–i and N-alkyl α-amino-gem-bisphosphonic acids 9a–i.
The mechanism of this three-component reaction has been investigated by Krutikov and Kafarski [69,70]. The first step is the condensation reaction of the amine with the orthoformate, in which imine-type intermediates A or B may be formed, followed by the nucleophilic addition of diethyl phosphite to the C=N bond of the imines, giving the α-aminophosphonates derivatives C and D, respectively. Then, the elimination of ethanol or amine molecules, gave the iminophosphonate E, which, by the addition of another unit of diethyl phosphite, produced the N-substituted α-amino-gem-bisphosphonates (Scheme 3).
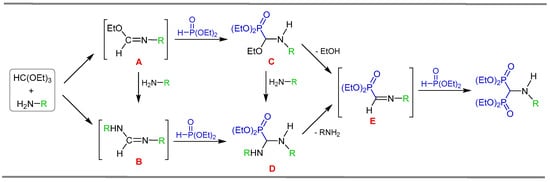
Scheme 3.
Mechanistic pathway for the formation of N-substituted α-amino-gem-bisphosphonates via three-component reaction.
Tortorella et al. [32] reported the reaction of triethyl orthoformate with several arylamines and diethyl phosphite at 160 °C, obtaining the N-aryl α-amino-gem-bisphosphonates 10a–v in 32 to 89% yield, which, by the hydrolysis of diethyl esters mediated by trimethylsilyl bromide (TMSBr) in anhydrous acetonitrile followed by treatment with methanol, gave the corresponding N-aryl amino-gem-bisphosphonic acids 11a–v in 21 to 98% yield (Scheme 4). These compounds were tested against metalloproteinases, such as MMP-2, MMP-8, MMP-9 and MMP-14.
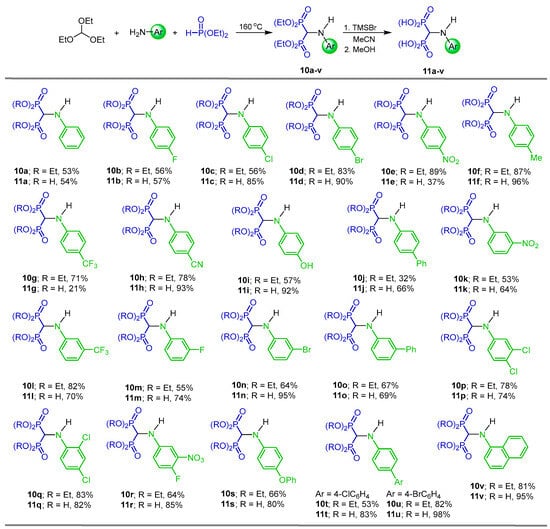
Scheme 4.
Synthesis of N-aryl α-amino-gem-bisphosphonates 10a–v and N-aryl α-amino-gem-bisphosphonic acids 11a–v.
N-sulfonamide gem-bisphosphonates 12a–h were obtained in 29 to 92% yield by a three-component reaction of triethyl orthoformate with the appropriate aryl sulfonamide and diethyl phosphite at 150 °C. Hydrolysis of 12a–h with 6 N HCl or BBr3 produced the N-sulfonamide gem-bisphosphonic acids 13a–h in 55 to 98% yield (Scheme 5). All compounds obtained were tested against metalloproteinases, such as MMP-2, MMP-8, MMP-9 and MMP-14 [33].
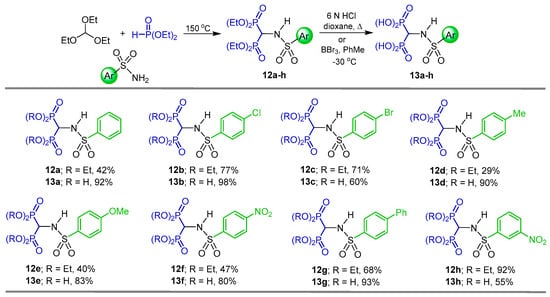
Scheme 5.
Preparation of N-sulfonamide gem-bisphosphonates 12a–h and N-sulfonamide gem-bisphosphonic acids 13a–h.
Bochno and Berlicki [71] carried out the three-component reaction of triethyl orthoformate with several amines and ethyl(diethoxymethyl)phosphonate at reflux, to obtain the N-substituted α-amino-gem-(diethoxymethyl)phosphinates 14a–h in 7 to 50% yield. Hydrolysis of 14a,b with HBr/AcOH followed by treatment with NaHCO3 afforded the N-substituted α-amino-gem-bisphosphonic acids 11a and 11w in excellent yield (Scheme 6).
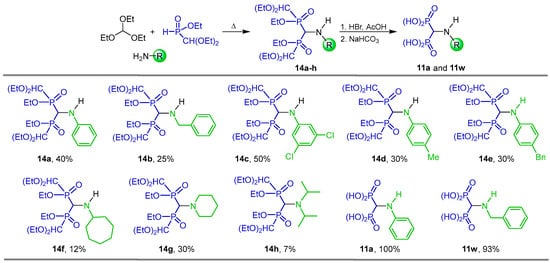
Scheme 6.
Synthesis of N-substituted α-amino-gem-(diethoxymethyl)phosphinates 14a–h and N-substituted α-amino-gem-bisphosphonic acids 11a and 11w.
In a similar way, Kafarski et al. [31] reported the synthesis of N-substituted α-amino-gem-bisphosphonic acids 11a, 11l and 15a in 30 to 64% yield through a three-component reaction of triethyl orthoformate, aromatic amines and diethyl phosphite followed by hydrolysis of diethyl esters mediated by TMSBr and subsequent treatment with methanol (Scheme 7). The N-substituted α-amino-gem-bisphosphonic acids 11a, 11l and 15a obtained were evaluated as effective inhibitors of Mycobacterium tuberculosis glutamine synthetase.
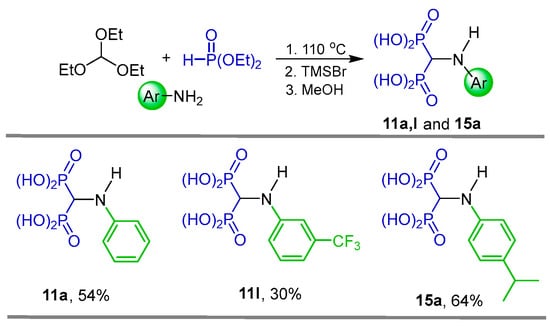
Scheme 7.
Synthesis of N-substituted α-amino-gem-bisphosphonic acids 11a, 11l and 15a.
Three-component reaction of triethyl orthoformate, aromatic amines and diethyl phosphite at 125 °C followed by hydrolysis with 6 N HCl afforded the N-aryl α-amino-gem-bisphosphonic acids 11l and 16a–j in 9 to 97% yield, against which their potential antiproliferative activity was evaluated using mouse macrophage-like J774E cells (Scheme 8) [34].
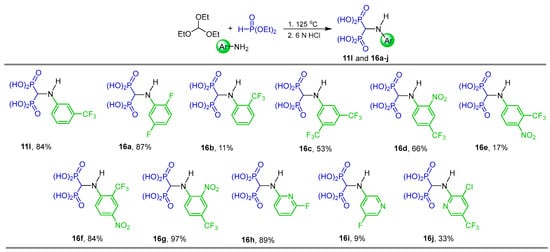
Scheme 8.
Synthesis of N-aryl α-amino-gem-bisphosphonic acids 11l and 16a–j.
Three-component reaction between triethyl orthoformate, N,N-dibenzylamine and diethyl phosphite followed by the cleavage of N-Bn bond under hydrogenolysis afforded the α-amino-gem-bisphosphonate 17, which is a key intermediate for the synthesis of 1,2,3,4-tetrahydroisoquinoline-3-phosphonic acid (±)-17a [72]. Wu et al. [35] used the α-amino-gem-bisphosphonate as a key intermediate for the synthesis of amides 17b and 17c, which were tested as a new class of potential bone-targeting reagents for bone tumors, showing a high affinity to hydroxyapatite in vitro. Additionally, α-amino-gem-bisphosphonate 17 was transformed into ligand DOTA-Bn-SCN-BP 17d for 68Ga- and 153Sm complexes, and tested as radiopharmaceuticals for both imaging skeletal metastases and palliation of pain arising out of it in cancer patients [73]. α-amino-gem-bisphosphonate 17 was also used to obtain the biotin derivative 17e used as a model linker for protein attachment to bone [74] (Scheme 9).
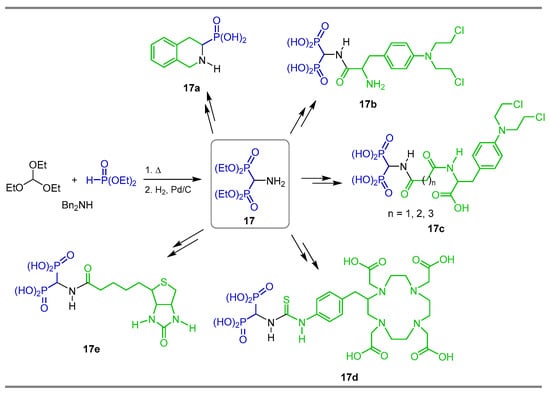
Scheme 9.
Synthesis of α-amino-gem-bisphosphonate 17 and its application as a key intermediate in the preparation of bioactive derivatives 17a–e.
The reaction of triethyl orthoformate with 3-amino-1,2,4-triazole and diethyl phosphite under heating, followed by hydrolysis with HCl, produced 1,2,4-triazoly-3-yl-amino-gem-bisphosphonic acid 18 accompanied by the production of significant quantities of N-ethylated derivative 19. 1,2,4-triazoly-3-yl-amino-gem-bisphosphonic acid 18 showed interesting activity in anti-osteolytic therapy, as a powerful inhibitor of the activity of J774E cells, and is equipotent to the popular drug Zoledronate and exhibits higher activity than the drug Incadronate (Scheme 10) [36].
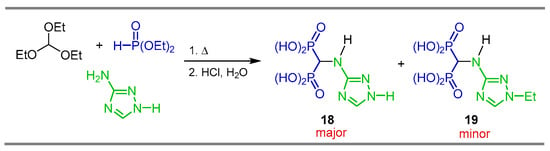
Scheme 10.
Synthesis of 1,2,4-triazolyl-3-yl-amino-gem-bisphosphonic acid 18 and N-ethylated derivative 19.
Tsantrizos et al. [37,38] carried out the three-component reaction of triethyl orthoformate with 2-(methylthio)thieno[2,3-d]pyrimidin-4-amine and diethyl phosphite in dry toluene at 130 °C, obtaining the tetraethyl ((2-(methylthio)thieno[2,3-d]pyrimidin-4-yl)amino)-gem-bisphosphonate 20 in 40% yield, used as a key intermediate in the synthesis of N-substituted α-amino-gem-bisphosphonic acid derivatives 21a–g, which are identified to exhibit toxicity in various cancer cell lines and are particularly toxic to human myeloma cells, blocking prenylation and inducing apoptosis (Scheme 11).
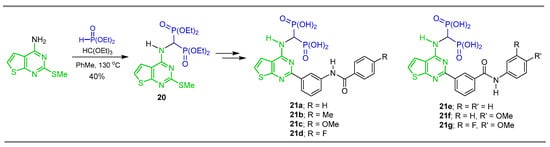
Scheme 11.
Synthesis of N-substituted α-amino-gem-bisphosphonate 20 and N-substituted α-amino-gem-bisphosphonic acids 21a–g.
The same research group carried out the three-component reaction of triethyl orthoformate with 6-bromothieno[2,3-d]pyrimidin-4-amine and diethyl phosphite in toluene at 130 °C, obtaining tetraethyl ((6-bromothieno[2,3-d]pyrimidin-4-yl)amino)-gem-bisphosphonate 22 in 75% yield, which was used as a starting material in the Suzuki cross-coupling reactions to obtain several N-substituted α-amino-gem-bisphosphonic acid derivatives 23a–j in 17 to 71% yield (Scheme 12). The compounds obtained were evaluated as inhibitors of human geranylgeranyl pyrophosphate synthase (hGGPPS) [75].
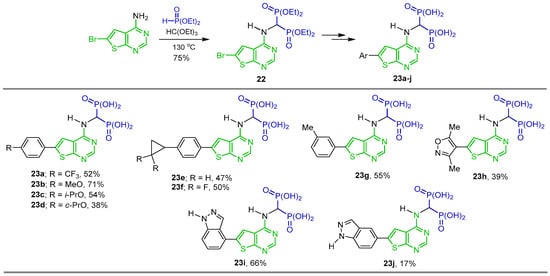
Scheme 12.
Synthesis of N-substituted α-amino-gem-bisphosphonate 22 and N-substituted α-amino-gem-bisphosphonic acids 23a–j.
Tsantrizos et al. [39] carried out the reaction of triethyl orthoformate with diethyl phosphite and the amines 25a,b obtained from 24a,b, at 130 °C, to give the α-amino-gem-bisphosphonates 26a,b in 50 and 13% yield, respectively, which were used as key intermediates in the Suzuki coupling reaction for the synthesis of the gem-bisphosphonates 27a–f (Scheme 13). The gem-bisphosphonates obtained were evaluated as inhibitors of the human geranylgeranyl pyrophosphate synthase and their evaluation as antitumor efficacy in multiple myeloma, pancreatic ductal adenocarcinoma and colorectal cancer cells.
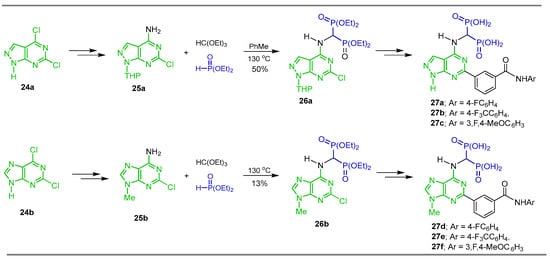
Scheme 13.
Synthesis of N-substituted α-amino-gem-bisphosphonates 26a,b and N-substituted α-amino-gem-bisphosphonic acids 27a–f.
Abdou et al. [40] synthesized several N-heterocyclic α-amino-gem-bisphosphonic acids 28a–g in 70 to 74% yield, reacting ethyl orthoformate with the corresponding amine, diethyl phosphite and metallic sodium in toluene followed by hydrolysis of diethyl ester intermediates with 1 N HCl (Scheme 14). The compounds obtained were tested as potential anti-inflammatory agents.
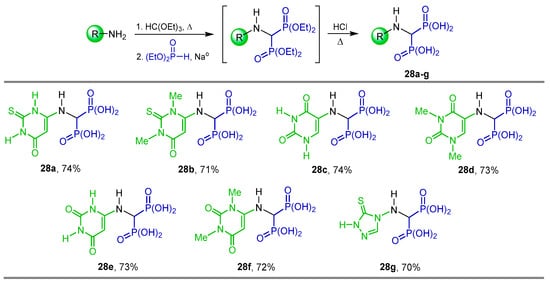
Scheme 14.
Synthesis of N-heterocyclic α-amino-gem-bisphosphonic acids 28a–g.
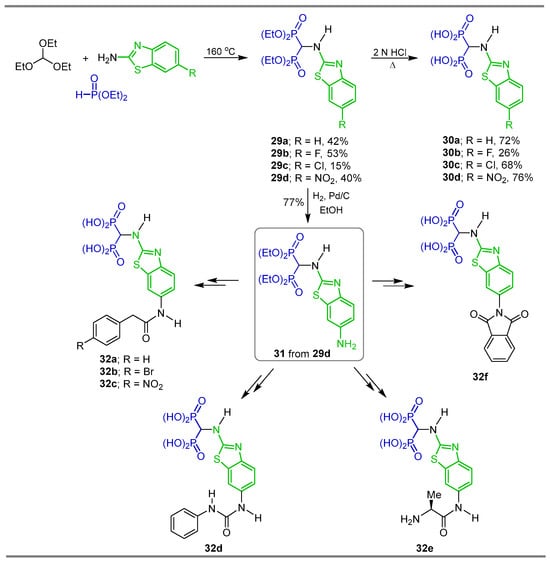
Three-component reaction of triethyl orthoformate with the appropriate 2-aminobenzothiazole and diethyl phosphite at 160 °C afforded the N-heterocyclic α-amino-gem-bisphosphonates 29a–d in 15 to 53% yield, which, by treatment with 2 N HCl, gave the α-amino-gem-bisphosphonic acids 30a–d in 26 to 76% yield. Additionally, N-heterocyclic α-amino-gem-bisphosphonates 29d, under hydrogenation of the nitro group using Pd/C, produced the N-substituted α-amino-gem-bisphosphonate 31 in 77% yield, which was used as a key intermediate in the synthesis of the α-amino-gem-bisphosphonic acids 32a–f, whose biological activities were evaluated as MMP-13 inhibitors (Scheme 15) [41].
Scheme 15.
Synthesis of N-heterocyclic α-amino-gem-bisphosphonates 29a–d and their conversion into α-amino-gem-bisphosphonic acids 30a–d and 32a–f.
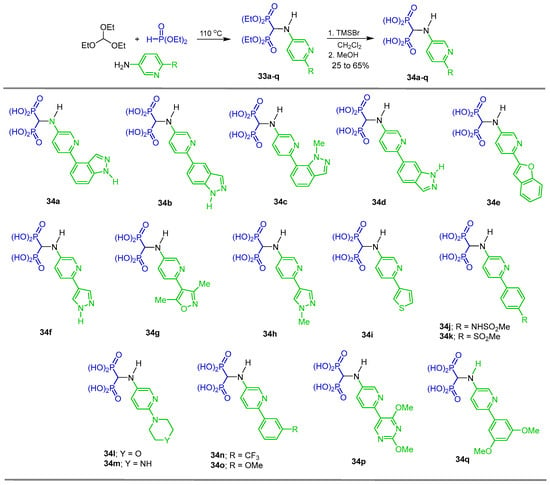
Three-component reactions of triethyl orthoformate with several substituted 3-aminopyridines and diethyl phosphite at 110 °C afforded the N-substituted α-amino-gem-bisphosphonates 33a–q, which, by O-Et bond cleavage with TMSBr in dichloromethane followed by treatment with methanol, gave the corresponding N-substituted α-amino-gem-bisphosphonic acids 34a–q in 25 to 65% yield, which were evaluated as inhibitors of human farnesyl pyrophosphate synthase (hFPPS) (Scheme 16) [42].
Scheme 16.
Synthesis of N-substituted α-amino-gem-bisphosphonates 33a–q and N-substituted α-amino-gem-bisphosphonic acids 34a–q.
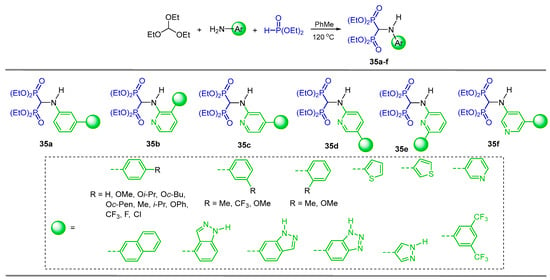
The one-pot reaction of triethyl orthoformate with several arylamines and diethyl phosphite at 130 °C gave the corresponding N-substituted α-amino-gem-bisphosphonates 35a–f. The compounds obtained induce cytotoxicity and apoptosis in human multiple myeloma cell lines and down-regulate the intended intracellular target in these cells (Scheme 17) [43].
Scheme 17.
Synthesis of N-substituted α-amino-gem-bisphosphonates 35a–f.
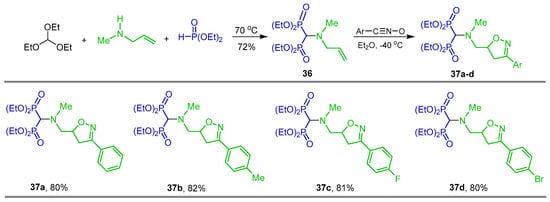
Brel [76] carried out the three-component reaction of triethyl orthoformate with N-allyl-N-methylamine and diethyl phosphite at 70 °C to obtain N-substituted α-amino-gem-bisphosphonate 36 in 72% yield, which, by [3 + 2] cycloaddition with nitrile oxides to the double bond at −40 °C, gave the 4,5-dihydroisoxazoles 37a–d in good yield which may be useful for drug design and fine organic synthesis (Scheme 18).
Scheme 18.
Synthesis of N-substituted α-amino-gem-bisphosphonate 36 and 4,5-dihydroisoxazoles 37a–d.
Chmielewska et al. [44,77] carried out a detailed study of the reaction of diamines with triethyl orthoformate and diethyl phosphite followed by hydrolysis with HCl. In the case of trans-cyclohexane-1,4-diamine 38 and trans-cyclohexane-1,3-diamine 40, which gave cyclohexane-1,4- and cyclohexane-1,3-di(aminomethylenebisphosphonic) acids trans-39 and trans-41 in 52 and 51% yield, respectively, derived from the reaction of the two amino groups. On the other hand, when (1R,2R)-cyclohexane-1,3-diamine 42 and (1S,2S)-cyclopentane-1,2-diamine 44 were used as starting reagents, (1R,2R)-4-cyclohexane-1-amino-2-aminomethylenebisphosphonic acid (1R,2R)-43 and (1S,2S)-cyclopentane-1-amino-2-aminomethylenebisphosphonic acid (1S,2S)-45 were obtained in 19 and 17% yield, respectively, derived from the reaction of the only one amino group (Scheme 19). Other diamines were also used in the study. The antiproliferative action of these di(aminomethylenebisphosphonic) acids and 1-amino-2-amino-gem-bisphosphonic acids obtained were evaluated against mouse macrophage-like RAW 264.7.
