
Synthesis of Amino-gem-Bisphosphonate Derivatives and Their Application as Synthons for the Preparation of Biorelevant Compounds


Abstract

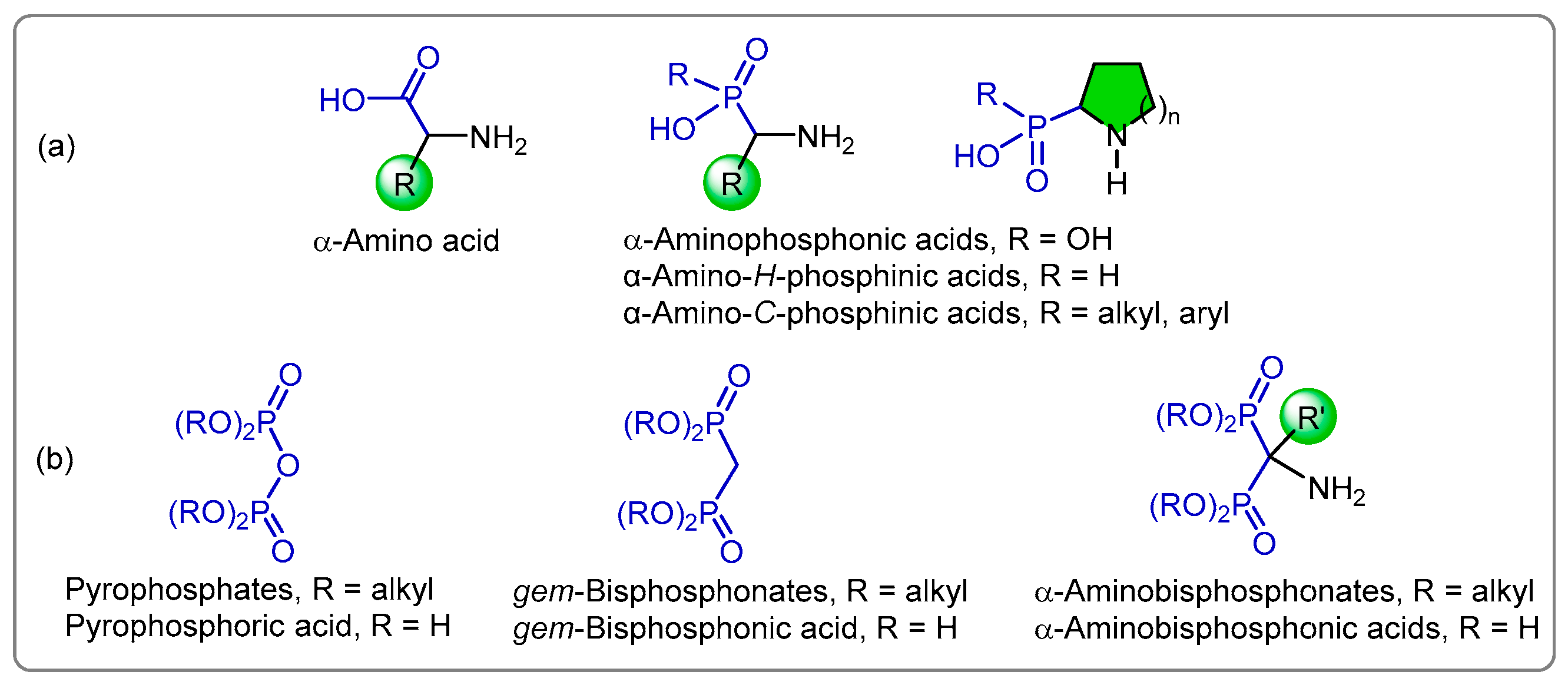
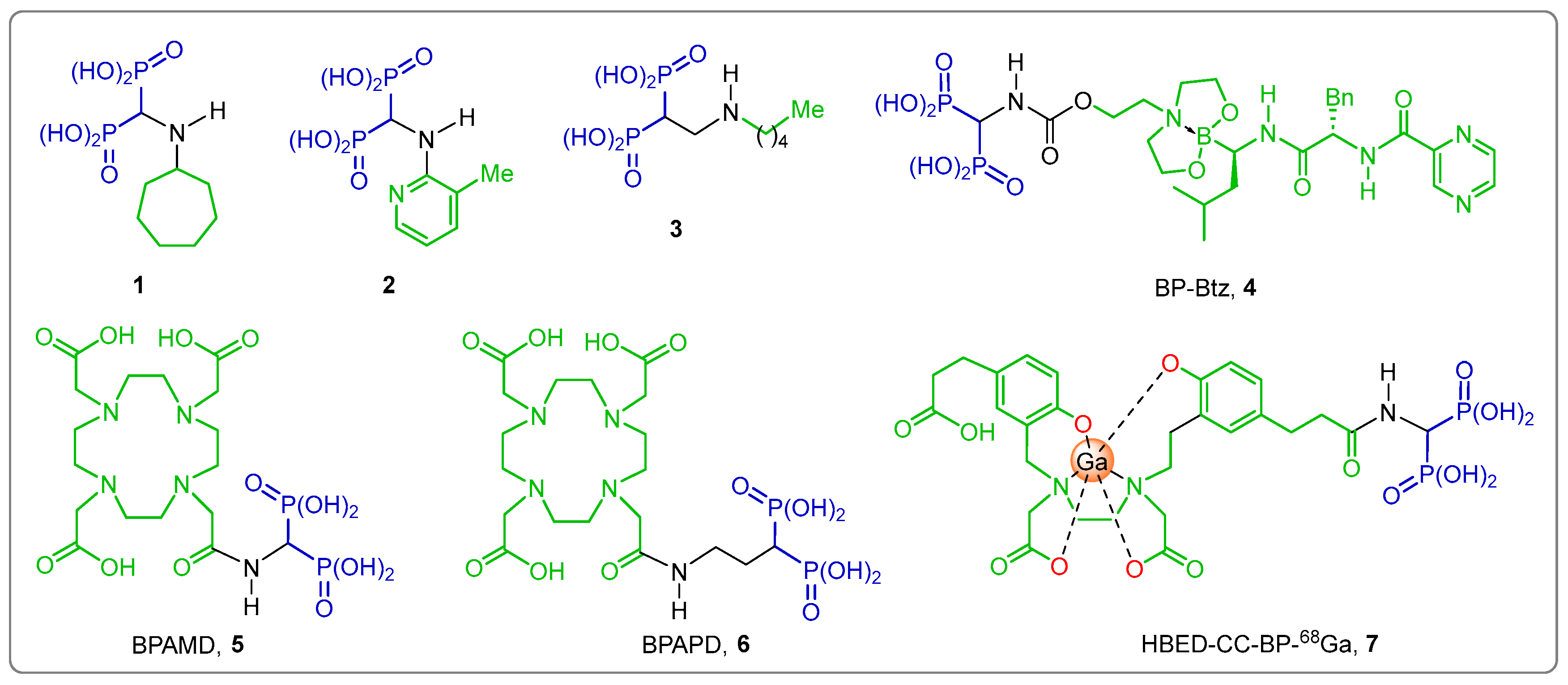
1. Introduction
2. Synthesis of α-Amino-gem-Bisphosphonate Derivatives
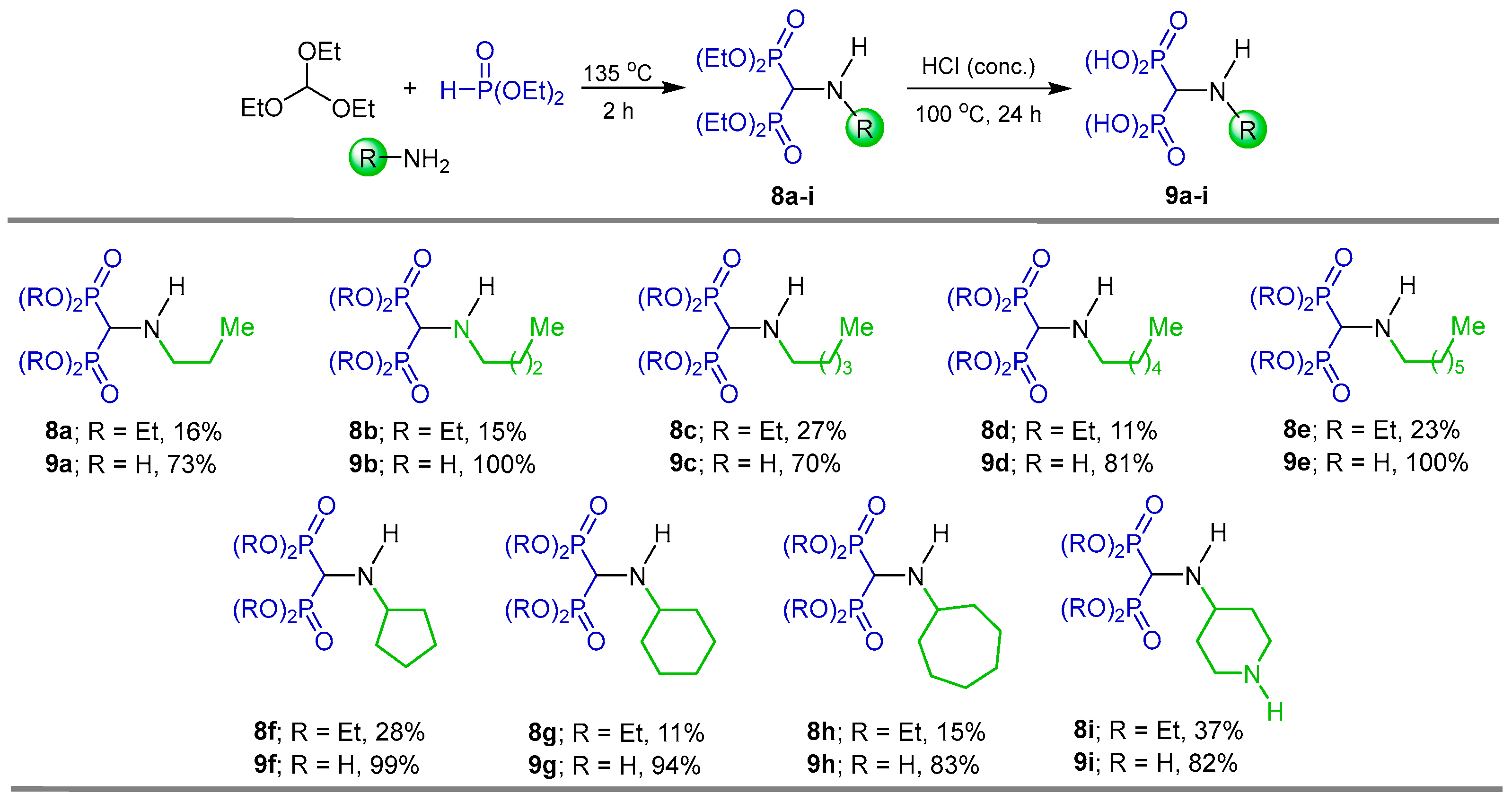
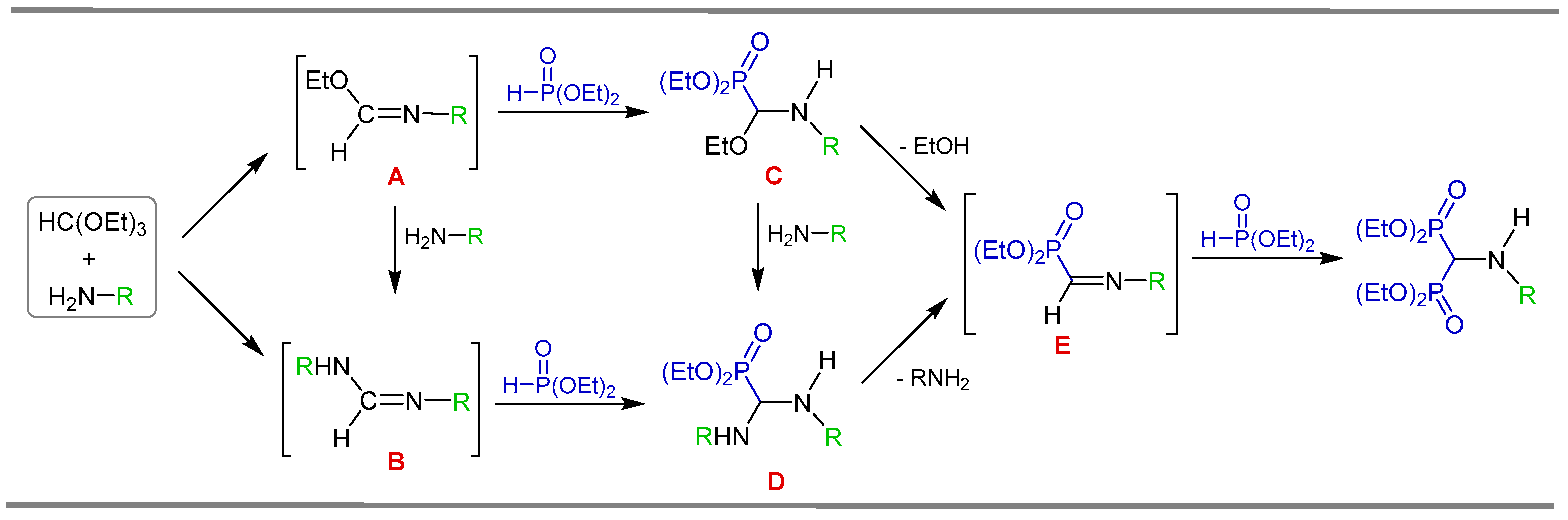
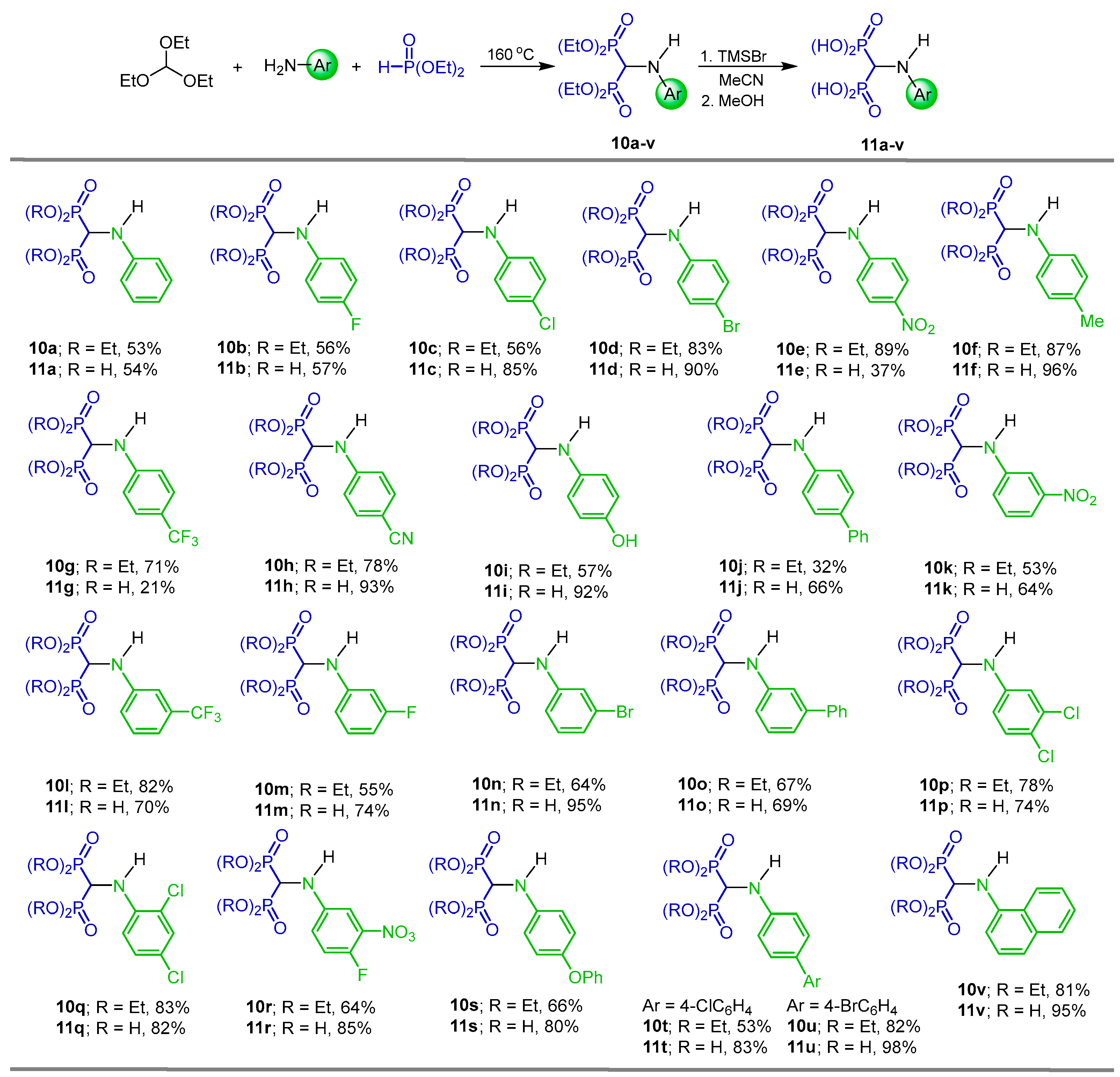
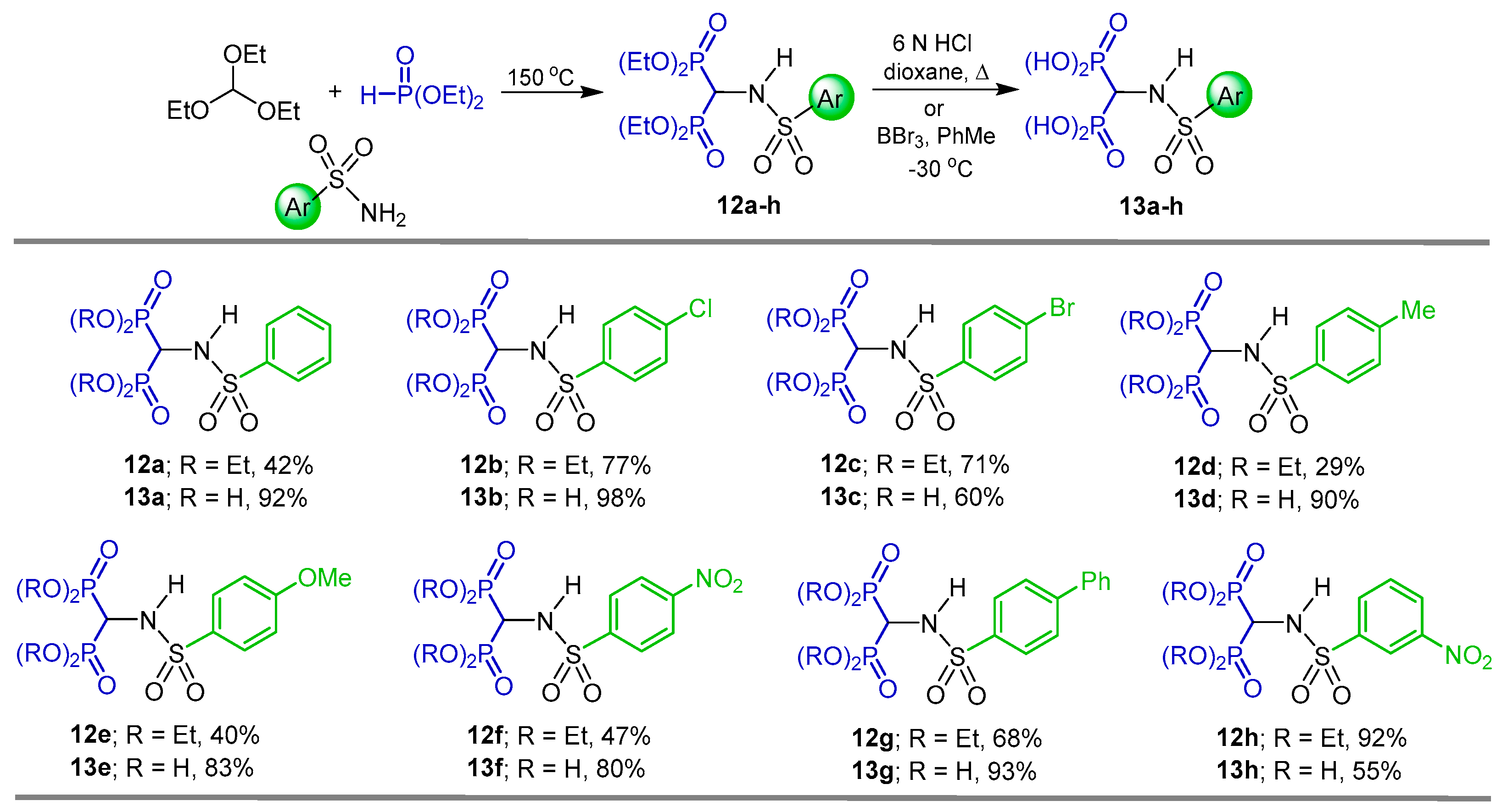
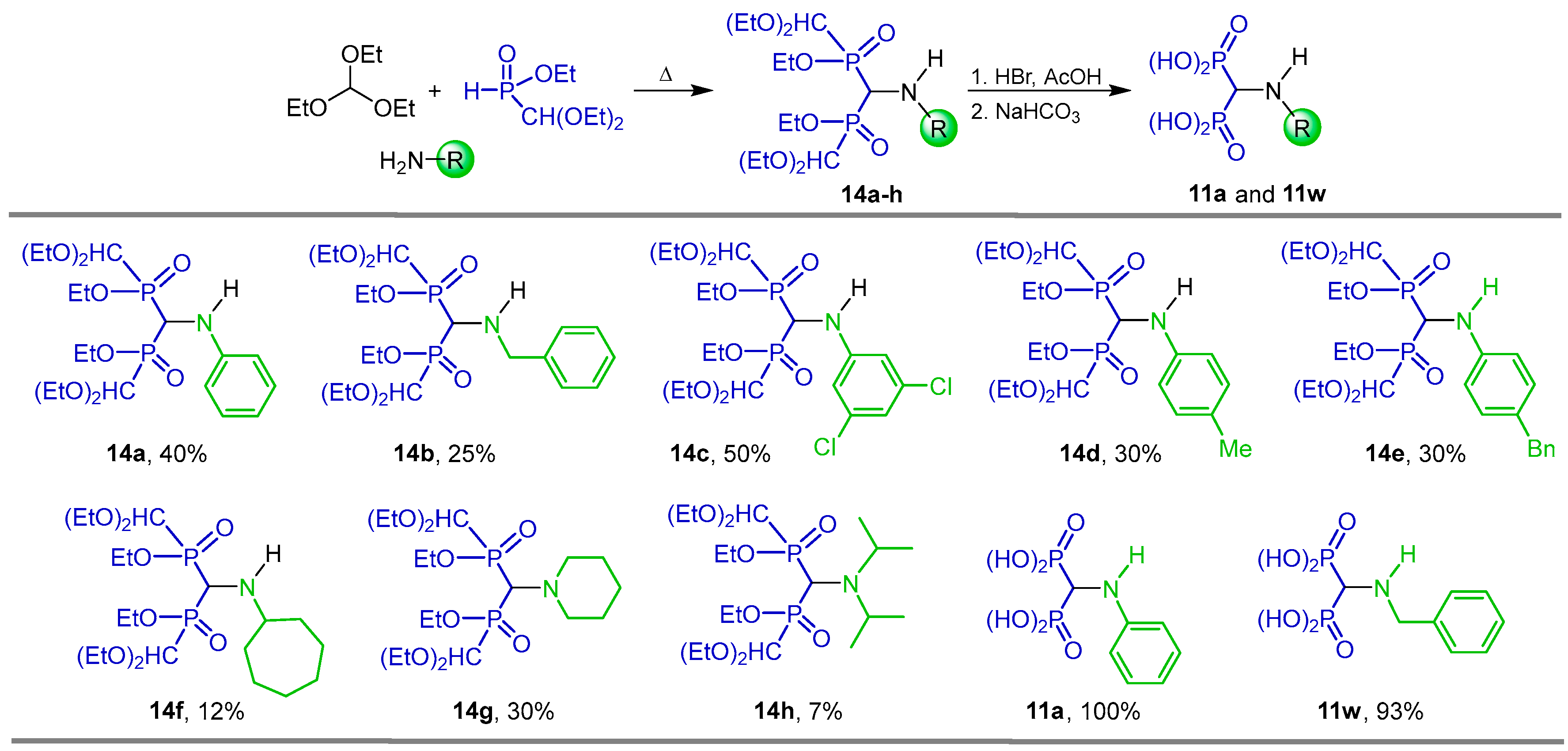
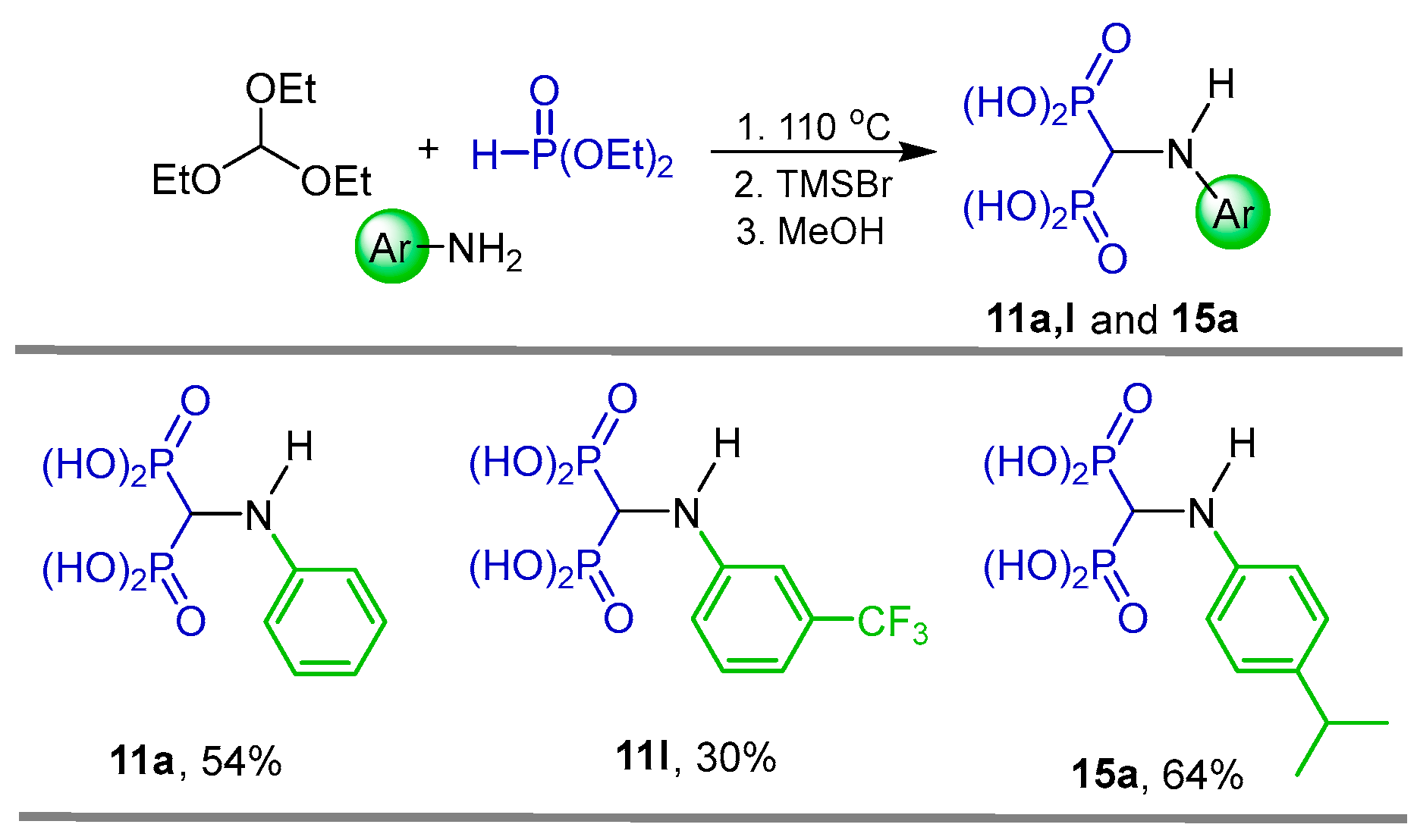
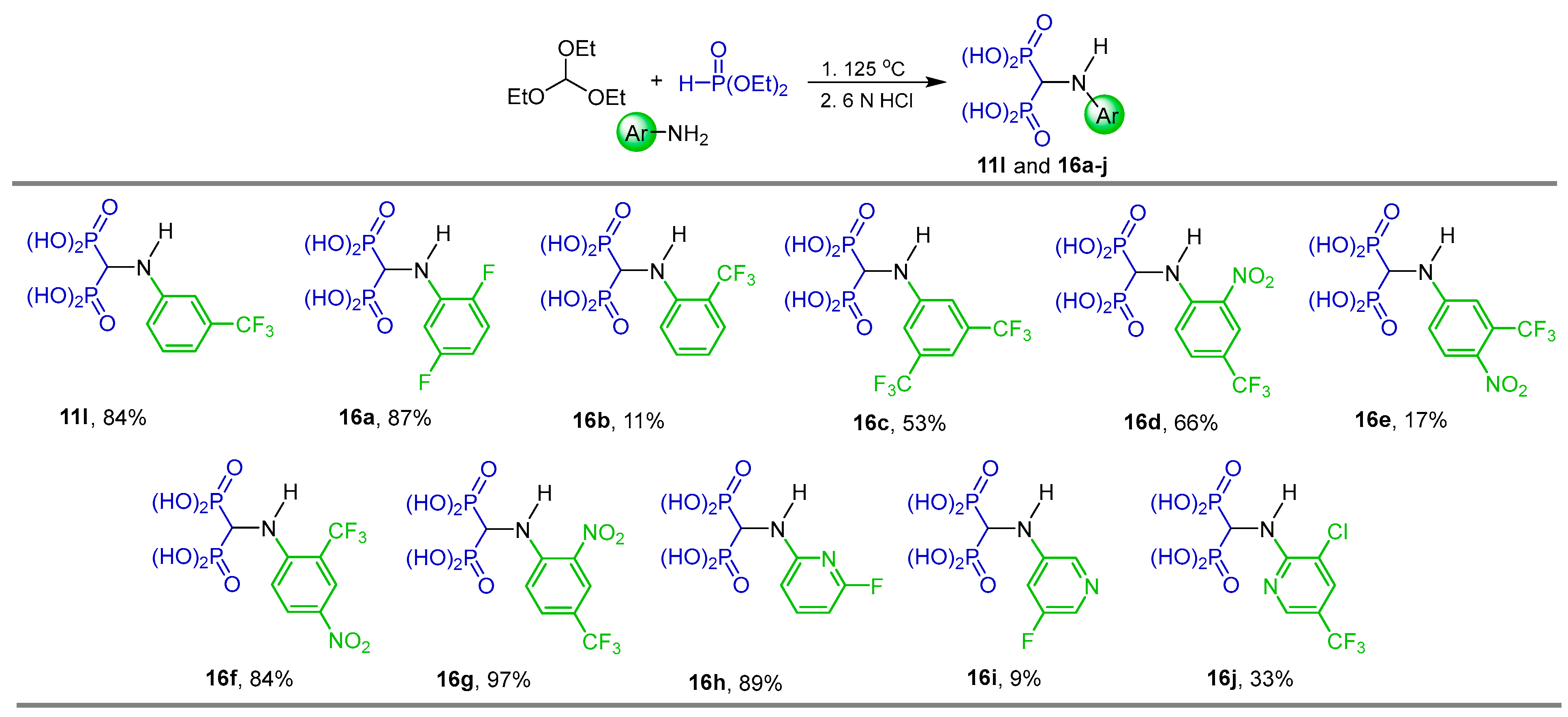
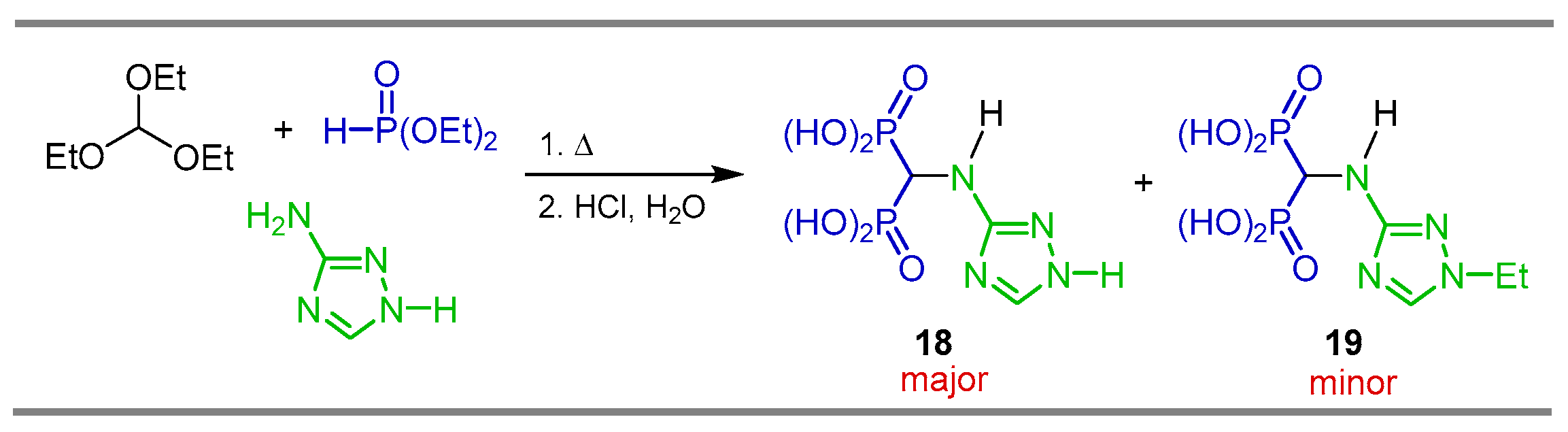
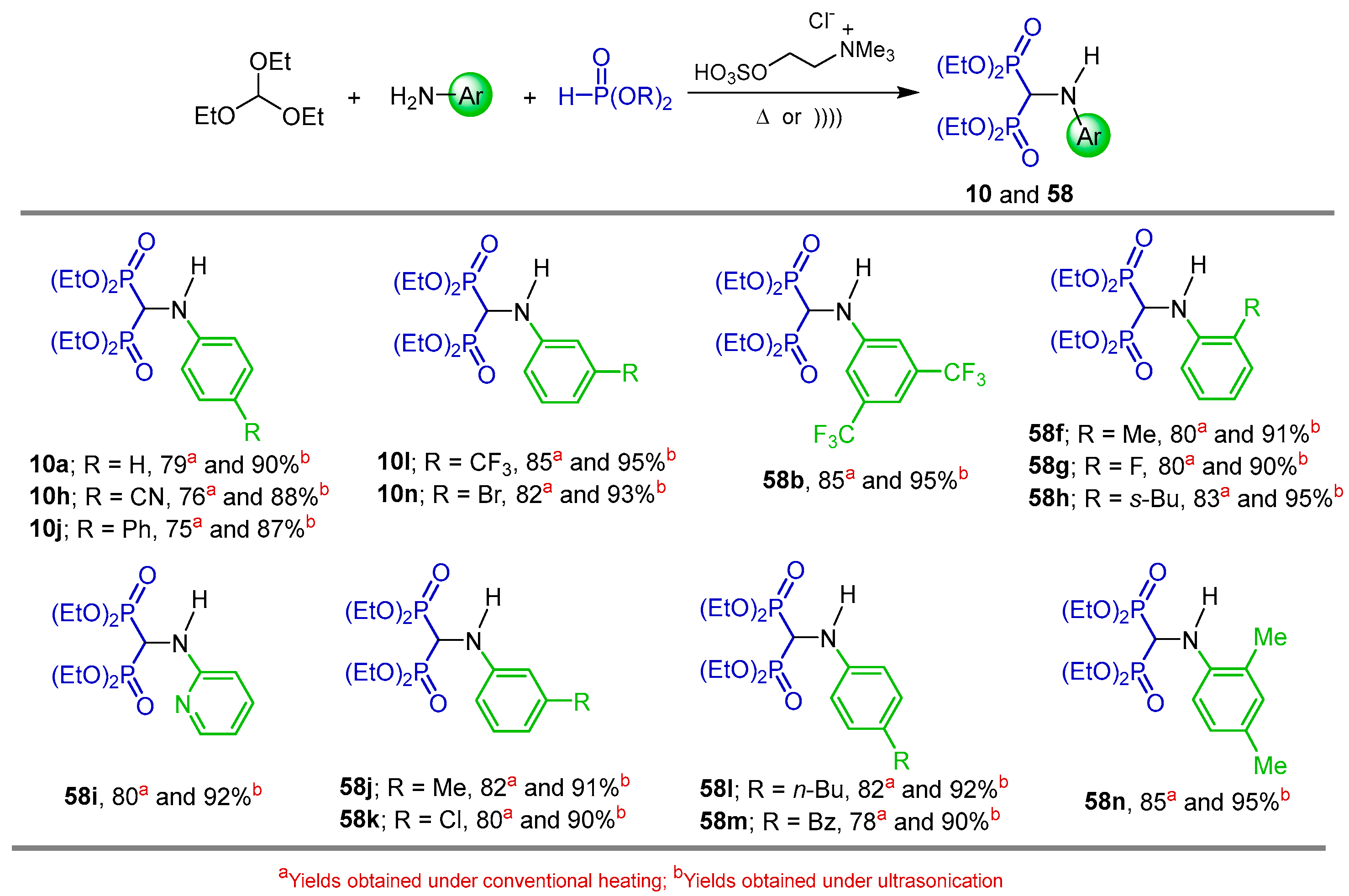
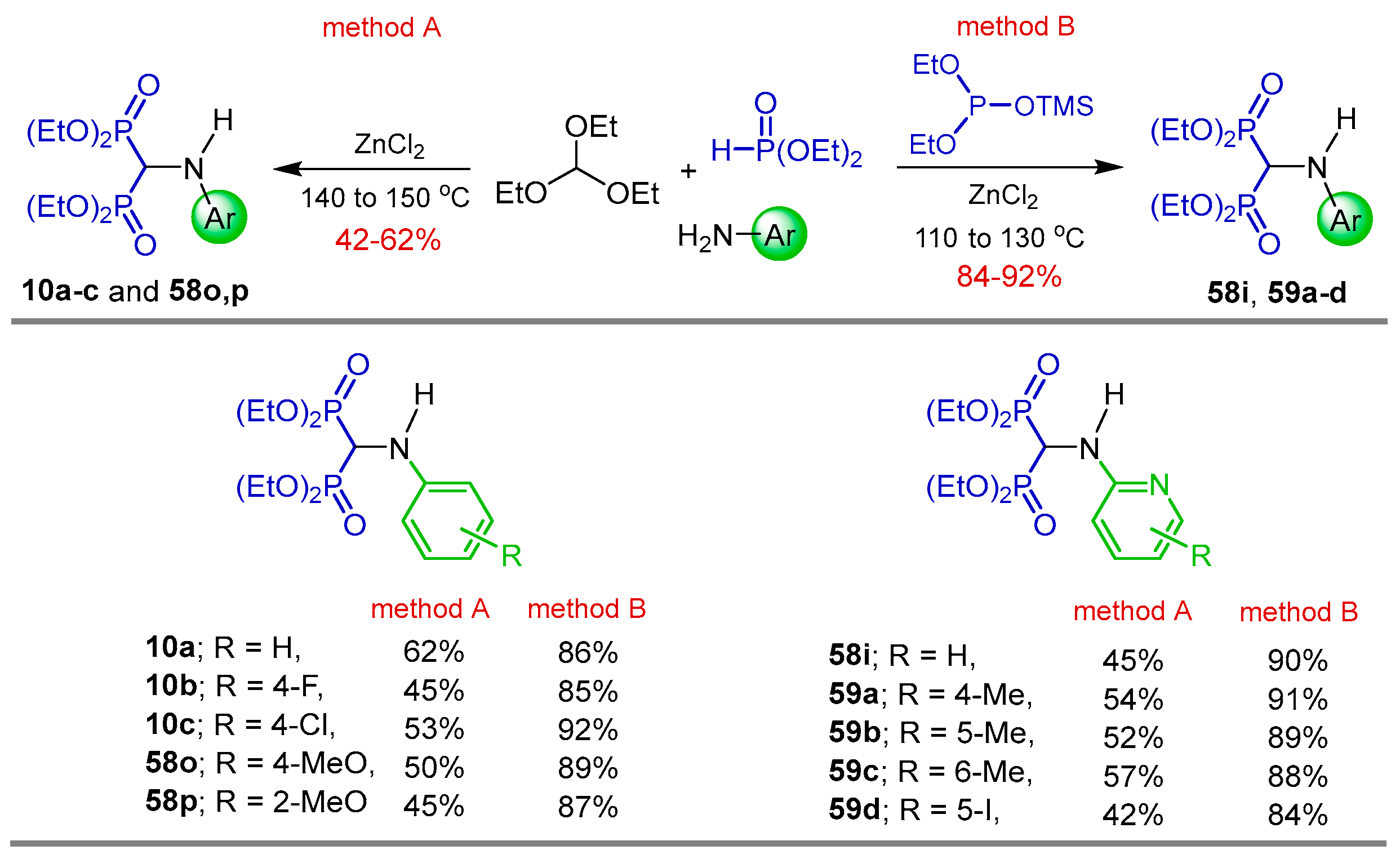
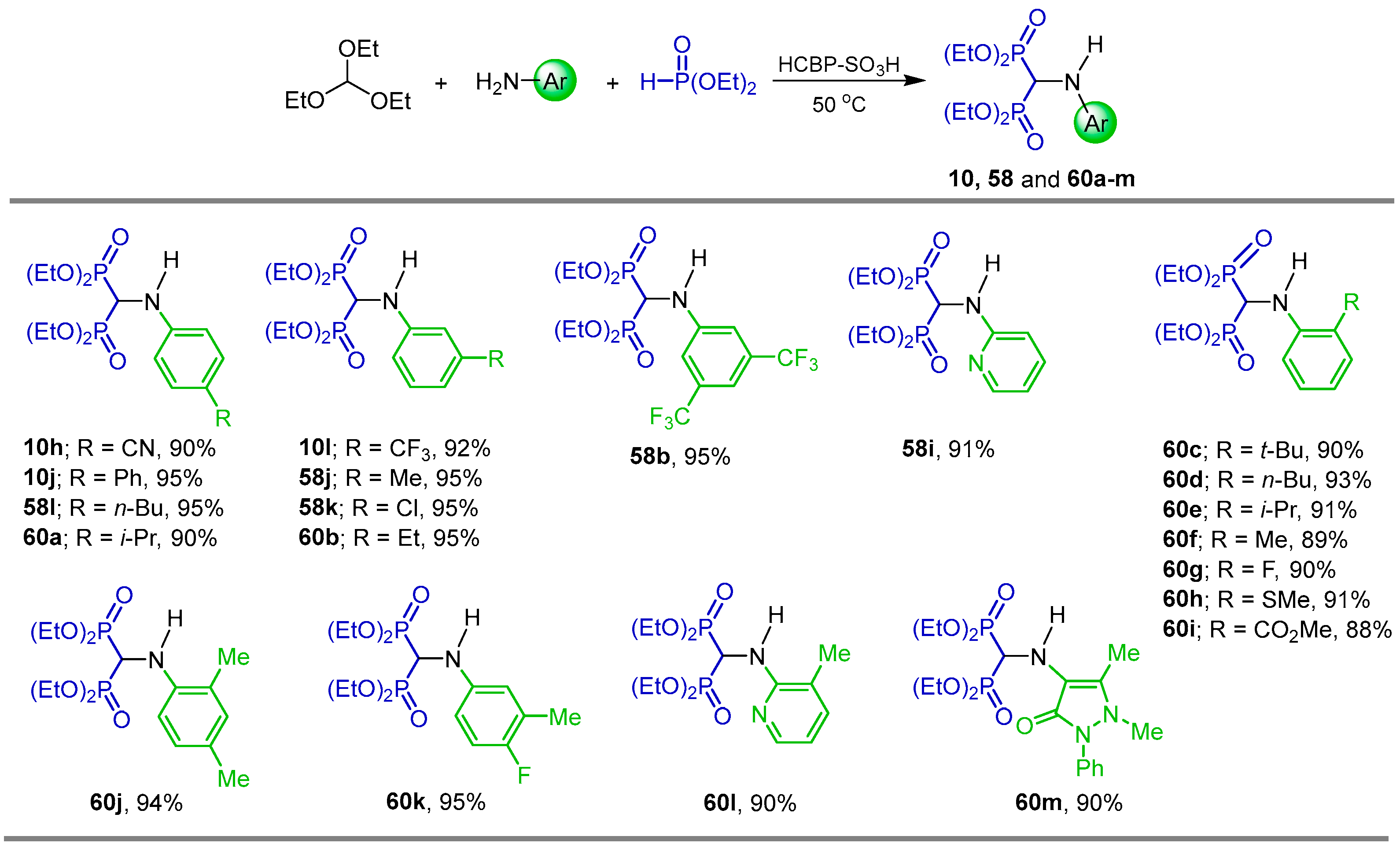
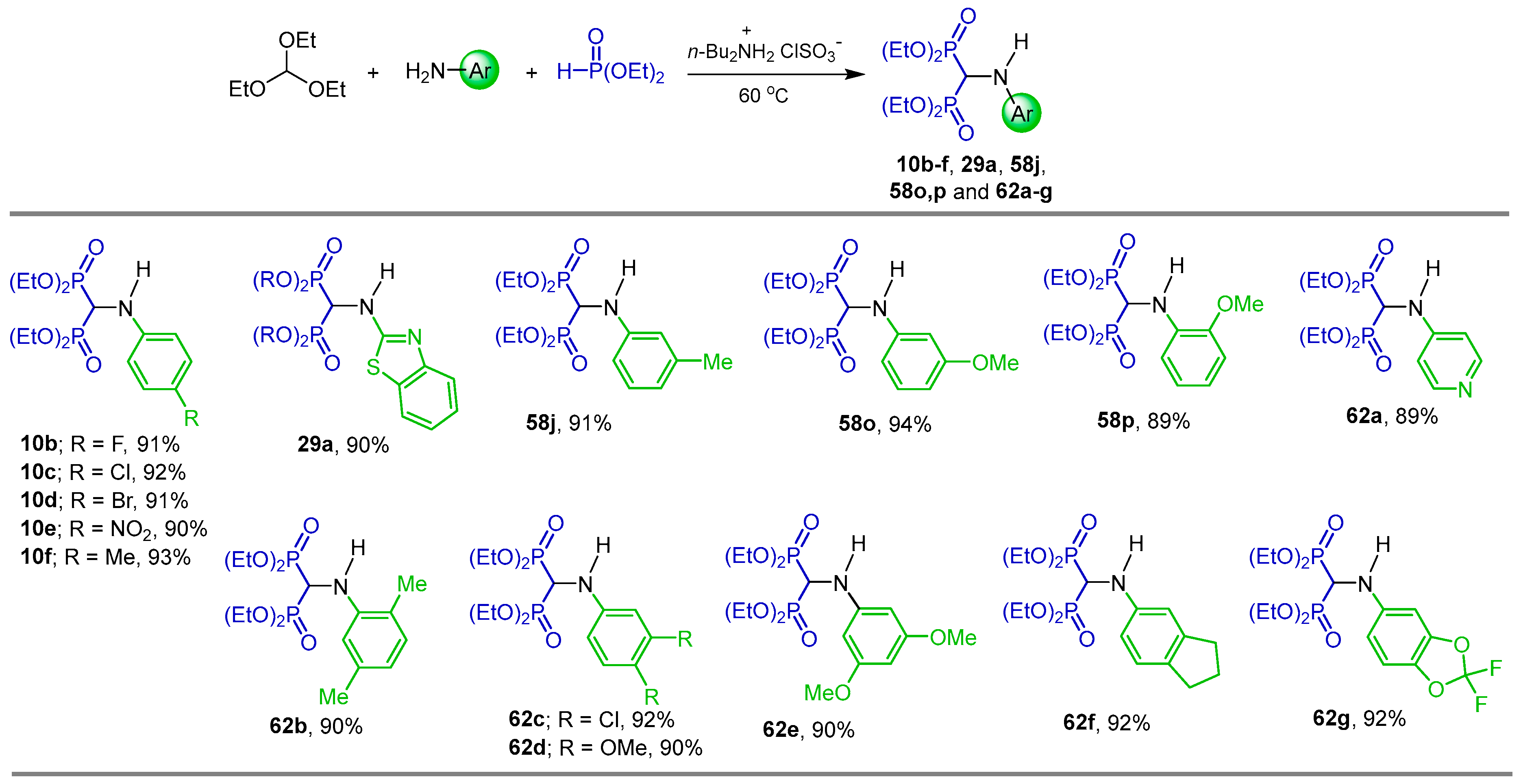
2.1. Three-Component Reaction of Orthoformates, Amines and Dialkyl Phosphites
2.2. Phosphonylation of Imidates
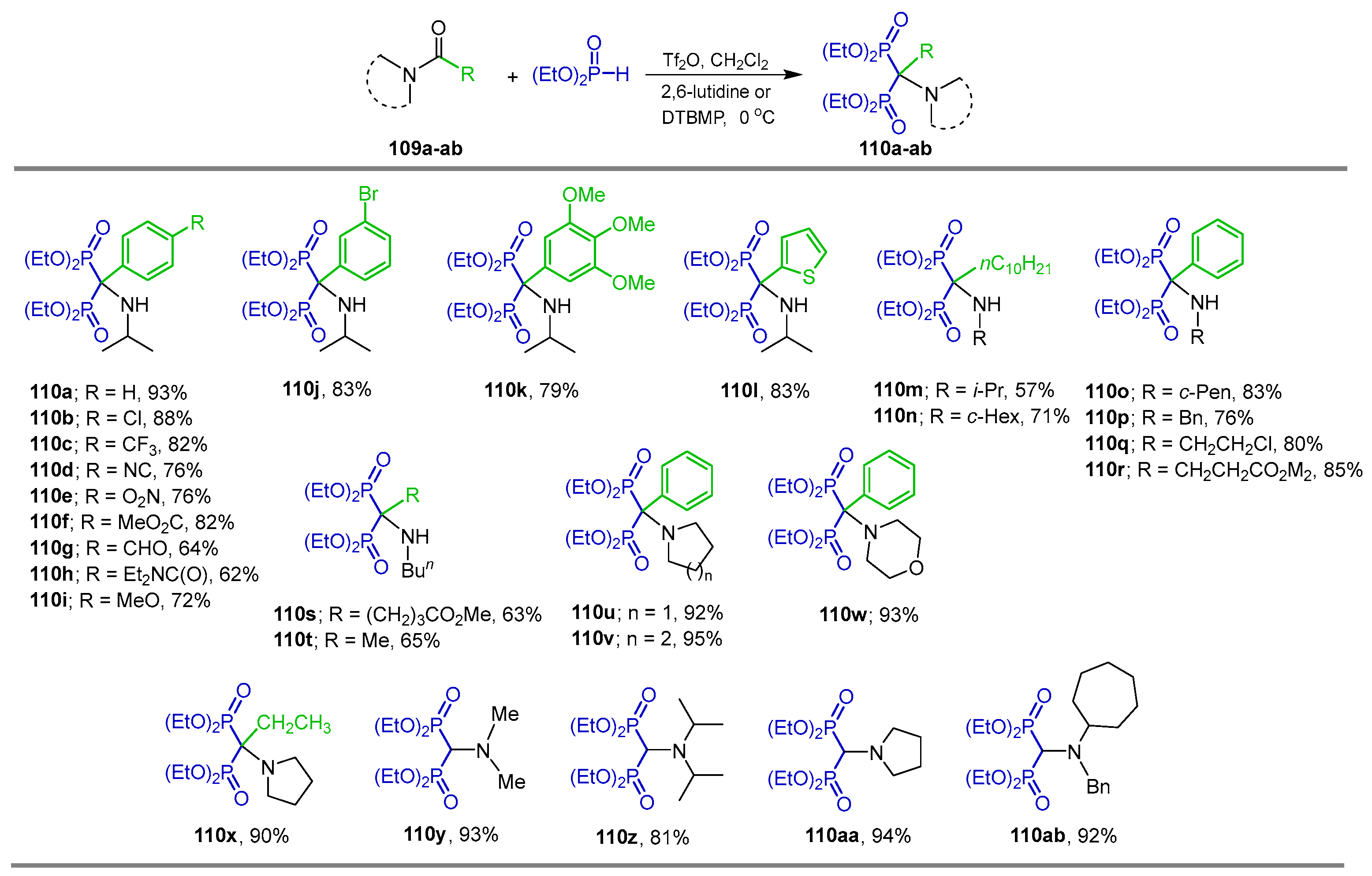
2.3. Phosphonylation of Amides
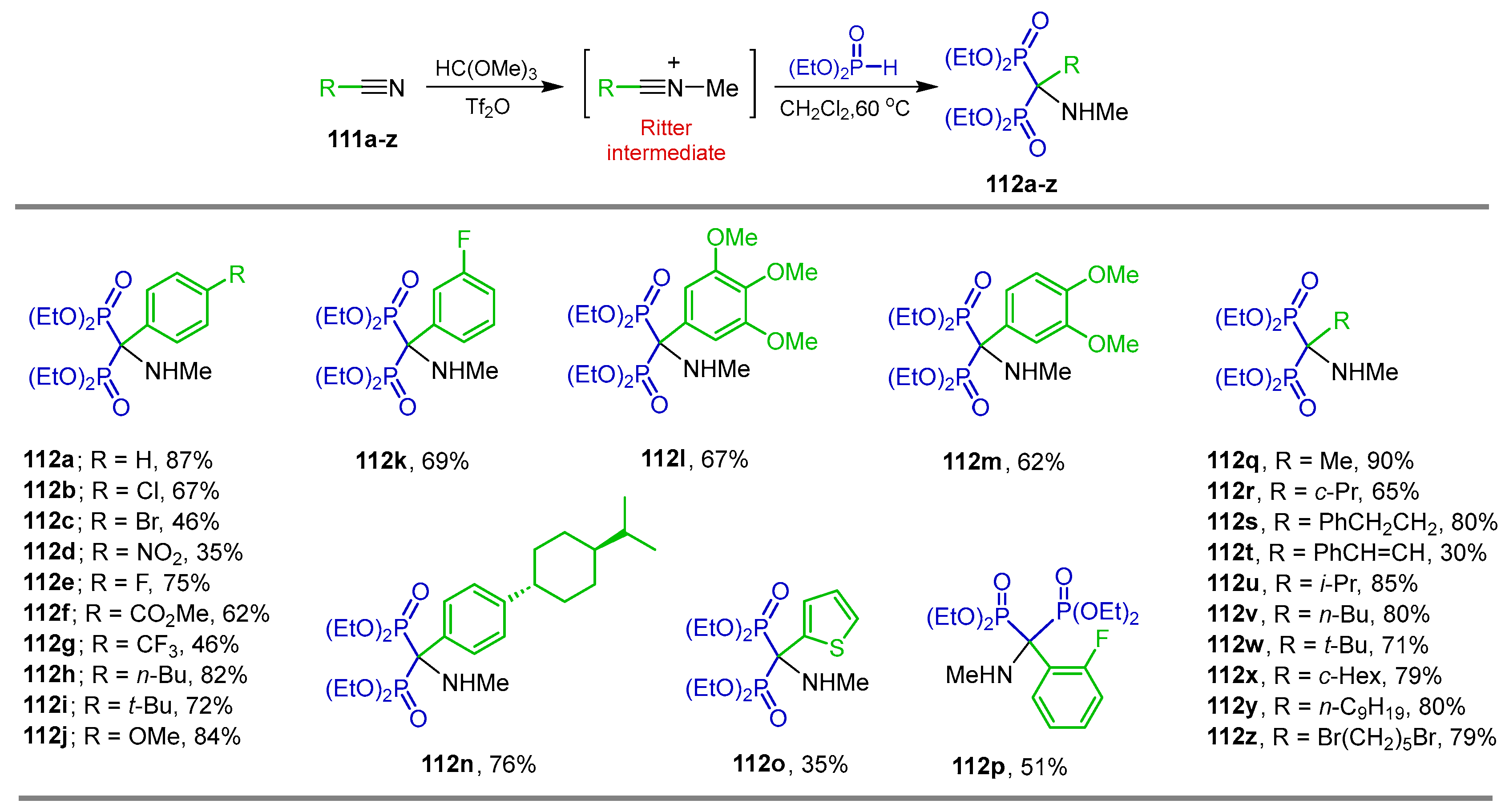
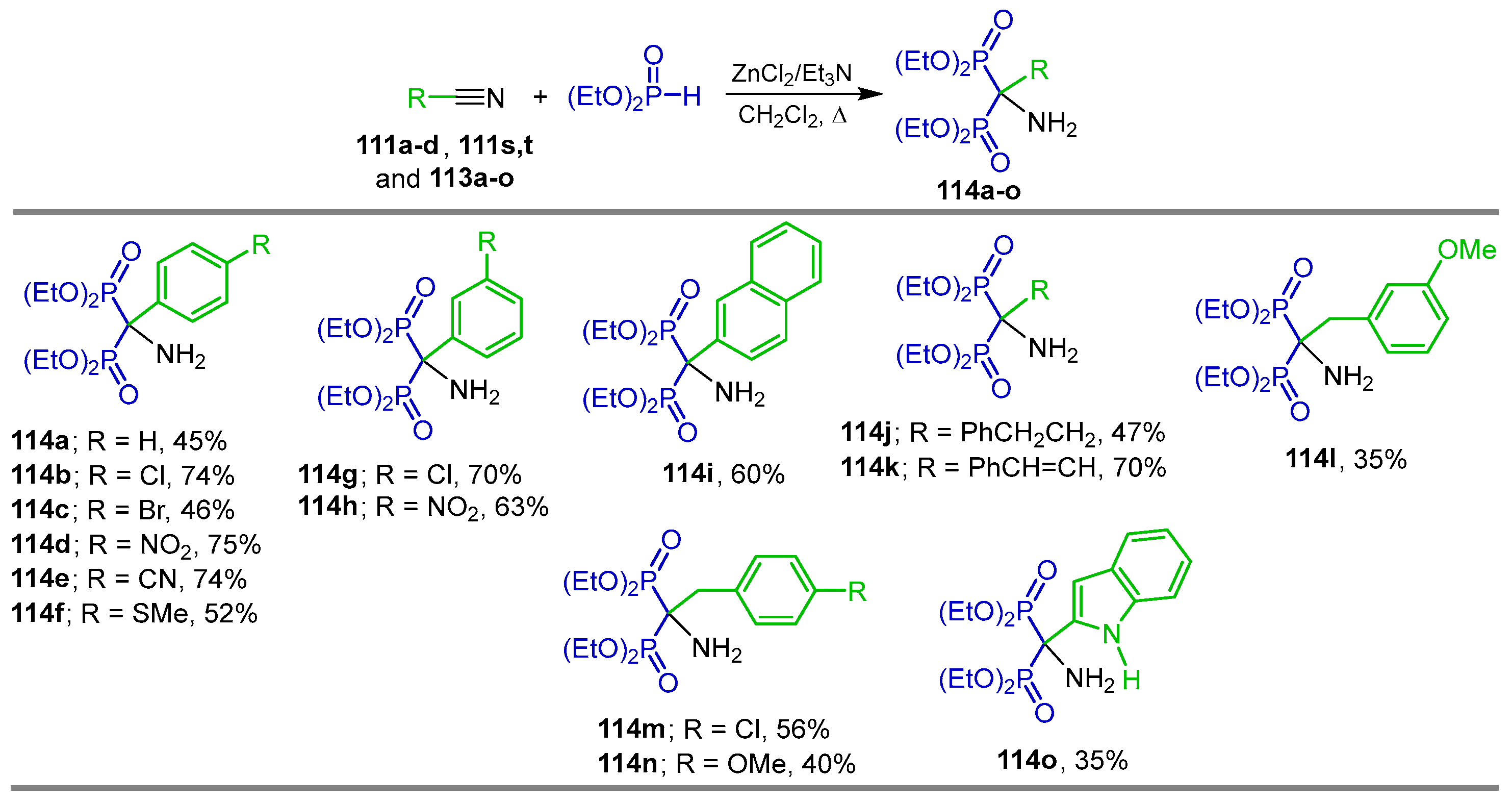
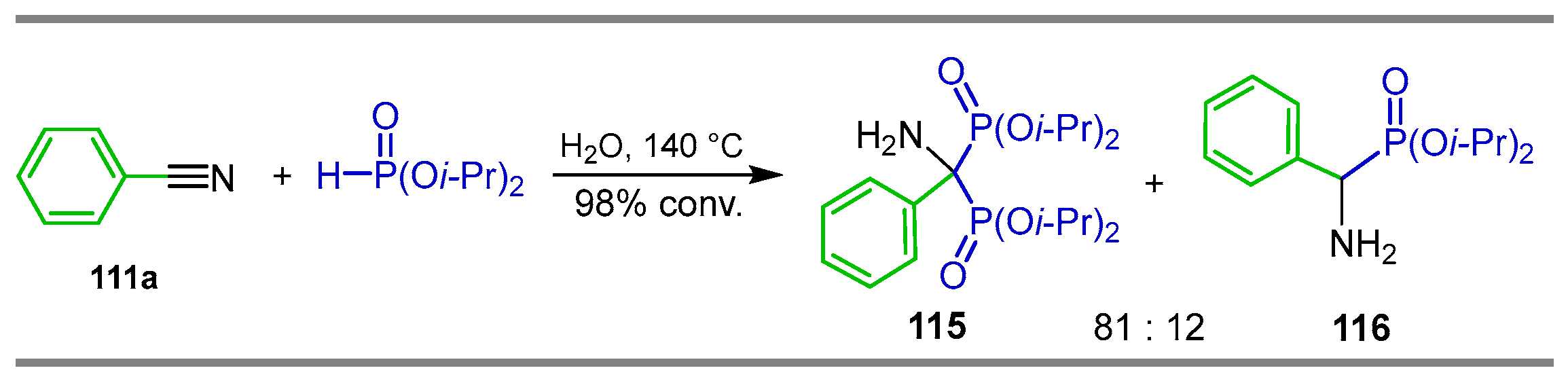
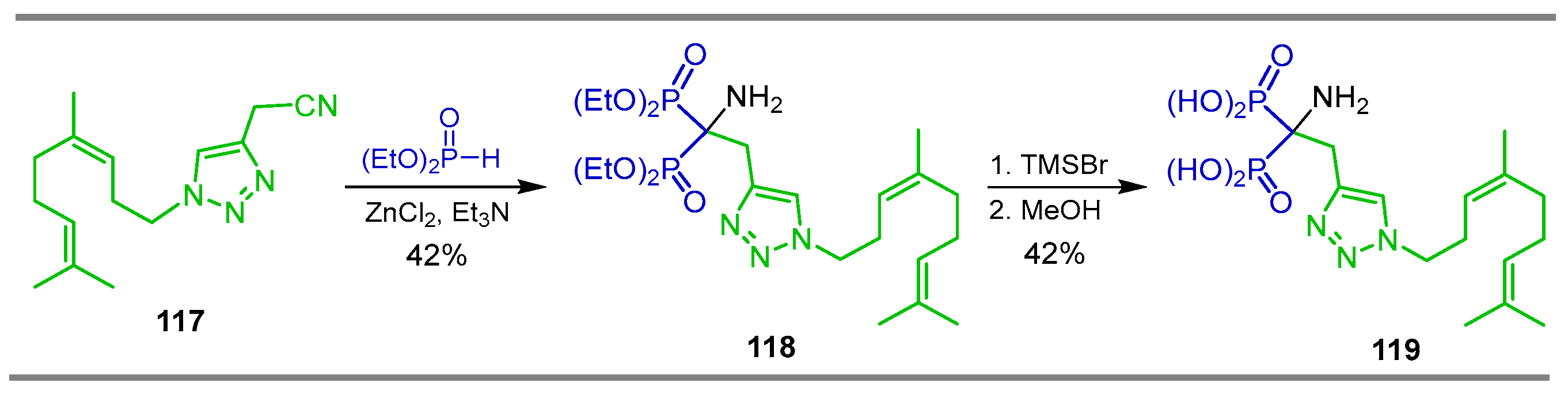
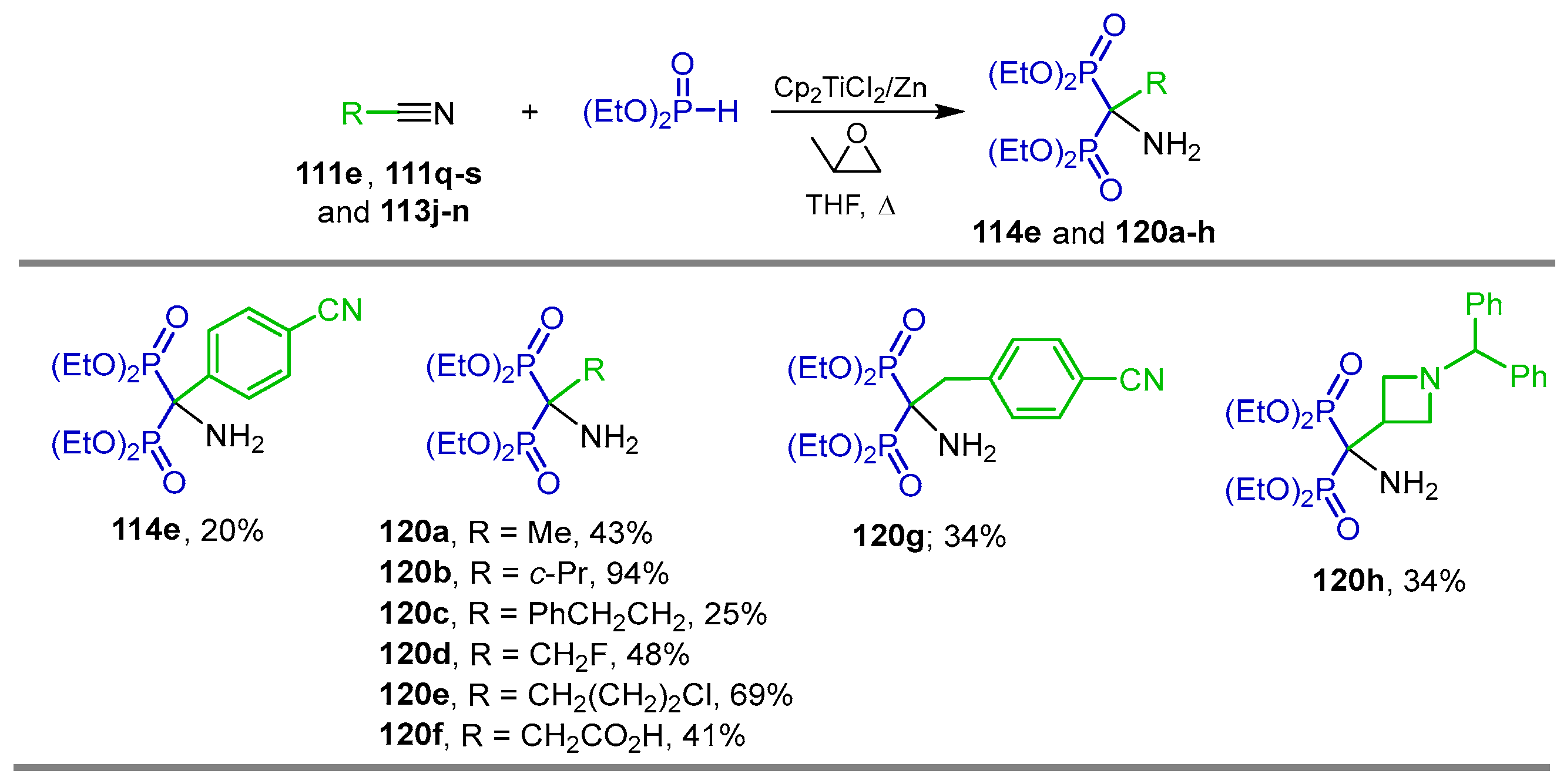
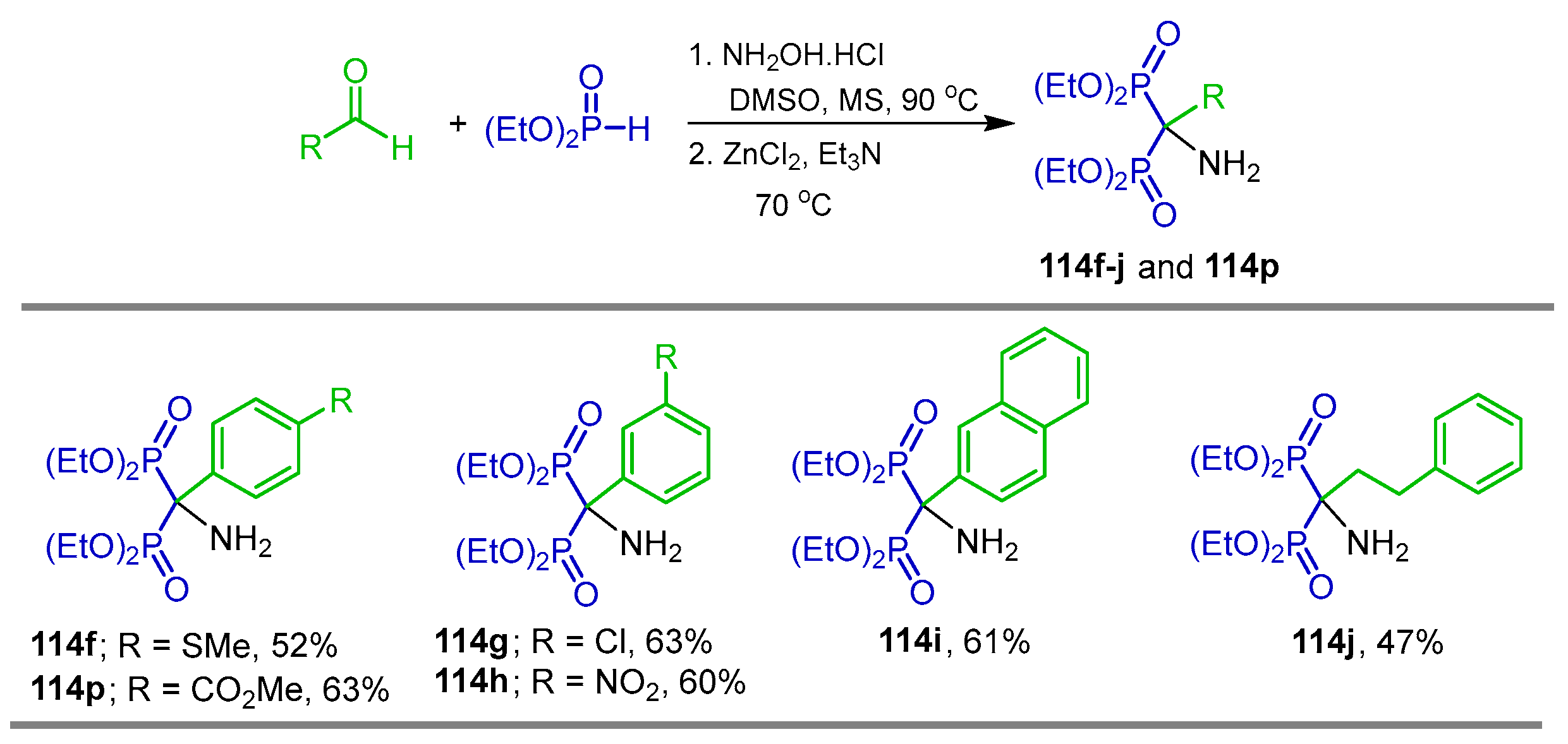
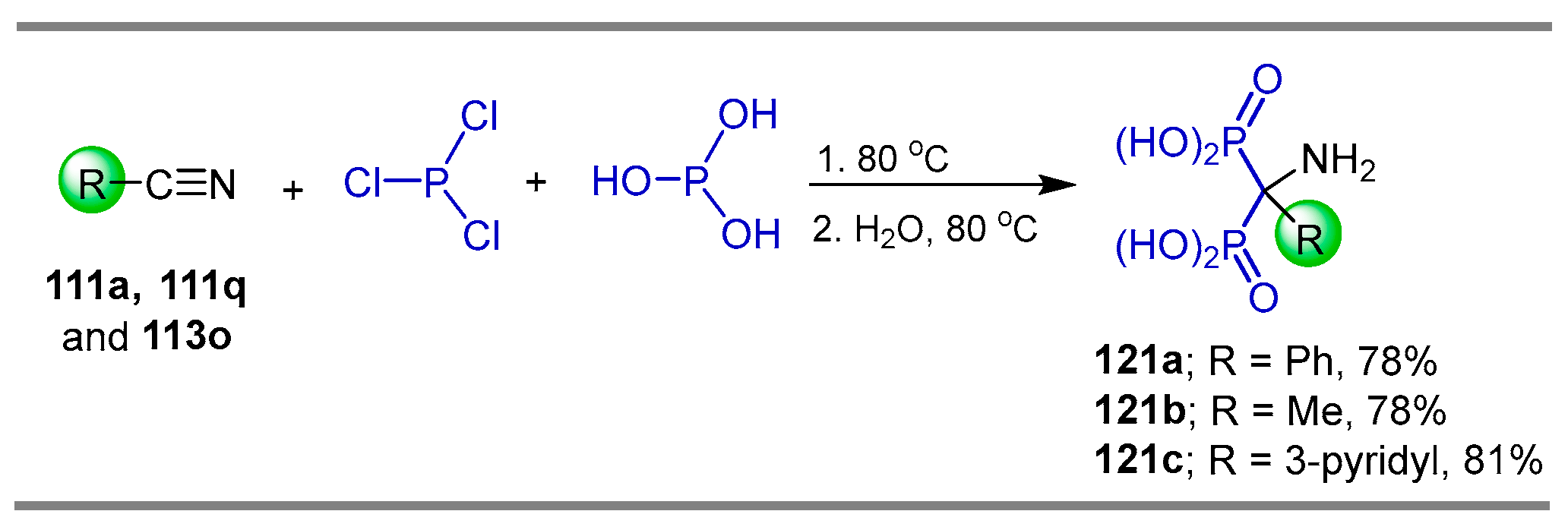
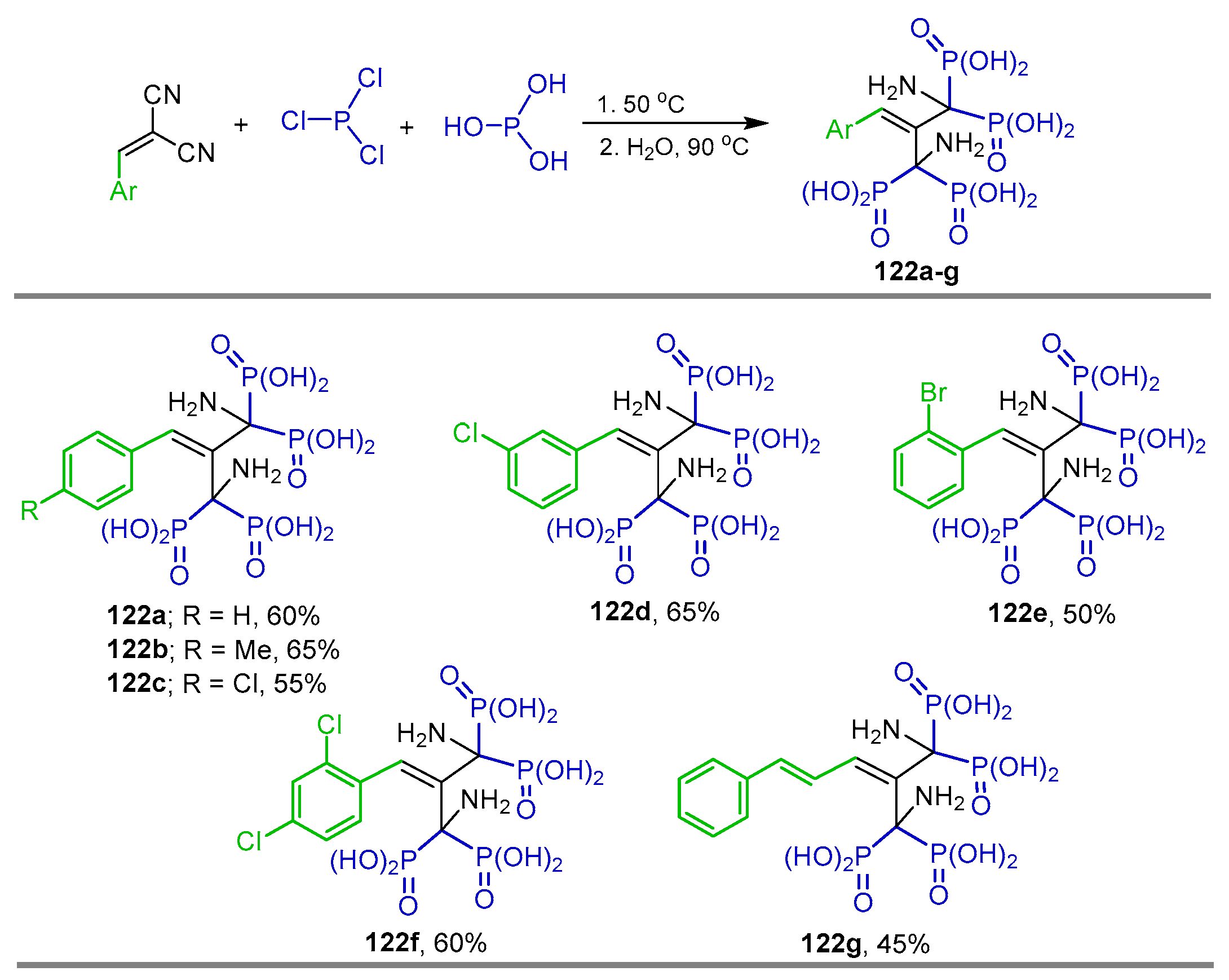
2.4. Phosphonylation of Nitriles
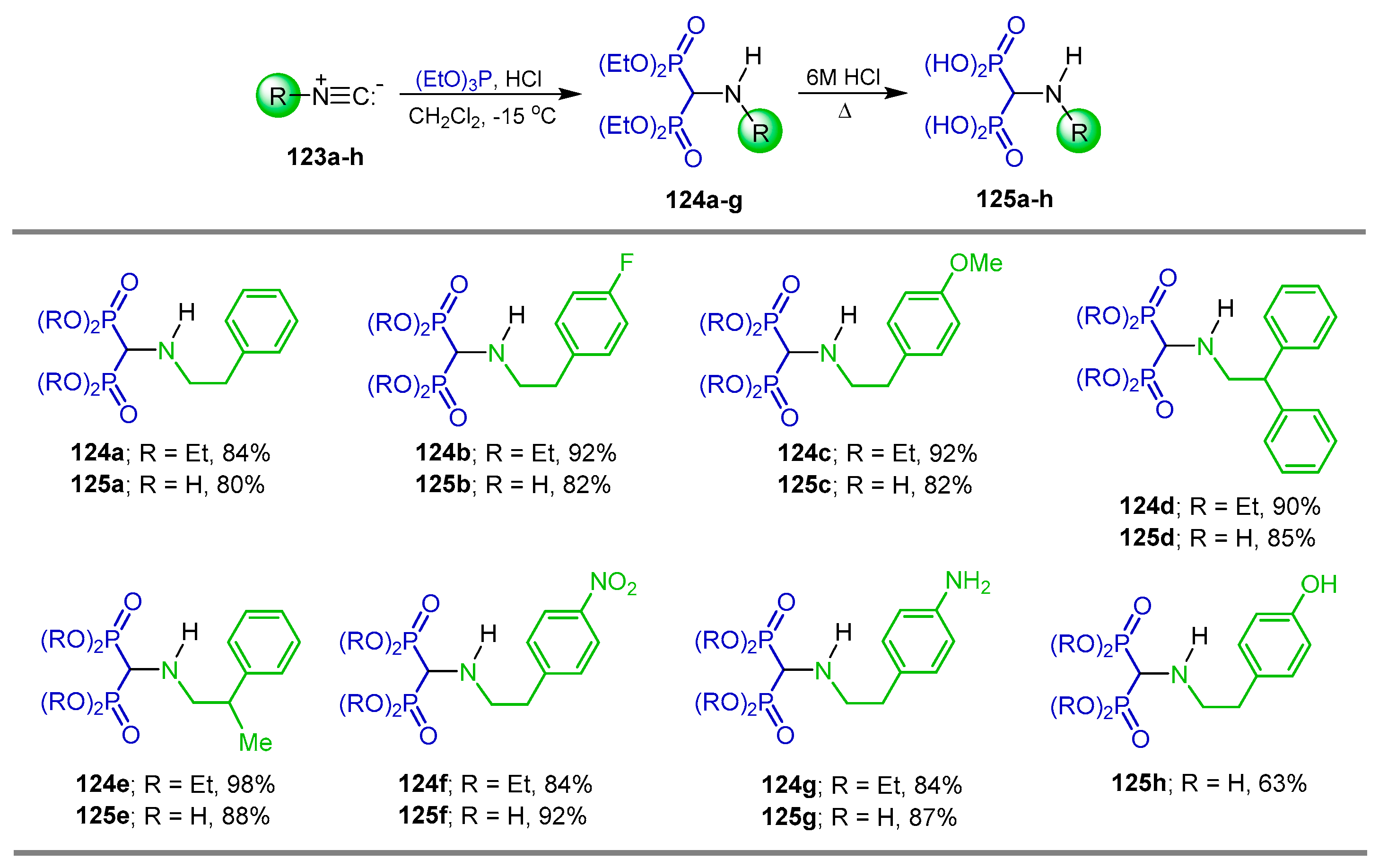
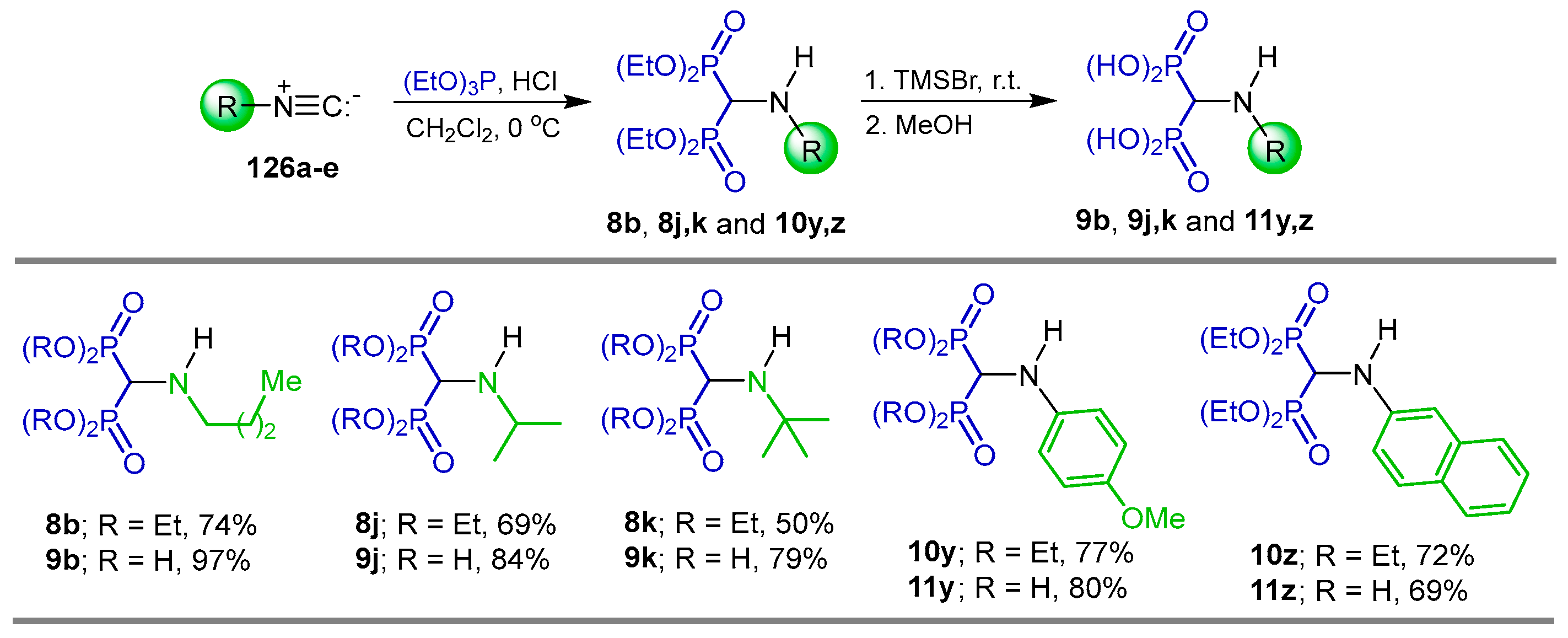
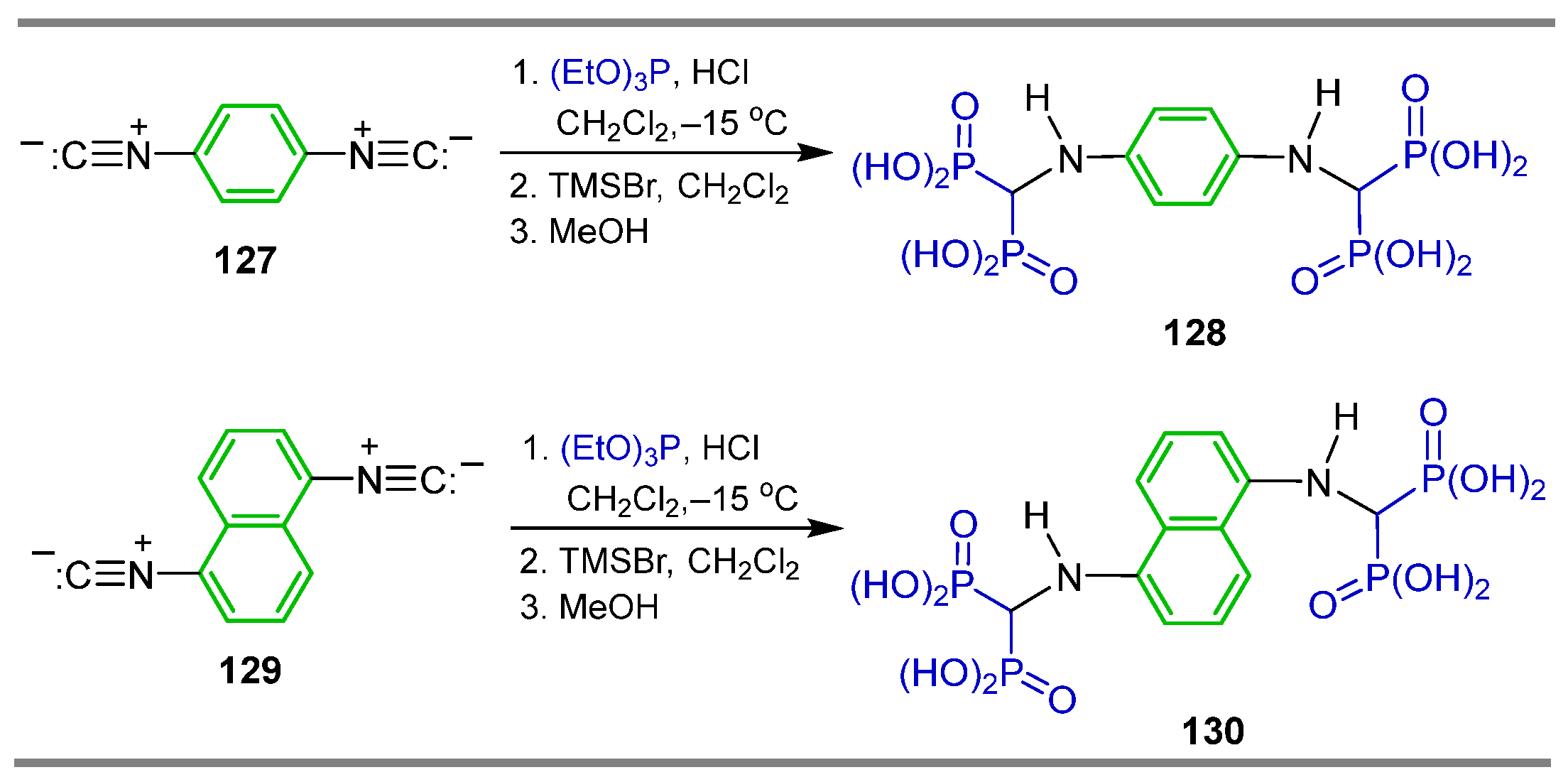
2.5. Phosphonylation of Isonitriles
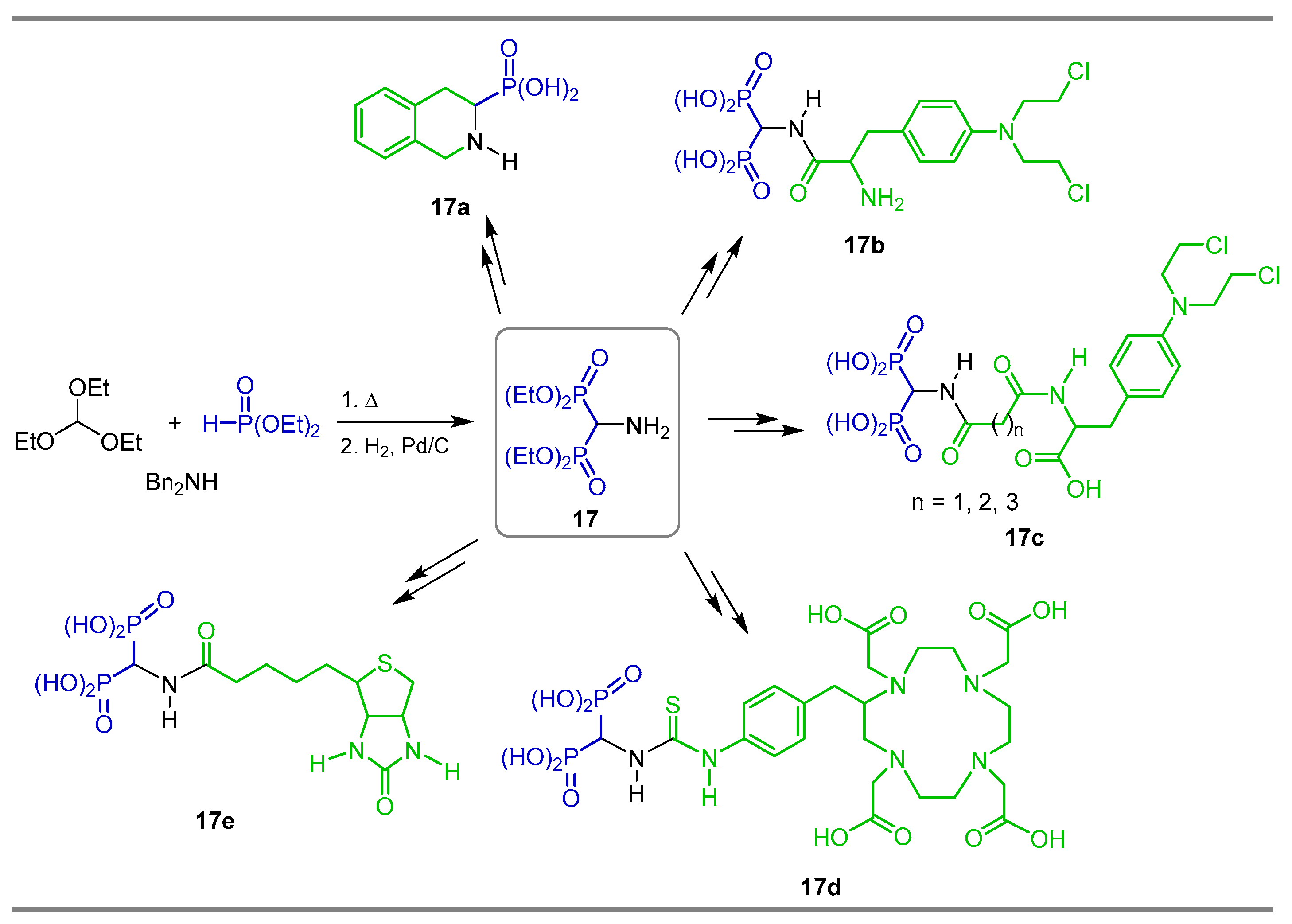
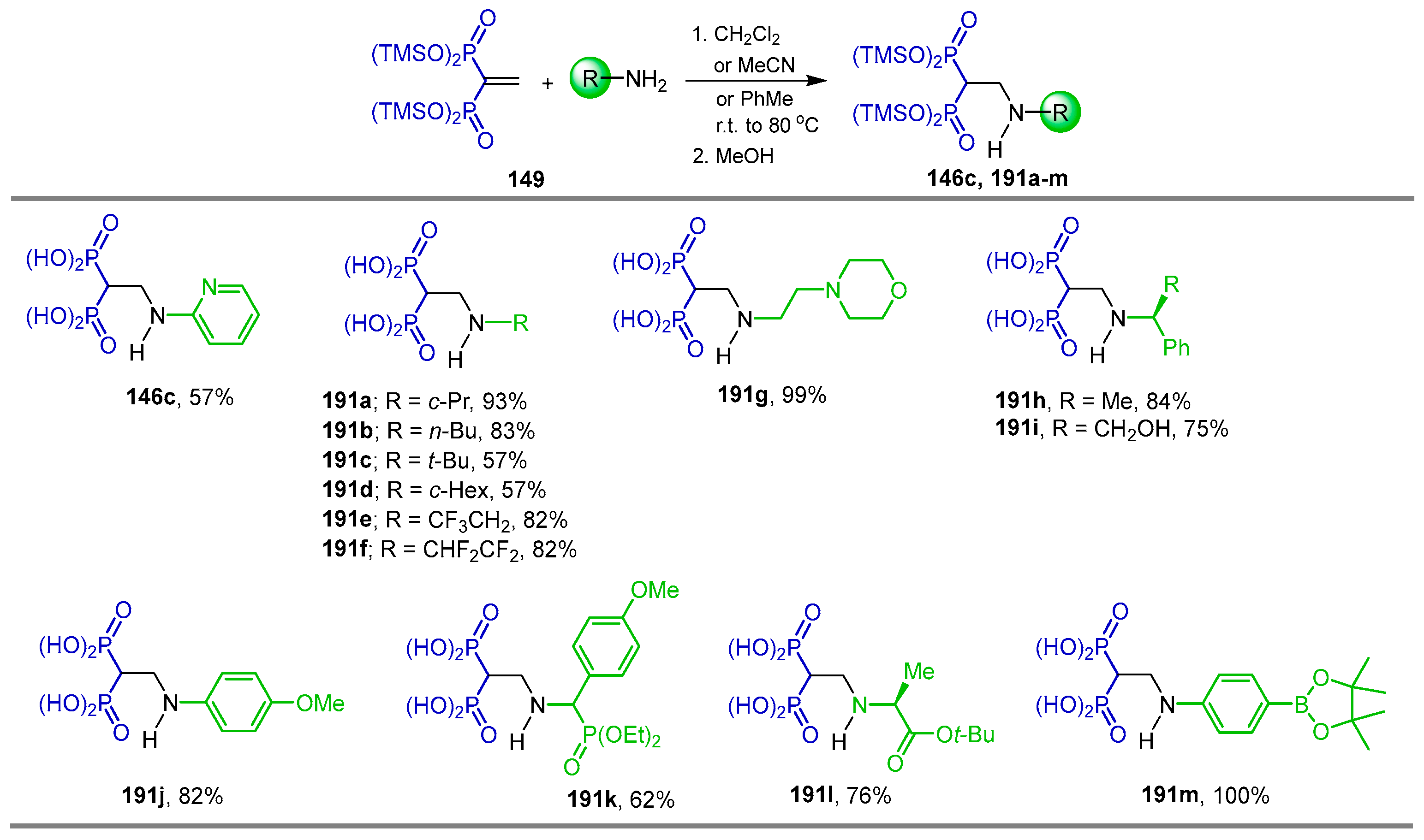
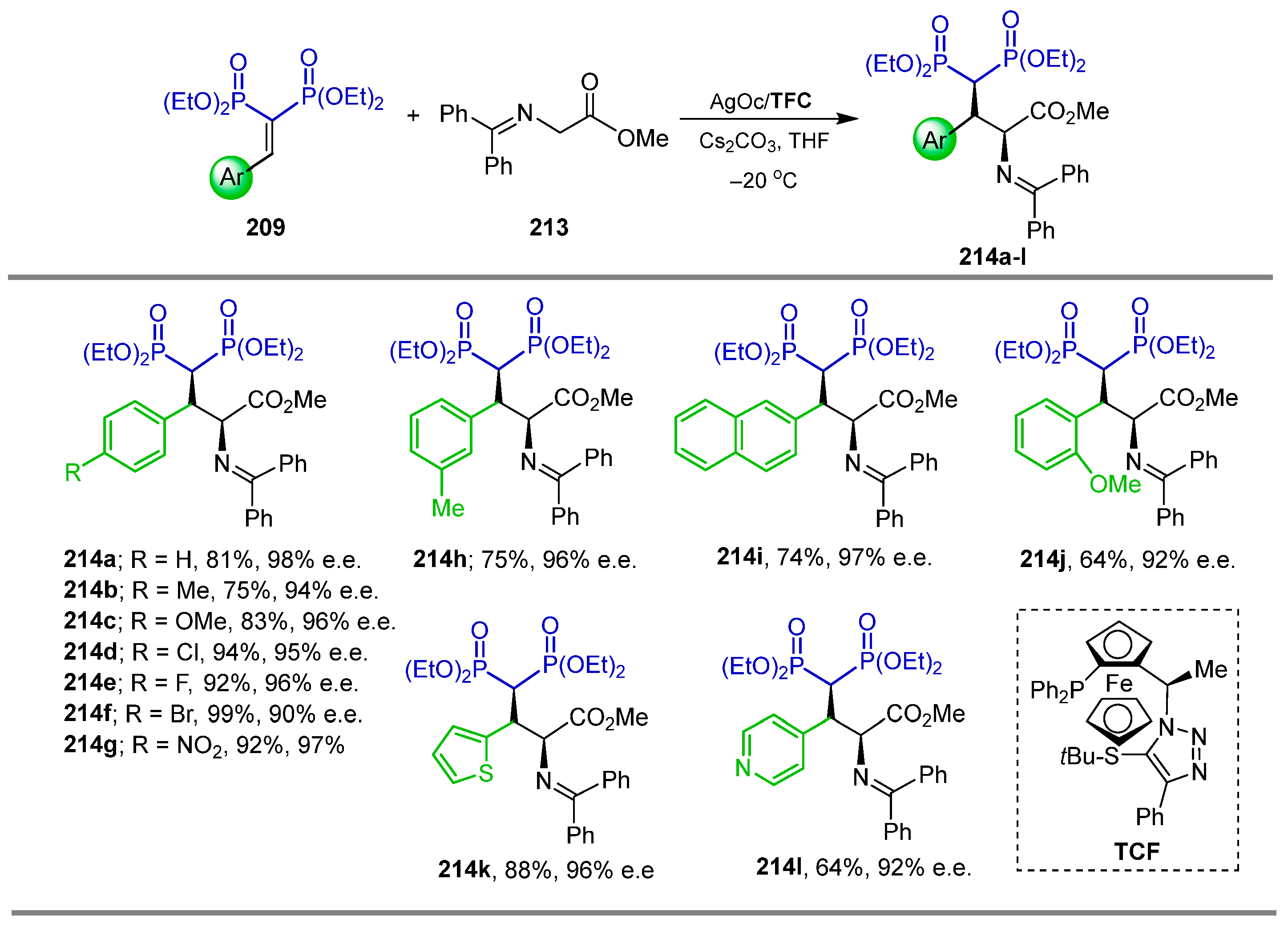
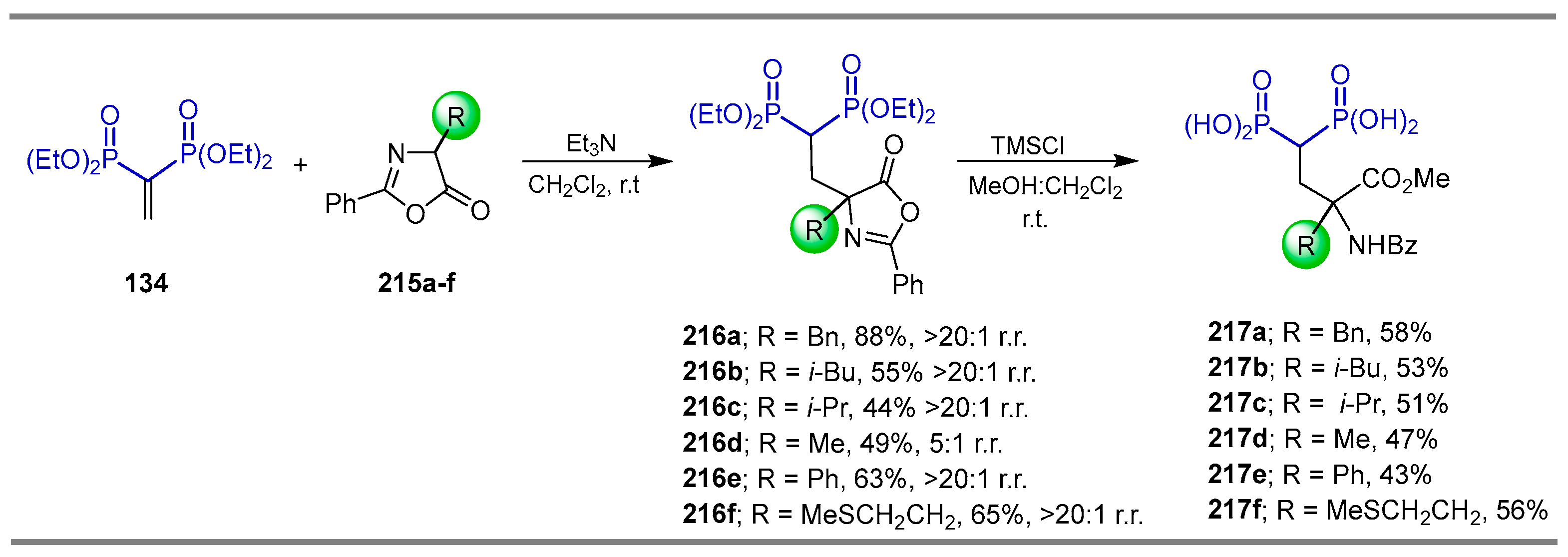
2.6. Electrophilic Amination of gem-Bisphosphonates
3. Synthesis of β-Amino-gem-Bisphosphonate Derivatives
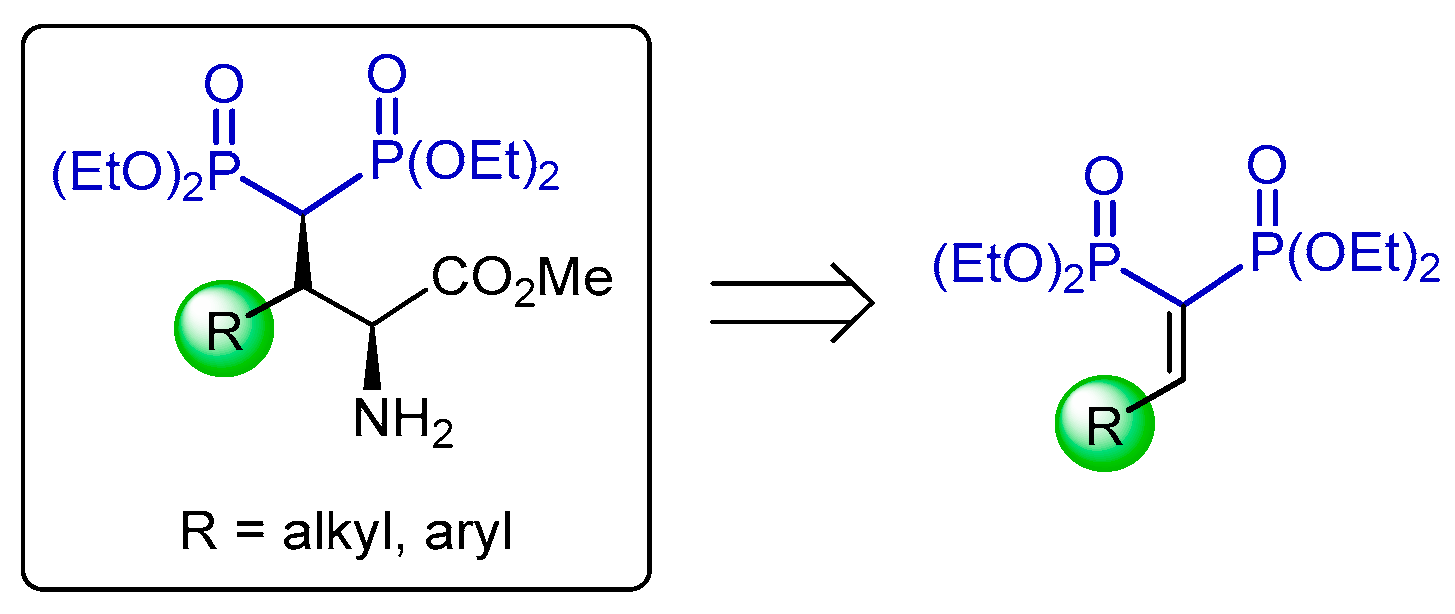
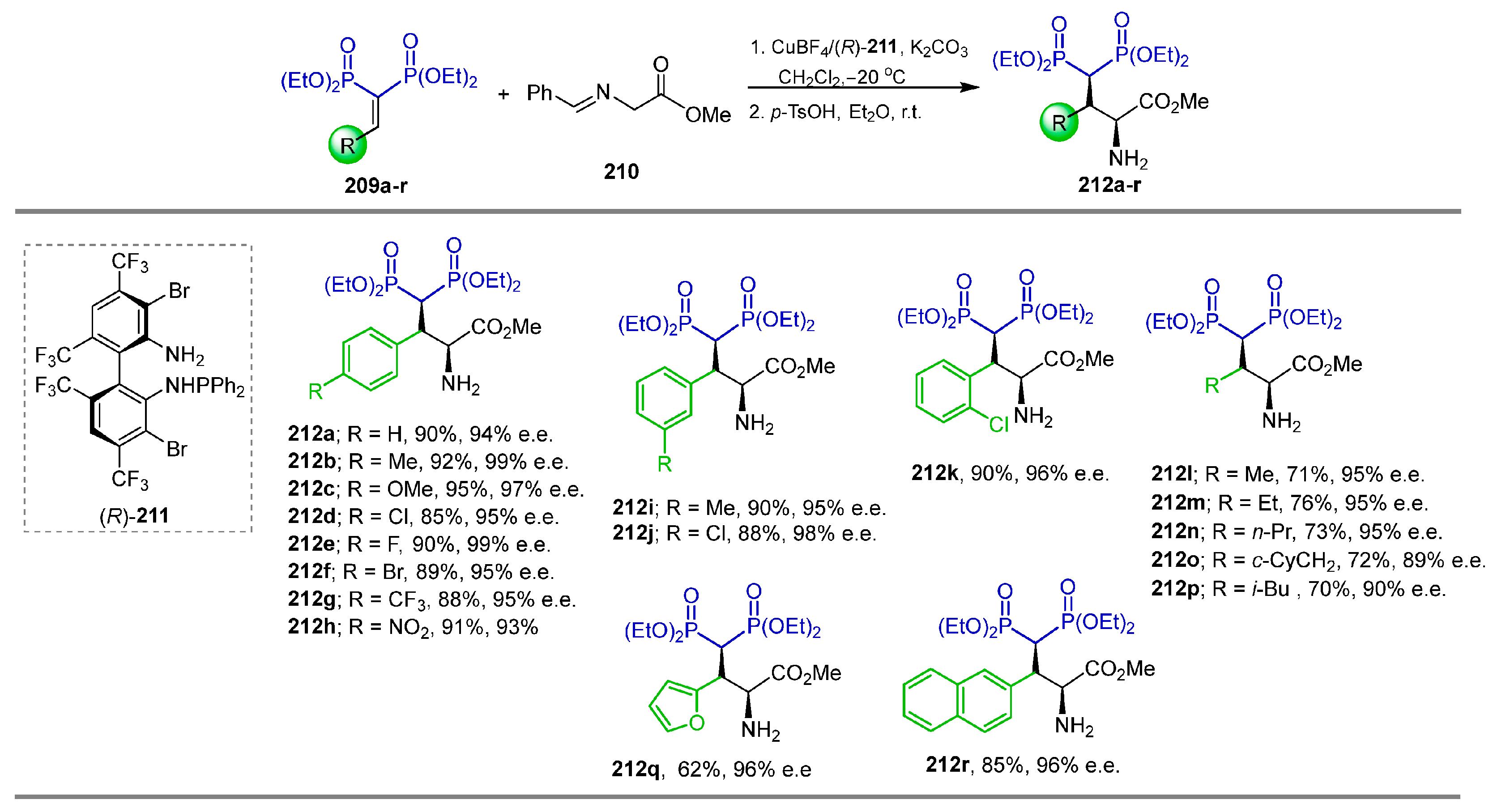
3.1. Michael-Type Addition of Amines to Vinyl gem-Bisphosphonates
3.2. Addition of gem-Bisphosphonates to Imines
4. Synthesis of γ-Amino-gem-Bisphosphonate Derivatives
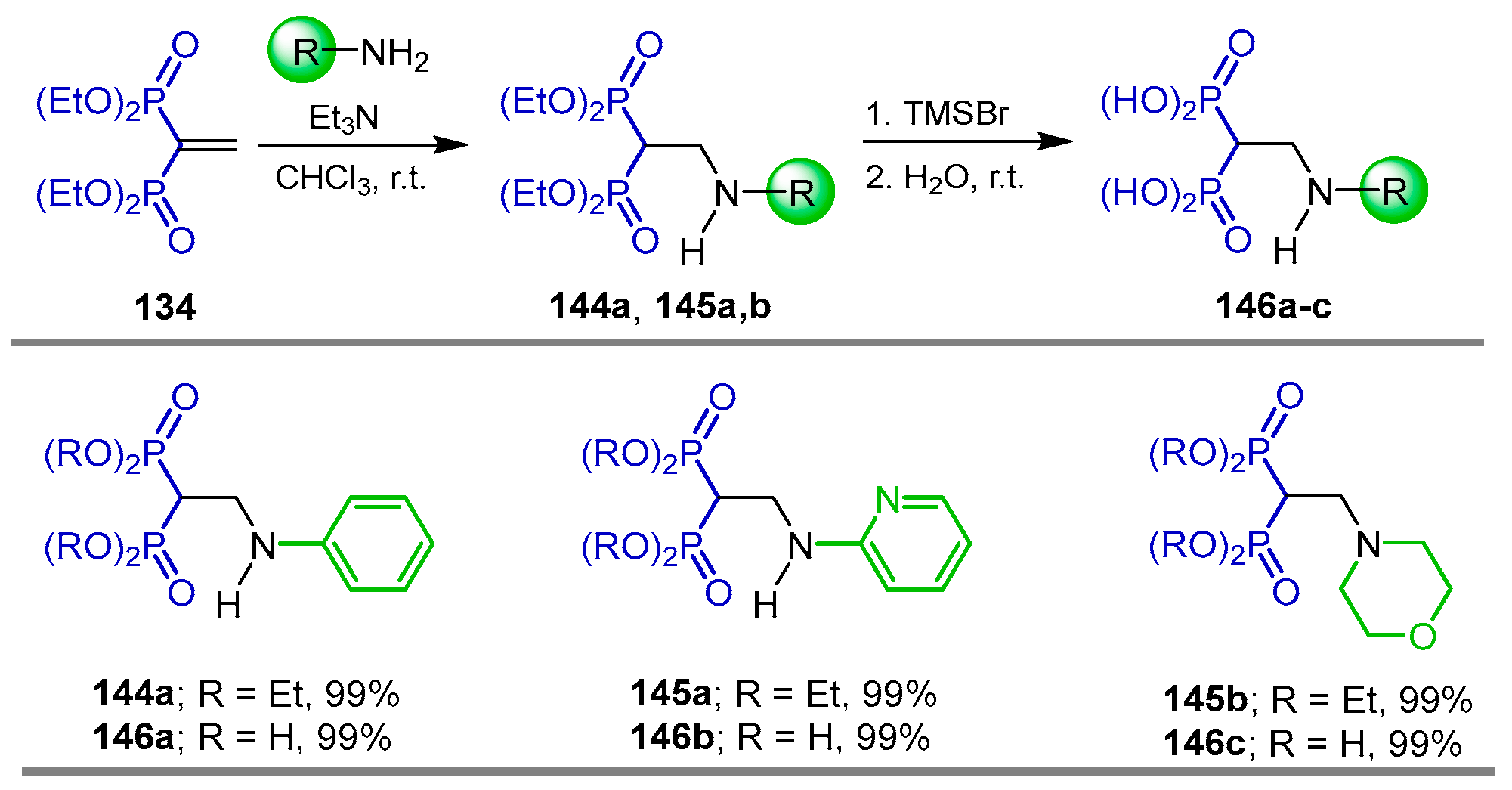
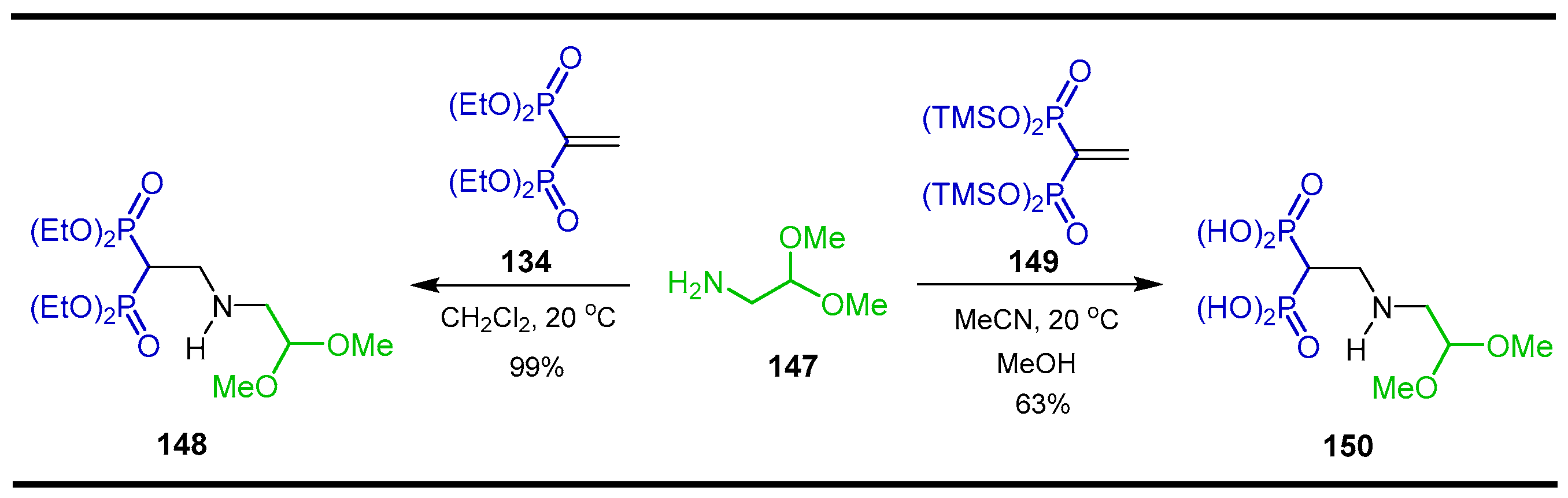
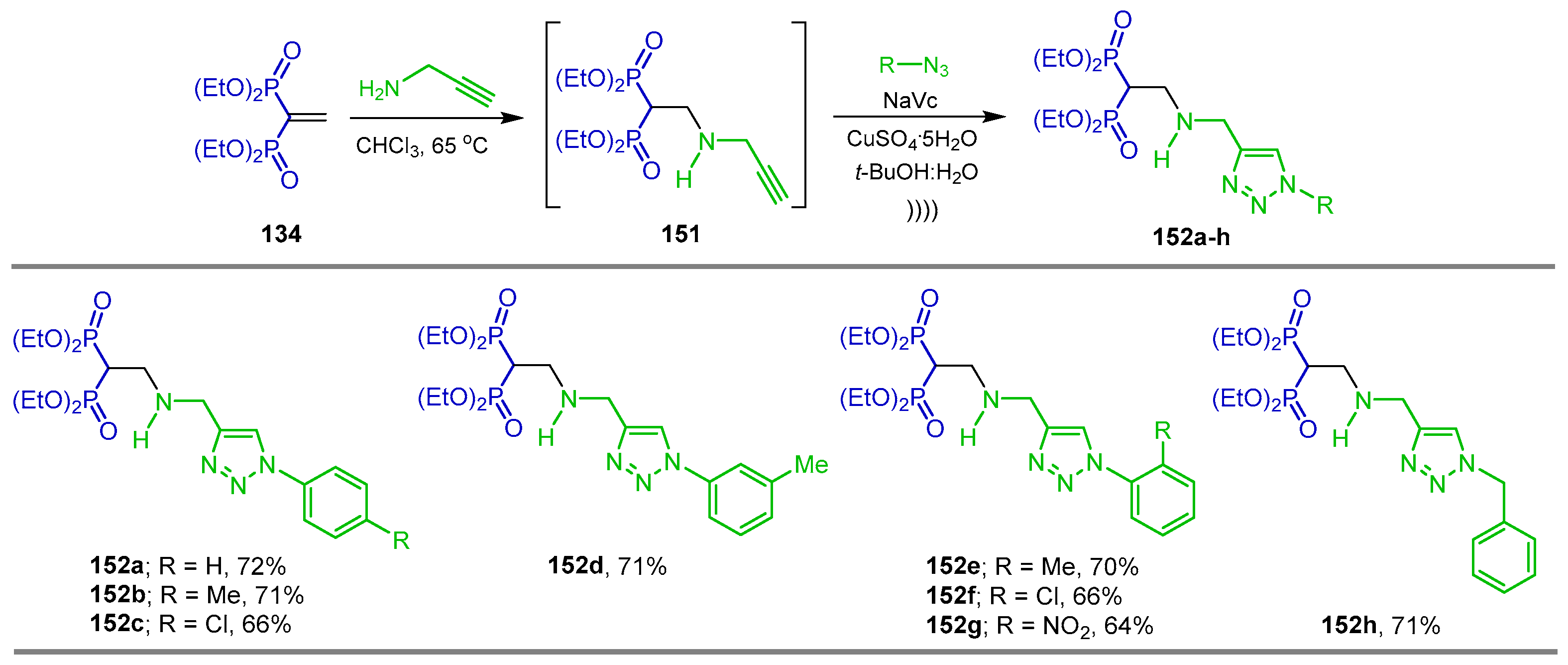
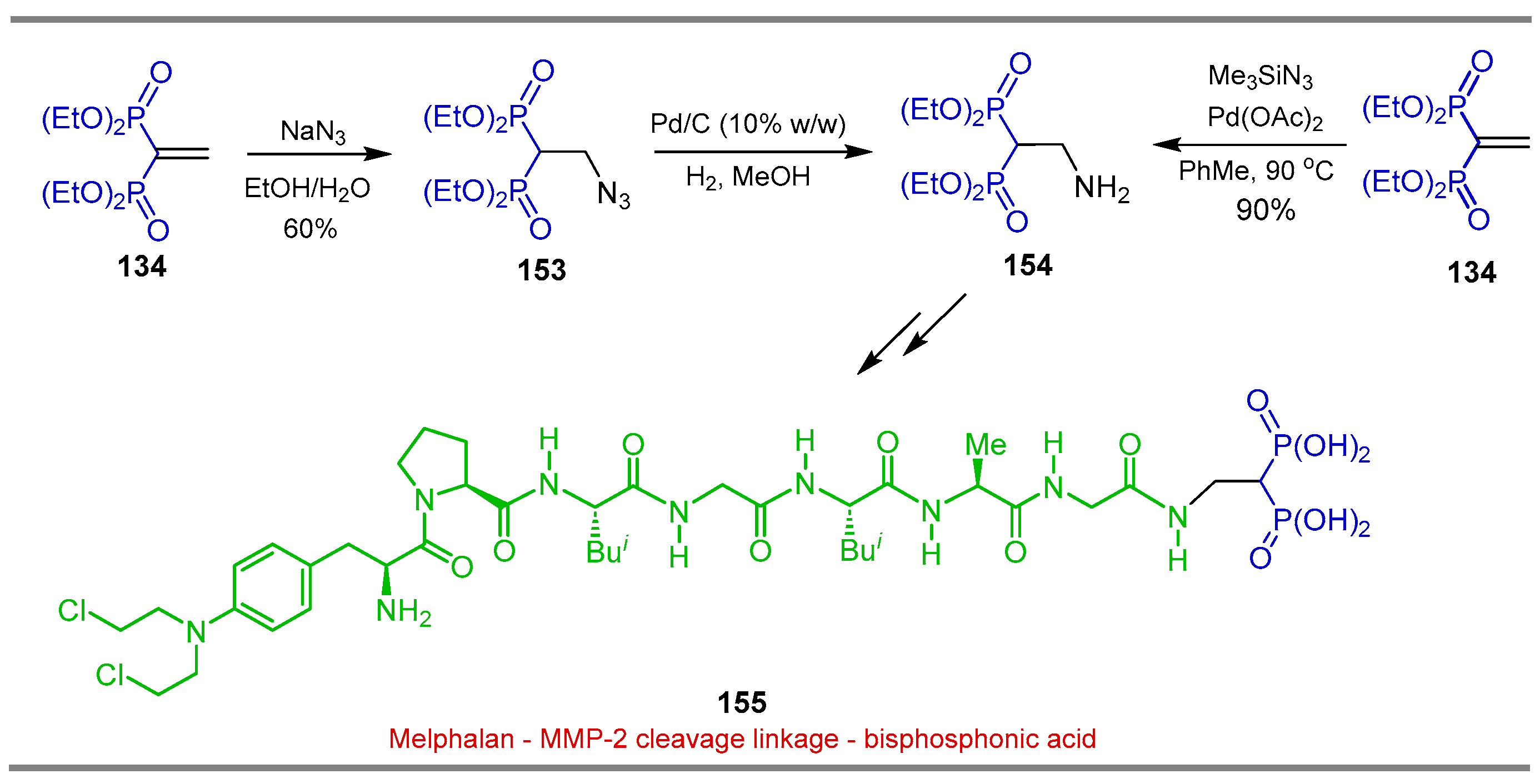
Synthesis from Vinyl gem-Bisphosphonates
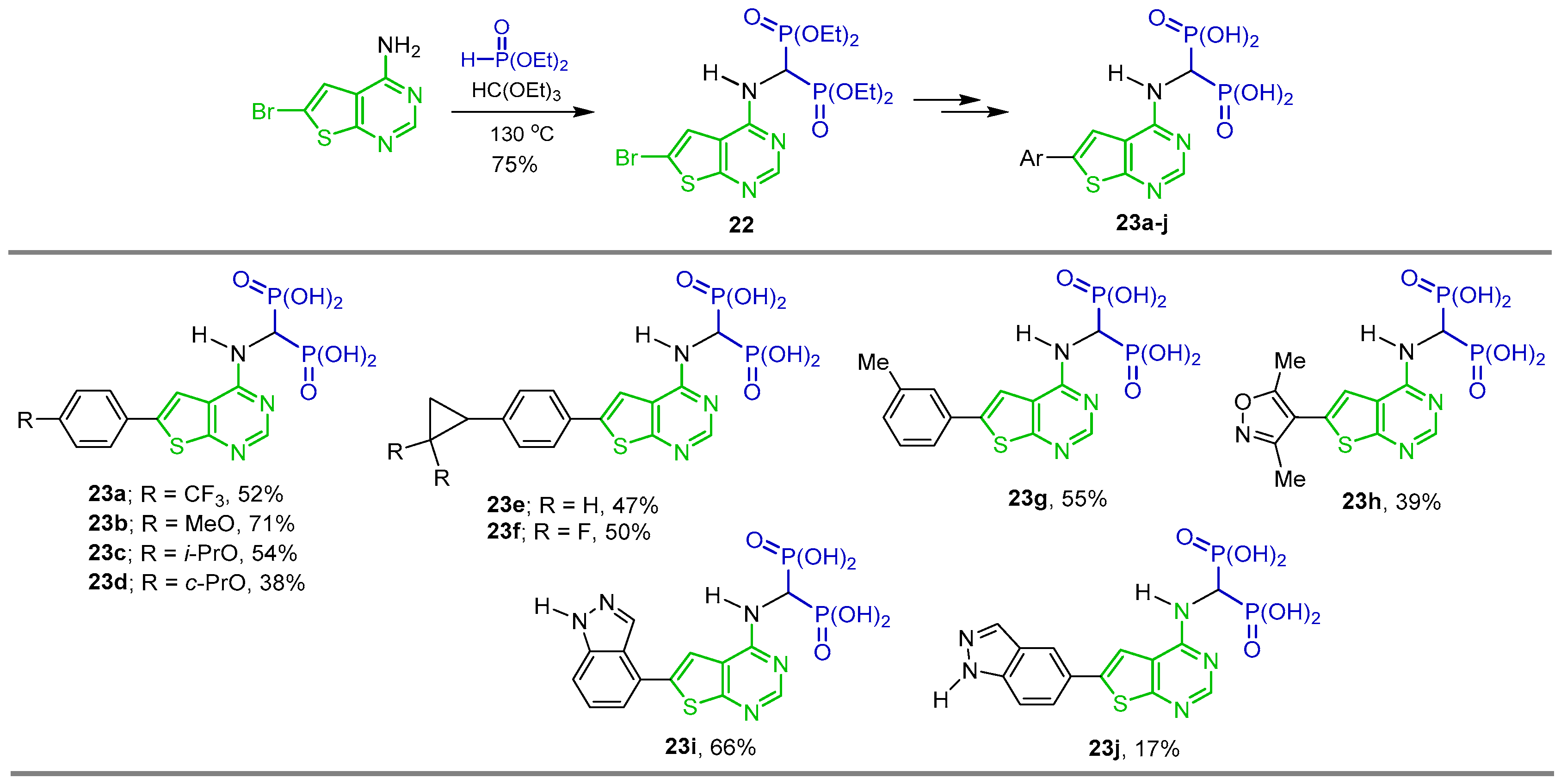
5. Synthesis of Heterocyclic α-Amino-gem-Bisphosphonate Derivatives
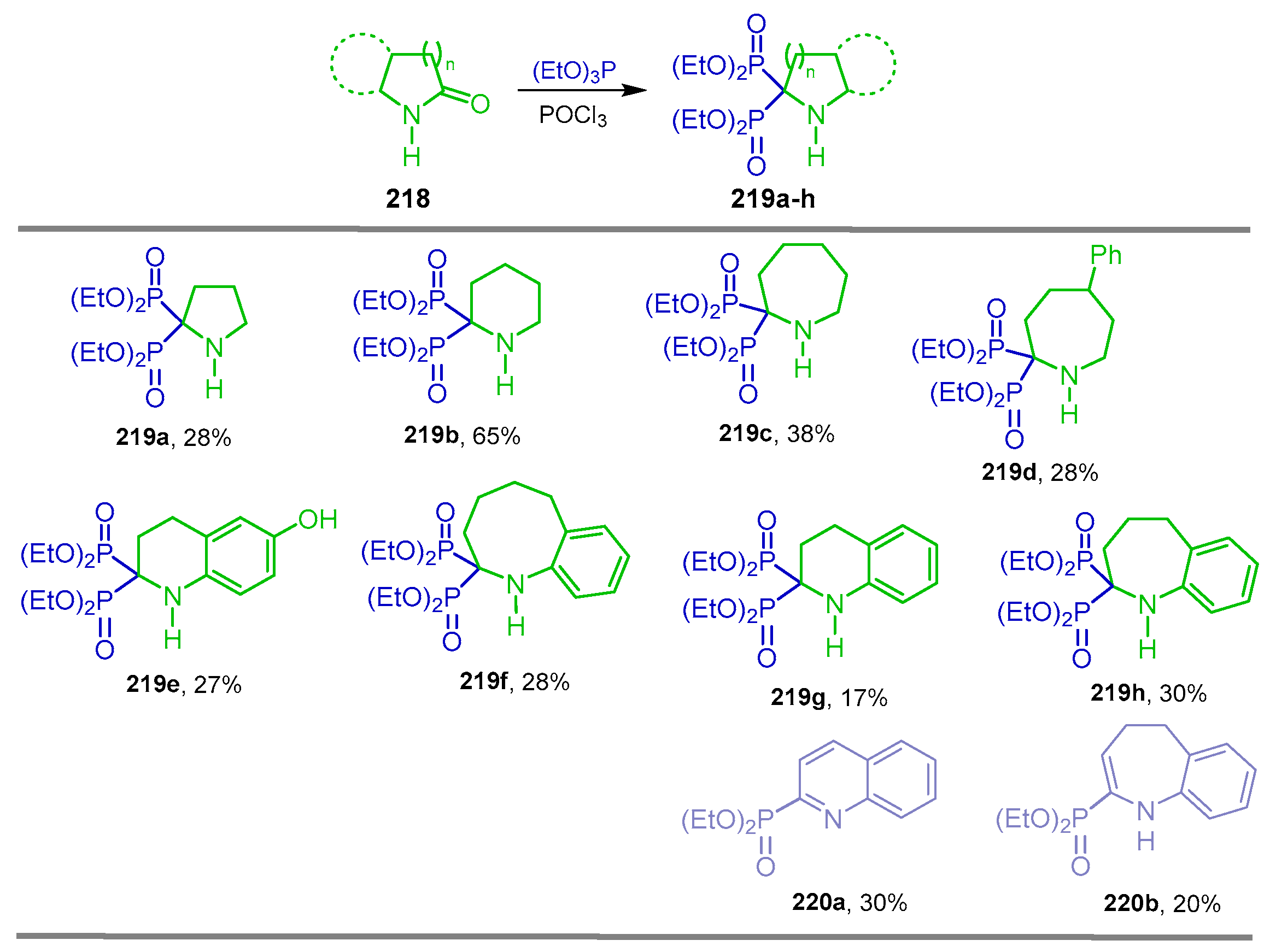
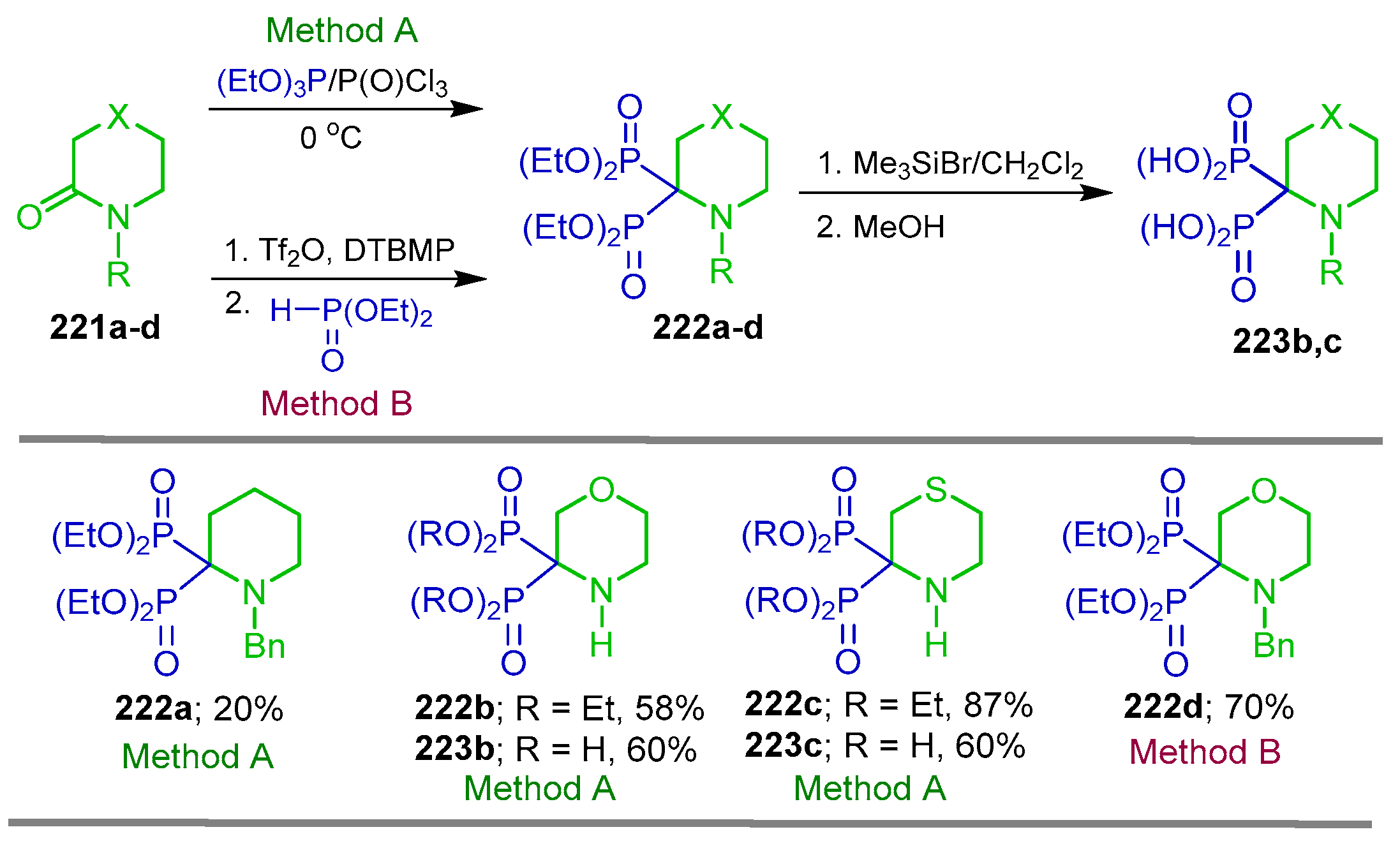
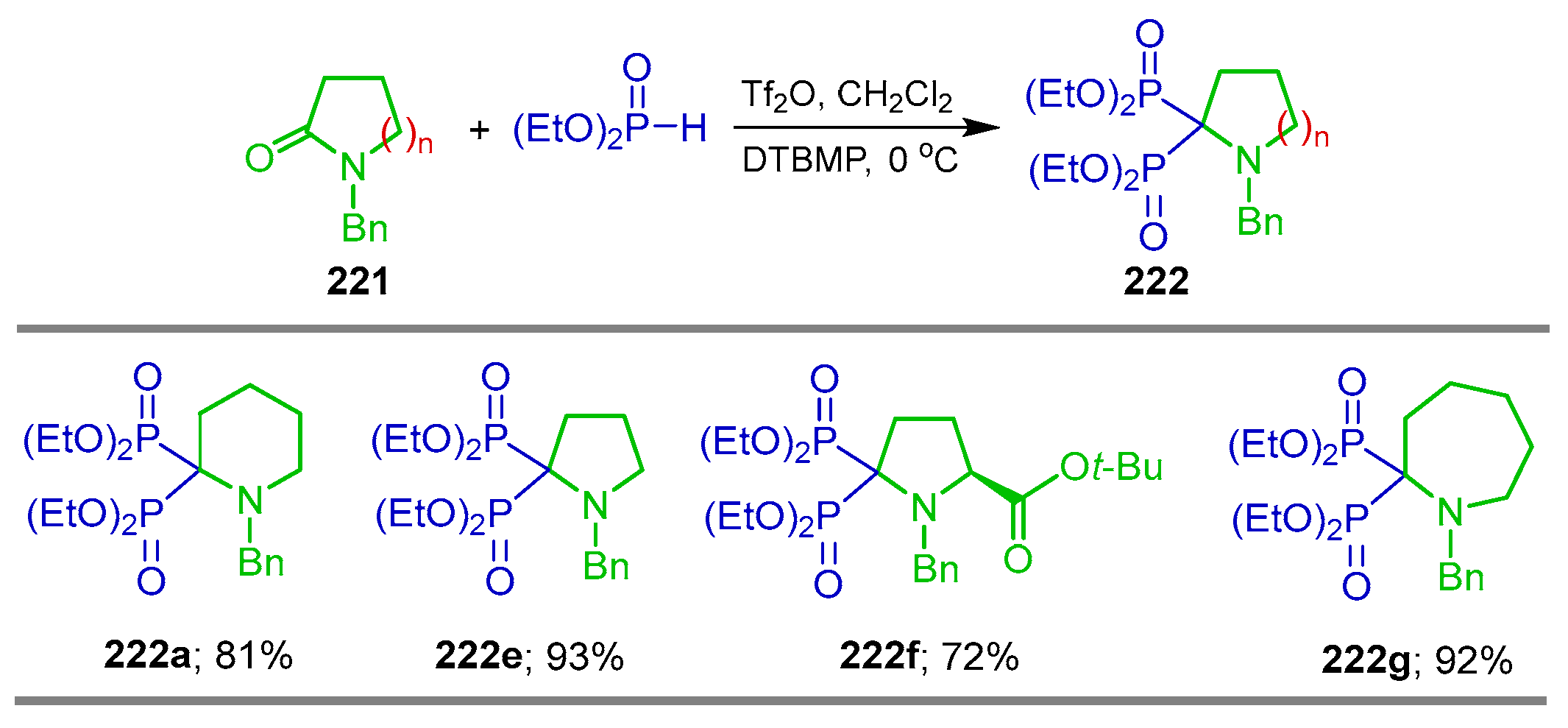
5.1. Phosphonylation of Lactams
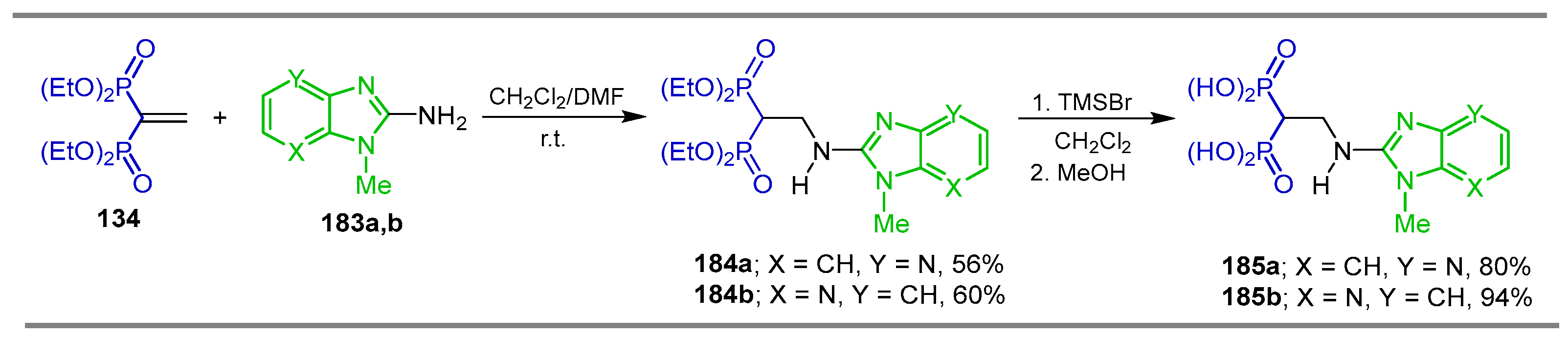
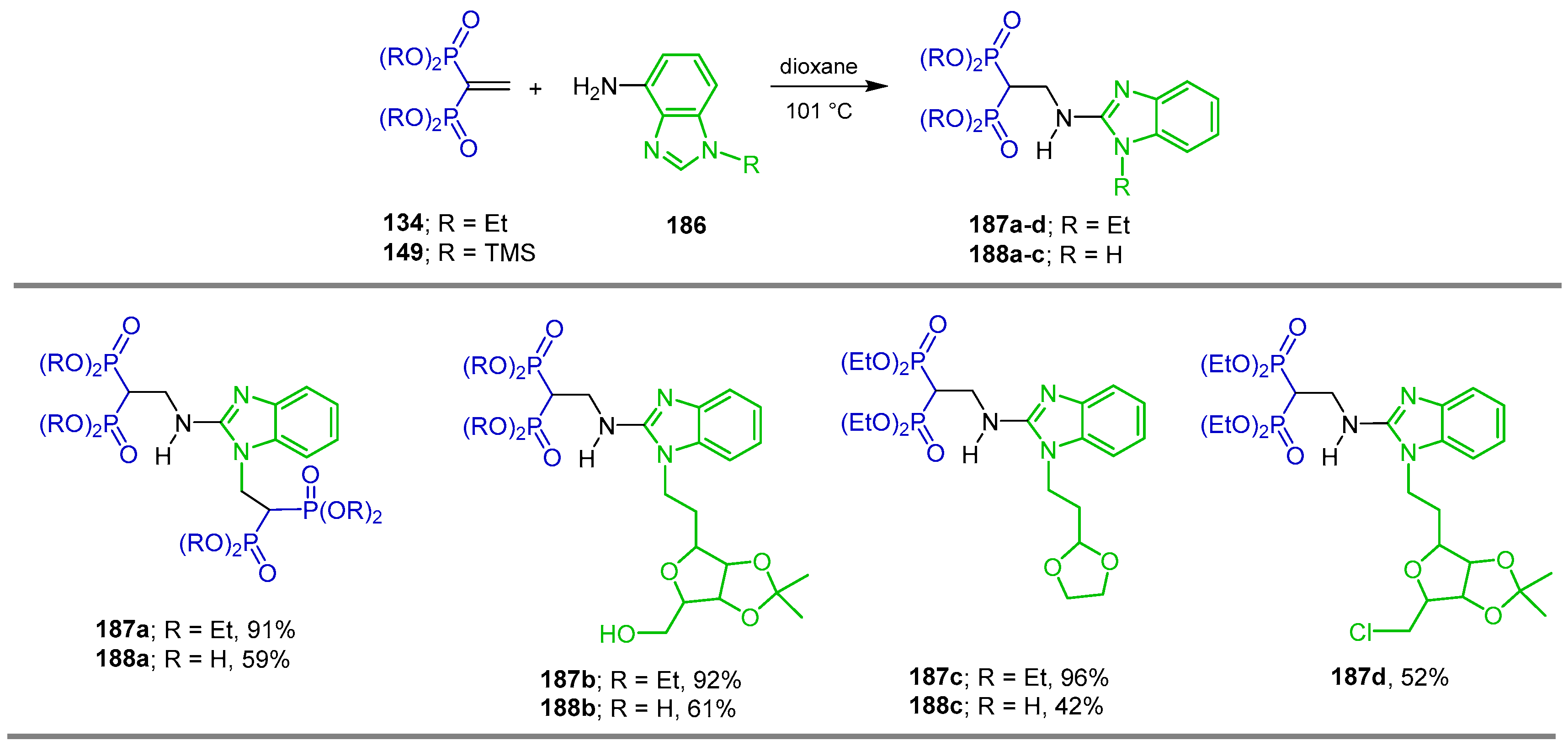
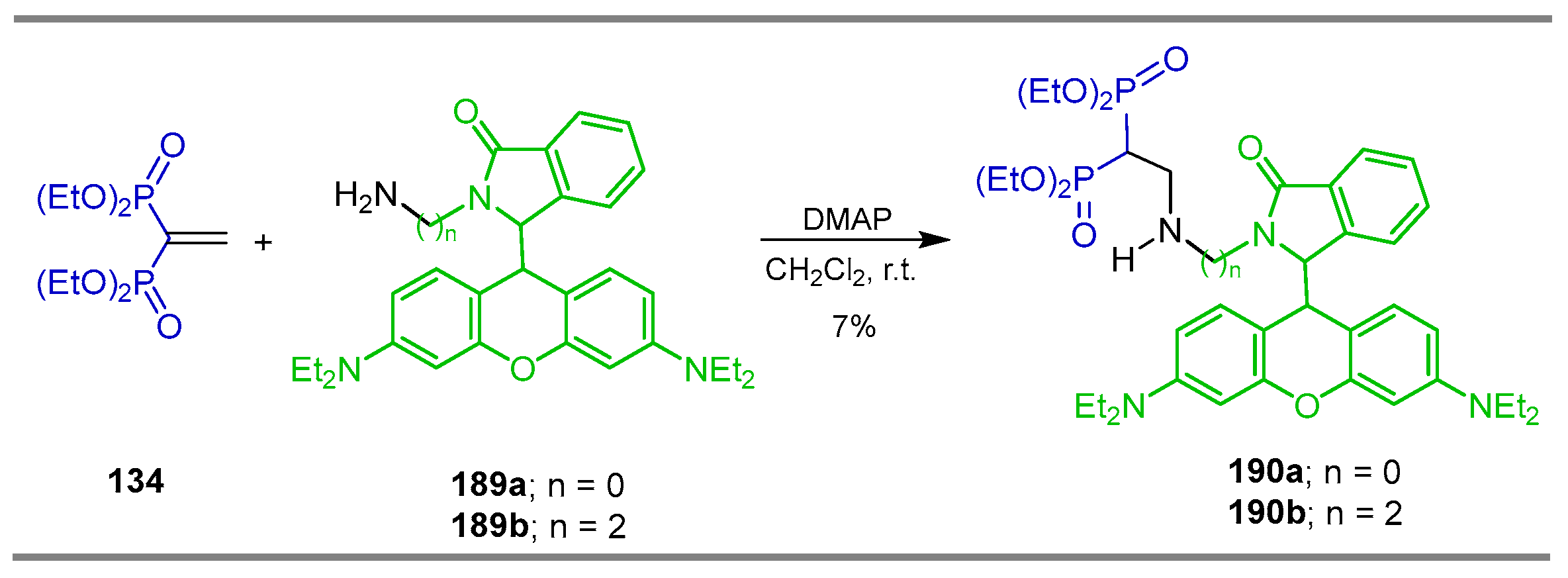
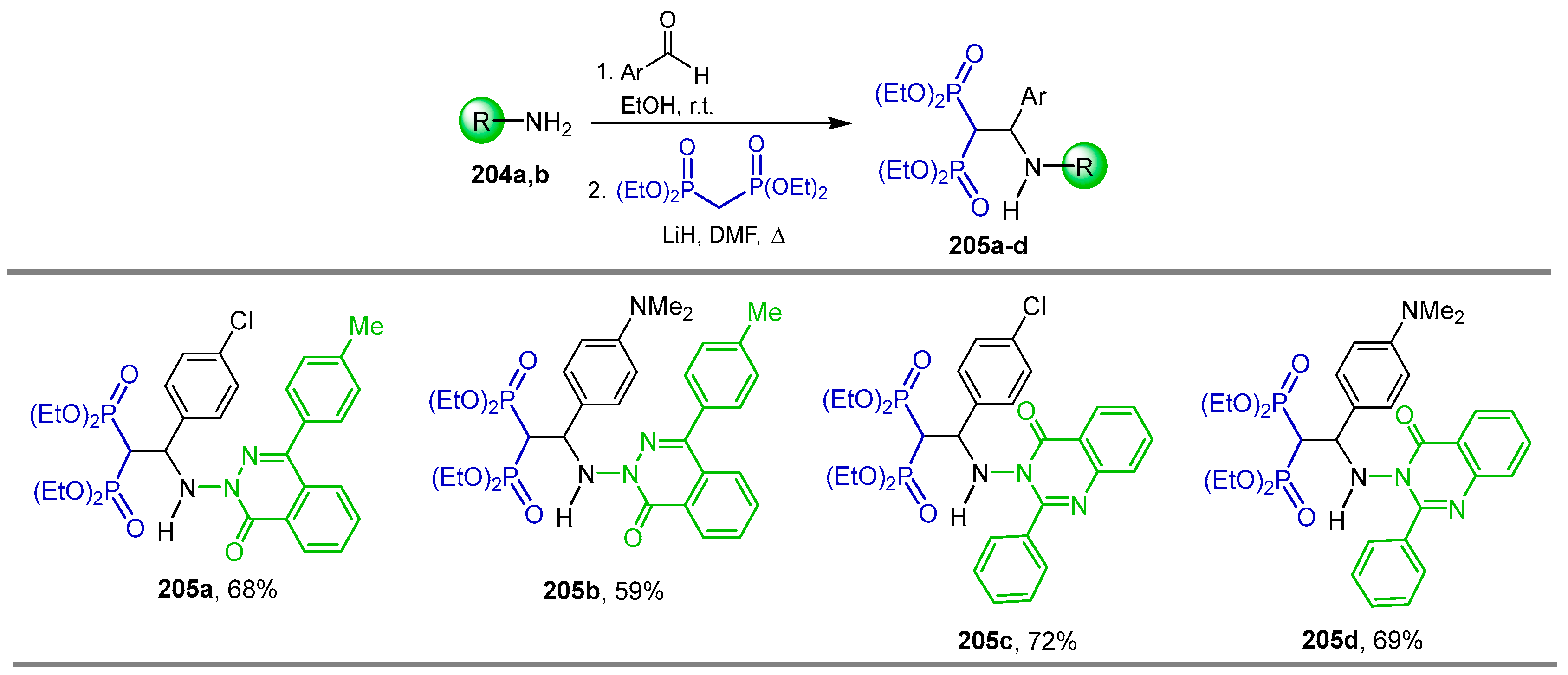
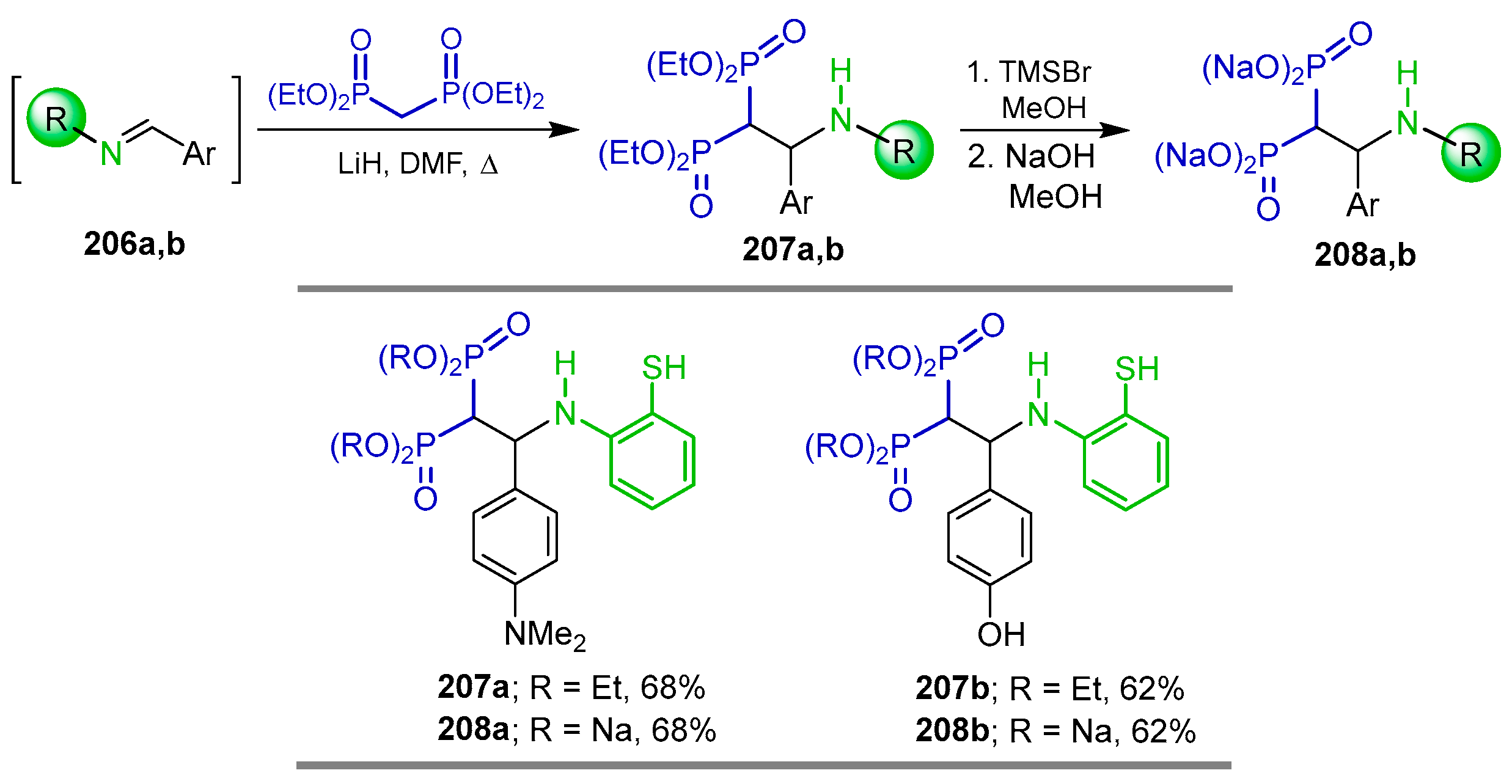
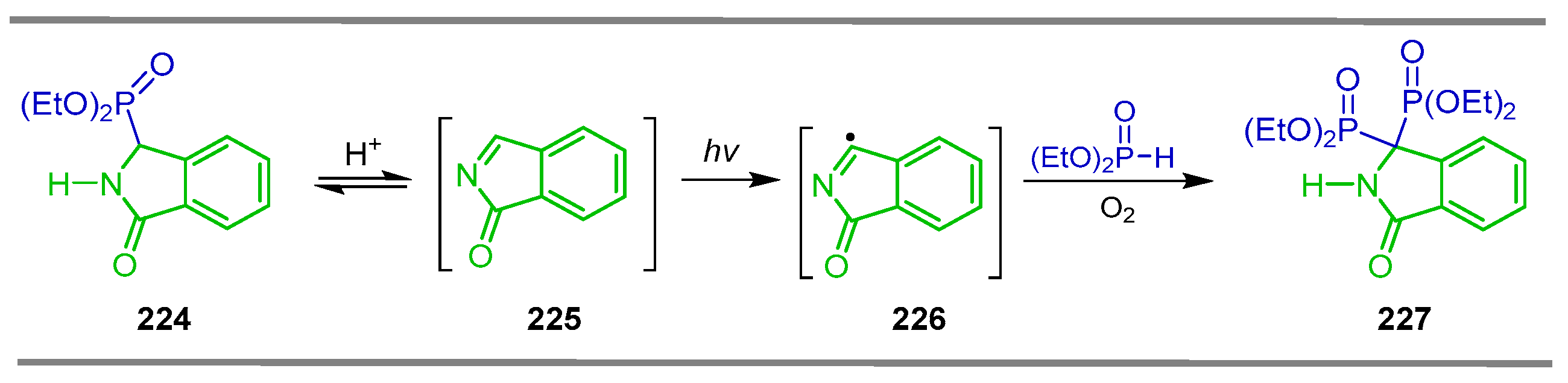
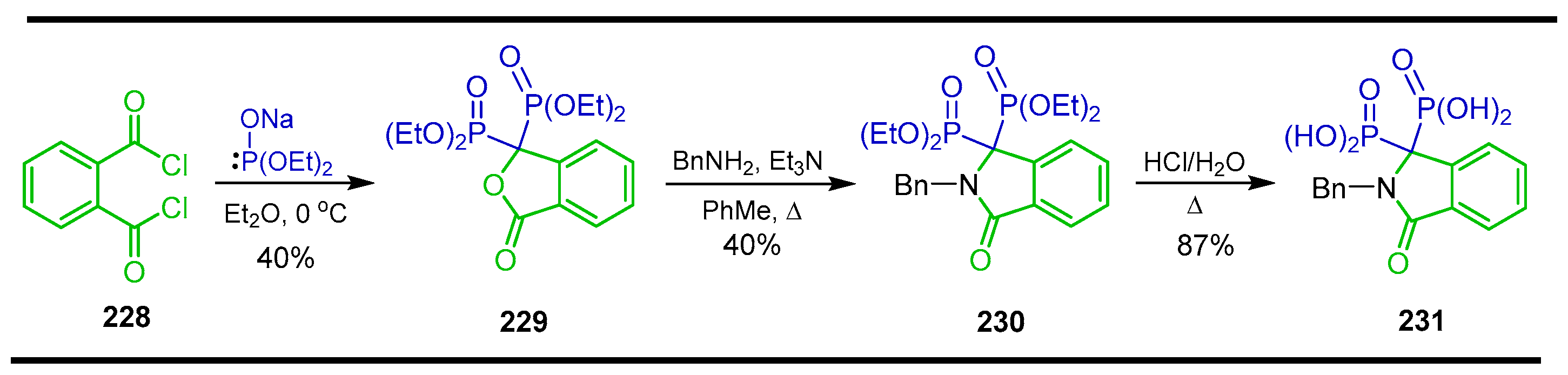
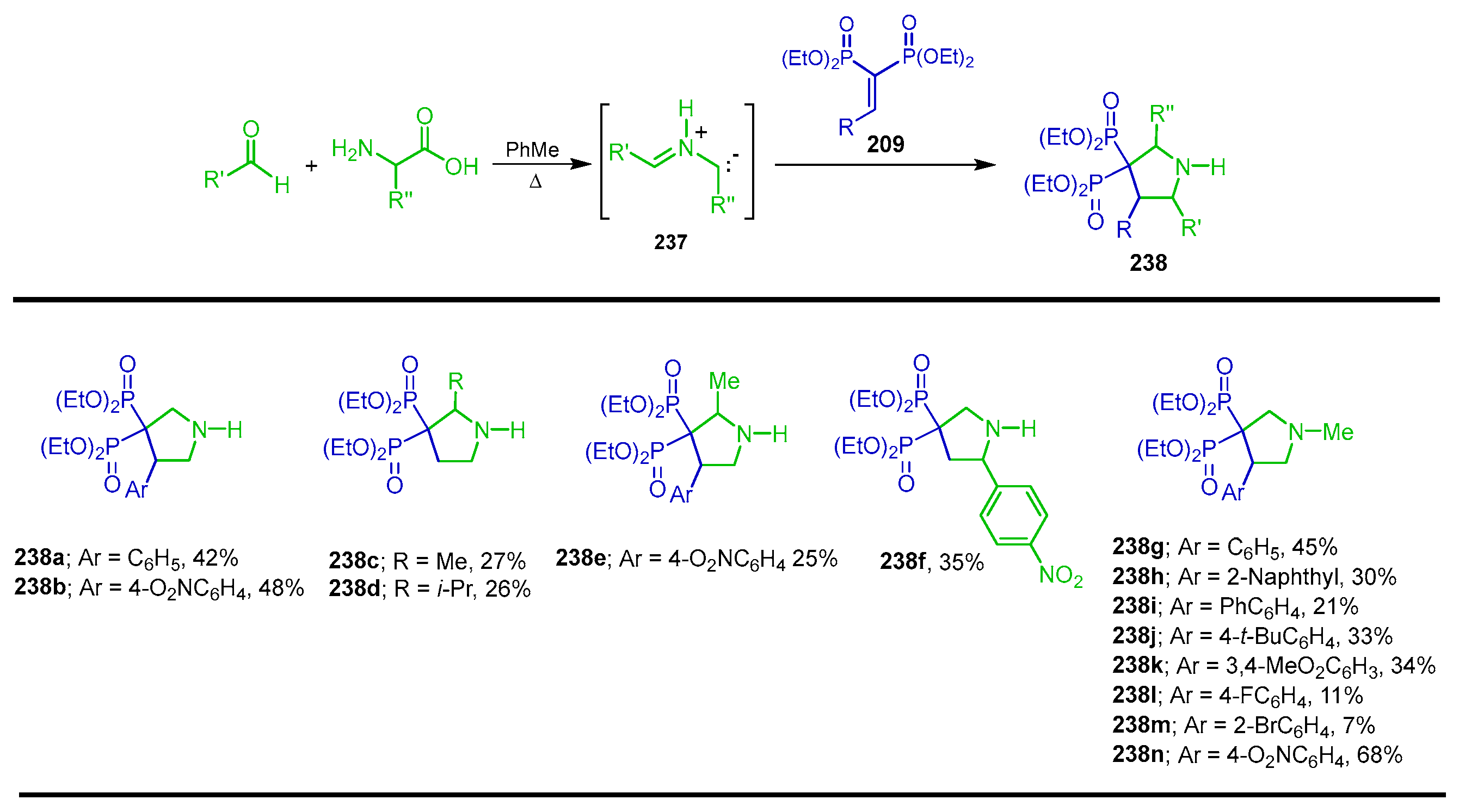
5.2. Miscellaneous
6. Concluding Remarks
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mucha, A.; Kafarski, P.; Berlicki, L. Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Sello, G.; Sisti, M. Aminophosphonic Acids and Derivatives. Synthesis and Biological Applications. Curr. Med. Chem. 2010, 17, 264–289. [Google Scholar] [CrossRef] [PubMed]
- Naydenova, E.D.; Todorov, P.T.; Troev, K.D. Recent synthesis of aminophosphonic acids as potential biological importance. Amino Acids 2010, 38, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Gluza, K.; Kafarski, P. Chap. 12 Transition state analogues of enzymatic reaction as potential drugs. In Drug Discovery; El-Shemy, H., Ed.; InTech: Rijeka, Croatia, 2013; pp. 325–372. [Google Scholar]
- Lejczak, B.; Kafarski, P. Aminated (Cyclopropylmethyl)Phosphonates: Synthesis and Anti-Pancreatic Cancer Activity. Top. Heterocycl. Chem. 2009, 20, 31–63. [Google Scholar]
- Ordoñez, M.; Viveros-Ceballos, J.L.; Romero-Estudillo, I. Chap. 6 Stereoselective synthesis of α-aminophosphonic acids through Pudovik and Kabachnik Fields-reaction. In Amino Acid-New Insights and Roles in Plant and Animal; Asao, T., Asaduzzaman, M.D., Eds.; InTech: Rijeka, Croatia, 2017; pp. 127–151. [Google Scholar]
- Ordoñez, M.; Viveros-Ceballos, J.L.; Cativiela, C.; Sayago, F.J. An update on the stereoselective synthesis of α-aminophosphonic acids and derivatives. Tetrahedron 2015, 71, 1745–1784. [Google Scholar] [CrossRef]
- Ordoñez, M.; Viveros-Ceballos, J.L.; Cativiela, C.; Arizpe, A. Stereoselective Synthesis of α-Aminophosphonic Acids Analogs of the 20 Proteinogenic α-Amino Acids. Curr. Org. Synth. 2012, 9, 310–341. [Google Scholar] [CrossRef]
- Ordoñez, M.; Rojas-Cabrera, A.; Cativiela, C. An overview of stereoselective synthesis of α-aminophosphonic acids and derivatives. Tetrahedron 2009, 65, 17–49. [Google Scholar] [CrossRef] [PubMed]
- Mayorquín-Torres, M.C.; Simoens, A.; Bonneure, E.; Stevens, C.V. Synthetic Methods for Azaheterocyclic Phosphonates and Their Biological Activity: An Update. 2004–2024. Chem. Rev. 2024, 124, 7907–7975. [Google Scholar] [CrossRef] [PubMed]