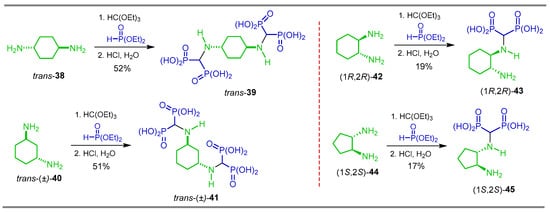
Scheme 19.
Synthesis of cyclohexane- and cyclopentane di(aminomethylenebisphosphonic) acids 39 an 41 and α-amino-gem-bisphosphonic acids 43 and 45.
Three-component reaction of triethyl orthoformate with primary and secondary amines and diethyl phosphite under microwave (MW) irradiation in the absence of catalyst and solvent produced the corresponding N-substituted and N-disubstituted α-amino-gem-bisphosphonates 8b, 8g, 10a and 46a–g in 61 to 86% yield (Scheme 20) [78].
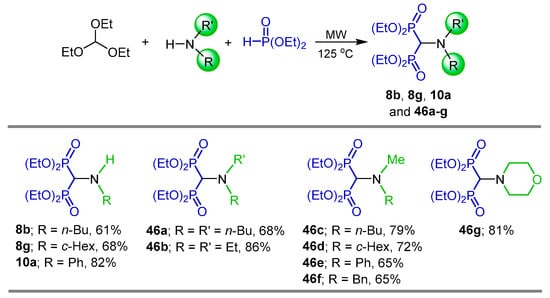
Scheme 20.
Synthesis of N-substituted and N-disubstituted α-amino-gem-bisphosphonates 8b, 8g, 10a, and 46a–g.
Additionally, the three-component reaction of triethyl orthoformate with aminoadamantanes 47a,b and 48 and diethyl phosphite under microwave irradiation (400 W, 150 °C) without catalyst and under solvent-free conditions gave the α-amino-gem-bisphosphonates 49a,b and 50 containing a biologically active adamantyl fragment. Reaction of α-amino-gem-bisphosphonates 49a,b with TMSBr followed by treatment with methanol and propylene oxide at room temperature produced α-amino-gem-bisphosphonic acid 51a,b in good yield (Scheme 21) [79].
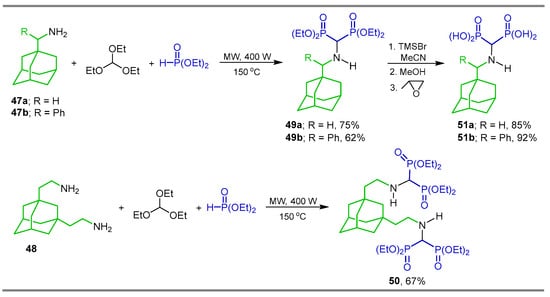
Scheme 21.
Synthesis of α-amino-gem-bisphosphonates 49a,b and 50 and α-amino-gem-bisphosphonic acids 51a,b.bearing adamantyl fragment.
In a similar way, Cirandur et al. [45] reported a simple, effective and green procedure for the synthesis of α-amino-gem-bisphosphonates. In this context, the one-pot reaction of triethyl orthoformate with aromatic amines and diethyl or methyl phosphite without catalyst or solvent under microwave irradiation at 600 W afforded the corresponding α-amino-gem-bisphosphonates 29a, 29e–g and 52a–f in 72 to 93% yield. (Scheme 22). The compounds obtained were tested in vitro for their antibacterial, antifungal and antioxidant activity. Molecular docking studies were also performed.
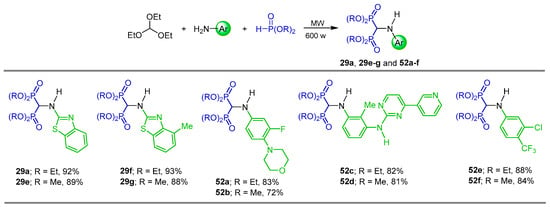
Scheme 22.
Synthesis of α-amino-gem-bisphosphonates 29a, 29e–g, and 52a–f.
Three-component reaction of triethyl orthoformate with appropriate sulfamides and diethyl phosphite under microwave irradiation at 150 °C and 500 W produced the corresponding α-amino-gem-bisphosphonates 12a,b and 53a–d in 50 to 65% yield, which were tested in vitro for their anti-inflammatory activity, showing moderate inhibition compared with Diclofenac. Furthermore, to rationalize the observed biological data, several in silico approaches were used to explain the structure and activity (Scheme 23) [48].
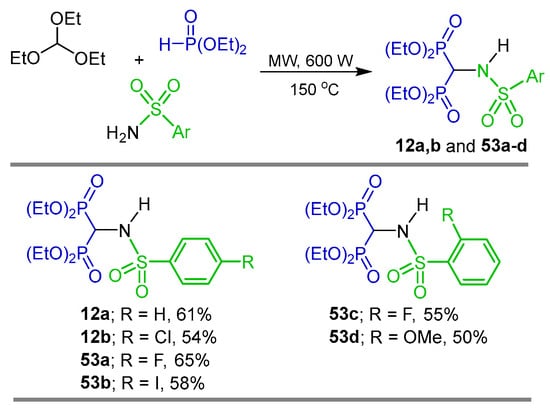
Scheme 23.
Synthesis of α-amino-gem-bisphosphonates 12a,b and 53a–d.
Reaction of triethyl orthoformate with 9-(5-amino-5-deoxy-2,3-O-isopropylidene-β-D-ribofuranosyl)-6-(2,5-dimethylpyrrol-1-yl)purine 54 and diethyl phosphite under microwave irradiation at 125 °C produced the corresponding N-substituted α-amino-gem-bisphosphonate 55 in 34% yield, which was transformed into 5′-deoxy-5′-N-(methylene bisphosphonate)adenosine (tetra sodium salts) 56 and evaluated for CD73 inhibition in a cell-based assay (MDA-MB-231) and towards the purified recombinant protein (Scheme 24) [30].
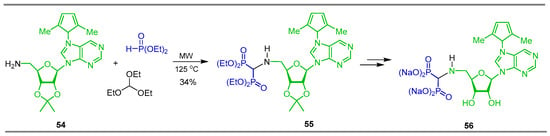
Scheme 24.
Synthesis of N-substituted α-amino-gem-bisphosphonate 55 and its conversion to 5′-deoxy-5′-N-(methylene bisphosphonate)adenosine 56.
Cirandur et al. [46] carried out a detailed study of the three-component reaction of triethyl orthoformate with arylamines and diethyl phosphite in the presence of various catalysts such as FeCl3, AlCl3, LaCl3, ZnCl2, NiCl2, CuCl2, CuBr2, BF3.SiO2, Fe3O4 and TiO2; however, under these conditions no favorable yields of α-amino-gem-bisphosphonates were obtained. The best yields were obtained when the reaction of triethyl orthoformate with several aryl, heteroaryl amines and diethyl phosphite was carried out in the presence of CuO nanoparticles (NPs) as catalysts under microwave irradiation at 400 W and solvent-free conditions (various solvents and watts were also evaluated), obtaining the corresponding N-substituted α-amino-gem-bisphosphonates 29e, 57a–i in 92 to 96% yield, which exhibited significant antioxidant and considerable antimicrobial activities (Scheme 25).
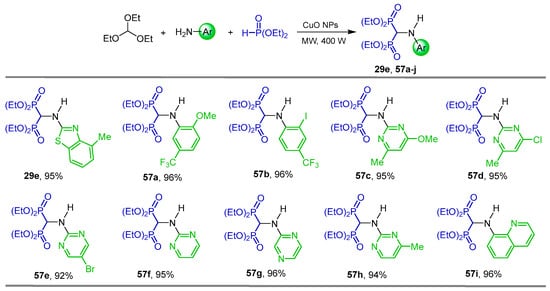
Scheme 25.
Synthesis of N-substituted α-amino-gem-bisphosphonates 29e and 57a–i.
Reddy et al. [47] carried out a detailed study of the three-component reaction of triethyl orthoformate with arylamines and diethyl phosphite in the presence of various catalysts such as FeCl3, ZnCl2, I2, CuCl2, AlCl3, p-TSA, BF3.SiO2, Amberlyst 15 and nano ZnO at 100 °C, finding that nano ZnO was the most effective catalyst. Thus, the three-component reaction of triethyl orthoformate with arylamines and diethyl phosphite in the presence of a catalytic amount of nano ZnO as an environmentally benign and heterogeneous catalyst under solvent-free and microwave irradiation at 400 W produced the corresponding α-amino-gem-bisphosphonates 57c–e and 57j–p in excellent yield (Scheme 26). The obtained compounds were evaluated in five cancer cell lines, including human breast (MCF7), prostate (DU-145), osteosarcoma (MG-63), fibrosarcoma (HT-1080) and multiple myeloma (RPMI-8226), showing promising cytotoxic activity in the five cell lines. Compound 57j was two times more active than Adriamycin in all five cancer cell lines.
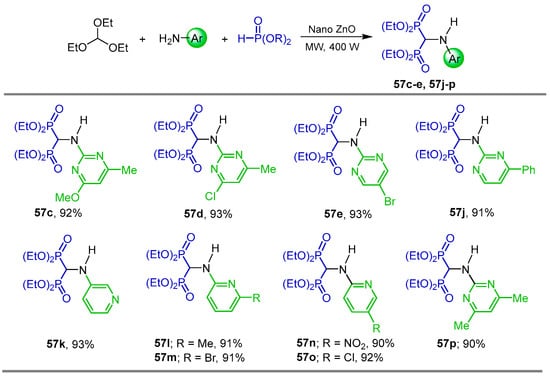
Scheme 26.
Synthesis of α-amino-gem-bisphosphonates 57c–e and 57j–p.
In order to develop a clean, eco-friendly, environmentally benign and economical sustainable protocol for the synthesis of α-amino-gem-bisphosphonates, Cirandur et al. [80] carried out a detailed study of the three-component reaction of triethyl orthoformate with arylamines and diethyl phosphite in the presence of various catalysts such as ZnCl2, FeCl3, AlCl3, NiCl2, Ni(acac)2, TiO2, LaCl3, Rh(OAc)4, CuCl2, CuBr2 and rGO-SO3H at room temperature to 100 °C, finding that rGO-SO3H was the better catalyst. Thus, the one-pot reaction of triethyl orthoformate with several anilines and diethyl phosphite in the presence of sulfonated reduced graphene oxide (rGO-SO3H) as a heterogeneous reusable catalyst under microwave irradiation and solvent-free conditions afforded the N-substituted-substituted α-amino-gem-bisphosphonates 10b, 10g, 10l, 57a and 58a–e in 85 to 94% yield (Scheme 27). The compounds obtained were evaluated for their anticancer activity against human breast cancer in MCF-7 cell lines, and molecular docking studies were also carried out against human estrogen receptor alpha (ERα).
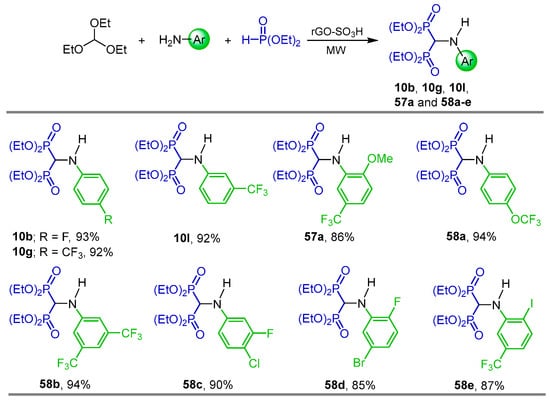
Scheme 27.
Synthesis of N-substituted α-amino-gem-bisphosphonates 10b, 10g, 10l, 57a and 58a–e.
In a similar way, the three-component reaction of triethyl orthoformate with aryl or 2-pyridylamine, diethyl phosphite and sulfated choline ionic liquid (SCIL) as a recyclable catalyst at room temperature, under conventional heating (Method A) or under ultrasonication (Method B), afforded the N-substituted α-amino-gem-bisphosphonates 10, 58b, 58f–n in 75 to 85% yield (Method A). Higher yields (87 to 95%) were obtained when the reaction was carried out under ultrasonication at room temperature (Scheme 28) [81].
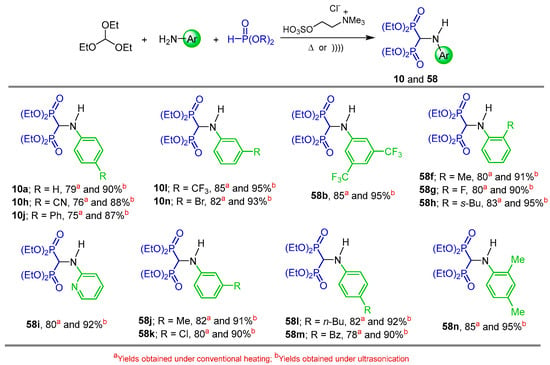
Scheme 28.
Synthesis of N-substituted α-amino-gem-bisphosphonates 10 and 58f–n.
On the other hand, Prishchenko et al. [82] carried out the reaction of triethyl orthoformate with several 4-aryl or 2-pyridylamines and diethyl phosphite in the presence of ZnCl2 at 140 to 150 °C under solvent free conditions, obtaining the N-substituted α-amino-gem-bisphosphonates 10a–c, 58i, 58o,p, 59a–d in 42 to 62% yield (Method A). Higher yields (84 to 92%) were obtained when the four-component reaction of triethyl orthoformate, 4-aryl or 2-pyridylamines, diethyl phosphite and diethyl (trimethylsilyl) phosphite was carried out (Method B) (Scheme 29).
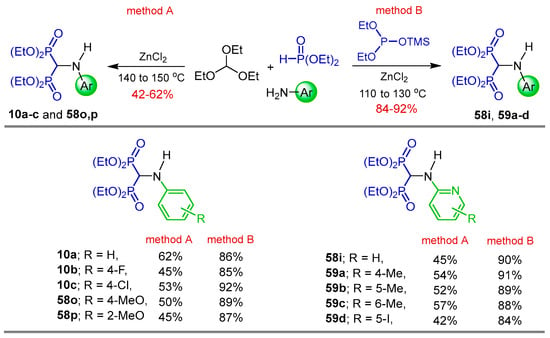
Scheme 29.
Synthesis of N-substituted α-amino-gem-bisphosphonates 10a–c, 58i, 58o,p, and 59a–d.
After several experiments, Kim et al. [83] found that the one-pot reaction of triethyl orthoformate, arylamines and diethyl phosphite in the presence of sulfonated micro-porous hyper-cross-linked 2,2′-biphenol polymer (HCBP-SO3H) as a catalyst in a vial sealed at 50 °C gave the corresponding N-substituted α-amino-gem-bisphosphonates 10h, 10j, 10l, 58b, 58i–l and 60a–m in 88 to 95% yield (Scheme 30).
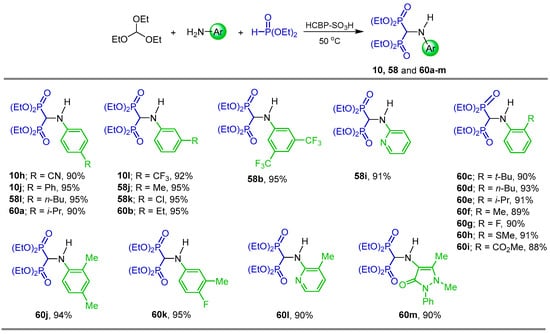
Scheme 30.
Synthesis of N-substituted α-amino-gem-bisphosphonates 10h, 10j, 10l, 58b, 58i–l, and 60a–m.
Cirandur et al. [84] carried out the reaction of triethyl orthoformate, various amines and diethyl phosphite in the presence of amberlyst-15 as the catalyst at room temperature under solvent free conditions to obtain the N-substituted α-amino-gem-bisphosphonates 10b, 10e, 10p, 29a and 61a–f in 70 to 95% yields. These compounds exhibited significant antioxidant properties in nitric oxide method inhibitory potency and antimicrobial activity (Scheme 31).
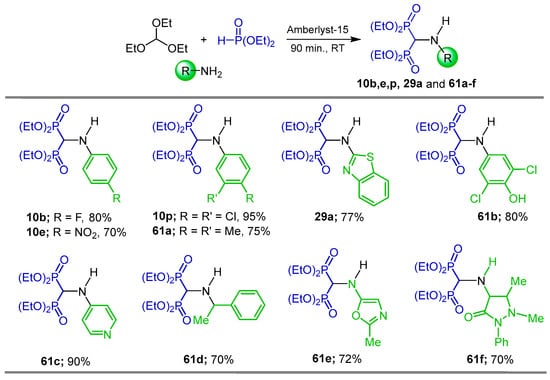
Scheme 31.
Synthesis of N-substituted α-amino-gem-bisphosphonates 10b, 10e, 10p, 29a, and 61a–f.
Ionic liquids (ILs) have attracted the attention of various researchers to reduce or eliminate the production and use of harmful and toxic chemicals, which has led to the development of much more efficient, improved and environmentally friendly processes and products [85,86,87,88]. For example, Jeong et al. [89], after comparison of several ionic liquids, found that the di-n-butyl ammonium ionic liquid (DIBA IL) as a recyclable ionic liquid is a good catalyst in the three-component reaction of triethyl orthoformate, arylamines and diethyl phosphite at 60 °C, producing the corresponding N-substituted α-amino-gem-bisphosphonates 10b–f, 29a, 58j, 58o,p and 62a–g in 89 to 94% yield (Scheme 32).
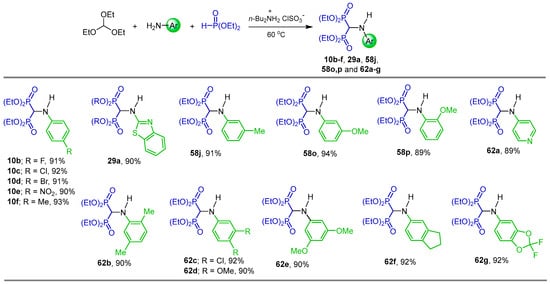
Scheme 32.
Synthesis of N-substituted α-amino-gem-bisphosphonates 10b–f, 29a, 58j, 58o,p, and 62a–g.
In a similar way, after a detailed study using several catalysts, it was found that the three-component reaction of triethyl orthoformate with 4-aryl substituted thiazol-2-amines 63a–l and dialkyl or aryl phosphites in the presence of catalytic amounts of Ag nanoparticles (NPs) at 60 °C under solvent free conditions afforded the N-substituted α-amino-gem-bisphosphonates 64a–l in 85 to 94% yield (Scheme 33). Computational docking methods were used to predict how these α-amino-gem-bisphosphonates compete against the inhibitor BPH-1330 at the crystal enzyme structure of the 4H3A protein active site and how the substituent influences their binding ability [90].
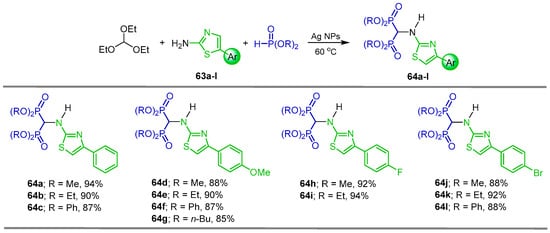
Scheme 33.
Synthesis of N-substituted α-amino-gem-bisphosphonates 64a–l.
On the other hand, the reaction of triethyl orthoformate with various α-amino acids, diethyl and dimethyl phosphite in a EtOH/H2O mixture at reflux afforded the α-amino acid-substituted α-amino-gem-bisphosphonates 65 and 66 in 54 to 77% yield (Method A). The α-amino acid-substituted α-amino-gem-bisphosphonates 65 and 66 were obtained in higher yields (72 to 85%) when the reaction was catalyzed by Tween 20 in aqueous medium at 70 °C (Method B) (Scheme 34) [91]. The α-amino-gem-bisphosphonates obtained showed in vitro antibacterial activity against clinically isolated bacteria Klebsiella pneumonia, Pseudomonas aeruginosa (Gram+) and Staphylococcus aureus, Bacillus subtilis (Gram−). Molecular docking studies against the bacterial target enzyme Type IIA topoisomerase were also carried out to establish the protein–ligand interactions.
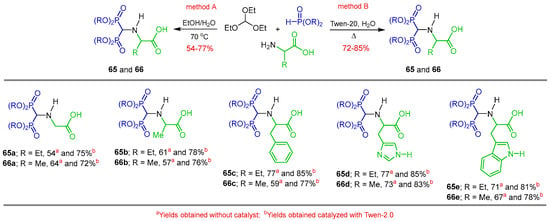
Scheme 34.
Synthesis of α-amino-gem-bisphosphonates 65 and 66 bearing α-amino acids.
2.2. Phosphonylation of Imidates
Phosphonylation of readily accessible alkyl imidate hydrochlorides is another methodology for the synthesis of tri- and tetrasubstituted α-amino-gem-bisphosphonates. The three-step synthesis involves acylation of the imidate hydrochloride, the addition of diethyl phosphite to the N-acylimidate and subsequent nucleophilic substitution of the ethoxy group in the 1-ethoxyphosphonate derivative with triphenylphosphonium tetrafluoroborate [92,93]. Under this method, Kuźnik et al. [94,95] carried out the reaction of ethyl imidate hydrochlorides 67a–g with benzyl chloroformate and Hünig’s base, obtaining the ethyl N-(benzyloxycarbonyl)phenylacetimidates 68a–g, which were reacted with diethyl phosphite, potassium carbonate and 18-crown-6 in acetonitrile at 70 °C, affording the diethyl 1-(N-benzyloxycarnonylamino)-1-ethoxyalkylphosphonates 69a–g. Subsequently, the reaction of 69a–g with triphenylphosphonium tetrafluoroborate followed by treatment with triethyl phosphite, produced the corresponding tetrasubstituted N-Cbz-α-amino-gem-bisphosphonates 71a–f in 40 to 95% yield via the 1-(N-benzyloxycarbonylamino)-1-triphenylphosphonium-methylphosphonate tetrafluoro-borate salts 70a–f. Additionally, the reaction of triphenylphosphonium-methyl-phosphonate tetrafluoroborate salts 70a–g with diethyl phenylphosphonite at 40 °C, gave the tetrasubstituted phosphonyl/phosphinyl derivatives 72a–g in 62 to 95% yield, with 1:1 to 1:1.7 diastereomeric ratio (Scheme 35).
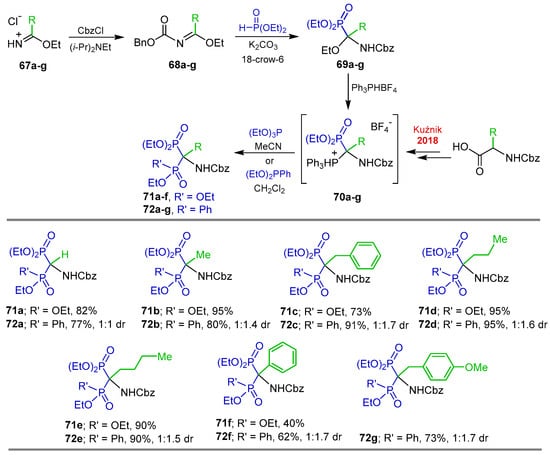
Scheme 35.
Synthesis of tetrasubstituted N-Cbz-α-amino-gem-bisphosphonates 71a–f and phosphonyl/phosphinyl derivatives 72a–g Kuźnik et al. 2018 [93].
Following the same procedure described above, the ethyl formidate hydrochloride was transformed into N-Cbz-α-amino-gem-bisphosphonate 73, which, by hydrogenolysis using Pd(OH)2/C, afforded α-amino-gem-bisphosphonate 17, used in the synthesis of bisphosphonate/betulin derivatives 74 and 75 and evaluated as a cytotoxic agent (Scheme 36) [96].
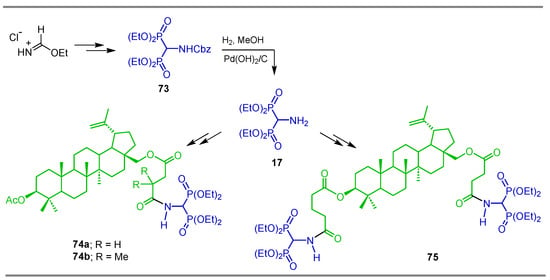
Scheme 36.
Synthesis of α-amino-gem-bisphosphonate 73 and its conversion into bisphosphonate/betulin derivatives 74 and 75.
Reaction of N-aryl ethyl imidates 76a,b with diethyl phosphite in the presence of ZnCl2 under heating afforded the corresponding N-aryl-α-amino-gem-bisphosphonates 10a,b in 57 and 62% yield, respectively. Additionally, the three-component reaction of N-aryl ethyl imidates 76a,b with bis(trimethylsilyl) phosphite and tris(trimethylsilyl) phosphite catalyzed by ZnCl2 under heating, produced the trimethylsilyl N-aryl-α-amino-gem-bisphosphonates 77a,b in 85 and 83% yield, respectively; which, by treatment with MeOH, gave the N-aryl-α-amino-gem-bisphosphonic acids 11a,b in 97 and 96% yield, respectively. (Scheme 37) [45].
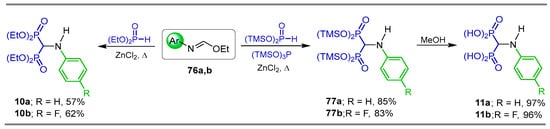
Scheme 37.
Synthesis of N-aryl-α-amino-gem-bisphosphonates 10a,b and N-aryl-α-amino-gem-bisphosphonic acids 11a,b.
N,O-Bis(trimethylsilyl)acetamides are also excellent electrophiles towards the attack of nucleophilic reagents such as silylated phosphonites for the synthesis of α-amino-gem-bisphosphonic acids. For example, the reaction of the amides 78a–i with trimethylsilyltrifloromethanesulfonate (TMSOTf) in an anhydrous pentane/dichloromethane mixture at room temperature gave the imidates 79a–i, which, by reaction with anhydrous hypophosphorous acid in the presence of ZnI2 in THF at 0 °C, produced the corresponding tetrasubstituted α-amino-gem-bisphosphinic acid derivatives as disodium salts 80a–i in 35 to 79% yield, through bis(trimethylsilyl)phosphonite and N-silylacetamide intermediates (Scheme 38) [97].
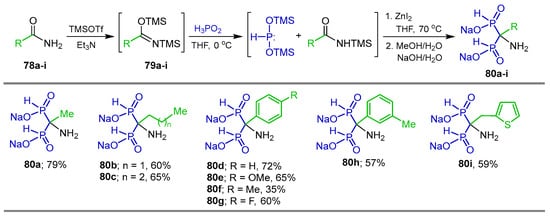
Scheme 38.
Synthesis of tetrasubstituted α-amino-gem-bisphosphinic acid derivatives 80a–i.
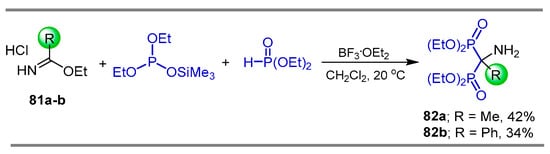
Petrosyan et. al. [98] carried out the reaction of imidates 81a,b with diethyl phosphite and diethyl (trimethylsilyl) phosphite, obtaining the α-amino-gem-bisphosphonates 82a,b in 42 and 34% yield, respectively (Scheme 39).
Scheme 39.
Synthesis of α-amino-gem-bisphosphonates 82a,b.
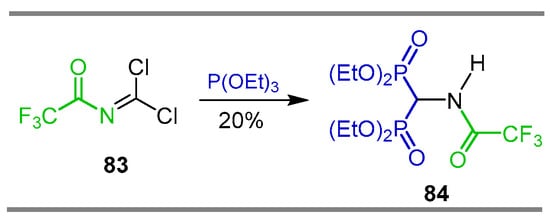
The Arbuzov reaction of triethyl phosphite with N-dichloromethylenetrifloroacetamide 83 obtained from the photochemical chlorination of N-methyltrifluoroacetamide gave the N-trifluoroacetyl α-amino-gem-bisphosphonate 84 in 20% yield, derived from the substitution of both chlorine atoms in 83 (Scheme 40) [99].
Scheme 40.
Synthesis of N-trifluoroacetyl α-amino-gem-bisphosphonate 84 via Arbuzov reaction.
2.3. Phosphonylation of Amides
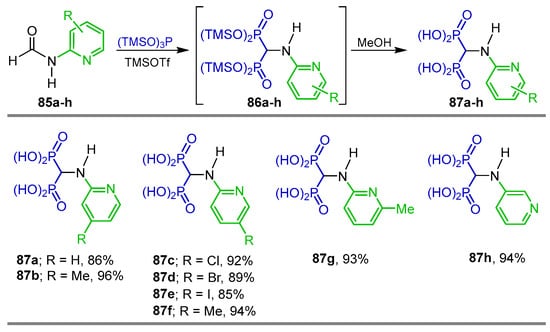
Phosphonylation of amides is another method for the synthesis of α-amino-gem-bisphosphonates. For example, the reaction of 2-(N-formyl)-aminopyridines 85a–h with tris(trimethylsilyl)phosphine and trimethylsilyltrifloro-methanesulfonate as activating agents gave the corresponding tetra(trimethylsilyl) α-amino-gem-bisphosphonates 86a–h, which, without further purification, were treated with MeOH to give the N-pyridyl-α-amino-gem-bisphosphonic acids 87a–h in 85 to 96% yield (Scheme 41) [45].
Scheme 41.
Synthesis of N-pyridyl-α-amino-gem-bisphosphonic acids 87a–h.
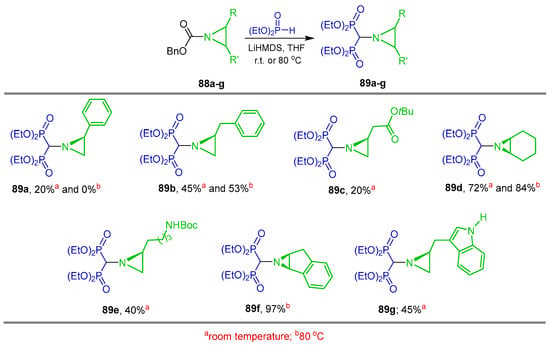
Cheviet and Peyrottes [100] used N-Cbz aziridines 88a–g as the starting material for the preparation of brand gem-bisphosphonylaziridine derivatives. Thus, the reaction of diethyl phosphite with LiHMDS followed by the addition N-Cbz aziridines 88a–g in THF at r.t. or 80 °C afforded the gem-bisphosphonylaziridines 89a–g (abbreviated as AzbisPs) in 0 to 97% yield (Scheme 42).
Scheme 42.
Synthesis of gem-bisphosphonylaziridines 89a–g from N-Cbz aziridines 88a–g.
Cativiela et al. [101] reported the reaction of formamide with phosphorus acid and phosphorus trichloride at 70 °C followed by treatment with water, obtaining the α-amino-gem-bisphosphonic acid 90 in 53% yield, which was used as a key intermediate in the synthesis of α-amino-gem-bisphosphonate 91 incorporating L-leucine (Scheme 43).
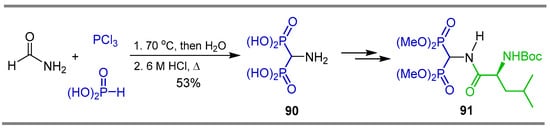
Scheme 43.
Synthesis of α-amino-gem-bisphosphonic acid 90 and α-amino-gem-bisphosphonate 91.
On the other hand, the reaction of N-Boc N-(3-aminopropyl)acetamide 92 with phosphorus oxychloride and triethylphosphite at room temperature produced the N-substituted α-amino-gem-bisphosphonate 93 in 80% yield, which, by treatment with TMSBr in dichloromethane followed by the addition of methanol, gave the corresponding α-amino-gem-bisphosphonic acid 94 in 52% yield. Additionally, the reaction of N-substituted α-amino-gem-bisphosphonate 93 with trifluoroacetic acid afforded the N-substituted α-amino-gem-bisphosphonate 95 in 94% yield (Scheme 44). The compounds obtained were used for the preparation of organometallic complexes and their cytotoxicity was assessed in vitro using various histologically different cell line models [102].
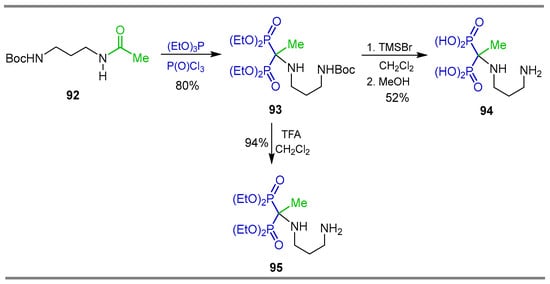
Scheme 44.
Synthesis of N-substituted α-amino-gem-bisphosphonates 93,95 and α-amino-gem-bisphosphonic acid 94.
In a similar way, the reaction of N-(2-aminoethyl)acetamide hydrochloride 96 with phosphorus oxychloride and triethyl phosphite at room temperature gave the corresponding N-substituted α-amino-gem-bisphosphonate 97 in 61% yield, which was transformed into acetyl ursolic acid derivative 98. Additionally, the reaction of acetamide 99 derived from acetyl ursolyl chloride, with phosphorus oxychloride and triethyl phosphite at room temperature, produced also the acetyl ursolic acid derivative 98 in 71% yield (Scheme 45) [103].
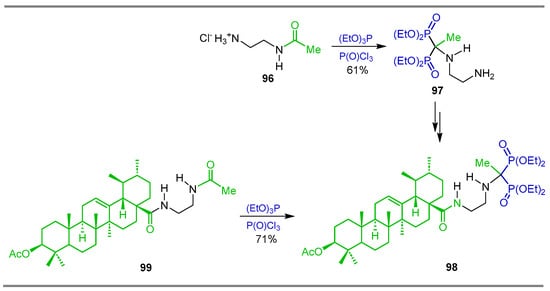
Scheme 45.
Synthesis of N-substituted α-amino-gem-bisphosphonate derivative 98.
The reaction of N-formyl heterocyclic derivatives 100a–f with tris(trimethylsilyl)-phosphite and TMSOTf as an activating agent in dichloromethane at 90 °C produced the corresponding tetra(trimethylsilyl) α-amino-gem-bisphosphonates 102a–f via the highly reactive trimethylsilyl phosphonates intermediates 101a–f. Treatment of tetra(trimethylsilyl) α-amino-gem-bisphosphonates 102a–f with methanol gave the corresponding α-amino-gem-bisphosphonic acids 103a–f (Scheme 46) [104].
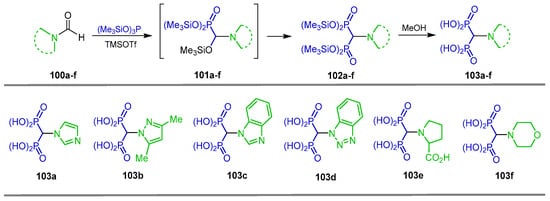
Scheme 46.
Synthesis of α-amino-gem-bisphosphonic acids 103a–f.
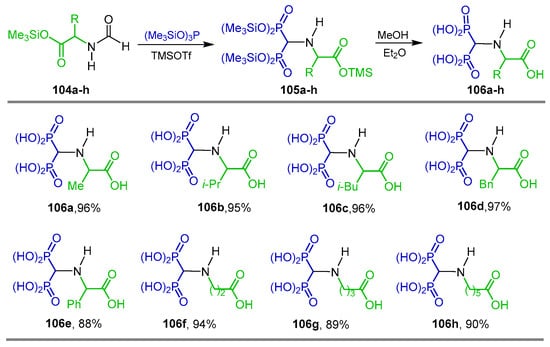
In a similar way, the reaction of N-formyl amino acid derivatives 104a–h with tris(trimethylsilyl)phosphite and TMSOTf as activating agents in dichloromethane at 20 °C produced the corresponding tetra(trimethylsilyl) α-amino-gem-bisphosphonates 105a–h, which, by treatment with methanol, gave the α-amino-gem-bisphosphonic acids 106a–h in 88 to 97% yield (Scheme 47) [105,106].
Scheme 47.
Synthesis of α-amino-gem-bisphosphonic acids 106a–h.
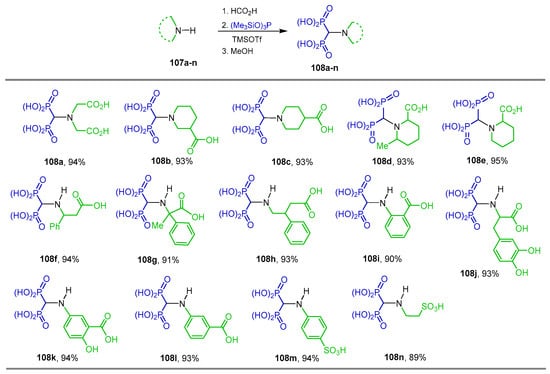
Additionally, the reaction of amines 107a–n with formic acid followed by treatment with N-tris(trimethylsilyl)phosphite and TMSOTf in dichloromethane at 20 °C and subsequent treatment with methanol afforded the N-substituted α-amino-gem-bisphosphonic acids 108a–n in excellent yield (Scheme 48) [106].
Scheme 48.
Synthesis of N-substituted α-amino-gem-bisphosphonic acids 108a–n.
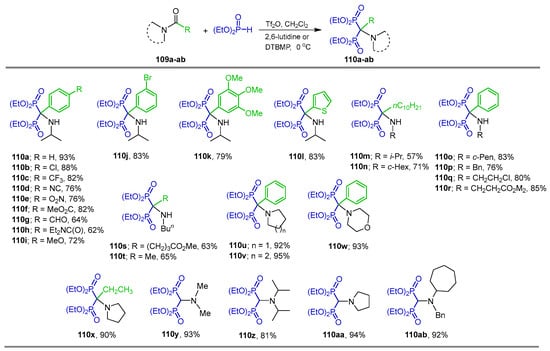
Phosphonylation of both secondary and tertiary amides activated with trifluoromethanesulfonic anhydride (Tf2O) is also a mild and general method for the synthesis of N-substituted α-amino-gem-bisphosphonates. For example, the reaction of secondary and tertiary amides 109a–ab with diethyl phosphite in the presence of Tf2O and 2,6-lutidine or 2,6-di-tert-butyl-4-methylpyridine (DTBMP) at 0 °C produced the corresponding α-amino-gem-bisphosphonates 110a–ab in 57 to 94% yield (Scheme 49) [107].
Scheme 49.
Synthesis of N-substituted α-amino-gem-bisphosphonates 110a–ab.
2.4. Phosphonylation of Nitriles
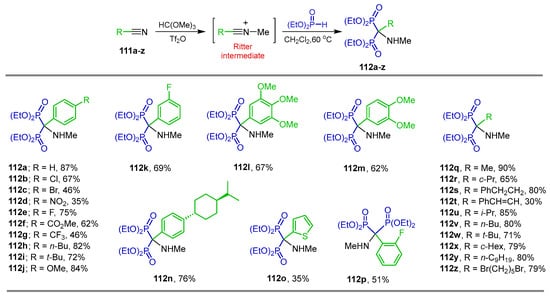
In the Ritter reaction, the reactive intermediate nitrilium has been used in the synthesis of a wide variety of compounds of chemical and pharmacological interest [108,109]. For example, the reaction of trimethyl orthoformate and the nitriles 111a–z in the presence of catalytic amounts of Tf2O gave the reactive Ritter intermediate nitrilium, which, by the addition of diethyl phosphite at 60 °C, produced the α-amino-gem-bisphosphonates 112a–z in 35 to 90% yield (Scheme 50) [110].
Scheme 50.
Synthesis of α-amino-gem-bisphosphonates 112a–z.
Kaboudin et al. [111] carried out a detailed study of the reaction of phosphonylation of nitriles with diethyl phosphite in the presence of various catalyst such as FeCl3, BiCl3, Sc(OTf)3, TiO2, ZnO and Zn(OAc)2; however, under these conditions, no favorable yield of the α-amino-gem-bisphosphonates was obtained. The best yields were obtained when the reaction of nitriles 111a–d, 111s,t and 113a–i with diethyl phosphite and triethylamine was carried out in the presence of catalytic amounts of ZnCl2 in dichloromethane at reflux, obtaining the α-amino-gem-bisphosphonates 114a–o in 35 to 75% yield (Scheme 51).
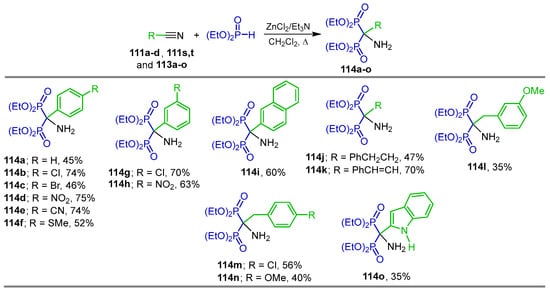
Scheme 51.
Synthesis of α-amino-gem-bisphosphonates 114a–o.
Islas and García [112] carried out a detailed study of the reaction of diisopropyl phosphite with benzonitrile 111a in the presence of various catalysts such as, NiCl2.6H2O, B(CH(Me)(Et))3, AlCl3, BF3.OEt2 and Et3B; however, in any case, α-amino-gem-bisphosphonate was produced. Additionally, when the reaction of diisopropyl phosphite was carried out with benzonitrile catalyzed with NiCl2.6H2O at 140 °C, it gave the α-amino-gem-bisphosphonate 115 and α-aminophosphonate 116 with a conversion of 98% and 81:12 ratio (Scheme 52).
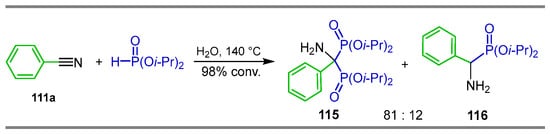
Scheme 52.
Synthesis of α-amino-gem-bisphosphonate 115.
Wiemer et al. [49] carried out the reaction of nitrile 117 with diethyl phosphite, ZnCl2 and triethylamine to obtain the corresponding α-amino-gem-bisphosphonate 118 in 42% yield, which, by treatment with TMSBr followed by the addition of methanol, gave the α-amino-gem-bisphosphonic acid 119 bearing a triazole fragment in 42% yield (Scheme 53). This compound exhibited an inhibitory activity against geranylgeranyl diphosphate synthase (GGDPS).
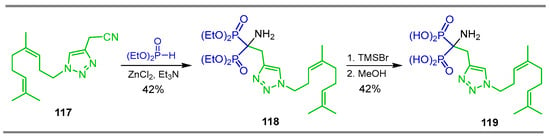
Scheme 53.
Synthesis α-amino-gem-bisphosphonate 118 and α-amino-gem-bisphosphonic acid 119.