- Amira, A.; Aouf, Z.; K’tir, H.; Chemam, Y.; Ghodbane, R.; Zerrouki, R.; Aouf, N.-E. Recent Advances in the Synthesis of α-Aminophosphonates: A Review. ChemistrySelect 2021, 6, 6137–6149. [Google Scholar] [CrossRef]
- Varga, P.R.; Keglevich, G. Synthesis of α-Aminophosphonates and Related Derivatives; The Last Decade of the Kabachnik–Fields Reaction. Molecules 2021, 26, 2511. [Google Scholar] [CrossRef] [PubMed]
- Herrera, R.P. Organocatalytic Hydrophosphonylation Reaction of Carbonyl Groups. Chem. Rec. 2017, 17, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.E.; Abdel-Kariem, S.M. Methods for the synthesis of α-heterocyclic/heteroaryl-α-aminophosphonic acids and their esters. Arkivoc 2015, 6, 246–287. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Kudzin, M.H.; Drabowicz, J.; Stevens, C.V. Aminophosphonic Acids—Phosphorus Analogues of Natural Amino Acids. Part 1: Syntheses of α-Aminophosphonic Acids. Curr. Org. Chem. 2011, 15, 2015–2071. [Google Scholar] [CrossRef]
- Merino, P.; Marqués-López, E.; Herrera, R.P. Catalytic Enantioselective Hydrophosphonylation of Aldehydes and Imines. Synth. Catal. 2008, 350, 1195–1208. [Google Scholar] [CrossRef]
- Ordoñez, M.; Viveros-Ceballos, J.L.; Sayago, F.J.; Cativiela, C. Stereoselective Synthesis of α-Amino-H-phosphinic Acids and Derivatives. Synthesis 2017, 49, 987–997. [Google Scholar] [CrossRef]
- Viveros-Ceballos, J.L.; Ordoñez, M.; Sayago, F.J.; Cativiela, C. Stereoselective Synthesis of α-Amino-C-phosphinic Acids and Derivatives. Molecules 2016, 21, 1141. [Google Scholar] [CrossRef]
- Takiguchi, S.; Nishino, Y.; Inoue, K.; Ikeda, M.; Kataoka, Y.; Matsusue, K.; Nishiyama, K.; Iguchi, H. The bisphosphonate incadronate inhibits intraperitoneal dissemination in an in vivo pancreatic cancer model. Oncol. Rep. 2012, 28, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, S.; Iguchi, K.; Usui, S.; Hirano, K. Incadronate Induces Cell Detachment and Apoptosis in Prostatic PC-3 Cells. Anticancer Res. 2007, 27, 927–932. [Google Scholar] [PubMed]
- Koizumi, M.; Kobayashi, M.; Furukawa, M.; Yamashita, T.; Ogata, E. The bisphosphonate incadronate for bone metastases of breast cancer. Int. J. Clin. Oncol. 2000, 5, 241–246. [Google Scholar] [CrossRef]
- Kafarski, P.; Lejczak, B.; Forlani, G. Herbicidally active aminomethylenebisphosphonic acids. Heteroat. Chem. 2000, 11, 449–453. [Google Scholar] [CrossRef]
- Rosso, V.S.; Szajnman, S.H.; Malayil, L.; Galizzi, M.; Moreno, S.N.J.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of new 2-alkylaminoethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii targeting farnesyl diphosphate synthase. Bioorg. Med. Chem. 2011, 19, 2211–2217. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xiao, L.; Tao, J.; Srinivasan, V.; Boyce, B.F.; Ebetino, F.H.; Oyajobi, B.O.; Boeckman, R.K., Jr.; Xing, L. Synthesis of a Bone-Targeted Bortezomib with In Vivo Anti-Myeloma Effects in Mice. Pharmaceutics 2018, 10, 154. [Google Scholar] [CrossRef]
- Holub, J.; Meckel, M.; KubíčeK, V.; Rösch, F.; Hermann, P. Gallium(III) complexes of NOTA-bis (phosphonate) conjugates as PET radiotracers for bone imaging. Contrast Media Mol. Imaging 2015, 10, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Vitha, T.; KubíčeK, V.; Kotek, J.; Hermann, P.; Elst, L.V.; Muller, T.N.; Lukeš, I.; Peters, J.A. Gd(III) complex of a monophosphinate-bis(phosphonate) DOTA analogue with a high relaxivity; Lanthanide(III) complexes for imaging and radiotherapy of calcified tissues. Dalton Trans. 2009, 17, 3204–3214. [Google Scholar] [CrossRef] [PubMed]
- Zha, Z.; Wu, Z.; Choi, S.R.; Ploessl, K.; Smith, M.; Alexoff, D.; Zhu, L.; Kung, H.F. A New [68Ga]Ga-HBED-CC-Bisphosphonate as a Bone Imaging Agent. Mol. Pharm. 2020, 17, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Pospiech, M.; Owens, S.E.; Miller, D.J.; Austin-Muttitt, K.; Mullins, J.G.L.; Cronin, J.G.; Allemann, R.K.; Sheldon, I.M. Bisphosphonate inhibitors of squalene synthase protect cells against cholesterol-dependent cytolysins. FASEB J. 2021, 35, e21640. [Google Scholar] [CrossRef] [PubMed]
- Galaka, T.; Falcone, B.N.; Li, C.; Szajnman, S.H.; Moreno, S.N.J.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 1-alkylaminomethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii. Bioorg. Med. Chem. 2019, 27, 3663–3673. [Google Scholar] [CrossRef] [PubMed]
- Ghoteimi, R.; Nguyen Van, T.; Rahimova, R.; Grosjean, F.; Cros-Perrial, E.; Uttaro, J.-P.; Mathé, C.; Chaloin, L.; Jordheim, L.P.; Peyrottes, S. Synthesis of Substituted 5′-Aminoadenosine Derivatives and Evaluation of Their Inhibitory Potential toward CD73. ChemMedChem 2019, 14, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Kosikowska, P.; Bochno, M.; Macegoniuk, K.; Forlani, G.; Kafarski, P.; Berlicki, Ł. Bisphosphonic acids as effective inhibitors of Mycobacterium tuberculosis glutamine synthetase. J. Enzyme Inhib. Med. Chem. 2016, 31, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Tauro, M.; Laghezza, A.; Loiodice, F.; Agamennone, M.; Campestre, C.; Tortorella, P. Arylamino methylene bisphosphonate derivatives as bone seeking matrix metalloproteinase inhibitors. Bioorg. Med. Chem. 2013, 21, 6456–6465. [Google Scholar] [CrossRef] [PubMed]