Titanium-mediated double phosphonylation of nitriles is another procedure for the preparation of α-amino-gem-bisphosphonates. For example, the reaction of several nitriles 111e,v111q–s and 113j–m with diethyl phosphite catalyzed by bis(cyclopentadienyl)titanium dichloride (Cp2TiCl2) activated Zn powder and propylene oxide in THF at reflux, affording the corresponding α-amino-gem-bisphosphonates 114e and 120a–h in 20 to 94% yield (Scheme 54) [113].
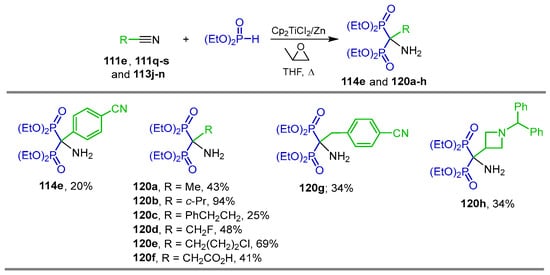
Scheme 54.
Synthesis of α-amino-gem-bisphosphonates 114e and 120a–h.
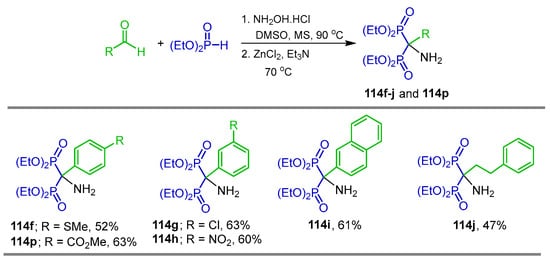
The reaction of aldehydes with hydroxylamine hydrochloride in DMSO at 90 °C afforded the corresponding nitrile, which, without further purification or separation, reacted with diethyl phosphite in the presence of ZnCl2 and Et3N at 70 °C, obtaining the α-amino-gem-bisphosphonates 114f–j and 114p in 47 to 63% yield (Scheme 55) [111].
Scheme 55.
Synthesis of α-amino-gem-bisphosphonates 114f–j and 114p.
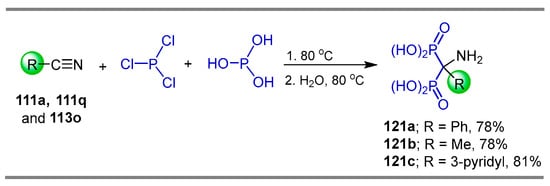
The reaction of nitriles 111a, 111q and 113o with phosphorus acid (H3PO3) in the presence of phosphorus trichloride (PCl3) at 80 °C followed by treatment with water at 80 °C produced the corresponding α-amino-gem-bisphosphonic acids 121a–c in good yield (Scheme 56) [98].
Scheme 56.
Synthesis of α-amino-gem-bisphosphonic acids 121a–c.
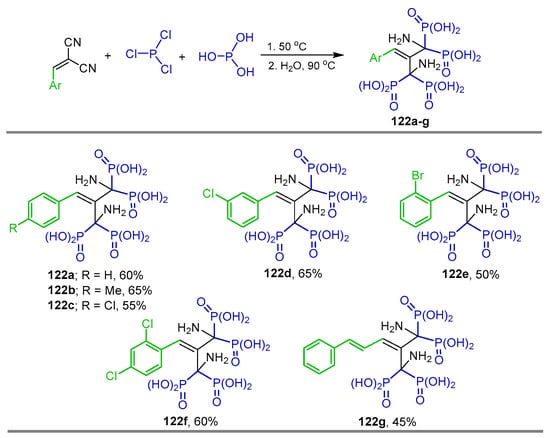
Ewies et al. [50] reported the synthesis of tetrakisphosphonic acid derivatives 122a–g in 45 to 65% yield by reacting the dinitriles with phosphorus acid and phosphorus trichloride at 50 °C (Scheme 57). The obtained compounds were evaluated for their farnesyl pyrophosphate synthase (FPPS) inhibitory activity and anti-osteoclastogenic properties in vitro using the MTT assay and the tartrate-resistant acid phosphatase (TRAP) staining test.
Scheme 57.
Synthesis tetrakisphosphonic acid derivatives 122a–g.
2.5. Phosphonylation of Isonitriles
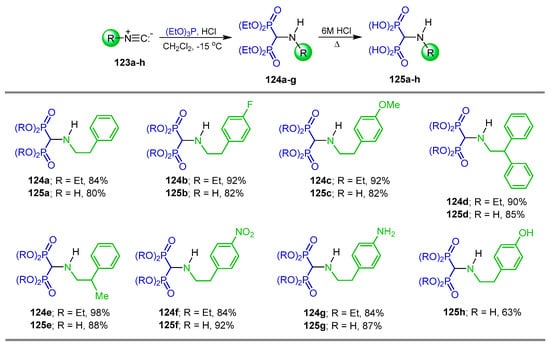
Isonitriles are also starting reagents for the synthesis of α-amino-gem-bisphosphonates. For example, the reaction of isonitriles 123a–h with triethyl phosphite in the presence of hydrochloric acid in dichloromethane at −15 °C afforded the corresponding N-substituted α-amino-gem-bisphosphonates 124a–g in 84 to 98% yield, which, by hydrolysis with 6 M hydrochloric acid at reflux, produced the α-amino-gem-bisphosphonic acids 125a–h in 63 to 92% yield (Scheme 58). These compounds were evaluated for their antiproliferative effect on MCF-7 human breast cancer cells, J774E mouse macrophages cells and HL-60 human promyelocytic leukemia cells [51,114].
Scheme 58.
Synthesis of N-substituted α-amino-gem-bisphosphonates 124a–g and α-amino-gem-bisphosphonic acids 125a–h.
Sheldon et al. [28] carried out the reaction of isonitriles 126a–e with triethyl phosphite and 4 M HCl in dichloromethane at 0 °C, obtaining the N-substituted α-amino-gem-bisphosphonates 8b, 8j, 8k and 10y, 10z in 50 to 77% yield, which, by treatment with TMSBr followed by the addition of methanol, gave the N-substituted α-amino-gem-bisphosphonic acids 9b, 9j, 9k and 11y, 11z in 69 to 97% yield (Scheme 59). These compounds provided cytoprotection against cholesterol-dependent cytolysins. In the work, Sheldon reported also the synthesis of α-amino-gem-bisphosphonic acids from triethyl orthoformate.
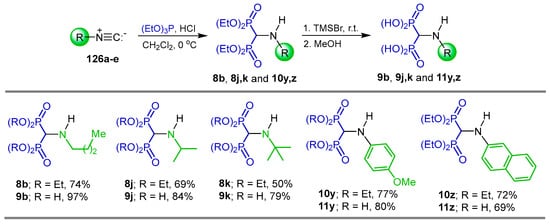
Scheme 59.
Synthesis of N-substituted α-amino-gem-bisphosphonates 8b, 8j, 8k, 10y, 10z and their N-substituted α-amino-gem-bisphosphonic acids 9b, 9j, 9k, 11y, 11z.
On the other hand, the reaction of 1,4-diisocyanobenzene 127 and 1,5-diisocyanonaphthalene 129 with triethyl phosphite in the presence of hydrochloric acid in dichloromethane at −15 °C, followed by treatment with TMSBr in dichloromethane and the subsequent addition of methanol, produced benzene-1,4-bis[amino methylidene(bisphosphonic)] acid 128 and naphthalene-1,5-bis[amino methylidene(bisphosphonic)] acid 130 (Scheme 60). The antiproliferative activity of these α-amino-gem-bisphosphonic acids in combination with doxorubicin and cisplatin toward J774E cells (a model of osteoclast precursors in vitro) was evaluated [52,115].
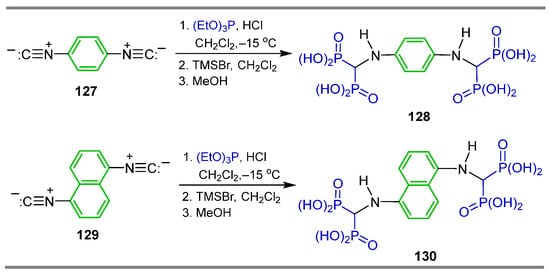
Scheme 60.
Synthesis of naphthalene bis[amino methylene(bisphosphonic)] acids 128 and 130.
2.6. Electrophilic Amination of gem-Bisphosphonates
The electrophilic amination is a powerful approach for the synthesis of useful intermediates such as α-amino aldehydes, α-amino ketones and α-amino acids [116,117,118]. In this context, the electrophilic amination of gem-bisphosphonates is also another method for the synthesis of α-amino-gem-bisphosphonates. For example, the reaction of tetraallyl methylenebisphosphonate 131 with sodium hydride in DMF followed by the addition of O-diphenylphosphinylhydroxylamine (DPPH) gave the α-amino-gem-bisphosphonate 132 in 59% yield, which was a key intermediate in the preparation of α-amino-gem-bisphosphonate glycopeptide derivative 133, used as prodrugs in the treatment of osteomyelitis (Scheme 61) [119].
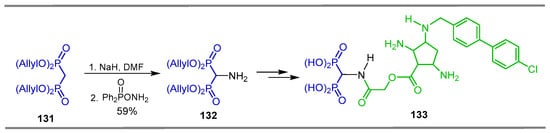
Scheme 61.
Synthesis of α-amino-gem-bisphosphonate 132.
3. Synthesis of β-Amino-gem-Bisphosphonate Derivatives
The synthesis of β-amino-gem-bisphosphonate derivatives can be obtained by two routes: (1) Michael-type addition of amines to vinyl gem-bisphosphonates and (2) addition of bisphosphonates to imines (Scheme 62).
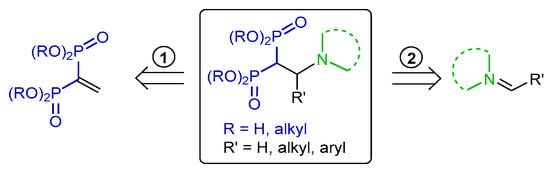
Scheme 62.
Retrosynthetic analysis for preparation of β-amino-gem-bisphosphonate derivatives.
3.1. Michael-Type Addition of Amines to Vinyl gem-Bisphosphonates
The Michael-type addition of cycloalkylamines to tetraethyl vinyl gem-bisphosphonate 134 at room temperature in CH2Cl2 afforded the corresponding N-cycloalkyl β-amino-gem-bisphosphonates 135a–c in excellent yields, which, by hydrolysis with hydrochloric acid, produced the N-cycloalkyl β-amino-gem-bisphosphonic acids 136a–c in 73 to 88% yield (Scheme 63). The compounds obtained were evaluated as effective inhibitors of Mycobacterium tuberculosis glutamine synthetase [29].
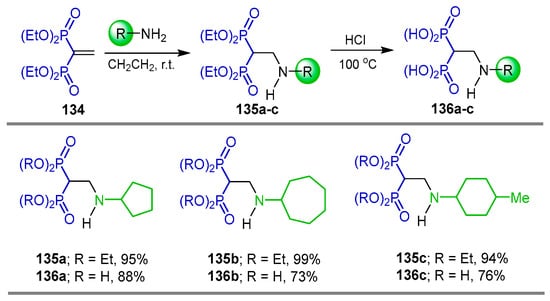
Scheme 63.
Synthesis of N-cycloalkyl β-amino-gem-bisphosphonates 135a–c and N-cycloalkyl β-amino-gem-bisphosphonic acids 136a–c.
In a similar way, the Michael-type addition of alkyl amines to tetraethyl vinyl gem-bisphosphonate 134 produced the N-alkyl β-amino-gem-bisphosphonates 137a–c in 82 to 97% yield, which, by treatment with hydrochloric acid at reflux, gave the N-alkyl β-amino-gem-bisphosphonic acids 138a–c in 53 to 81% yield (Scheme 64). The activity against Toxoplasma gondii proliferation at sub-micromolar levels of the obtained compounds was tested [53].
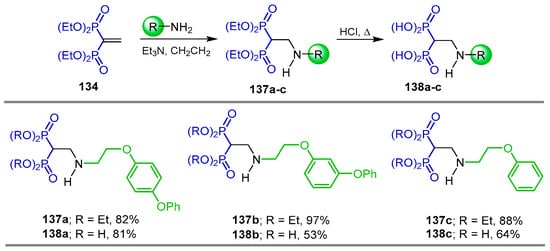
Scheme 64.
Synthesis of N-alkyl β-amino-gem-bisphosphonates 137a–c and N-alkyl β-amino-gem-bisphosphonic acids 138a–c.
Grigor’ev et al. [54] carried out the addition of 2,2,6,6-tetramethylpiperidine 139 to tetraethyl vinyl gem-bisphosphonate 134 to obtain N-alkyl β-amino-gem-bisphosphonate 140 in 85% yield, and antitumor and cytotoxic activity was tested (Scheme 65).
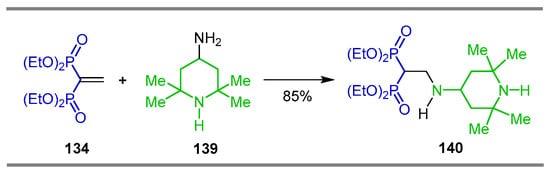
Scheme 65.
Synthesis of N-alkyl β-amino-gem-bisphosphonate 140.
The Michael-type addition of alkyl amines to tetraethyl vinyl gem-bisphosphonate 134 produced the N-alkyl β-amino-gem-bisphosphonates 141a–h, which, by treatment with TMSBr in dichloromethane, gave the N-alkyl β-amino-gem-bisphosphonic acids 142a–h (Scheme 66). The activity against Trypanosoma cruzzi, the etiologic agent of American trypanosomiasis (Chagas’ disease), and against tachyzoites of Taxoplasma gondii was evaluated for the obtained compounds [23].
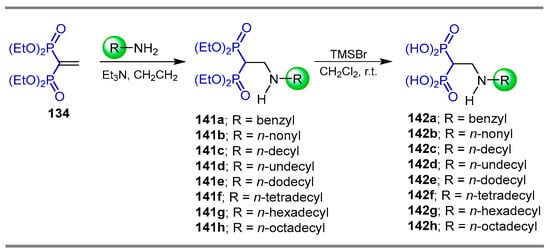
Scheme 66.
Synthesis of N-alkyl β-amino-gem-bisphosphonates 141a–h and N-alkyl β-amino-gem-bisphosphonic acids 142a–h.
Berlicki et al. [31] carried out the additional reaction of aromatic amines to tetraethyl vinyl gem-bisphosphonate 134 at room temperature followed by treatment with TMSBr and the subsequent addition of MeOH, obtaining the N-aryl β-amino-gem-bisphosphonic acids 143a–g in 3 to 60% yield, which are effective inhibitors of Arabidopsis thaliana δ1-pyrroline-5-carboxylate reductase and Mycobacterium tuberculosis glutamine synthetase (Scheme 67).
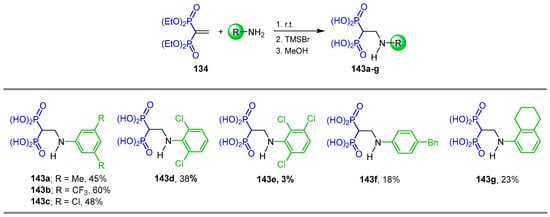
Scheme 67.
Synthesis of N-aryl β-amino-gem-bisphosphonic acids 143a–g.
After optimizing the solvent and temperature, Lv et al. [120] carried out the addition of various aromatic amines to tetraethyl vinyl gem-bisphosphonate 134 in CHCl3 at 60 °C, obtaining the corresponding N-aryl β-amino-gem-bisphosphonates 144a–j in 46 to 91% yield (Scheme 68).
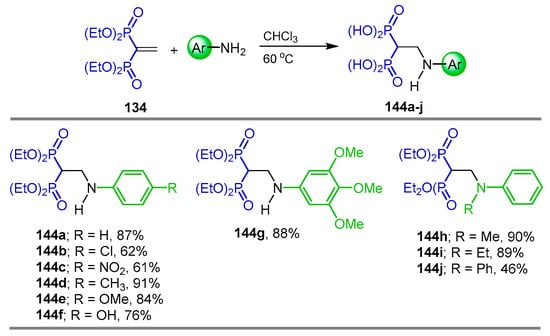
Scheme 68.
Synthesis of N-aryl β-amino-gem-bisphosphonates 144a–j.
Strukul et al. [55] carried out the addition reaction of amines to tetraethyl vinyl gem-bisphosphonate 134 in the presence of Et3N in chloroform at room temperature, obtaining the N-substituted β-amino-gem-bisphosphonates 144a and 145a,b in quantitative yield, which, by treatment with TMSBr followed by the addition of H2O, produced the N-substituted β-amino-gem-bisphosphonic acids 146a–c in quantitative yield, which are effective inhibitors of Arabidopsis thaliana δ1-pyrroline-5-carboxylate reductase and Mycobacterium tuberculosis glutamine synthetase (Scheme 69).
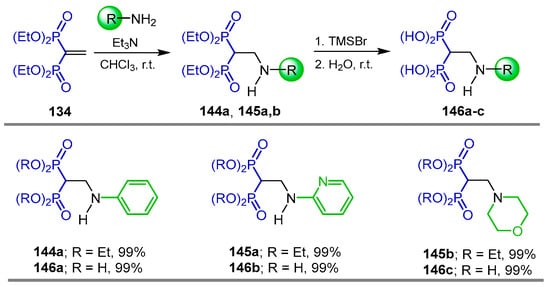
Scheme 69.
Synthesis of N-substituted β-amino-gem-bisphosphonates 144a, 145a,b and N-substituted β-amino-gem-bisphosphonic acids 146a–c.
The addition reactional of amino acetaldehyde dimethyl acetal 147 with tetraethyl vinyl gem-bisphosphonate 134 in dichloromethane produced the N-substituted β-amino-gem-bisphosphonate 148 in 99% yield, while the amino acetaldehyde dimethyl acetal 147 reacted with tetrakis(trimethylsilyl) vinyl gem-bisphosphonate 149 in acetonitrile at 20 °C followed by treatment with H2O afforded the N-substituted β-amino-gem-bisphosphonic acid 150 in 63% yield (Scheme 70) [121].
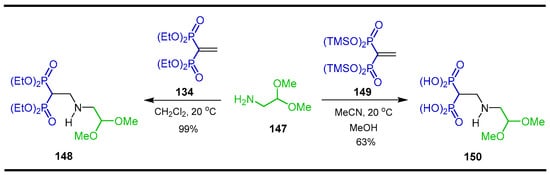
Scheme 70.
Synthesis of N-substituted β-amino-gem-bisphosphonate 148 and N-substituted β-amino-gem-bisphosphonic acid 150.
The reaction of tetraethyl vinyl gem-bisphosphonate 134 with propargylamine in chloroform at 65 °C afforded the β-amino-gem-bisphosphonates 151, which without further purification was reacted under 1,3-dipolar click cycloaddition with the corresponding azides, sodium ascorbate (NaVc) and CuSO4·5H2O in t-BuOH:H2O mixture assisted by ultrasound irradiation, obtaining the 1,2,3-triazole-amino-bisphosphonates derivatives 152a–h in 64 to 72% yield (Scheme 71) [122].
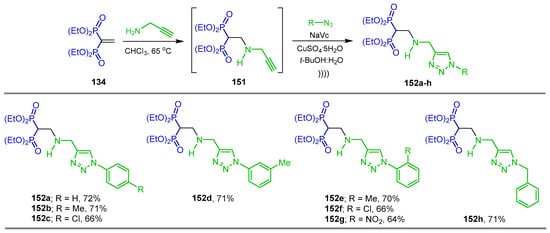
Scheme 71.
Synthesis -β-amino-gem-bisphosphonates 152a–h bearing 1,2,3-triazole ring.
Xu et al. [123] carried out the reaction of tetraethyl vinyl gem-bisphosphonate 134 and sodium azide in EtOH/H2O, obtaining the corresponding azide intermediate 153 in 60% yield, which, by hydrogenation using Pd/C in anhydrous MeOH, produced the β-amino-gem-bisphosphonate 154, that without further purification was used in the synthesis of melphalan—MMP-2-linkage—bisphosphonic acid 155, evaluated as an anticancer prodrug. Additionally, the reaction of tetraethyl vinyl gem-bisphosphonate 134 with trimethylsilyl azide in the presence of Pd(OAc)2 in toluene at 90 °C, also gives the β-amino-gem-bisphosphonate 154 in 90% yield in only one step (Scheme 72) [124].
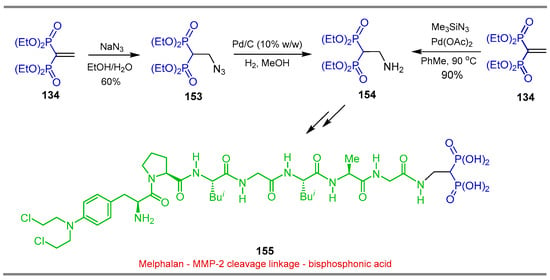
Scheme 72.
Synthesis of β-amino-gem-bisphosphonate 154.
Chen et al. [125] developed a practical and efficient one-pot strategy for the synthesis of N-attached 1,2,3-triazole-containing bisphosphonates, integrating two key transformations into a single operation. Thus, the Michael addition of sodium azide to tetraethyl vinyl gem-bisphosphonate 134 in AcOH/H2O under mild conditions and ultrasonication gave the corresponding azide derivative 153, which, by a copper-catalyzed 1,3-dipolar, was reacted with terminal alkynes in the presence of CuSO4·5H2O and sodium ascorbate (NaVc) to obtain the N-triazole-functionalized gem-bisphosphonates 156a–m in 73 to 84% yield (Scheme 73).
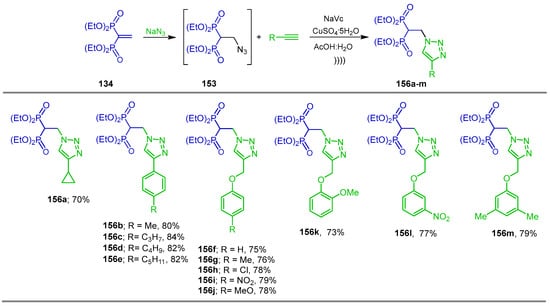
Scheme 73.
Synthesis of β-amino-gem-bisphosphonates 156a–m bearing 1,2,3-triazole ring.
The reaction of the gem-bisphosphonate 157 with NaH followed by the addition of N-(bromomethyl)phthalimide gave the alkylated product 158 in only low yield. Best results were obtained when the conjugate addition of phthalimide to tetraethyl vinyl diphosphonate 134 in dimethylformamide was carried out, obtaining the N-substituted β-amino-gem-bisphosphonate 159 in 73% yield, which, by alkylation with propargyl bromide using NaH as a base, produced the acetylene derivative 160 in 80% yield. The 1,3-dipolar click cycloaddition with geranyl azide afforded the corresponding triazole derivative 158 in 40% yield, used as a key intermediate for the synthesis of α-modified triazole bisphosphonic salts 161, which were evaluated for their activity as GGDPS inhibitors in both enzyme and cell-based assays (Scheme 74) [126].
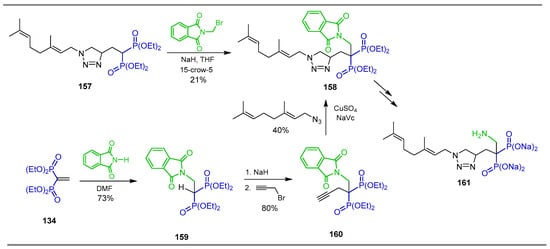
Scheme 74.
Synthesis of N-substituted β-amino-gem-bisphosphonates 158, 159 and 160.
The additional reaction of thiourea arylamines derived from dehydroabietic acid 162 to tetraethyl vinyl gem-bisphosphonate 134 in the presence of dimethylaminopyridine (DMAP) in dichloromethane at room temperature produced the corresponding N-substituted β-amino-gem-bisphosphonates 163a–k in 73 to 93% yield. These compounds exhibited potent antitumor activity against the SK-OV-3, BEL-7404, A549, HCT-116 and NCI-H460 tumor cell lines in vitro; especially, the β-amino-gem-bisphosphonate 163d exhibited the best anticancer activity against the SK-OV-3 cell line (Scheme 75) [127].
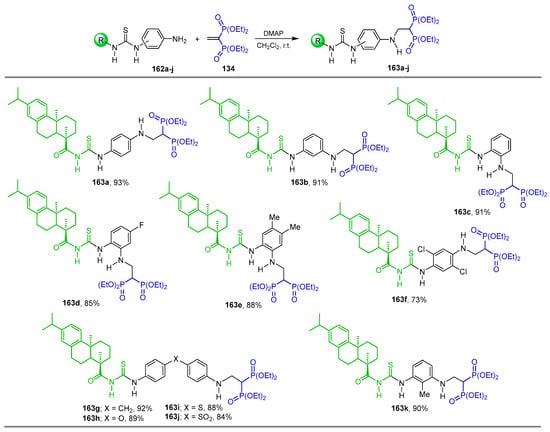
Scheme 75.
Synthesis of N-substituted β-amino-gem-bisphosphonates 163a–k.
The Aza-Michael addition of O-benzylhydroxylamine to tetraethyl or tetrabenzyl vinyl gem-bisphosphonates 134 and 164 in CH2Cl2 or Et3N in CH2Cl2 at room temperature produced the corresponding β-amino-gem-bisphosphonate 165a,b fosmidomycin analogs in 40% yield, which were transformed into amides 166a,b. The pro-herbicide activity of these compounds was evaluated on model plants, having considerable herbicidal activity (Scheme 76) [56].
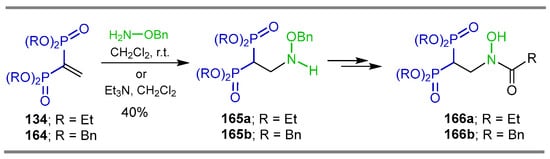
Scheme 76.
Synthesis of β-amino-gem-bisphosphonate 165a,b fosmidomycin analogs and their amides 166a,b.
Rodriguez et al. [57], through molecular design, found that compounds 168 and 171 could act against Trypanosoma cruzi (amastigotes), Toxoplasma gondii (tachyzoites), TcFPPS and TgFPPS. With these results, they carried out the additional reaction of 2-methylallylamine to tetraethyl vinyl gem-bisphosphonate 134 in methylene chloride at room temperature, obtaining the β-amino-gem-bisphosphonate 167 in 91% yield, which, by reaction with TMSBr in dichloromethane followed by treatment with methanol, produced the β-amino-gem-bisphosphonic acid 168 in 83% yield. Additionally, the Aza-Michael addition of (E)-2,6-dimethylhepta-2,5-dien-1-amine to tetraethyl vinyl diphosphonate 134 produced the β-amino-gem-bisphosphonate 169 in quantitative yield. However, after several attempts of hydrolysis of phosphonic esters, it was not possible to obtain β-amino-gem-bisphosphonic acid 170, therefore β-amino-gem-bisphosphonate 169 was treated with acetic anhydride to give acetylated β-amino-gem-bisphosphonate 171 in 63% yield (Scheme 77). Compound 168 was inactive against Trypanosoma cruzi and Toxoplasma gondii cells but exhibited a marginal activity against the target enzymes TcFPPS and TgFPPS.
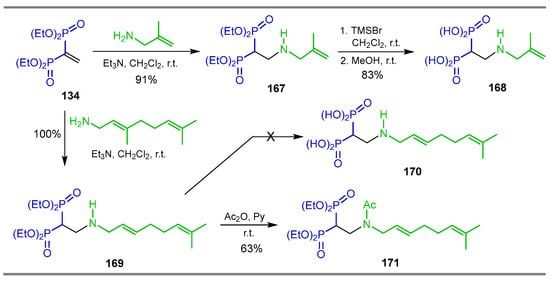
Scheme 77.
Synthesis of β-amino-gem-bisphosphonic acids 168 and 171.
The reaction of tetraethyl vinyl gem-bisphosphonate 134 with NH-3,5-bis(arylidene)piperid-4-one 172 and triethylamine in dichloromethane at room temperature gave the corresponding N-substituted β-amino-gem-bisphosphonates 173a–g in 72 to 99% yield. The reaction of the N-substituted β-amino-gem-bisphosphonates 173b–d with TMSBr in CHCl3 at room temperature followed by treatment with methanol gave the N-substituted β-amino-gem-bisphosphonic acids 174a–c in 56 to 99% yield (Scheme 78). The synthesized compounds displayed high inhibitory properties towards Caov3, A549, PC3 and KB 3-1 human carcinoma cell lines, among those, compounds bearing 4-cyano-phenyl 173d and 3-pyridinyl 173f, identified as the most active drug candidates possessing fluorescence properties that could be of interest for the visualization of BP skeletal distribution and cellular uptake in bones and other tissues [58].
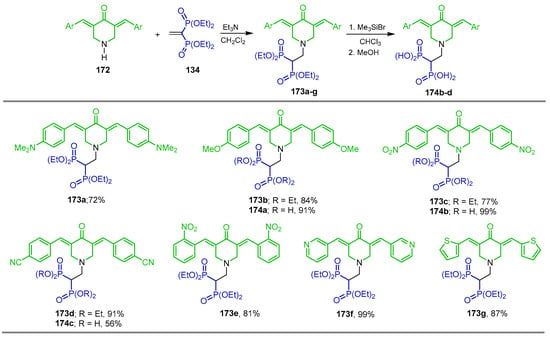
Scheme 78.
Synthesis of N-substituted β-amino-gem-bisphosphonates 173a–g and N-substituted β-amino-gem-bisphosphonic acids 174a–c.
The Michael-type addition of N-Boc diamines to tetra isopropyl vinyl gem-bisphosphonate 175 in toluene at reflux gave the β-amino-gem-bisphosphonates 176a,b in 78 and 81% yield, respectively, which, by hydrolysis with 4 M HCl, produced the β-amino-gem-bisphosphonic acids 177a,b in 71 and 76% yield, respectively. In a similar way, the reaction of tetraamine with tetra isopropyl vinyl gem-bisphosphonate 175 gave the β-amino-gem-bisphosphonate 178 in 83% yield, which, by hydrolysis with 4 M HCl, afforded the β-amino-gem-bisphosphonic acid 179 in 70% yield (Scheme 79). The affinity of these compounds to hydroxyapatite was also determined by using the 99mTc procedure [59].
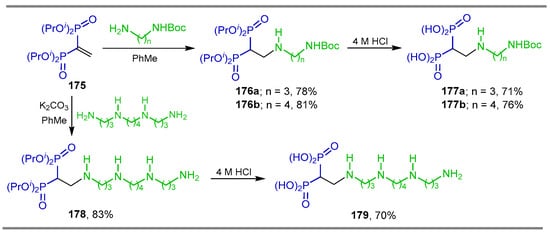
Scheme 79.
Synthesis of β-amino-gem-bisphosphonates 176a,b, 178 and β-amino-gem-bisphosphonic acids 177a,b and 179.
The Michael-type addition of 2-amino-6-chlorobenzothiazole 180 to tetraethyl vinyl gem-bisphosphonate 134 in CHCl3 at 40 °C produced the N-substituted β-amino-gem-bisphosphonate 181 in 65% yield, which, by hydrolysis with 3 N HCl, afforded the N-substituted β-amino-gem-bisphosphonic acid 182 in 96% yield (Scheme 80). Compound 182 was evaluated in an enzyme inhibition assay against MMP-2, MMP-8, MMP-9 and MMP-13 [41].
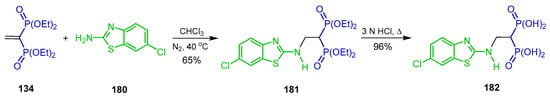
Scheme 80.
Synthesis of N-substituted β-amino-gem-bisphosphonate 181 and N-substituted β-amino-gem-bisphosphonic acid 182.
Tsantrizos et al. [128] carried out the reaction of 1-aminoimidazoles 183a,b with tetraethyl vinyl gem-bisphosphonate 134 in a CH2Cl2/DMF mixture at room temperature to obtain the corresponding β-amino-gem-bisphosphonates 184a,b in 56% and 60% yield, respectively, which, by treatment with TMSBr in dichloromethane followed by the addition of methanol, produced the N-substituted α-amino-gem-bisphosphonic acids 185a,b in 80% and 94% yield, respectively (Scheme 81). They also explored the interactions of these compounds with both the active site and an allosteric pocket of human farnesyl pyrophosphate synthase (hFPPS) to evaluate their potential utility in the treatment of cancer and neurodegenerative diseases.
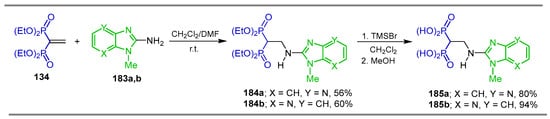
Scheme 81.
Synthesis of β-amino-gem-bisphosphonates 184a,b and α-amino-gem-bisphosphonic acids 185a,b bearing 1-aminoimidazole ring.
Vagapova et al. [129,130] carried out the reaction of either tetraethyl vinyl gem-bisphosphonate 134 or tetrakis(trimethylsilyl) vinyl gem-bisphosphonate 149 with amine 186 in dioxane at 100 °C, obtaining the N-substituted α-amino-gem-bisphosphonates 187a–d in 52 to 96% yield and N-substituted α-amino-gem-bisphosphonic acids 188a–c in 42 to 61% yield (Scheme 82). These compounds showed a potential antiresorptive and antiproliferative activity.
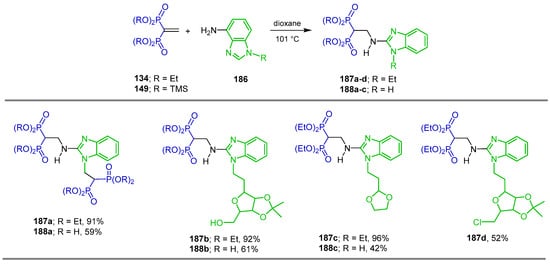
Scheme 82.
Synthesis of N-substituted α-amino-gem-bisphosphonates 187a–d and N-substituted α-amino-gem-bisphosphonic acids 188a–c.
The reaction of tetraethyl vinyl gem-bisphosphonate 134 with Rhodamine B derivatives 189a,b in the presence of DMAP in CH2Cl2 at room temperature afforded the β-amino-gem-bisphosphonates 190a,b both in 7% yield (Scheme 83). The fluorescent probes of these compounds were evaluated for theranostic applications, showing promising properties such as rapid response, optical stability, hydroxyapatite sensitivity and low toxicity [131].
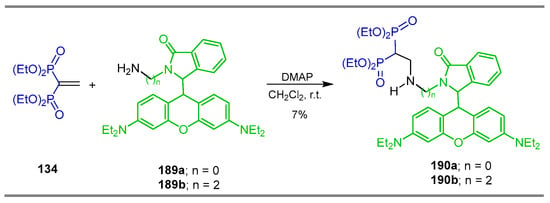
Scheme 83.
Synthesis of β-amino-gem-bisphosphonates 190a,b.
The reaction of tetrakis(trimethylsilyl) vinyl gem-bisphosphonate 149 with several alkyl or aryl amines and α-amino acids was carried out in dichloromethane or acetonitrile or toluene at room temperature or 80 °C depending on the amine used, followed by treatment with methanol, giving the N-substituted β-amino-gem-bisphosphonic acids 146c, and 191a–m in 57 to 93% yield (Scheme 84) [60].
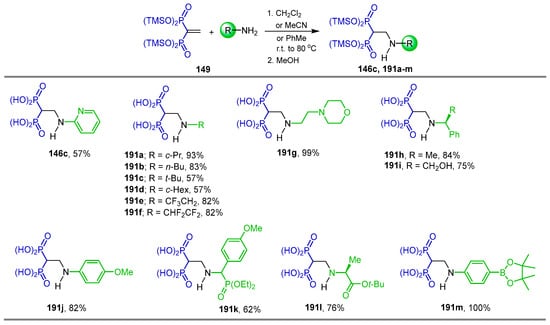
Scheme 84.
Synthesis of N-substituted β-amino-gem-bisphosphonic acids 146c and 191a–m.
The additional reaction of S-trityl cysteamine to tetraethyl vinyl gem-bisphosphonate 134 in CH2Cl2 at room temperature produced tetraethyl (2-((2-(tritylthio)ethyl)-amino)ethane-1,1-diyl) bis(phosphonate) 192 in 95% yield, which, by reaction with triethylsilane and trifluoroacetic acid at room temperature, gave the N-substituted β-amino-gem-bisphosphonate 193 in 93% yield (Scheme 85). This compound is a key intermediate for the synthesis of potential anti-resorption bone drugs [132].
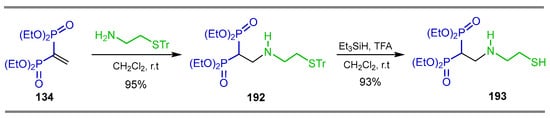
Scheme 85.
Synthesis of N-substituted β-amino-gem-bisphosphonates 192 and 193.
Tessmar et al. [133] carried out the reaction of 3,5-diaminobenzoic acid 194 with tetraethyl vinyl gem-bisphosphonate 134 in THF at 60 °C, obtaining 3,5-di(tetraethylamino-2,2-bisphosphonate)benzoic acid 195 in 85% yield, which, by treatment with TMSBr followed by the addition of methanol, produced β-amino-gem-bisphosphonic acid 196, which was incorporated into PEG-based polymer 197, enabling the formation of stable gold nanoparticle (GNP) coatings with a strong affinity for hydroxyapatite, making them promising candidates for bone-targeted delivery systems (Scheme 86).
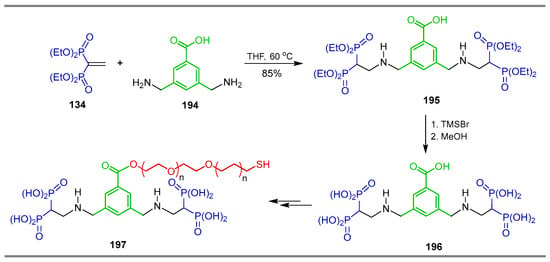
Scheme 86.
Synthesis of β-amino-gem-bisphosphonate 195 and β-amino-gem-bisphosphonic acid 196.
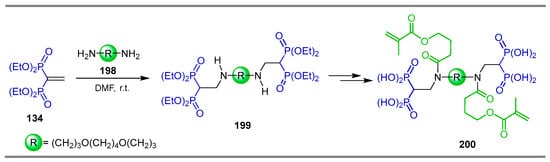
The reaction of 4,9-dioxa-1,12-dodecanediamine 198 and tetraethyl vinyl gem-bisphosphonate 134 in dry DMF at room temperature gave gem-bi(bisphosphonate) derivative 199 in 80% yield. This compound was transformed into the cross-linker 200 and incorporated into hydrogels to evaluate their properties (Scheme 87) [134,135].
Scheme 87.
Synthesis of gem-bi(bisphosphonate) derivative 199.
3.2. Addition of gem-Bisphosphonates to Imines
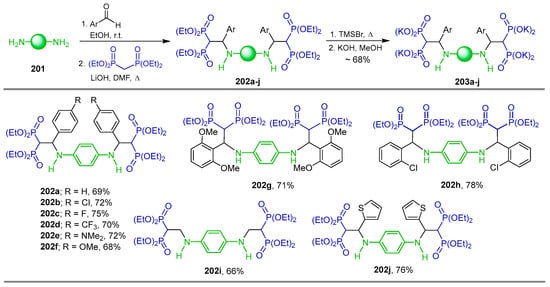
The addition of carbanion derived from gem-bisphosphonates to imines of Schiff bases is another method for the synthesis of β-amino-gem-bisphosphonates. For example, Abdou et al. [136] carried out the reaction of amine 201 with several aldehydes to obtain the corresponding imine; in situ, these were reacted with tetraethyl gem-bisphosphonate and LiOH in DMF at reflux, affording the tetraethyl gem-bi(bisphophonates) 202a–j in 66 to 78% yield, which, by treatment with TMSBr followed by reaction with KOH in methanol, gave the N-substituted β-bis(amino-gem-bisphosphonic) acids as potassium salt 203a–j in ~68% yield (Scheme 88). Cytotoxic properties were evaluated for the compounds obtained against five malignant melanoma cell lines that originated from different categories of malignant melanoma primary stage (I/II), histologically advanced stage (III/IV) and metastasized malignancy.
Scheme 88.
Synthesis of N-substituted β-bis(amino-gem-bisphosphonates) 202a–j and N-substituted β-bis(amino-gem-bisphosphonic acids) 203a–j.
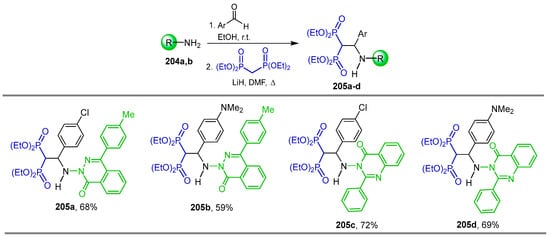
The reaction of Schiff bases derived from 4-(4-methylphenyl)-2,3-benzoxazin-1-one 204a or 3-phenyl-2,4-benzoxazin-1-one 204b and aromatic aldehydes, with tetraethyl gem-bisphosphonate and LiH in DMF at room temperature, gave the N-substituted β-amino-gem-bisphosphonates 205a–d in 59 to 72% yield, (Scheme 89). The cytogenetic activity in normal human lymphocyte cultures of the obtained compounds was evaluated [61].
Scheme 89.
Synthesis of N-substituted β-amino-gem-bisphosphonates 205a–d.
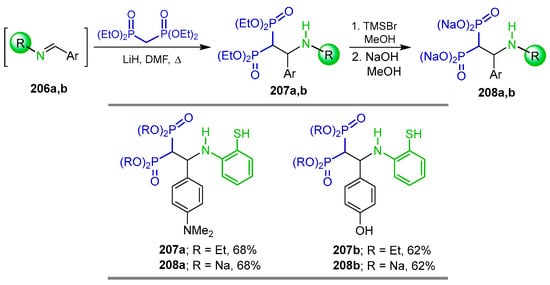
The Schiff bases 206a,b derived from 2-aminobenzenethiol and aromatic aldehydes were reacted with tetraethyl gem-bisphosphonate and LiH in DMF at reflux, obtaining the corresponding gem-bisphosphonates 207a,b in 68 and 62% yield, respectively, which, by treatment with TMSBr followed by reaction with NaOH in methanol, gave the N-substituted β-amino-gem-bisphosphonic acids as sodium salt 208a,b in 68 and 62% yield, respectively (Scheme 90). The compounds obtained were evaluated in a mouse model of antigen-induced arthritis and the delayed-type hypersensitivity garanuloma reaction (DTH-GRA) for chronic inflammation [137].
Scheme 90.
Synthesis of N-substituted β-amino-gem-bisphosphonic acids 208a,b.
4. Synthesis of γ-Amino-gem-Bisphosphonate Derivatives
For the synthesis of γ-amino-gem-bisphosphonates, this review described only the addition of carbanions derived from amino acids to vinyl gem-bisphosphonates (Scheme 91).