- Rubino, M.T.; Agamennone, M.; Campestre, C.; Campiglia, P.; Cremasco, V.; Faccio, R.; Laghezza, A.; Loiodice, F.; Maggi, D.; Panza, E.; et al. Biphenyl Sulfonylamino Methyl Bisphosphonic Acids as Inhibitors of Matrix Metalloproteinases and Bone Resorption. ChemMedChem 2011, 6, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Chmielewska, E.; Mazur, Z.; Kempińska, K.; Wietrzyk, J.; Piątek, A.; Kuryszko, J.J.; Kiełbowicz, Z.; Kafarski, P. N-Arylaminomethylenebisphosphonates Bearing Fluorine Atoms: Synthesis and Antiosteoporotic Activity. Phosphorus Sulfur. 2015, 190, 2164–2172. [Google Scholar] [CrossRef]
- Jiang, Q.; Yang, L.; Hai, L.; Wu, Y. Synthesis of Melphalan-Gem-Bisphosphonate Conjugation to Bone Tumors and Study of Affinity to Hydroxyapatite In Vitro. Lett. Org. Chem. 2008, 5, 229–233. [Google Scholar] [CrossRef]
- Miszczyk, P.; Wieczorek, D.; Gałęzowska, J.; Dziuk, B.; Wietrzyk, J.; Chmielewska, E. Reaction of 3-Amino-1,2,4-Triazole with Diethyl Phosphite and Triethyl Orthoformate: Acid-Base Properties and Antiosteoporotic Activities of the Products. Molecules 2017, 22, 254. [Google Scholar] [CrossRef] [PubMed]
- Lacbay, C.M.; Waller, D.D.; Park, J.; Gómez Palou, M.; Vincent, F.; Huang, J.F.; Ta, V.; Berghuis, A.M.; Sebag, M.; Tsantrizos, Y.S. Unraveling the Prenylation–Cancer Paradox in Multiple Myeloma with Novel Geranylgeranyl Pyrophosphate Synthase (GGPPS) Inhibitors. J. Med. Chem. 2018, 61, 6904–6917. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-F.; Lacbay, C.M.; Boutin, R.; Matralis, A.N.; Park, J.; Waller, D.D.; Guan, T.L.; Sebag, M.; Tsantrizos, Y.S. Synthesis and Evaluation of Structurally Diverse C-2-Substituted Thienopyrimidine-Based Inhibitors of the Human Geranylgeranyl Pyrophosphate Synthase. J. Med. Chem. 2022, 65, 2471–2496. [Google Scholar] [CrossRef] [PubMed]
- Boutin, R.; Lee, H.-F.; Guan, T.L.; Nguyen, T.T.; Huang, J.F.; Waller, D.D.; Lu, J.; Chio, I.I.C.; Michel, R.P.; Sebag, M.; et al. Discovery and Evaluation of C6-Substituted Pyrazolopyrimidine-Based Bisphosphonate Inhibitors of the Human Geranylgeranyl Pyrophosphate Synthase and Evaluation of Their Antitumor Efficacy in Multiple Myeloma, Pancreatic Ductal Adenocarcinoma, and Colorectal Cancer. J. Med. Chem. 2023, 66, 15776–15800. [Google Scholar] [PubMed]
- Shaddy, A.A.; Kamel, A.A.; Abdou, W.M. Synthesis, Quantitative Structure—Activity Relationship, and Anti-Inflammatory Profiles of Substituted 5- and 6-N-Heterocycle Bisphosphonate Esters. Synth. Commun. 2013, 43, 236–252. [Google Scholar] [CrossRef]
- Laghezza, A.; Piemontese, L.; Brunetti, L.; Caradonna, A.; Agamennone, M.; Loiodice, F.; Tortorella, P. (2-Aminobenzothiazole)-Methyl-1,1-Bisphosphonic Acids: Targeting Matrix Metalloproteinase 13 Inhibition to the Bone. Pharmaceuticals 2021, 14, 85. [Google Scholar] [CrossRef] [PubMed]
- De Schutter, J.W.; Zaretsky, S.; Welbourn, S.; Pause, A.; Tsantrizos, Y.S. Novel bisphosphonate inhibitors of the human farnesyl pyrophosphate synthase. Bioorg. Med. Chem. Lett. 2010, 20, 5781–5786. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-S.; Park, J.; De Schutter, J.W.; Huang, J.F.; Berghuis, A.M.; Sebag, M.; Tsantrizos, Y.S. Design and Synthesis of Active Site Inhibitors of the Human Farnesyl Pyrophosphate Synthase: Apoptosis and Inhibition of ERK Phosphorylation in Multiple Myeloma Cells. J. Med. Chem. 2012, 55, 3201–3215. [Google Scholar] [CrossRef] [PubMed]
- Gałęzowska, J.; Czapor-Irzabek, H.; Chmielewska, E.; Kafarski, P.; Janeka, T. Aminobisphosphonates based on cyclohexane backbone as coordinating agents for metal ions. Thermodynamic, spectroscopic and biological studies. New J. Chem. 2018, 42, 7723–7736. [Google Scholar] [CrossRef]
- Shaik, M.S.; Nadiveedhi, M.R.; Gundluru, M.; Katike, U.; Obulam, V.S.R.; Cirandur, S.R. Efficient catalyst free green synthesis and in vitro antimicrobial, antioxidant and molecular docking studies of α-substituted aromatic/heteroaromatic aminomethylene bisphosphonates. Synth. Commun. 2021, 51, 747–764. [Google Scholar] [CrossRef]
- Tellamekala, S.; Gundluru, M.; Sudileti, M.; Sarva, S.; Putta, C.R.K.; Cirandur, S.R. Green one-pot synthesis of N-bisphosphonates as antimicrobial and antioxidant agents. Monatsh. Chem. 2020, 151, 251–260. [Google Scholar] [CrossRef]
- Mohan, G.; Santhisudha, S.; Reddy, N.M.; Sreekanth, T.; Murali, S.; Reddy, C.S. Nano ZnO catalyzed green synthesis and cytotoxic assay of pyridinyl and pyrimidinyl bisphosphonates. Monatsh. Chem. 2017, 148, 1843–1851. [Google Scholar] [CrossRef]
- Amira, A.; K’tir, H.; Aouf, Z.; Khaldi, T.; Bentoumi, H.; Khattabi, L.; Zerrouki, R.; Ibrahim-Ouali, M.; Aouf, N.-E. One-Pot Microwave-Assisted Synthesis, in Vitro Anti-inflammatory Evaluation and Computer-Aided Molecular Design of Novel Sulfamide-Containing Bisphosphonates Derivatives. ChemitrySelect 2022, 7, e202201889. [Google Scholar] [CrossRef]