Scheme 91.
Retrosynthetic analysis for the preparation of γ-amino-gem-bisphosphonates.
Synthesis from Vinyl gem-Bisphosphonates
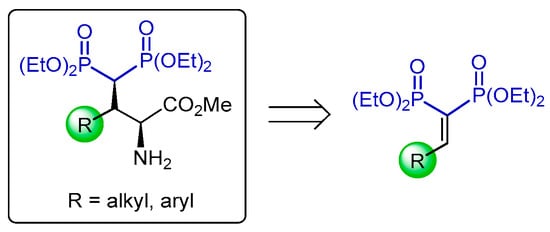
Wang et al. [62] carried out a detailed study of the catalytic asymmetric Michael addition for the synthesis of optically active γ-amino-gem-bisphosphonates containing two adjacent stereogenic centers. In this context, the reaction of vinyl gem-bisphosphonates 209a–r with N-benzylidene glycine methyl ester 210 in the presence of catalytic amounts of (R)-211, CuBF4 and K2CO3 in dichloromethane at −20 °C followed by treatment with p-toluenesulfonic acid at room temperature afforded the γ-amino-gem-bisphosphonates 212a–r in 62 to 95% yield and 89 to 99% enantiomeric excess (Scheme 92).
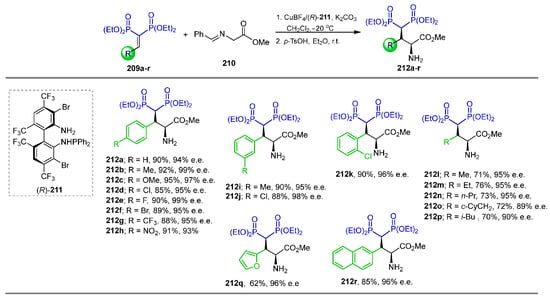
Scheme 92.
Synthesis of optically active γ-amino-gem-bisphosphonates 212a–r.
In a similar way, Fukuzawa et al. [63] carried out the reaction of vinyl gem-bisphosphonates 209 with methyl N-diphenyleneglycinate 213 in the presence of catalytic amounts of ThioClickFerrophos (TCF), AgOAc and Cs2CO3 in THF at −20 °C, obtaining the γ-amino-gem-bisphosphonates 214a–l in 64 to 99% yield and 90 to 98% enantiomeric excess (Scheme 93).
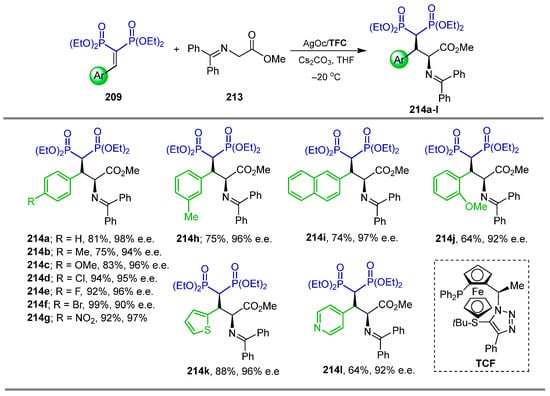
Scheme 93.
Synthesis of γ-amino-gem-bisphosphonates 214a–l.
On the other hand, the reaction of vinyl gem-bisphosphonate 134 with α-substituted azlactones 215a–f and Et3N in dichloromethane at room temperature afforded the compounds 216a–f in 44 to 88% yield and 5:1 to >20:1 regioisomeric ratio, which, by treatment with chlorotrimethylsilane in MeOH:CH2Cl2 at room temperature, gave the corresponding γ-amino-gem-bisphosphonic acids 217a–f in 43 to 58% yield (Scheme 94) [138]. In this work, the stereoselective version with α-substituted azlactone 215 and different chiral Brønsted bases was evaluated.
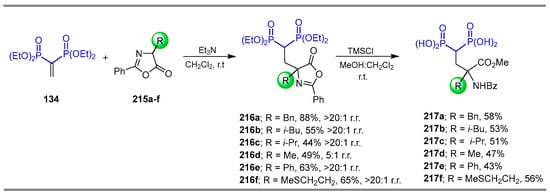
Scheme 94.
Synthesis of γ-amino-gem-bisphosphonates 216a–f and γ-amino-gem-bisphosphonic acids 217a–f.
5. Synthesis of Heterocyclic α-Amino-gem-Bisphosphonate Derivatives
5.1. Phosphonylation of Lactams
The bisphosphonylation of lactams with dialkyl or trialkyl phosphites using activating agents such as PCl3, P(O)Cl3 and Tf2O is an efficient and general method for the synthesis of heterocyclic α-amino-gem-bisphosphonate derivatives. For example, Kafarski et al. [139] carried out the reaction of γ-, δ- and ε-lactams 218a–f with triethyl phosphite and phosphoryl chloride at 0 °C, obtaining the heterocyclic α-amino-gem-bisphosphonates 219a–f in 28 to 65% yield. Additionally, the reaction of benzoannulated lactams 218g,h under identical conditions gave the corresponding heterocyclic α-amino-gem-bisphosphonates 219g,h in 17 to 30% yield, accompanied by benzo-fused monophosphonates 220a,b in 30 and 20% yield, respectively (Scheme 95).
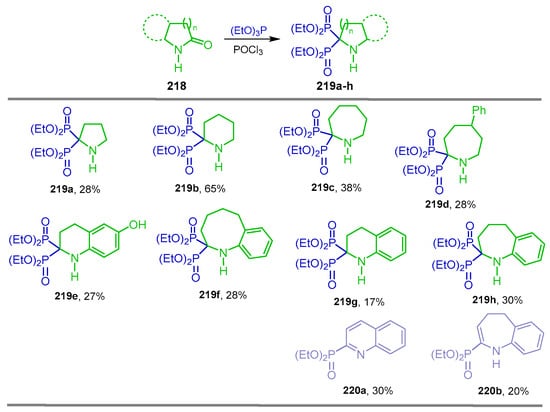
Scheme 95.
Synthesis of heterocyclic α-amino-gem-bisphosphonates 219a–h.
We developed a versatile methodology for the synthesis of heterocyclic α-amino-gem-bisphosphonates from lactams using two distinct approaches. In the first strategy (Method A), the N-benzyl piperidin-2-one 221a, morpholin-3-one 221b and thiomorpholin-3-one 221c were reacted with triethyl phosphite and phosphoryl chloride, affording the corresponding heterocyclic α-amino-gem-bisphosphonates 222a–c in 20 to 87% yield. Treatment of 222b,c with TMSBr followed by the addition of methanol gave the heterocyclic α-amino-gem-bisphosphonic acids 223b,c in 60% yield. Additionally, the reaction N-benzylated thiomorpholin-3-one 221d with triethyl phosphite Tf2O DTBMP (Method B) gave the heterocyclic α-amino-gem-bisphosphonate 222d in 70% yield. These heterocyclic α-amino-gem-bisphosphonates constitute valuable building blocks for medicinal chemistry, particularly due to their potential bone-targeting properties and structural relevance to bioactive bisphosphonates (Scheme 96) [140].
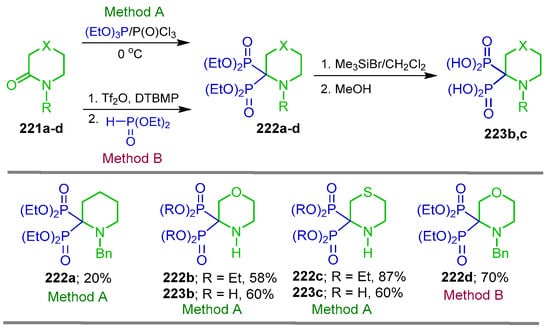
Scheme 96.
Synthesis of heterocyclic α-amino-gem-bisphosphonates 222a–d and heterocyclic α-amino-gem-bisphosphonic acids 223b,c.
Wang et al. [107] reported a mild and efficient bisphosphonylation protocol of lactams based on the activation with Tf2O. Thus, the reaction of N-benzyl lactams 221 with Tf2O, diethyl phosphite and DTBMP at 0 °C produced the desired heterocyclic α-amino-gem-bisphosphonates 222 in 72 to 93% yield. These reaction conditions proceed with excellent chemoselectivity and tolerate the presence of tert-butoxycarbonyl groups (Scheme 97).
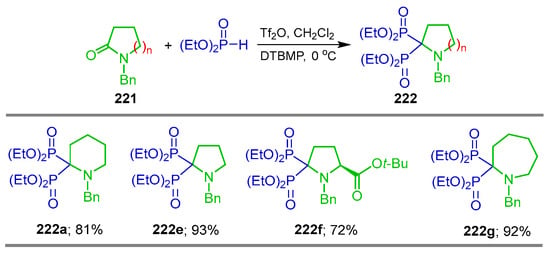
Scheme 97.
Mild bisphosphonylation of N-benzyl lactams to synthesis of heterocyclic α-amino-gem-bisphosphonates 222a–g.
5.2. Miscellaneous
Kolodiazhnyi et al. [141] found that, during the treatment of ethyl isoindolin-1-one-3-yl-phosphonate 224 in acidic conditions, this compound undergoes a retro-Michael reaction, releasing diethyl phosphite and generating imine 225, which is susceptible to autoxidation and diphosphonylation with diethyl phosphite to give the heterocyclic α-amino-gem-bisphosphonate 227, an analog of benzo[c]pyroglutamic acid via the carbon-centered radical 226 (Scheme 98).
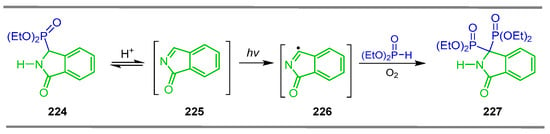
Scheme 98.
Synthesis of heterocyclic α-amino-gem-bisphosphonate 227 via radical pathway.
The reaction of o-phthaloyl chloride 228 with the sodium salt of diethyl phosphite in diethyl ether at 0 °C afforded 3,3-bis(diethylphosphono)-1(3H)-isobenzofuranone 229 in 40% yield, which, by reaction with benzylamine and triethylamine in toluene at reflux, produced tetraethyl (2-benzyl-3-oxoisoindolyl-1,1-diyl)-bisphosphonate 230 in 41% yield. The reaction of 230 with HCl at reflux gave 2-benzyl-3-oxoisoindolin-1,1-dyil-bisphosphonic acid 231 in 87% yield (Scheme 99) [142,143].
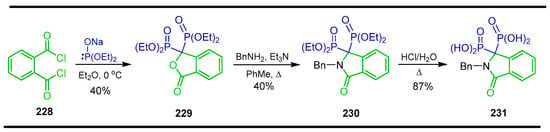
Scheme 99.
Synthesis of heterocyclic α-amino-gem-bisphosphonate 230 and heterocyclic α-amino-gem-bisphosphonic acid 231.
On the other hand, the reaction of tetraethyl methylenebisphosphonate with 2H-3,1-benzothiazine-2,4(1H)-dithiones 232a,b and sodium ethoxide in ethanol at reflux followed by treatment with HCl at −5 °C gave the 3-thioxoindoline-2,2-diyldiphosphonates 234a,b in 41 and 40% yield, respectively, via the intermediates 233a,b (Scheme 100). Similar results were obtained in the reaction of 1H-benzo[d][1,3]-oxazin-2,4-diones 235a,b with tetraethyl methylenebisphosphonate, affording the tetraethyl 3-oxoindoline-2,2-diyldiphosphonates 236a,b in 58 and 54% yield, respectively. These compounds exhibited remarkable antitumor activity against four tested carcinoma cell lines and showed also significant to moderate anti-inflammatory activity capable of inhibiting polyarthritis [144], and their cytogenetic activity in normal human lymphocyte cultures was also evaluated [61].
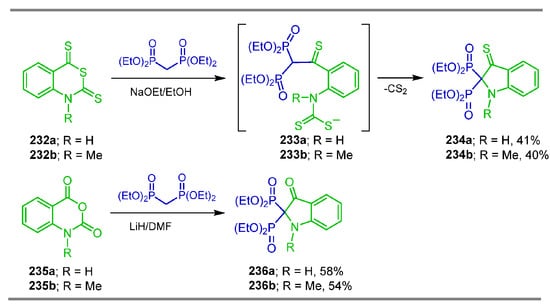
Scheme 100.
Synthesis of 3-thioxoindoline-2,2-diyldiphosphonates 234a,b and 3-oxoindoline-2,2-diyldiphosphonates 236a,b.
Scarso et al. [145] reported the synthesis of a large series of substituted pyrrolidine containing bisphosphonates through 1,3-dipolar cycloaddition of azomethine ylides to vinyl gem-bisphosphonates as dipolarophiles. Thus, the 1,3-dipolar cycloaddition of azomethine ylides 237 derived from the condensation/decarboxylation of aldehydes with the corresponding α-amino acids or sarcosine, and the tetraethyl vinyl gem-bisphosphonates 209, afforded the (pyrrolidine-3,3-diyl)bis(phosphonate) esters 238a–n in 7 to 68% yields. This strategy represents a straightforward and modular approach to access structurally diverse N-heterocyclic gem-bisphosphonates, with potential application in bone resorption diseases such as osteoporosis (Scheme 101).
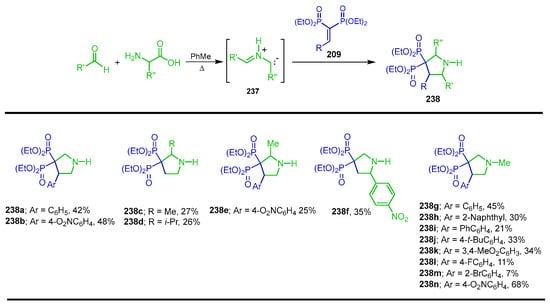
Scheme 101.
Synthesis of (pyrrolidine-3,3-diyl)bis(phosphonates) 238a–n.
Finally, the 1,3-dipolar cycloaddition of azomethine ylides derived from the condensation/decarboxylation of aldehydes with diethyl 2-aminomalonate 239 and tetraethyl vinyl gem-bisphosphonate 134 afforded the diethyl 4,4-bis(diethoxyphosphoryl)-5-pentylpyrrolidine-2,2-dicarboxylates 240a–c in 41 to 68% yield (Scheme 102) [146].
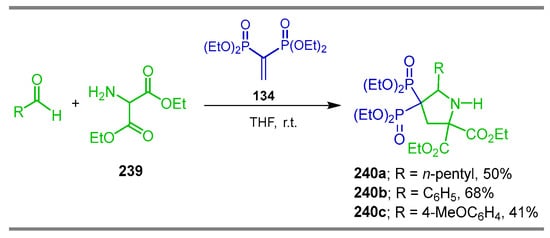
Scheme 102.
Synthesis of pyrrolidine-2,2-dicarboxylates 240a–c.
6. Concluding Remarks
In this review, we have covered the last fifteen years in the development of new synthetic methodologies for the synthesis of acyclic and heterocyclic amino-gem-bisphosphonic acids and derivatives; the biological activities have also been discussed.
All these methodologies give the synthetic organic chemist the opportunity to select the most appropriate way to obtain the desired acyclic and heterocyclic amino-gem-bisphosphonic acids and derivatives.
We propose that continued efforts should be made to search for and improve synthetic procedures for the preparation of new amino-gem-bisphosphonates, and that the exploration of new chemical and biological applications of these interesting compounds will be a very rewarding task for researchers in this area and necessary for the coming years.
Funding
This research was funded by Secretaría de Ciencia, Humanidades, Tecnología e Innovación (SECIHTI), for financial support through projects 256985 and 286614.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The authors thank Victoria Labastida-Galván, José Luis Patiño Carbajal and Sebastian Vázquez Aguirre for their valuable technical support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mucha, A.; Kafarski, P.; Berlicki, L. Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Sello, G.; Sisti, M. Aminophosphonic Acids and Derivatives. Synthesis and Biological Applications. Curr. Med. Chem. 2010, 17, 264–289. [Google Scholar] [CrossRef] [PubMed]
- Naydenova, E.D.; Todorov, P.T.; Troev, K.D. Recent synthesis of aminophosphonic acids as potential biological importance. Amino Acids 2010, 38, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Gluza, K.; Kafarski, P. Chap. 12 Transition state analogues of enzymatic reaction as potential drugs. In Drug Discovery; El-Shemy, H., Ed.; InTech: Rijeka, Croatia, 2013; pp. 325–372. [Google Scholar]
- Lejczak, B.; Kafarski, P. Aminated (Cyclopropylmethyl)Phosphonates: Synthesis and Anti-Pancreatic Cancer Activity. Top. Heterocycl. Chem. 2009, 20, 31–63. [Google Scholar]
- Ordoñez, M.; Viveros-Ceballos, J.L.; Romero-Estudillo, I. Chap. 6 Stereoselective synthesis of α-aminophosphonic acids through Pudovik and Kabachnik Fields-reaction. In Amino Acid-New Insights and Roles in Plant and Animal; Asao, T., Asaduzzaman, M.D., Eds.; InTech: Rijeka, Croatia, 2017; pp. 127–151. [Google Scholar]
- Ordoñez, M.; Viveros-Ceballos, J.L.; Cativiela, C.; Sayago, F.J. An update on the stereoselective synthesis of α-aminophosphonic acids and derivatives. Tetrahedron 2015, 71, 1745–1784. [Google Scholar] [CrossRef]
- Ordoñez, M.; Viveros-Ceballos, J.L.; Cativiela, C.; Arizpe, A. Stereoselective Synthesis of α-Aminophosphonic Acids Analogs of the 20 Proteinogenic α-Amino Acids. Curr. Org. Synth. 2012, 9, 310–341. [Google Scholar] [CrossRef]
- Ordoñez, M.; Rojas-Cabrera, A.; Cativiela, C. An overview of stereoselective synthesis of α-aminophosphonic acids and derivatives. Tetrahedron 2009, 65, 17–49. [Google Scholar] [CrossRef] [PubMed]
- Mayorquín-Torres, M.C.; Simoens, A.; Bonneure, E.; Stevens, C.V. Synthetic Methods for Azaheterocyclic Phosphonates and Their Biological Activity: An Update. 2004–2024. Chem. Rev. 2024, 124, 7907–7975. [Google Scholar] [CrossRef] [PubMed]
- Amira, A.; Aouf, Z.; K’tir, H.; Chemam, Y.; Ghodbane, R.; Zerrouki, R.; Aouf, N.-E. Recent Advances in the Synthesis of α-Aminophosphonates: A Review. ChemistrySelect 2021, 6, 6137–6149. [Google Scholar] [CrossRef]
- Varga, P.R.; Keglevich, G. Synthesis of α-Aminophosphonates and Related Derivatives; The Last Decade of the Kabachnik–Fields Reaction. Molecules 2021, 26, 2511. [Google Scholar] [CrossRef] [PubMed]
- Herrera, R.P. Organocatalytic Hydrophosphonylation Reaction of Carbonyl Groups. Chem. Rec. 2017, 17, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.E.; Abdel-Kariem, S.M. Methods for the synthesis of α-heterocyclic/heteroaryl-α-aminophosphonic acids and their esters. Arkivoc 2015, 6, 246–287. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Kudzin, M.H.; Drabowicz, J.; Stevens, C.V. Aminophosphonic Acids—Phosphorus Analogues of Natural Amino Acids. Part 1: Syntheses of α-Aminophosphonic Acids. Curr. Org. Chem. 2011, 15, 2015–2071. [Google Scholar] [CrossRef]
- Merino, P.; Marqués-López, E.; Herrera, R.P. Catalytic Enantioselective Hydrophosphonylation of Aldehydes and Imines. Synth. Catal. 2008, 350, 1195–1208. [Google Scholar] [CrossRef]
- Ordoñez, M.; Viveros-Ceballos, J.L.; Sayago, F.J.; Cativiela, C. Stereoselective Synthesis of α-Amino-H-phosphinic Acids and Derivatives. Synthesis 2017, 49, 987–997. [Google Scholar] [CrossRef]
- Viveros-Ceballos, J.L.; Ordoñez, M.; Sayago, F.J.; Cativiela, C. Stereoselective Synthesis of α-Amino-C-phosphinic Acids and Derivatives. Molecules 2016, 21, 1141. [Google Scholar] [CrossRef]
- Takiguchi, S.; Nishino, Y.; Inoue, K.; Ikeda, M.; Kataoka, Y.; Matsusue, K.; Nishiyama, K.; Iguchi, H. The bisphosphonate incadronate inhibits intraperitoneal dissemination in an in vivo pancreatic cancer model. Oncol. Rep. 2012, 28, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, S.; Iguchi, K.; Usui, S.; Hirano, K. Incadronate Induces Cell Detachment and Apoptosis in Prostatic PC-3 Cells. Anticancer Res. 2007, 27, 927–932. [Google Scholar] [PubMed]
- Koizumi, M.; Kobayashi, M.; Furukawa, M.; Yamashita, T.; Ogata, E. The bisphosphonate incadronate for bone metastases of breast cancer. Int. J. Clin. Oncol. 2000, 5, 241–246. [Google Scholar] [CrossRef]
- Kafarski, P.; Lejczak, B.; Forlani, G. Herbicidally active aminomethylenebisphosphonic acids. Heteroat. Chem. 2000, 11, 449–453. [Google Scholar] [CrossRef]
- Rosso, V.S.; Szajnman, S.H.; Malayil, L.; Galizzi, M.; Moreno, S.N.J.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of new 2-alkylaminoethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii targeting farnesyl diphosphate synthase. Bioorg. Med. Chem. 2011, 19, 2211–2217. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xiao, L.; Tao, J.; Srinivasan, V.; Boyce, B.F.; Ebetino, F.H.; Oyajobi, B.O.; Boeckman, R.K., Jr.; Xing, L. Synthesis of a Bone-Targeted Bortezomib with In Vivo Anti-Myeloma Effects in Mice. Pharmaceutics 2018, 10, 154. [Google Scholar] [CrossRef]
- Holub, J.; Meckel, M.; KubíčeK, V.; Rösch, F.; Hermann, P. Gallium(III) complexes of NOTA-bis (phosphonate) conjugates as PET radiotracers for bone imaging. Contrast Media Mol. Imaging 2015, 10, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Vitha, T.; KubíčeK, V.; Kotek, J.; Hermann, P.; Elst, L.V.; Muller, T.N.; Lukeš, I.; Peters, J.A. Gd(III) complex of a monophosphinate-bis(phosphonate) DOTA analogue with a high relaxivity; Lanthanide(III) complexes for imaging and radiotherapy of calcified tissues. Dalton Trans. 2009, 17, 3204–3214. [Google Scholar] [CrossRef] [PubMed]
- Zha, Z.; Wu, Z.; Choi, S.R.; Ploessl, K.; Smith, M.; Alexoff, D.; Zhu, L.; Kung, H.F. A New [68Ga]Ga-HBED-CC-Bisphosphonate as a Bone Imaging Agent. Mol. Pharm. 2020, 17, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Pospiech, M.; Owens, S.E.; Miller, D.J.; Austin-Muttitt, K.; Mullins, J.G.L.; Cronin, J.G.; Allemann, R.K.; Sheldon, I.M. Bisphosphonate inhibitors of squalene synthase protect cells against cholesterol-dependent cytolysins. FASEB J. 2021, 35, e21640. [Google Scholar] [CrossRef] [PubMed]
- Galaka, T.; Falcone, B.N.; Li, C.; Szajnman, S.H.; Moreno, S.N.J.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 1-alkylaminomethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii. Bioorg. Med. Chem. 2019, 27, 3663–3673. [Google Scholar] [CrossRef] [PubMed]
- Ghoteimi, R.; Nguyen Van, T.; Rahimova, R.; Grosjean, F.; Cros-Perrial, E.; Uttaro, J.-P.; Mathé, C.; Chaloin, L.; Jordheim, L.P.; Peyrottes, S. Synthesis of Substituted 5′-Aminoadenosine Derivatives and Evaluation of Their Inhibitory Potential toward CD73. ChemMedChem 2019, 14, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Kosikowska, P.; Bochno, M.; Macegoniuk, K.; Forlani, G.; Kafarski, P.; Berlicki, Ł. Bisphosphonic acids as effective inhibitors of Mycobacterium tuberculosis glutamine synthetase. J. Enzyme Inhib. Med. Chem. 2016, 31, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Tauro, M.; Laghezza, A.; Loiodice, F.; Agamennone, M.; Campestre, C.; Tortorella, P. Arylamino methylene bisphosphonate derivatives as bone seeking matrix metalloproteinase inhibitors. Bioorg. Med. Chem. 2013, 21, 6456–6465. [Google Scholar] [CrossRef] [PubMed]
- Rubino, M.T.; Agamennone, M.; Campestre, C.; Campiglia, P.; Cremasco, V.; Faccio, R.; Laghezza, A.; Loiodice, F.; Maggi, D.; Panza, E.; et al. Biphenyl Sulfonylamino Methyl Bisphosphonic Acids as Inhibitors of Matrix Metalloproteinases and Bone Resorption. ChemMedChem 2011, 6, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Chmielewska, E.; Mazur, Z.; Kempińska, K.; Wietrzyk, J.; Piątek, A.; Kuryszko, J.J.; Kiełbowicz, Z.; Kafarski, P. N-Arylaminomethylenebisphosphonates Bearing Fluorine Atoms: Synthesis and Antiosteoporotic Activity. Phosphorus Sulfur. 2015, 190, 2164–2172. [Google Scholar] [CrossRef]
- Jiang, Q.; Yang, L.; Hai, L.; Wu, Y. Synthesis of Melphalan-Gem-Bisphosphonate Conjugation to Bone Tumors and Study of Affinity to Hydroxyapatite In Vitro. Lett. Org. Chem. 2008, 5, 229–233. [Google Scholar] [CrossRef]
- Miszczyk, P.; Wieczorek, D.; Gałęzowska, J.; Dziuk, B.; Wietrzyk, J.; Chmielewska, E. Reaction of 3-Amino-1,2,4-Triazole with Diethyl Phosphite and Triethyl Orthoformate: Acid-Base Properties and Antiosteoporotic Activities of the Products. Molecules 2017, 22, 254. [Google Scholar] [CrossRef] [PubMed]
- Lacbay, C.M.; Waller, D.D.; Park, J.; Gómez Palou, M.; Vincent, F.; Huang, J.F.; Ta, V.; Berghuis, A.M.; Sebag, M.; Tsantrizos, Y.S. Unraveling the Prenylation–Cancer Paradox in Multiple Myeloma with Novel Geranylgeranyl Pyrophosphate Synthase (GGPPS) Inhibitors. J. Med. Chem. 2018, 61, 6904–6917. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-F.; Lacbay, C.M.; Boutin, R.; Matralis, A.N.; Park, J.; Waller, D.D.; Guan, T.L.; Sebag, M.; Tsantrizos, Y.S. Synthesis and Evaluation of Structurally Diverse C-2-Substituted Thienopyrimidine-Based Inhibitors of the Human Geranylgeranyl Pyrophosphate Synthase. J. Med. Chem. 2022, 65, 2471–2496. [Google Scholar] [CrossRef] [PubMed]
- Boutin, R.; Lee, H.-F.; Guan, T.L.; Nguyen, T.T.; Huang, J.F.; Waller, D.D.; Lu, J.; Chio, I.I.C.; Michel, R.P.; Sebag, M.; et al. Discovery and Evaluation of C6-Substituted Pyrazolopyrimidine-Based Bisphosphonate Inhibitors of the Human Geranylgeranyl Pyrophosphate Synthase and Evaluation of Their Antitumor Efficacy in Multiple Myeloma, Pancreatic Ductal Adenocarcinoma, and Colorectal Cancer. J. Med. Chem. 2023, 66, 15776–15800. [Google Scholar] [PubMed]
- Shaddy, A.A.; Kamel, A.A.; Abdou, W.M. Synthesis, Quantitative Structure—Activity Relationship, and Anti-Inflammatory Profiles of Substituted 5- and 6-N-Heterocycle Bisphosphonate Esters. Synth. Commun. 2013, 43, 236–252. [Google Scholar] [CrossRef]
- Laghezza, A.; Piemontese, L.; Brunetti, L.; Caradonna, A.; Agamennone, M.; Loiodice, F.; Tortorella, P. (2-Aminobenzothiazole)-Methyl-1,1-Bisphosphonic Acids: Targeting Matrix Metalloproteinase 13 Inhibition to the Bone. Pharmaceuticals 2021, 14, 85. [Google Scholar] [CrossRef] [PubMed]
- De Schutter, J.W.; Zaretsky, S.; Welbourn, S.; Pause, A.; Tsantrizos, Y.S. Novel bisphosphonate inhibitors of the human farnesyl pyrophosphate synthase. Bioorg. Med. Chem. Lett. 2010, 20, 5781–5786. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-S.; Park, J.; De Schutter, J.W.; Huang, J.F.; Berghuis, A.M.; Sebag, M.; Tsantrizos, Y.S. Design and Synthesis of Active Site Inhibitors of the Human Farnesyl Pyrophosphate Synthase: Apoptosis and Inhibition of ERK Phosphorylation in Multiple Myeloma Cells. J. Med. Chem. 2012, 55, 3201–3215. [Google Scholar] [CrossRef] [PubMed]
- Gałęzowska, J.; Czapor-Irzabek, H.; Chmielewska, E.; Kafarski, P.; Janeka, T. Aminobisphosphonates based on cyclohexane backbone as coordinating agents for metal ions. Thermodynamic, spectroscopic and biological studies. New J. Chem. 2018, 42, 7723–7736. [Google Scholar] [CrossRef]
- Shaik, M.S.; Nadiveedhi, M.R.; Gundluru, M.; Katike, U.; Obulam, V.S.R.; Cirandur, S.R. Efficient catalyst free green synthesis and in vitro antimicrobial, antioxidant and molecular docking studies of α-substituted aromatic/heteroaromatic aminomethylene bisphosphonates. Synth. Commun. 2021, 51, 747–764. [Google Scholar] [CrossRef]
- Tellamekala, S.; Gundluru, M.; Sudileti, M.; Sarva, S.; Putta, C.R.K.; Cirandur, S.R. Green one-pot synthesis of N-bisphosphonates as antimicrobial and antioxidant agents. Monatsh. Chem. 2020, 151, 251–260. [Google Scholar] [CrossRef]
- Mohan, G.; Santhisudha, S.; Reddy, N.M.; Sreekanth, T.; Murali, S.; Reddy, C.S. Nano ZnO catalyzed green synthesis and cytotoxic assay of pyridinyl and pyrimidinyl bisphosphonates. Monatsh. Chem. 2017, 148, 1843–1851. [Google Scholar] [CrossRef]
- Amira, A.; K’tir, H.; Aouf, Z.; Khaldi, T.; Bentoumi, H.; Khattabi, L.; Zerrouki, R.; Ibrahim-Ouali, M.; Aouf, N.-E. One-Pot Microwave-Assisted Synthesis, in Vitro Anti-inflammatory Evaluation and Computer-Aided Molecular Design of Novel Sulfamide-Containing Bisphosphonates Derivatives. ChemitrySelect 2022, 7, e202201889. [Google Scholar] [CrossRef]
- Gehrke, N.R.; Feng, D.; Ali, M.A.; Maalouf, M.A.; Holstein, S.A.; Weimer, D.F. α-Amino bisphosphonate triazoles serve as GGDPS inhibitors. Bioorg. Med. Chem. Lett. 2024, 102, 129659. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.M.A.; El-Hussieny, M.; El-Sayed, N.F.; Fouad, M.A.; Ewies, F.E.; Ezzat, M.A.F. Synthesis and characterization of new tetrakisphosphonic acid derivatives as FPPS inhibitors and evaluation of their anti-osteoclastogenic potential for prevention of osteoporosis. Med. Chem. Res. 2024, 33, 1167–1177. [Google Scholar] [CrossRef]
- Goldeman, W.; Nasulewicz-Goldeman, A. Synthesis and biological evaluation of aminomethylidenebisphosphonic derivatives of β-arylethylamines. Tetrahedron 2015, 71, 3282–3289. [Google Scholar] [CrossRef]
- Nasulewicz-Goldeman, A.; Goldeman, W.; Mrówczyńska, E.; Wietrzyk, J. Biological effects of aromatic bis[aminomethylidenebis(phosphonic)] acids in osteoclast precursors in vitro. Chem. Biol. Drug Des. 2019, 94, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.N.; Li, C.; Storey, M.; Falcone, B.N.; Szajnman, S.H.; Bonesi, S.M.; Docampo, R.; Moreno, S.N.J.; Rodriguez, J.B. Activity of Fluorine-Containing Analogues of WC-9 and Structurally Related Analogues against Two Intracellular Parasites: Trypanosoma cruzi and Toxoplasma gondii. ChemMedChem 2016, 11, 2690–2702. [Google Scholar] [CrossRef] [PubMed]
- Grigor’ev, I.A.; Morozova, D.A.; Svyatchenkoc, V.A.; Kiselevc, N.N.; Loktevc, V.B.; Luk’yanetse, E.A.; Vorozhtsove, G.N. Synthesis and Study of Antitumor Activity of Tetraethyl 2-(2′,2′,6′,6′-Tetramethylpiperidin-4′-ylamino)ethylidene-1,1-bisphosphonate. Dokl. Chem. 2014, 457, 137–140. [Google Scholar] [CrossRef]
- Granchi, D.; Scarso, A.; Bianchini, G.; Chiminazzo, A.; Minto, A.; Sgarbossa, P.; Michelin, R.A.; Di Pompo, G.; Avnet, S.; Strukul, G. Low toxicity and unprecedented anti-osteoclast activity of a simple sulfur-containing gem-bisphosphonate: A comparative study. Eur. J. Med. Chem. 2013, 65, 448–455. [Google Scholar] [CrossRef]
- Wu, X.; Yang, Z.; Song, C.; Bu, M.; Li, W.; Duan, J.; Yang, G.-F.; Zhang, A. Hydroxamate-Containing Bisphosphonates as Fosmidomycin Analogues: Design, Synthesis, and Proherbicide Activity. J. Agric. Food Chem. 2024, 72, 7684–7693. [Google Scholar] [CrossRef] [PubMed]
- Galaka, T.; Casal, M.F.; Storey, M.; Li, C.; Chao, M.N.; Szajnman, S.H.; Docampo, R.; Moreno, S.N.J.; Rodriguez, J.B. Antiparasitic Activity of Sulfur- and Fluorine-Containing Bisphosphonates against Trypanosomatids and Apicomplexan Parasites. Molecules 2017, 22, 82. [Google Scholar] [CrossRef] [PubMed]
- Makarov, M.V.; Leonova, E.S.; Rybalkina, E.Y.; Khrustalev, V.N.; Shepel, N.E.; Röschenthaler, G.-V.; Timofeeva, T.V.; Odinets, I.L. Methylenebisphosphonates with Dienone Pharmacophore: Synthesis, Structure, Antitumor and Fluorescent Properties. Arch. Pharm. Chem. Life Sci. 2012, 345, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Sankala, E.; Weisell, J.M.; Huhtala, T.; Närvänen, A.T.O.; Vepsäläinen, J.J. Synthesis of novel bisphosphonate polyamine conjugates and their affinity to hydroxyapatite. Arkivoc 2012, 4, 233–241. [Google Scholar] [CrossRef]
- Shevchuk, M.V.; Sotiropoulos, J.-M.; Miqueu, K.; Romanenko, V.D.; Kukhar, V.P. Tetrakis(trimethylsilyl) Ethenylidene-1,1-bisphosphonate: A Mild and Convenient Michael Acceptor for the Synthesis of 2-Aminoethylidene-1,1-bisphosphonic Acids and Their Potassium Salts. Synlett 2011, 2011, 1370–1374. [Google Scholar]
- Abdou, W.M.; Kamel, A.A.; Khidre, R.E.; Geronikaki, A.; Ekonomopoulou, M.T. Synthesis of 5- and 6-N-heterocyclic Methylenebisphosphonate Derivatives and Evaluation of their Cytogenetic Activity in Normal Human Lymphocyte Cultures. Chem. Biol. Drug Des. 2012, 79, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.-Y.; Li, Q.-H.; Tao, H.-Y.; Wang, C.-J. A Facile Cu(I)/TF-BiphamPhos-Catalyzed Asymmetric Approach to Unnatural α-Amino Acid Derivatives Containing gem-Bisphosphonates. J. Am. Chem. Soc. 2011, 133, 11757–11765. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Tada, A.; Tokoro, Y.; Fukuzawa, S.I. Silver-catalyzed asymmetric Michael addition of azomethine ylide to arylidene diphosphonates using ThioClickFerrophos ligand. Tetrahedron Lett. 2015, 56, 2251–2253. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I.; Kolodiazhna, A.O. Stereoselective Syntheses of Organophosphorus Compounds. Symmetry 2024, 16, 342. [Google Scholar] [CrossRef]
- Kaboudin, B.; Daliri, P.; Faghih, S.; Esfandiari, H. Hydroxy- and Amino-Phosphonates and -Bisphosphonates: Synthetic Methods and Their Biological Applications. Front. Chem. 2022, 10, 890696. [Google Scholar] [CrossRef] [PubMed]
- Popov, K.; Oshchepkov, M.; Tkachenko, S.; Sergienko, V.; Oshchepkov, A. Bisphosphonates: Synthesis, structures, properties, medical and industrial applications. J. Mol. Liq. 2022, 351, 118619. [Google Scholar] [CrossRef]
- Romanenko, V.D. α-Heteroatom-substituted gem-Bisphosphonates: Advances in the Synthesis and Prospects for Biomedical Application. Curr. Org. Chem. 2019, 23, 530–615. [Google Scholar] [CrossRef]
- Romanenko, V.D.; Kukhar, V.P. 1-Amino-1,1-bisphosphonates. Fundamental syntheses and new developments. Arkivoc 2012, 4, 127–166. [Google Scholar] [CrossRef]
- Krutikov, V.I.; Erkin, A.V.; Pautov, P.A.; Zolotukhina, M.M. Heteryl- and Arylaminomethylenebisphosphonates: Synthesis and Biologic Activity. Russ. J. Gen. Chem. 2003, 73, 187–191. [Google Scholar] [CrossRef]
- Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. Insight into the mechanism of three component condensation leading to aminomethylenebisphosphonates. J. Organomet. Chem. 2009, 694, 3806–3813. [Google Scholar] [CrossRef]
- Bochno, M.; Berlicki, Ł. A three-component synthesis of aminomethylenebis-H-phosphinates. Tetrahedron Lett. 2014, 55, 219–223. [Google Scholar] [CrossRef]
- Viveros-Ceballos, J.L.; Matías-Valdez, L.A.; Sayago, F.J.; Cativiela, C.; Ordoñez, M. New approaches towards the synthesis of 1,2,3,4-tetrahydro isoquinoline-3-phosphonic acid (TicP). Amino Acids 2021, 53, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Goswami, D.; Chakravarty, R.; Mohammed, K.S.; Sarma, H.D.; Dash, A. Syntheses and evaluation of 68Ga- and 153Sm-labeled DOTA-conjugated bisphosphonate ligand for potential use in detection of skeletal metastases and management of pain arising from skeletal metastases. Chem. Biol. Drug Des. 2018, 92, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Ehrick, R.S.; Capaccio, M.; Puleo, D.A.; Bachas, L.G. Ligand-Modified Aminobisphosphonate for Linking Proteins to Hydroxyapatite and Bone Surface. Bioconj. Chem. 2008, 19, 315–321. [Google Scholar] [CrossRef]
- Leung, C.Y.; Park, J.; De Schutter, J.W.; Sebag, M.; Berghuis, A.M.; Tsantrizos, Y.S. Thienopyrimidine Bisphosphonate (ThPBP) Inhibitors of the Human Farnesyl Pyrophosphate Synthase: Optimization and Characterization of the Mode of Inhibition. J. Med. Chem. 2013, 56, 7939–7950. [Google Scholar] [CrossRef] [PubMed]