- Gehrke, N.R.; Feng, D.; Ali, M.A.; Maalouf, M.A.; Holstein, S.A.; Weimer, D.F. α-Amino bisphosphonate triazoles serve as GGDPS inhibitors. Bioorg. Med. Chem. Lett. 2024, 102, 129659. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.M.A.; El-Hussieny, M.; El-Sayed, N.F.; Fouad, M.A.; Ewies, F.E.; Ezzat, M.A.F. Synthesis and characterization of new tetrakisphosphonic acid derivatives as FPPS inhibitors and evaluation of their anti-osteoclastogenic potential for prevention of osteoporosis. Med. Chem. Res. 2024, 33, 1167–1177. [Google Scholar] [CrossRef]
- Goldeman, W.; Nasulewicz-Goldeman, A. Synthesis and biological evaluation of aminomethylidenebisphosphonic derivatives of β-arylethylamines. Tetrahedron 2015, 71, 3282–3289. [Google Scholar] [CrossRef]
- Nasulewicz-Goldeman, A.; Goldeman, W.; Mrówczyńska, E.; Wietrzyk, J. Biological effects of aromatic bis[aminomethylidenebis(phosphonic)] acids in osteoclast precursors in vitro. Chem. Biol. Drug Des. 2019, 94, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.N.; Li, C.; Storey, M.; Falcone, B.N.; Szajnman, S.H.; Bonesi, S.M.; Docampo, R.; Moreno, S.N.J.; Rodriguez, J.B. Activity of Fluorine-Containing Analogues of WC-9 and Structurally Related Analogues against Two Intracellular Parasites: Trypanosoma cruzi and Toxoplasma gondii. ChemMedChem 2016, 11, 2690–2702. [Google Scholar] [CrossRef] [PubMed]
- Grigor’ev, I.A.; Morozova, D.A.; Svyatchenkoc, V.A.; Kiselevc, N.N.; Loktevc, V.B.; Luk’yanetse, E.A.; Vorozhtsove, G.N. Synthesis and Study of Antitumor Activity of Tetraethyl 2-(2′,2′,6′,6′-Tetramethylpiperidin-4′-ylamino)ethylidene-1,1-bisphosphonate. Dokl. Chem. 2014, 457, 137–140. [Google Scholar] [CrossRef]
- Granchi, D.; Scarso, A.; Bianchini, G.; Chiminazzo, A.; Minto, A.; Sgarbossa, P.; Michelin, R.A.; Di Pompo, G.; Avnet, S.; Strukul, G. Low toxicity and unprecedented anti-osteoclast activity of a simple sulfur-containing gem-bisphosphonate: A comparative study. Eur. J. Med. Chem. 2013, 65, 448–455. [Google Scholar] [CrossRef]
- Wu, X.; Yang, Z.; Song, C.; Bu, M.; Li, W.; Duan, J.; Yang, G.-F.; Zhang, A. Hydroxamate-Containing Bisphosphonates as Fosmidomycin Analogues: Design, Synthesis, and Proherbicide Activity. J. Agric. Food Chem. 2024, 72, 7684–7693. [Google Scholar] [CrossRef] [PubMed]
- Galaka, T.; Casal, M.F.; Storey, M.; Li, C.; Chao, M.N.; Szajnman, S.H.; Docampo, R.; Moreno, S.N.J.; Rodriguez, J.B. Antiparasitic Activity of Sulfur- and Fluorine-Containing Bisphosphonates against Trypanosomatids and Apicomplexan Parasites. Molecules 2017, 22, 82. [Google Scholar] [CrossRef] [PubMed]
- Makarov, M.V.; Leonova, E.S.; Rybalkina, E.Y.; Khrustalev, V.N.; Shepel, N.E.; Röschenthaler, G.-V.; Timofeeva, T.V.; Odinets, I.L. Methylenebisphosphonates with Dienone Pharmacophore: Synthesis, Structure, Antitumor and Fluorescent Properties. Arch. Pharm. Chem. Life Sci. 2012, 345, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Sankala, E.; Weisell, J.M.; Huhtala, T.; Närvänen, A.T.O.; Vepsäläinen, J.J. Synthesis of novel bisphosphonate polyamine conjugates and their affinity to hydroxyapatite. Arkivoc 2012, 4, 233–241. [Google Scholar] [CrossRef]
- Shevchuk, M.V.; Sotiropoulos, J.-M.; Miqueu, K.; Romanenko, V.D.; Kukhar, V.P. Tetrakis(trimethylsilyl) Ethenylidene-1,1-bisphosphonate: A Mild and Convenient Michael Acceptor for the Synthesis of 2-Aminoethylidene-1,1-bisphosphonic Acids and Their Potassium Salts. Synlett 2011, 2011, 1370–1374. [Google Scholar]
- Abdou, W.M.; Kamel, A.A.; Khidre, R.E.; Geronikaki, A.; Ekonomopoulou, M.T. Synthesis of 5- and 6-N-heterocyclic Methylenebisphosphonate Derivatives and Evaluation of their Cytogenetic Activity in Normal Human Lymphocyte Cultures. Chem. Biol. Drug Des. 2012, 79, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.-Y.; Li, Q.-H.; Tao, H.-Y.; Wang, C.-J. A Facile Cu(I)/TF-BiphamPhos-Catalyzed Asymmetric Approach to Unnatural α-Amino Acid Derivatives Containing gem-Bisphosphonates. J. Am. Chem. Soc. 2011, 133, 11757–11765. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Tada, A.; Tokoro, Y.; Fukuzawa, S.I. Silver-catalyzed asymmetric Michael addition of azomethine ylide to arylidene diphosphonates using ThioClickFerrophos ligand. Tetrahedron Lett. 2015, 56, 2251–2253. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I.; Kolodiazhna, A.O. Stereoselective Syntheses of Organophosphorus Compounds. Symmetry 2024, 16, 342. [Google Scholar] [CrossRef]
- Kaboudin, B.; Daliri, P.; Faghih, S.; Esfandiari, H. Hydroxy- and Amino-Phosphonates and -Bisphosphonates: Synthetic Methods and Their Biological Applications. Front. Chem. 2022, 10, 890696. [Google Scholar] [CrossRef] [PubMed]
- Popov, K.; Oshchepkov, M.; Tkachenko, S.; Sergienko, V.; Oshchepkov, A. Bisphosphonates: Synthesis, structures, properties, medical and industrial applications. J. Mol. Liq. 2022, 351, 118619. [Google Scholar] [CrossRef]