- Brel, V.K. Synthesis of gem-bisphosphonates with (3-aryl-4,5-dihydroisoxazol-5-yl)methylamino moiety. Mendeleev Commun. 2015, 25, 234–235. [Google Scholar] [CrossRef]
- Petruczynik, P.; Kafarski, P.; Psurski, M.; Wietrzyk, J.; Kielbowicz, Z.; Kuryszko, J.; Chmielewska, E. Three-Component Reaction of Diamines with Triethyl Orthoformate and Diethyl Phosphite and Anti-Proliferative and Antiosteoporotic Activities of the Products. Molecules 2020, 25, 1424. [Google Scholar] [CrossRef] [PubMed]
- Bálint, E.; Tajti, A.; Dzielak, A.; Hägele, G.; Keglevich, G. Microwave-assisted synthesis of (aminomethylene)bisphosphine oxides and (aminomethylene)bisphosphonates by a three-component condensation. Beilstein J. Org. Chem. 2016, 12, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Minaeva, L.I.; Patrikeeva, L.S.; Kabachnik, M.M.; Beletskaya, I.P.; Orlinson, B.S.; Novakov, I.A. Synthesis of novel aminomethylenebisphosphonates and bisphosphonic acids, containing adamantyl fragment. Heteroatom Chem. 2011, 22, 55–58. [Google Scholar] [CrossRef]
- Sudileti, M.; Nagaripati, S.; Gundluru, M.; Chintha, V.; Aita, S.; Wudayagiri, R.; Chamarthi, N.; Cirandur, S.R. rGO-SO3H Catalysed Green Synthesis of Fluoro-Substituted Aminomethylene Bisphosphonates and their Anticancer, Molecular Docking studies. ChemitrySelect 2019, 4, 13006–13011. [Google Scholar] [CrossRef]
- Manjula, G.; Kalla, R.M.N.; Reddy, A.E.; Almutairi, T.M.; Lee, J. Ultrasonic-promoted synthesis of amino methylene bisphosphonates by sulfated choline ionic liquid. Res. Chem. Intermediat. 2025, 51, 1357–1370. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Synthesis of new functionalized aryl and pyridyl aminomethylenebisphosphonic acids and their derivatives via silicon-assisted methodology. J. Organomet. Chem. 2020, 912, 121177. [Google Scholar] [CrossRef]
- Reddy, S.S.; Kalla, R.M.N.; Varyambath, A.; Kim, I. Sulfonic acid functionalized hyper-cross-linked polymer: An efficient heterogeneous acid catalyst for the synthesis of N-containing bisphosphonates. Catal. Commun. 2019, 126, 15–20. [Google Scholar] [CrossRef]
- Kunda, U.M.R.; Balam, S.K.; Nemallapudi, B.R.; Chereddy, S.S.; Nayak, S.K.; Cirandur, S.R. Facile Synthesis, Antioxidant and Antimicrobial Activity of Amino Methylene Bisphosphonates. Chem. Pharm. Bull. 2012, 60, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Shahvelayati, A.S.; Tanha, A. A Review of Ionic Liquids: From Solvent Applications to Template-Assisted Synthesis of Metal Oxide Nanoparticles. Appl. Organomet. Chem. 2025, 39, e70026. [Google Scholar] [CrossRef]
- Kaur, G.; Kumar, H.; Singla, M. Diverse applications of ionic liquids: A comprehensive review. J. Mol. Liq. 2022, 351, 118556. [Google Scholar] [CrossRef]
- Kianfar, E.; Mafi, S. Ionic Liquids: Properties, Application, and Synthesis. Fine Chem. Eng. 2020, 2, 21–29. [Google Scholar] [CrossRef]
- Lei, Z.; Chen, B.; Koo, Y.-M.; MacFarlane, D.R. Introduction: Ionic Liquids. Chem. Rev. 2017, 117, 6633–6635. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.; Kalla, R.M.N.; Dong, L.S.; Jeong, T.J. Di-n-butyl ammonium chlorosulfonate as a highly efficient and recyclable ionic liquid for the synthesis of N-containing bisphosphonates. Catal. Commun. 2015, 61, 102–106. [Google Scholar] [CrossRef]
- Poola, S.; Gundluru, M.; Nadiveedhi, M.R.; Saddala, M.S.; Ptsrk, P.R.; Cirandur, S.R. Nano silver particles catalyzed synthesis, molecular docking and bioactivity of α-thiazolyl aminomethylene bisphosphonates. Phosphorus Sulfur. 2020, 195, 409–420. [Google Scholar] [CrossRef]
- Silva Prasad, S.; Jayaprakash, S.H.; Syamasundar, C.; Sreelakshmi, P.; Bhuvaneswar, C.; Bhaskar, B.V.; Rajendra, W.; Nayak, S.K.; Cirandur, S.R. Tween 20-/H2O Promoted Green Synthesis, Computational and Antibacterial Activity of Amino Acid Substituted Methylene Bisphosphonates. Phosphorus Sulfur. 2015, 190, 2040–2050. [Google Scholar] [CrossRef]
- Kuźnik, A.; Mazurkiewicz, R.; Grymel, M.; Zielińska, K.; Adamek, J.; Chmielewska, E.; Bochno, M.; Kubica, S. A new method for the synthesis of α-aminoalkylidenebisphosphonates and their asymmetric phosphonyl-phosphinyl and phosphonyl-phosphinoyl analogues. Beilstein J. Org. Chem. 2015, 11, 1418–1424. [Google Scholar] [CrossRef] [PubMed]
- Kuźnik, A.; Mazurkiewicz, R.; Zięba, M.; Erfurt, K. 1-(N-Acylamino)-1-triphenylphosphoniumalkylphosphonates: General synthesis and prospects for further synthetic applications. Tettrahedron Lett. 2018, 59, 3307–3320. [Google Scholar] [CrossRef]
- Kuźnik, A.; Kozicka, D.; Październiok-Holewa, A.; Dąbek, A.; Juszczak, K.; Sokołowska, G.; Erfur, K. A method for the synthesis of unsymmetric bisphosphoric analogs of α-amino acids. RSC Adv. 2023, 13, 18908. [Google Scholar] [CrossRef] [PubMed]
- Kuźnik, A.; Kozicka, D.; Hawranek, W.; Socha, K.; Erfurt, K. One-Pot and Catalyst-Free Transformation of N-Protected 1-Amino-1-Ethoxyalkylphosphonates into Bisphosphonic Analogs of Protein and Non-Protein α-Amino Acids. Molecules 2022, 27, 3571. [Google Scholar] [CrossRef] [PubMed]
- Kozicka, D.; Krzesniak, M.; Grymel, M.; Adamek, J.; Łasut-Szyszka, B.; Cichoń, T.; Kuznik, A. Ultrasound-assisted synthesis of new bisphosphonate–betulin conjugates and preliminary evaluation of their cytotoxic activity. RSC Adv. 2025, 15, 4086–4094. [Google Scholar] [CrossRef] [PubMed]
- Ayadi, N.; Descamps, A.; Legigan, T.; Dussart-Gautheret, J.; Monteil, M.; Migianu-Griffoni, E.; Ayed, T.B.; Deschamp, J.; Lecouvey, M. Synthesis of Aminobisphosphinates through a Cascade Reaction between Hypophosphorous Acid and Bis(trimethylsilyl)imidates Mediated by ZnI2. Molecules 2023, 28, 6226. [Google Scholar] [CrossRef] [PubMed]
- Prishchenko, A.A.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Averochkin, G.M.; Petrosyan, V.S. Synthesis of Substituted N-Formylaminomethylenediphosphonates and Their Derivatives. Heteroatom Chem. 2015, 26, 405–410. [Google Scholar] [CrossRef]
- Kolotilo, N.V.; Onys’ko, P.P. Cycloaddition and chlorotropic transformations in reactions of dichloromethylenetrifluoroacetamide with phosphites. Russ. J. Gen. Chem. 2016, 86, 534–538. [Google Scholar] [CrossRef]
- Cheviet, T.; Peyrottes, S. Synthesis of Aminomethylene-gem-bisphosphonates Containing an Aziridine Motif: Studies of the Reaction Scope and Insight into the Mechanism. J. Org. Chem. 2021, 86, 3107–3119. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Andreu, M.M.; Sayago, F.J.; Cativiela, C. An Improved Synthesis of the Antibiotic Dehydrophos. Eur. J. Org. Chem. 2018, 2018, 3965–3973. [Google Scholar] [CrossRef]
- Makris, G.; Tseligka, E.D.; Pirmettis, I.; Papadopoulos, M.S.; Vizirianakis, I.S.; Papagiannopoulou, D. Development and Pharmacological Evaluation of New Bone-Targeted 99mTc-Radiolabeled Bisphosphonates. Mol. Pharm. 2016, 13, 2301–2317. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.S.; Chukanov, N.V.; Grigor’ev, I.A. New Amino-Bisphosphonate Building Blocks in the Synthesis of Bisphosphonic Derivatives Based on Lead Compounds. Phosphorus Sulfur. 2015, 190, 1201–1212. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Tris(trimethylsilyl) phosphite as key synthon for convenient synthesis of new organosilicon(phosphorus)-containing N-heterocycles. J. Organomet. Chem. 2018, 867, 149–154. [Google Scholar] [CrossRef]
- Prishcenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novokova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Organosilicon based synthesis of new functionalized aminomethylenediphosphonates with moieties of amino acids. J. Organomet. Chem. 2018, 871, 36–39. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novikova, E.P.; Livantsova, L.I.; Petrosyan, V.S. A convenient catalytic silicon-assisted route towards new non-proteinogenic amino acids with methylenebisphosphonic acids moieties. J. Organomet. Chem. 2021, 949, 121950. [Google Scholar] [CrossRef]
- Wang, A.E.; Chang, Z.; Sun, W.-T.; Huang, P.-Q. General and Chemoselective Bisphosphonylation of Secondary and Tertiary Amides. Org. Lett. 2015, 17, 732–735. [Google Scholar] [CrossRef]
- Chen, M.-E.; Chen, X.-W.; Hu, Y.-H.; Ye, R.; Lv, J.-W.; Li, J.-W.; Zhnag, F.-M. Recent advances of Ritter reaction and its synthetic applications. Org. Chem. Front. 2021, 8, 4623–4664. [Google Scholar] [CrossRef]
- Guérinot, A.; Reymond, S.; Cossy, J. Ritter Reaction: Recent Catalytic Developments. Eur. J. Org. Chem. 2012, 2012, 19–28. [Google Scholar] [CrossRef]
- Hong, Y.-C.; Ye, J.-L.; Huang, P.-Q. One-Pot Synthesis of α-Amino Bisphosphonates from Nitriles via Tf2O/HC(OR)3-Mediated Interrupted Ritter-Type Reaction. J. Org. Chem. 2022, 87, 9044–9055. [Google Scholar] [CrossRef] [PubMed]
- Kaboudin, B.; Esfandiari, H.; Moradi, A.; Kazemi, F.; Aoyama, H. ZnCl2-Mediated Double Addition of Dialkylphosphite to Nitriles for the Synthesis of 1-Aminobisphosphonates. J. Org. Chem. 2019, 84, 14943–14948. [Google Scholar] [CrossRef] [PubMed]
- Islas, R.S.; García, J.J. Nickel-Catalyzed Hydrophosphonylation and Hydrogenation of Aromatic Nitriles Assisted by Lewis Acid. Chem. Cat. Chem. 2019, 14, 1337–1345. [Google Scholar] [CrossRef]
- Midrier, C.; Lantsoght, M.; Volle, J.-N.; Pirat, J.-L.; Virieux, D.; Stevens, C.S. Hydrophosphonylation of alkenes or nitriles by double radical transfer mediated by titanocene/propylene oxide. Tetrahedron Lett. 2011, 52, 6693–6696. [Google Scholar] [CrossRef]
- Goldeman, W.; Kluczyński, A.; Soroka, M. The preparation of N-substituted aminomethylidenebisphosphonates and their tetraalkyl esters via reaction of isonitriles with trialkyl phosphites and hydrogen chloride. Part 1. Tetrahedron Lett. 2012, 53, 5290–5292. [Google Scholar] [CrossRef]
- Goldeman, W.; Nasulewicz-Goldeman, A. Synthesis and antiproliferative activity of aromatic and aliphatic bis[aminomethylidene(bisphosphonic)] acids. Bioorg. Med. Chem. Lett. 2014, 24, 3475–3479. [Google Scholar] [CrossRef] [PubMed]
- Scarpi, D.; Visi, S.; Occhiato, E.G. Recent Advances in the α-Hydrazination (α-Amination) of Carbonyl Compounds. Eur. J. Org. Chem. 2025, 28, e202500049. [Google Scholar] [CrossRef]
- Lauren, G.; O’Neil, L.G.; Bower, J.E. Electrophilic Aminating Agents in Total Synthesis. Angew. Chem. Int. Ed. 2021, 60, 25640–25666. [Google Scholar]
- Erdik, E. Electrophilic α-amination of carbonyl compounds. Tetrahedron 2004, 60, 8747–8782. [Google Scholar] [CrossRef]
- Tanaka, K.S.E.; Dietrich, E.; Ciblat, S.; Métayer, C.; Arhin, F.F.; Sarmiento, I.; Moeck, G.; Parr, T.R., Jr.; Far, A.R. Synthesis and in vitro evaluation of bisphosphonated glycopeptide prodrugs for the treatment of osteomyelitis. Bioorg. Med. Chem. Lett. 2010, 20, 1355–1359. [Google Scholar] [CrossRef]
- Lv, M.; Wang, M.; Lu, K.; Peng, L.; Zhao, Y. An efficient synthesis of 2-Aminoethylidene-1,1-Bisphosphonates derivatives via Michael addition reaction. Phosphorus Sulfur. 2018, 193, 149–154. [Google Scholar] [CrossRef]
- Vagapova, L.I.; Makhrus, E.M.; Burilov, A.R.; Pudovik, M.A. Synthesis of new diarylmethanes on the basis of resorcinol derivatives and amino acetals containing an aminoethylidenebisphosphonate fragment. Russ. J. Gen. Chem. 2017, 87, 2103–2106. [Google Scholar] [CrossRef]
- Liu, S.; Bi, W.; Li, X.; Chen, X.; Qu, L.; Zhao, Y. A Practical Method to Synthesize 1,2,3-Triazole-Amino-Bisphosphonate Derivatives. Phosphorus Sulfur. 2015, 190, 1735–1742. [Google Scholar] [CrossRef]
- Chen, C.; Li, Y.; Yu, X.; Jiang, Q.; Yang, Q.; Xu, X.; Qian, Z. Bone-targeting melphalan prodrug with tumor-microenvironment sensitivity: Synthesis, in vitro and in vivo evaluation. Chin. Chem. Lett. 2018, 29, 1609–1612. [Google Scholar] [CrossRef]
- Chiminazzo, A.; Borsato, G.; Favero, A.; Fabbro, C.; McKenna, C.E.; Carbonare, L.G.D.; Valenti, M.T.; Fabrils, F.; Scarso, A. Diketopyrrolopyrrole Bis-Phosphonate Conjugate: A New Fluorescent Probe for In Vitro Bone Imaging. Chem. Eur. J. 2019, 25, 3617–3626. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, X.; Yuan, J.; Qu, L.; Wang, S.; Shi, H.; Tang, Y.; Duan, L. Simple, efficient one-pot method for synthesis of novel N-attached 1,2,3-triazole containing bisphosphonates. Tetrahedron 2013, 69, 4047–4052. [Google Scholar] [CrossRef]
- Fairweather, A.E.R.; Goetz, D.B.; Schroeder, C.M.; Bhuiyan, N.H.; Varney, M.L.; Wiemer, D.F.; Holstein, S.A. Impact of α-modifications on the activity of triazole bisphosphonates as geranylgeranyl diphosphate synthase inhibitors. Bioorg. Med. Chem. 2021, 44, 116307. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Huang, R.; Liao, Z.; Pan, Y.; Gou, S.; Wang, H. Synthesis and pharmacological evaluation of dehydroabietic acid thiourea derivatives containing bisphosphonate moiety as an inducer of apoptosis. Eur. J. Med. Chem. 2016, 108, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Gritzalis, D.; Park, J.; Chiu, W.; Cho, H.; Lin, Y.; De Schutter, J.W.; Lacbay, C.M.; Zielinski, M.; Berghuis, A.M.; Tsantrizos, Y.S. Probing the molecular and structural elements of ligands binding to the active site versus an allosteric pocket of the human farnesyl pyrophosphate synthase. Bioorg. Med. Chem. Lett. 2015, 25, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Vagapova, L.I.; Smolobochkin, A.V.; Gazizov, A.S.; Burilov, A.R.; Bogdanov, A.A.; Pudovik, M.A. Synthesis of new phosphorylated analogs of nucleotides containing adenine and ethylidene-1,1-bisphosphoryl moieties. Russ. J. Gen. Chem. 2017, 87, 2119–2121. [Google Scholar] [CrossRef]
- Vagapova, L.I.; Smolobochkin, A.V.; Gazizov, A.S.; Burilov, A.R.; Bogdanov, A.A.; Pudovik, M.A.; Sinyashin, O.G. Synthesis of new nucleoside analogs containing amino bisphosphonic groups. Russ. J. Org. Chem. 2016, 52, 1335–1338. [Google Scholar] [CrossRef]
- Ye, Y.; Guan, J.; Chen, X.; Yu, Y.; Xu, Z.; Zeng, S.; Wang, Z.; Wang, B.; Jiao, Q.; Zhu, H. A new fluorescently labeled bisphosphonate for theranostics in tumor bone metastasis. Talanta 2021, 235, 122796. [Google Scholar] [CrossRef] [PubMed]
- Massarenti, C.; Bortolini, O.; Fantin, G.; Cristofaro, D.; Ragno, D.; Perrone, D.; Marchesi, E.; Toniolo, G.; Massi, A. Fluorous-tag assisted synthesis of bile acid–bisphosphonate conjugates via orthogonal click reactions: An access to potential anti-resorption bone drugs. Org. Biomol. Chem. 2017, 15, 4907–4920. [Google Scholar] [CrossRef] [PubMed]
- Sayed, G.M.S.; Tessmar, J.K.V. Heterobifunctional Poly(ethylene glycol) Derivatives for the Surface Modification of Gold Nanoparticles Toward Bone Mineral Targeting. Macromol. Biosci. 2012, 12, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Guven, M.N.; Altuncu, M.S.; Bal, T.; Oran, D.C.; Gulyuz, U.; Kizilel, S.; Okay, O.; Avci, D. Bisphosphonic Acid-Functionalized Cross-Linkers to Tailor Hydrogel Properties for Biomedical Applications. ACS Omega 2018, 3, 8638–8647. [Google Scholar] [CrossRef] [PubMed]
- Guven, M.N.; Altuncu, M.S.; Duman, F.D.; Eren, T.M.; Acar, H.Y.; Avci, D. Bisphosphonate-functionalized poly(β-amino ester) network polymers. J. Biomed. Mater. Res. 2017, 105, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Bekheit, M.S.; Barghash, R.F.; Abdou, W.M. Computer-aided design, synthesis, and biological studies of anticological nitrogen-containing tetraphosphonic acids against melanoma. Monatsh. Chem. 2018, 149, 1481–1491. [Google Scholar] [CrossRef]
- Abdou, W.M.; Ganoub, N.A.; Ismail, M.A.H.; Sabry, E.; Barghash, R.F.; Geronikaki, A. Developing efficient protocols for synthesis, antiosteoarthritic, antiinflammatory assessments and docking studies of nitrogen-containing bisphosphonate derivatives. Arab. J. Chem. 2017, 10, 1084–1097. [Google Scholar] [CrossRef]
- Dzięgielewski, M.; Hejmanowska, J.; Albrecht, Ł. A Convenient Approach to a Novel Group of Quaternary Amino Acids Containing a Geminal Bisphosphonate Moiety. Synthesis 2014, 46, 3233–3238. [Google Scholar] [CrossRef]
- Chmielewska, E.; Miszczyk, P.; Kozlowska, J.; Prokopowicz, M.; Mlynarz, P.; Kafarski, P. Reaction of benzolactams with triethyl phosphite prompted by phosphoryl chloride affords benzoannulated monophosphonates instead of expected bisphosphonates. J. Organomet. Chem. 2015, 785, 84–91. [Google Scholar] [CrossRef]
- Arguëllo-Velasco, R.O.; Dziuk, B.; Zarychta, B.; Ordoñez, M.; Kafarski, P. Reactions of Piperazin-2-one, Morpholin-3-one, and Thiomorpholin-3-one with Triethyl Phosphite Prompted by Phosphoryl Chloride: Scope and Limitations. ACS Omega 2019, 4, 9056–9064. [Google Scholar] [CrossRef] [PubMed]
- Kachkovskyi, G.O.; Kolodiazhnyi, O.I. Synthesis of the Phosphonoanalogue of Benzo[c]pyroglutamic Acid. Phosphorus Sulfur. 2010, 185, 2441–2448. [Google Scholar] [CrossRef]
- Zemlianoi, V.N.; Chernega, A.; Gutov, A.V.; Kolodiazhna, O.; Kolodiazhnyi, O.I. Synthesis of 3,3-Bis(diethylphosphono)-1-(3H)-isobenzofuranone and Its Chemical Properties. Phosphorus Sulfur. 2011, 186, 481–488. [Google Scholar] [CrossRef]
- Zemilanoi, V.N.; Kolodyazhnyi, O.I. Synthesis of tetraethyl (2-benzyl-3-oxoisoindolyl-1,1-diyl)-bisphosphonate and its properties. Russ. J. General Chem. 2011, 81, 1105–1110. [Google Scholar] [CrossRef]
- Kamel, A.A.; Geronikaki, A.; Abdou, W.M. Inhibitory effect of novel S,N-bisphosphonates on some carcinoma cell lines, osteoarthritis, and chronic inflammation. Eur. J. Med. Chem. 2012, 51, 239–249. [Google Scholar] [CrossRef] [PubMed]
- De Luca, L.; Chiminazzo, A.; Sperni, L.; Strukul, G.; Scarso, A. Pyrrolidine-Containing Bisphosphonates as Potential Anti-Resorption Bone Drugs. Chem. Eur. J. 2017, 23, 3474–3478. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Casal, M.; Barboza, A.P.; Szajnman, S.H.; Rodriguez, J.B. 1,3-Dipolar Cycloadditions of the Versatile Intermediate Tetraethyl Vinylidenebisphosphonate. Synthesis 2013, 45, 2397–2404. [Google Scholar] [CrossRef]
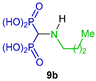
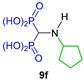
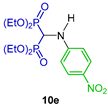
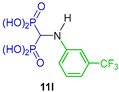
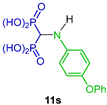
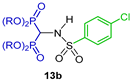
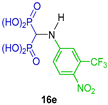
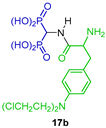
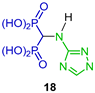
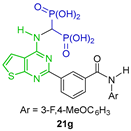
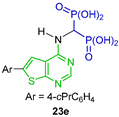
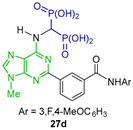
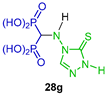
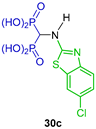
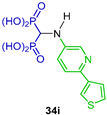
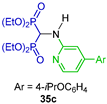
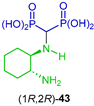
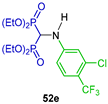
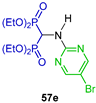
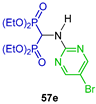
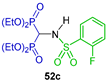
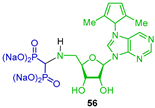
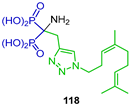
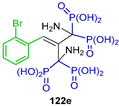
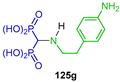
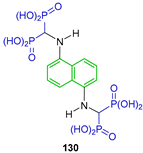
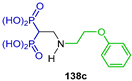
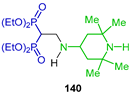
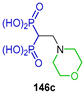
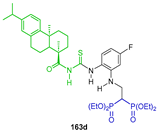
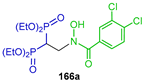
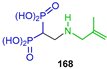
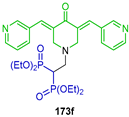
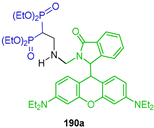
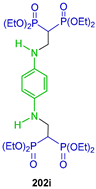
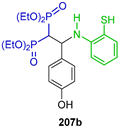
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).