- Romanenko, V.D. α-Heteroatom-substituted gem-Bisphosphonates: Advances in the Synthesis and Prospects for Biomedical Application. Curr. Org. Chem. 2019, 23, 530–615. [Google Scholar] [CrossRef]
- Romanenko, V.D.; Kukhar, V.P. 1-Amino-1,1-bisphosphonates. Fundamental syntheses and new developments. Arkivoc 2012, 4, 127–166. [Google Scholar] [CrossRef]
- Krutikov, V.I.; Erkin, A.V.; Pautov, P.A.; Zolotukhina, M.M. Heteryl- and Arylaminomethylenebisphosphonates: Synthesis and Biologic Activity. Russ. J. Gen. Chem. 2003, 73, 187–191. [Google Scholar] [CrossRef]
- Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. Insight into the mechanism of three component condensation leading to aminomethylenebisphosphonates. J. Organomet. Chem. 2009, 694, 3806–3813. [Google Scholar] [CrossRef]
- Bochno, M.; Berlicki, Ł. A three-component synthesis of aminomethylenebis-H-phosphinates. Tetrahedron Lett. 2014, 55, 219–223. [Google Scholar] [CrossRef]
- Viveros-Ceballos, J.L.; Matías-Valdez, L.A.; Sayago, F.J.; Cativiela, C.; Ordoñez, M. New approaches towards the synthesis of 1,2,3,4-tetrahydro isoquinoline-3-phosphonic acid (TicP). Amino Acids 2021, 53, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Goswami, D.; Chakravarty, R.; Mohammed, K.S.; Sarma, H.D.; Dash, A. Syntheses and evaluation of 68Ga- and 153Sm-labeled DOTA-conjugated bisphosphonate ligand for potential use in detection of skeletal metastases and management of pain arising from skeletal metastases. Chem. Biol. Drug Des. 2018, 92, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Ehrick, R.S.; Capaccio, M.; Puleo, D.A.; Bachas, L.G. Ligand-Modified Aminobisphosphonate for Linking Proteins to Hydroxyapatite and Bone Surface. Bioconj. Chem. 2008, 19, 315–321. [Google Scholar] [CrossRef]
- Leung, C.Y.; Park, J.; De Schutter, J.W.; Sebag, M.; Berghuis, A.M.; Tsantrizos, Y.S. Thienopyrimidine Bisphosphonate (ThPBP) Inhibitors of the Human Farnesyl Pyrophosphate Synthase: Optimization and Characterization of the Mode of Inhibition. J. Med. Chem. 2013, 56, 7939–7950. [Google Scholar] [CrossRef] [PubMed]
- Brel, V.K. Synthesis of gem-bisphosphonates with (3-aryl-4,5-dihydroisoxazol-5-yl)methylamino moiety. Mendeleev Commun. 2015, 25, 234–235. [Google Scholar] [CrossRef]
- Petruczynik, P.; Kafarski, P.; Psurski, M.; Wietrzyk, J.; Kielbowicz, Z.; Kuryszko, J.; Chmielewska, E. Three-Component Reaction of Diamines with Triethyl Orthoformate and Diethyl Phosphite and Anti-Proliferative and Antiosteoporotic Activities of the Products. Molecules 2020, 25, 1424. [Google Scholar] [CrossRef] [PubMed]
- Bálint, E.; Tajti, A.; Dzielak, A.; Hägele, G.; Keglevich, G. Microwave-assisted synthesis of (aminomethylene)bisphosphine oxides and (aminomethylene)bisphosphonates by a three-component condensation. Beilstein J. Org. Chem. 2016, 12, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Minaeva, L.I.; Patrikeeva, L.S.; Kabachnik, M.M.; Beletskaya, I.P.; Orlinson, B.S.; Novakov, I.A. Synthesis of novel aminomethylenebisphosphonates and bisphosphonic acids, containing adamantyl fragment. Heteroatom Chem. 2011, 22, 55–58. [Google Scholar] [CrossRef]
- Sudileti, M.; Nagaripati, S.; Gundluru, M.; Chintha, V.; Aita, S.; Wudayagiri, R.; Chamarthi, N.; Cirandur, S.R. rGO-SO3H Catalysed Green Synthesis of Fluoro-Substituted Aminomethylene Bisphosphonates and their Anticancer, Molecular Docking studies. ChemitrySelect 2019, 4, 13006–13011. [Google Scholar] [CrossRef]
- Manjula, G.; Kalla, R.M.N.; Reddy, A.E.; Almutairi, T.M.; Lee, J. Ultrasonic-promoted synthesis of amino methylene bisphosphonates by sulfated choline ionic liquid. Res. Chem. Intermediat. 2025, 51, 1357–1370. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Synthesis of new functionalized aryl and pyridyl aminomethylenebisphosphonic acids and their derivatives via silicon-assisted methodology. J. Organomet. Chem. 2020, 912, 121177. [Google Scholar] [CrossRef]
- Reddy, S.S.; Kalla, R.M.N.; Varyambath, A.; Kim, I. Sulfonic acid functionalized hyper-cross-linked polymer: An efficient heterogeneous acid catalyst for the synthesis of N-containing bisphosphonates. Catal. Commun. 2019, 126, 15–20. [Google Scholar] [CrossRef]
- Kunda, U.M.R.; Balam, S.K.; Nemallapudi, B.R.; Chereddy, S.S.; Nayak, S.K.; Cirandur, S.R. Facile Synthesis, Antioxidant and Antimicrobial Activity of Amino Methylene Bisphosphonates. Chem. Pharm. Bull. 2012, 60, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Shahvelayati, A.S.; Tanha, A. A Review of Ionic Liquids: From Solvent Applications to Template-Assisted Synthesis of Metal Oxide Nanoparticles. Appl. Organomet. Chem. 2025, 39, e70026. [Google Scholar] [CrossRef]
- Kaur, G.; Kumar, H.; Singla, M. Diverse applications of ionic liquids: A comprehensive review. J. Mol. Liq. 2022, 351, 118556. [Google Scholar] [CrossRef]
- Kianfar, E.; Mafi, S. Ionic Liquids: Properties, Application, and Synthesis. Fine Chem. Eng. 2020, 2, 21–29. [Google Scholar] [CrossRef]
- Lei, Z.; Chen, B.; Koo, Y.-M.; MacFarlane, D.R. Introduction: Ionic Liquids. Chem. Rev. 2017, 117, 6633–6635. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.; Kalla, R.M.N.; Dong, L.S.; Jeong, T.J. Di-n-butyl ammonium chlorosulfonate as a highly efficient and recyclable ionic liquid for the synthesis of N-containing bisphosphonates. Catal. Commun. 2015, 61, 102–106. [Google Scholar] [CrossRef]
- Poola, S.; Gundluru, M.; Nadiveedhi, M.R.; Saddala, M.S.; Ptsrk, P.R.; Cirandur, S.R. Nano silver particles catalyzed synthesis, molecular docking and bioactivity of α-thiazolyl aminomethylene bisphosphonates. Phosphorus Sulfur. 2020, 195, 409–420. [Google Scholar] [CrossRef]
- Silva Prasad, S.; Jayaprakash, S.H.; Syamasundar, C.; Sreelakshmi, P.; Bhuvaneswar, C.; Bhaskar, B.V.; Rajendra, W.; Nayak, S.K.; Cirandur, S.R. Tween 20-/H2O Promoted Green Synthesis, Computational and Antibacterial Activity of Amino Acid Substituted Methylene Bisphosphonates. Phosphorus Sulfur. 2015, 190, 2040–2050. [Google Scholar] [CrossRef]
- Kuźnik, A.; Mazurkiewicz, R.; Grymel, M.; Zielińska, K.; Adamek, J.; Chmielewska, E.; Bochno, M.; Kubica, S. A new method for the synthesis of α-aminoalkylidenebisphosphonates and their asymmetric phosphonyl-phosphinyl and phosphonyl-phosphinoyl analogues. Beilstein J. Org. Chem. 2015, 11, 1418–1424. [Google Scholar] [CrossRef] [PubMed]
- Kuźnik, A.; Mazurkiewicz, R.; Zięba, M.; Erfurt, K. 1-(N-Acylamino)-1-triphenylphosphoniumalkylphosphonates: General synthesis and prospects for further synthetic applications. Tettrahedron Lett. 2018, 59, 3307–3320. [Google Scholar] [CrossRef]
- Kuźnik, A.; Kozicka, D.; Październiok-Holewa, A.; Dąbek, A.; Juszczak, K.; Sokołowska, G.; Erfur, K. A method for the synthesis of unsymmetric bisphosphoric analogs of α-amino acids. RSC Adv. 2023, 13, 18908. [Google Scholar] [CrossRef] [PubMed]
- Kuźnik, A.; Kozicka, D.; Hawranek, W.; Socha, K.; Erfurt, K. One-Pot and Catalyst-Free Transformation of N-Protected 1-Amino-1-Ethoxyalkylphosphonates into Bisphosphonic Analogs of Protein and Non-Protein α-Amino Acids. Molecules 2022, 27, 3571. [Google Scholar] [CrossRef] [PubMed]
- Kozicka, D.; Krzesniak, M.; Grymel, M.; Adamek, J.; Łasut-Szyszka, B.; Cichoń, T.; Kuznik, A. Ultrasound-assisted synthesis of new bisphosphonate–betulin conjugates and preliminary evaluation of their cytotoxic activity. RSC Adv. 2025, 15, 4086–4094. [Google Scholar] [CrossRef] [PubMed]
- Ayadi, N.; Descamps, A.; Legigan, T.; Dussart-Gautheret, J.; Monteil, M.; Migianu-Griffoni, E.; Ayed, T.B.; Deschamp, J.; Lecouvey, M. Synthesis of Aminobisphosphinates through a Cascade Reaction between Hypophosphorous Acid and Bis(trimethylsilyl)imidates Mediated by ZnI2. Molecules 2023, 28, 6226. [Google Scholar] [CrossRef] [PubMed]
- Prishchenko, A.A.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Averochkin, G.M.; Petrosyan, V.S. Synthesis of Substituted N-Formylaminomethylenediphosphonates and Their Derivatives. Heteroatom Chem. 2015, 26, 405–410. [Google Scholar] [CrossRef]
- Kolotilo, N.V.; Onys’ko, P.P. Cycloaddition and chlorotropic transformations in reactions of dichloromethylenetrifluoroacetamide with phosphites. Russ. J. Gen. Chem. 2016, 86, 534–538. [Google Scholar] [CrossRef]
- Cheviet, T.; Peyrottes, S. Synthesis of Aminomethylene-gem-bisphosphonates Containing an Aziridine Motif: Studies of the Reaction Scope and Insight into the Mechanism. J. Org. Chem. 2021, 86, 3107–3119. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Andreu, M.M.; Sayago, F.J.; Cativiela, C. An Improved Synthesis of the Antibiotic Dehydrophos. Eur. J. Org. Chem. 2018, 2018, 3965–3973. [Google Scholar] [CrossRef]
- Makris, G.; Tseligka, E.D.; Pirmettis, I.; Papadopoulos, M.S.; Vizirianakis, I.S.; Papagiannopoulou, D. Development and Pharmacological Evaluation of New Bone-Targeted 99mTc-Radiolabeled Bisphosphonates. Mol. Pharm. 2016, 13, 2301–2317. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.S.; Chukanov, N.V.; Grigor’ev, I.A. New Amino-Bisphosphonate Building Blocks in the Synthesis of Bisphosphonic Derivatives Based on Lead Compounds. Phosphorus Sulfur. 2015, 190, 1201–1212. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Tris(trimethylsilyl) phosphite as key synthon for convenient synthesis of new organosilicon(phosphorus)-containing N-heterocycles. J. Organomet. Chem. 2018, 867, 149–154. [Google Scholar] [CrossRef]
- Prishcenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novokova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Organosilicon based synthesis of new functionalized aminomethylenediphosphonates with moieties of amino acids. J. Organomet. Chem. 2018, 871, 36–39. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novikova, E.P.; Livantsova, L.I.; Petrosyan, V.S. A convenient catalytic silicon-assisted route towards new non-proteinogenic amino acids with methylenebisphosphonic acids moieties. J. Organomet. Chem. 2021, 949, 121950. [Google Scholar] [CrossRef]
- Wang, A.E.; Chang, Z.; Sun, W.-T.; Huang, P.-Q. General and Chemoselective Bisphosphonylation of Secondary and Tertiary Amides. Org. Lett. 2015, 17, 732–735. [Google Scholar] [CrossRef]
- Chen, M.-E.; Chen, X.-W.; Hu, Y.-H.; Ye, R.; Lv, J.-W.; Li, J.-W.; Zhnag, F.-M. Recent advances of Ritter reaction and its synthetic applications. Org. Chem. Front. 2021, 8, 4623–4664. [Google Scholar] [CrossRef]
- Guérinot, A.; Reymond, S.; Cossy, J. Ritter Reaction: Recent Catalytic Developments. Eur. J. Org. Chem. 2012, 2012, 19–28. [Google Scholar] [CrossRef]
- Hong, Y.-C.; Ye, J.-L.; Huang, P.-Q. One-Pot Synthesis of α-Amino Bisphosphonates from Nitriles via Tf2O/HC(OR)3-Mediated Interrupted Ritter-Type Reaction. J. Org. Chem. 2022, 87, 9044–9055. [Google Scholar] [CrossRef] [PubMed]
- Kaboudin, B.; Esfandiari, H.; Moradi, A.; Kazemi, F.; Aoyama, H. ZnCl2-Mediated Double Addition of Dialkylphosphite to Nitriles for the Synthesis of 1-Aminobisphosphonates. J. Org. Chem. 2019, 84, 14943–14948. [Google Scholar] [CrossRef] [PubMed]
- Islas, R.S.; García, J.J. Nickel-Catalyzed Hydrophosphonylation and Hydrogenation of Aromatic Nitriles Assisted by Lewis Acid. Chem. Cat. Chem. 2019, 14, 1337–1345. [Google Scholar] [CrossRef]
- Midrier, C.; Lantsoght, M.; Volle, J.-N.; Pirat, J.-L.; Virieux, D.; Stevens, C.S. Hydrophosphonylation of alkenes or nitriles by double radical transfer mediated by titanocene/propylene oxide. Tetrahedron Lett. 2011, 52, 6693–6696. [Google Scholar] [CrossRef]
- Goldeman, W.; Kluczyński, A.; Soroka, M. The preparation of N-substituted aminomethylidenebisphosphonates and their tetraalkyl esters via reaction of isonitriles with trialkyl phosphites and hydrogen chloride. Part 1. Tetrahedron Lett. 2012, 53, 5290–5292. [Google Scholar] [CrossRef]
- Goldeman, W.; Nasulewicz-Goldeman, A. Synthesis and antiproliferative activity of aromatic and aliphatic bis[aminomethylidene(bisphosphonic)] acids. Bioorg. Med. Chem. Lett. 2014, 24, 3475–3479. [Google Scholar] [CrossRef] [PubMed]
- Scarpi, D.; Visi, S.; Occhiato, E.G. Recent Advances in the α-Hydrazination (α-Amination) of Carbonyl Compounds. Eur. J. Org. Chem. 2025, 28, e202500049. [Google Scholar] [CrossRef]
- Lauren, G.; O’Neil, L.G.; Bower, J.E. Electrophilic Aminating Agents in Total Synthesis. Angew. Chem. Int. Ed. 2021, 60, 25640–25666. [Google Scholar]
- Erdik, E. Electrophilic α-amination of carbonyl compounds. Tetrahedron 2004, 60, 8747–8782. [Google Scholar] [CrossRef]
- Tanaka, K.S.E.; Dietrich, E.; Ciblat, S.; Métayer, C.; Arhin, F.F.; Sarmiento, I.; Moeck, G.; Parr, T.R., Jr.; Far, A.R. Synthesis and in vitro evaluation of bisphosphonated glycopeptide prodrugs for the treatment of osteomyelitis. Bioorg. Med. Chem. Lett. 2010, 20, 1355–1359. [Google Scholar] [CrossRef]
- Lv, M.; Wang, M.; Lu, K.; Peng, L.; Zhao, Y. An efficient synthesis of 2-Aminoethylidene-1,1-Bisphosphonates derivatives via Michael addition reaction. Phosphorus Sulfur. 2018, 193, 149–154. [Google Scholar] [CrossRef]
- Vagapova, L.I.; Makhrus, E.M.; Burilov, A.R.; Pudovik, M.A. Synthesis of new diarylmethanes on the basis of resorcinol derivatives and amino acetals containing an aminoethylidenebisphosphonate fragment. Russ. J. Gen. Chem. 2017, 87, 2103–2106. [Google Scholar] [CrossRef]
- Liu, S.; Bi, W.; Li, X.; Chen, X.; Qu, L.; Zhao, Y. A Practical Method to Synthesize 1,2,3-Triazole-Amino-Bisphosphonate Derivatives. Phosphorus Sulfur. 2015, 190, 1735–1742. [Google Scholar] [CrossRef]
- Chen, C.; Li, Y.; Yu, X.; Jiang, Q.; Yang, Q.; Xu, X.; Qian, Z. Bone-targeting melphalan prodrug with tumor-microenvironment sensitivity: Synthesis, in vitro and in vivo evaluation. Chin. Chem. Lett. 2018, 29, 1609–1612. [Google Scholar] [CrossRef]
- Chiminazzo, A.; Borsato, G.; Favero, A.; Fabbro, C.; McKenna, C.E.; Carbonare, L.G.D.; Valenti, M.T.; Fabrils, F.; Scarso, A. Diketopyrrolopyrrole Bis-Phosphonate Conjugate: A New Fluorescent Probe for In Vitro Bone Imaging. Chem. Eur. J. 2019, 25, 3617–3626. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, X.; Yuan, J.; Qu, L.; Wang, S.; Shi, H.; Tang, Y.; Duan, L. Simple, efficient one-pot method for synthesis of novel N-attached 1,2,3-triazole containing bisphosphonates. Tetrahedron 2013, 69, 4047–4052. [Google Scholar] [CrossRef]
- Fairweather, A.E.R.; Goetz, D.B.; Schroeder, C.M.; Bhuiyan, N.H.; Varney, M.L.; Wiemer, D.F.; Holstein, S.A. Impact of α-modifications on the activity of triazole bisphosphonates as geranylgeranyl diphosphate synthase inhibitors. Bioorg. Med. Chem. 2021, 44, 116307. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Huang, R.; Liao, Z.; Pan, Y.; Gou, S.; Wang, H. Synthesis and pharmacological evaluation of dehydroabietic acid thiourea derivatives containing bisphosphonate moiety as an inducer of apoptosis. Eur. J. Med. Chem. 2016, 108, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Gritzalis, D.; Park, J.; Chiu, W.; Cho, H.; Lin, Y.; De Schutter, J.W.; Lacbay, C.M.; Zielinski, M.; Berghuis, A.M.; Tsantrizos, Y.S. Probing the molecular and structural elements of ligands binding to the active site versus an allosteric pocket of the human farnesyl pyrophosphate synthase. Bioorg. Med. Chem. Lett. 2015, 25, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Vagapova, L.I.; Smolobochkin, A.V.; Gazizov, A.S.; Burilov, A.R.; Bogdanov, A.A.; Pudovik, M.A. Synthesis of new phosphorylated analogs of nucleotides containing adenine and ethylidene-1,1-bisphosphoryl moieties. Russ. J. Gen. Chem. 2017, 87, 2119–2121. [Google Scholar] [CrossRef]
- Vagapova, L.I.; Smolobochkin, A.V.; Gazizov, A.S.; Burilov, A.R.; Bogdanov, A.A.; Pudovik, M.A.; Sinyashin, O.G. Synthesis of new nucleoside analogs containing amino bisphosphonic groups. Russ. J. Org. Chem. 2016, 52, 1335–1338. [Google Scholar] [CrossRef]
- Ye, Y.; Guan, J.; Chen, X.; Yu, Y.; Xu, Z.; Zeng, S.; Wang, Z.; Wang, B.; Jiao, Q.; Zhu, H. A new fluorescently labeled bisphosphonate for theranostics in tumor bone metastasis. Talanta 2021, 235, 122796. [Google Scholar] [CrossRef] [PubMed]
- Massarenti, C.; Bortolini, O.; Fantin, G.; Cristofaro, D.; Ragno, D.; Perrone, D.; Marchesi, E.; Toniolo, G.; Massi, A. Fluorous-tag assisted synthesis of bile acid–bisphosphonate conjugates via orthogonal click reactions: An access to potential anti-resorption bone drugs. Org. Biomol. Chem. 2017, 15, 4907–4920. [Google Scholar] [CrossRef] [PubMed]
- Sayed, G.M.S.; Tessmar, J.K.V. Heterobifunctional Poly(ethylene glycol) Derivatives for the Surface Modification of Gold Nanoparticles Toward Bone Mineral Targeting. Macromol. Biosci. 2012, 12, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Guven, M.N.; Altuncu, M.S.; Bal, T.; Oran, D.C.; Gulyuz, U.; Kizilel, S.; Okay, O.; Avci, D. Bisphosphonic Acid-Functionalized Cross-Linkers to Tailor Hydrogel Properties for Biomedical Applications. ACS Omega 2018, 3, 8638–8647. [Google Scholar] [CrossRef] [PubMed]
- Guven, M.N.; Altuncu, M.S.; Duman, F.D.; Eren, T.M.; Acar, H.Y.; Avci, D. Bisphosphonate-functionalized poly(β-amino ester) network polymers. J. Biomed. Mater. Res. 2017, 105, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Bekheit, M.S.; Barghash, R.F.; Abdou, W.M. Computer-aided design, synthesis, and biological studies of anticological nitrogen-containing tetraphosphonic acids against melanoma. Monatsh. Chem. 2018, 149, 1481–1491. [Google Scholar] [CrossRef]
- Abdou, W.M.; Ganoub, N.A.; Ismail, M.A.H.; Sabry, E.; Barghash, R.F.; Geronikaki, A. Developing efficient protocols for synthesis, antiosteoarthritic, antiinflammatory assessments and docking studies of nitrogen-containing bisphosphonate derivatives. Arab. J. Chem. 2017, 10, 1084–1097. [Google Scholar] [CrossRef]
- Dzięgielewski, M.; Hejmanowska, J.; Albrecht, Ł. A Convenient Approach to a Novel Group of Quaternary Amino Acids Containing a Geminal Bisphosphonate Moiety. Synthesis 2014, 46, 3233–3238. [Google Scholar] [CrossRef]
- Chmielewska, E.; Miszczyk, P.; Kozlowska, J.; Prokopowicz, M.; Mlynarz, P.; Kafarski, P. Reaction of benzolactams with triethyl phosphite prompted by phosphoryl chloride affords benzoannulated monophosphonates instead of expected bisphosphonates. J. Organomet. Chem. 2015, 785, 84–91. [Google Scholar] [CrossRef]
- Arguëllo-Velasco, R.O.; Dziuk, B.; Zarychta, B.; Ordoñez, M.; Kafarski, P. Reactions of Piperazin-2-one, Morpholin-3-one, and Thiomorpholin-3-one with Triethyl Phosphite Prompted by Phosphoryl Chloride: Scope and Limitations. ACS Omega 2019, 4, 9056–9064. [Google Scholar] [CrossRef] [PubMed]
- Kachkovskyi, G.O.; Kolodiazhnyi, O.I. Synthesis of the Phosphonoanalogue of Benzo[c]pyroglutamic Acid. Phosphorus Sulfur. 2010, 185, 2441–2448. [Google Scholar] [CrossRef]
- Zemlianoi, V.N.; Chernega, A.; Gutov, A.V.; Kolodiazhna, O.; Kolodiazhnyi, O.I. Synthesis of 3,3-Bis(diethylphosphono)-1-(3H)-isobenzofuranone and Its Chemical Properties. Phosphorus Sulfur. 2011, 186, 481–488. [Google Scholar] [CrossRef]
- Zemilanoi, V.N.; Kolodyazhnyi, O.I. Synthesis of tetraethyl (2-benzyl-3-oxoisoindolyl-1,1-diyl)-bisphosphonate and its properties. Russ. J. General Chem. 2011, 81, 1105–1110. [Google Scholar] [CrossRef]
- Kamel, A.A.; Geronikaki, A.; Abdou, W.M. Inhibitory effect of novel S,N-bisphosphonates on some carcinoma cell lines, osteoarthritis, and chronic inflammation. Eur. J. Med. Chem. 2012, 51, 239–249. [Google Scholar] [CrossRef] [PubMed]
- De Luca, L.; Chiminazzo, A.; Sperni, L.; Strukul, G.; Scarso, A. Pyrrolidine-Containing Bisphosphonates as Potential Anti-Resorption Bone Drugs. Chem. Eur. J. 2017, 23, 3474–3478. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Casal, M.; Barboza, A.P.; Szajnman, S.H.; Rodriguez, J.B. 1,3-Dipolar Cycloadditions of the Versatile Intermediate Tetraethyl Vinylidenebisphosphonate. Synthesis 2013, 45, 2397–2404. [Google Scholar] [CrossRef]



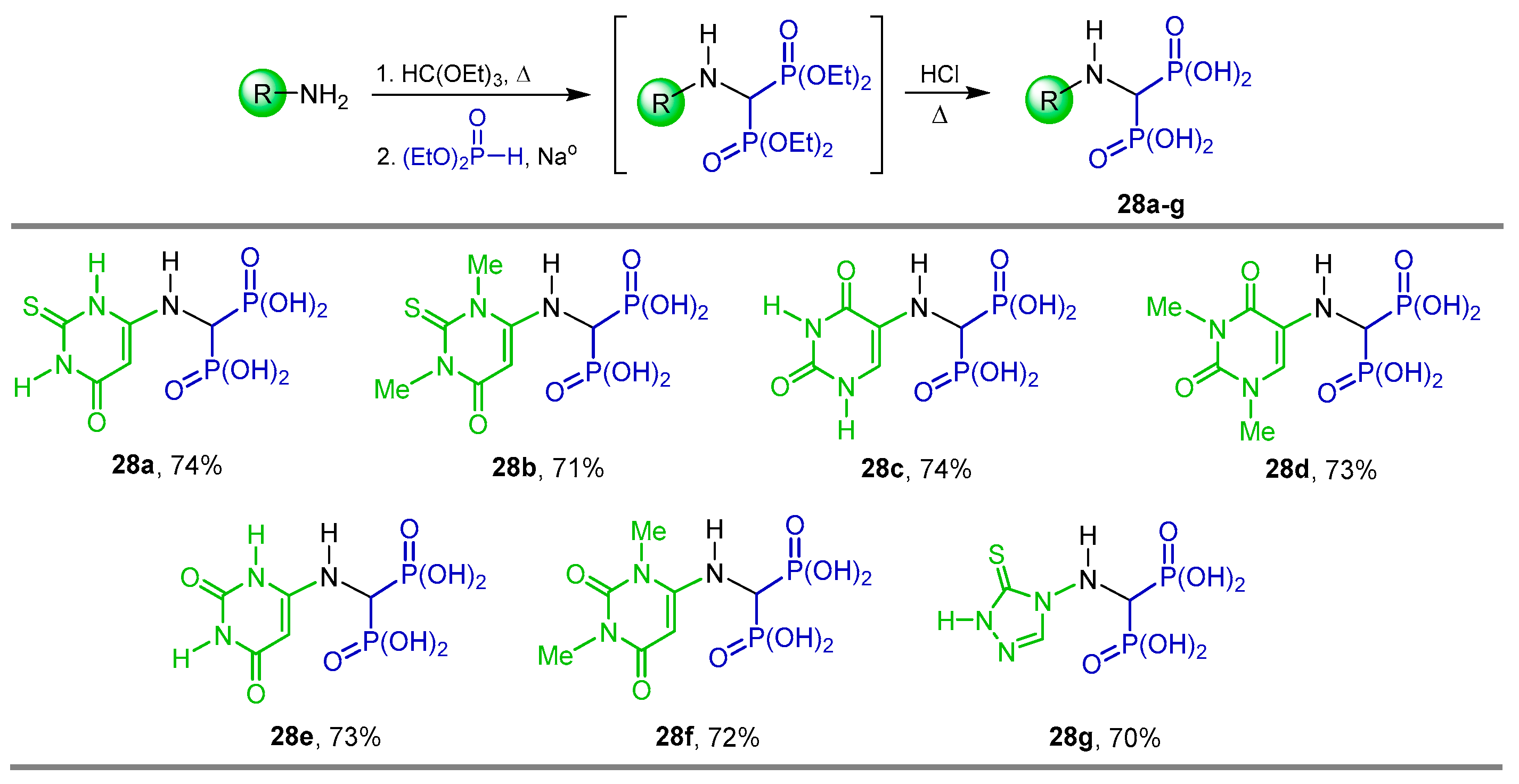
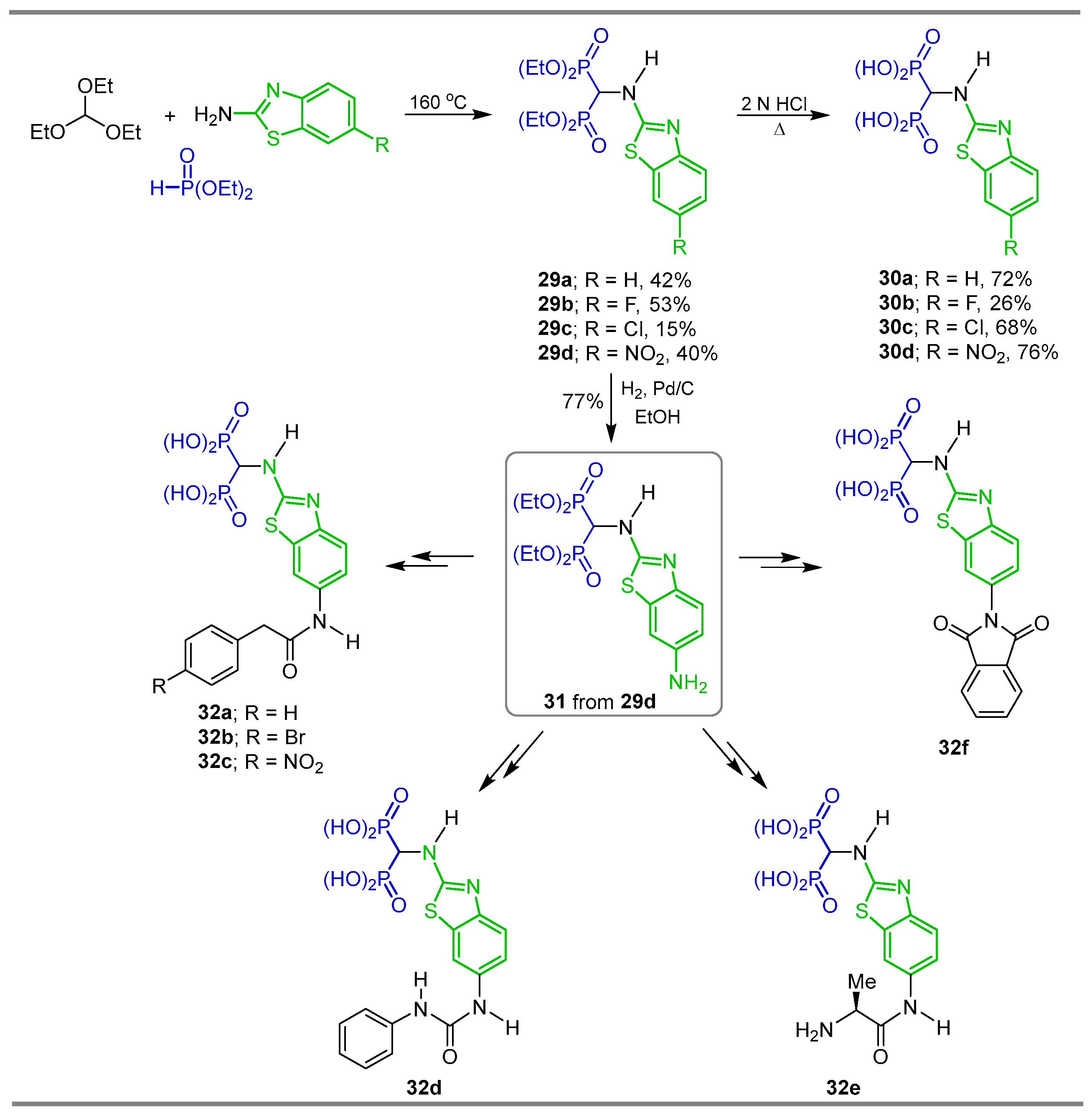
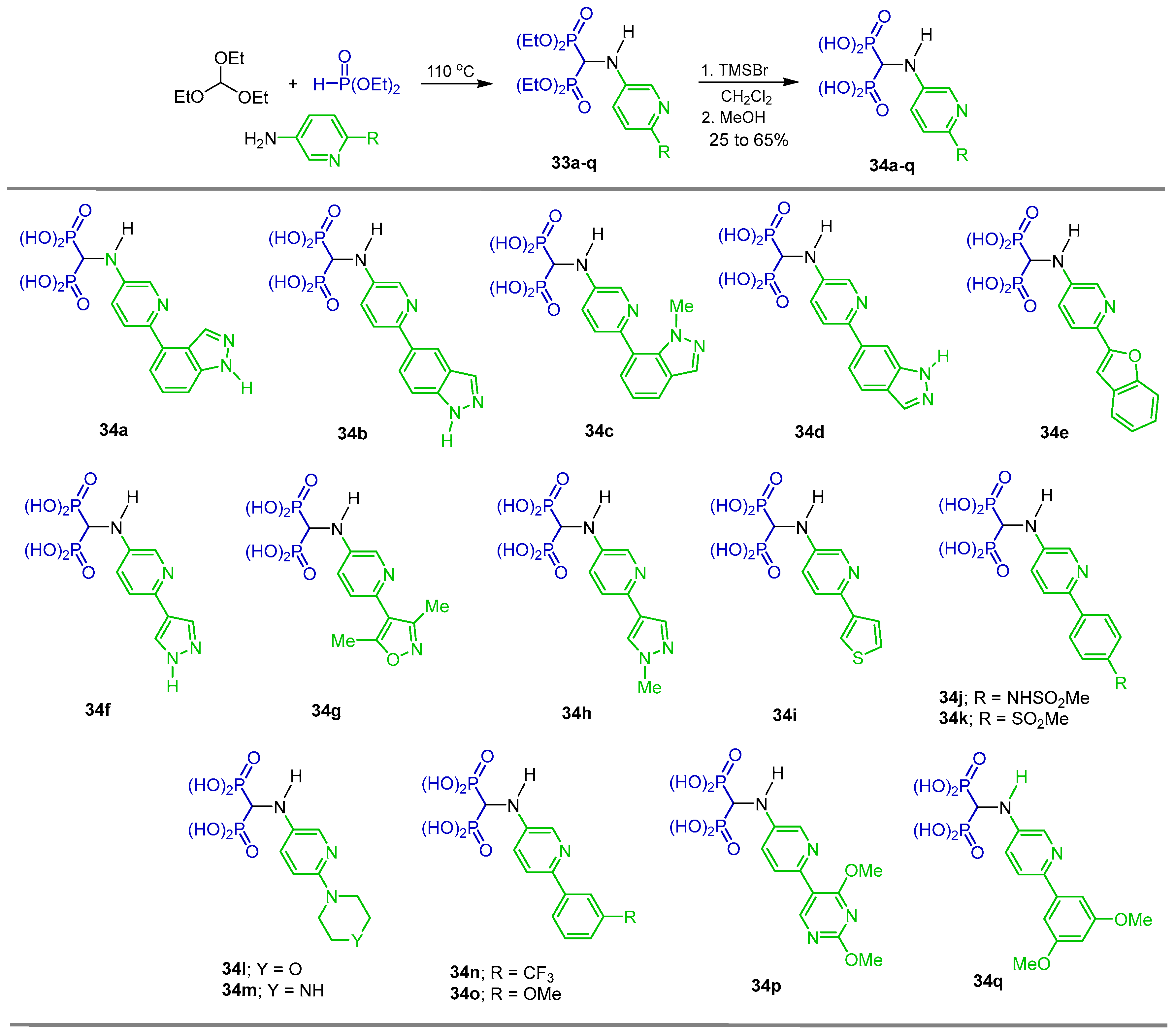
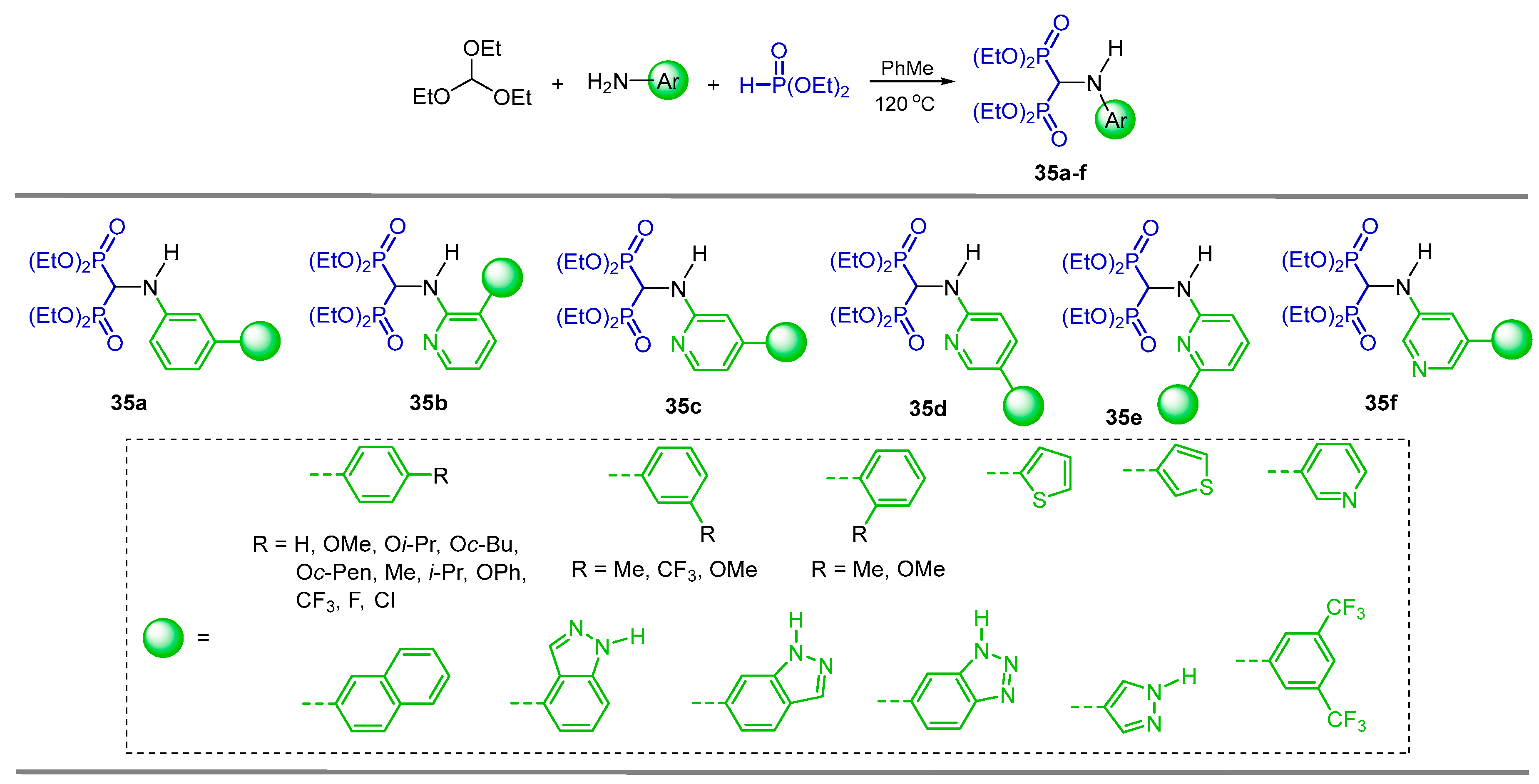
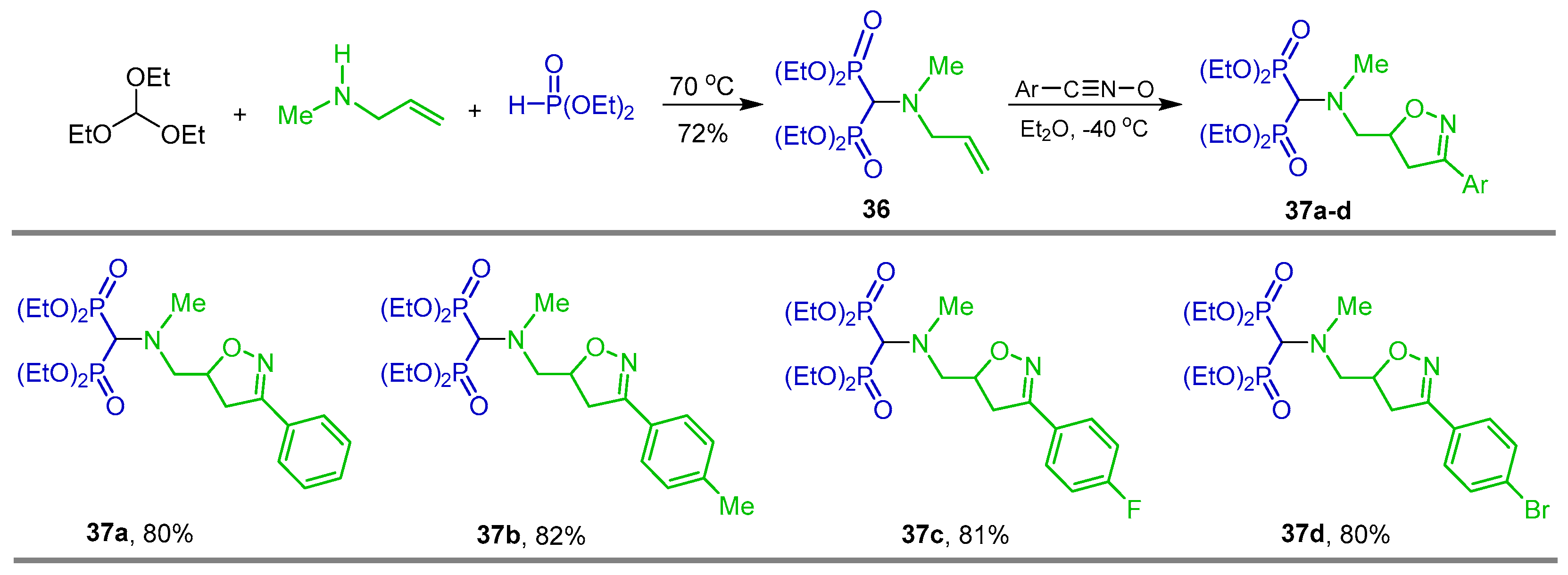
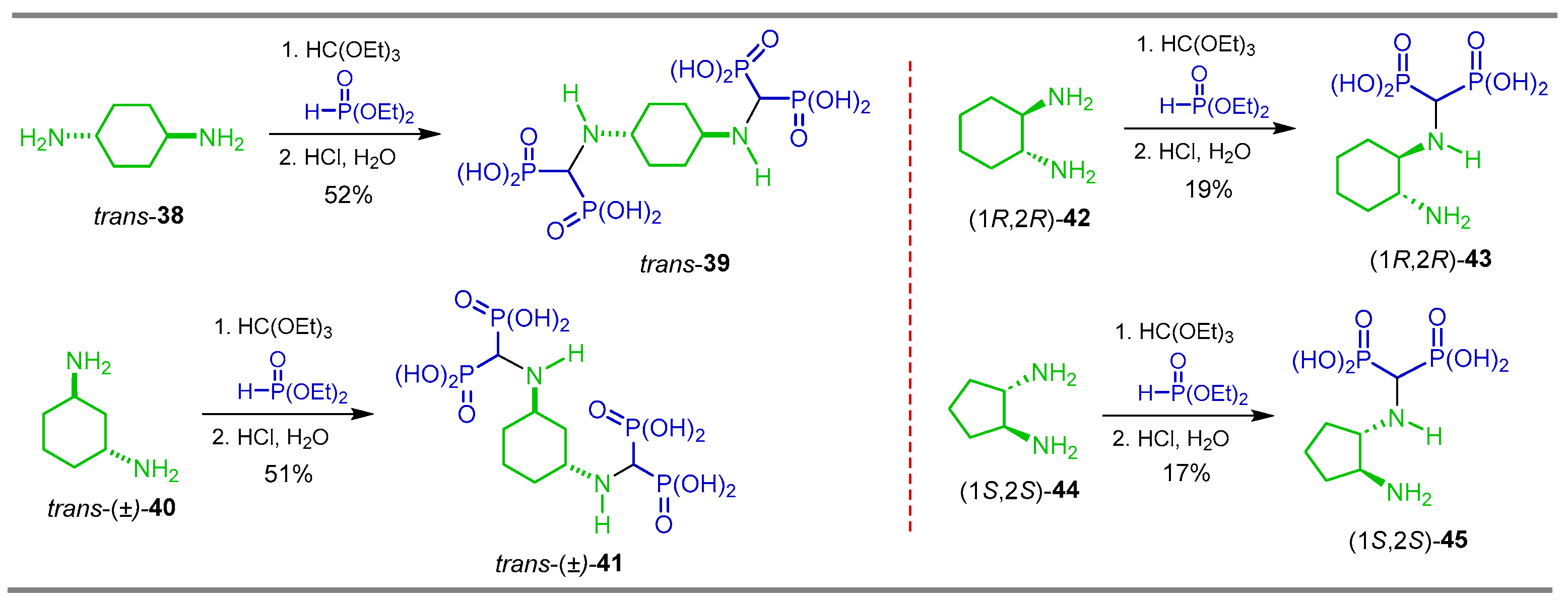

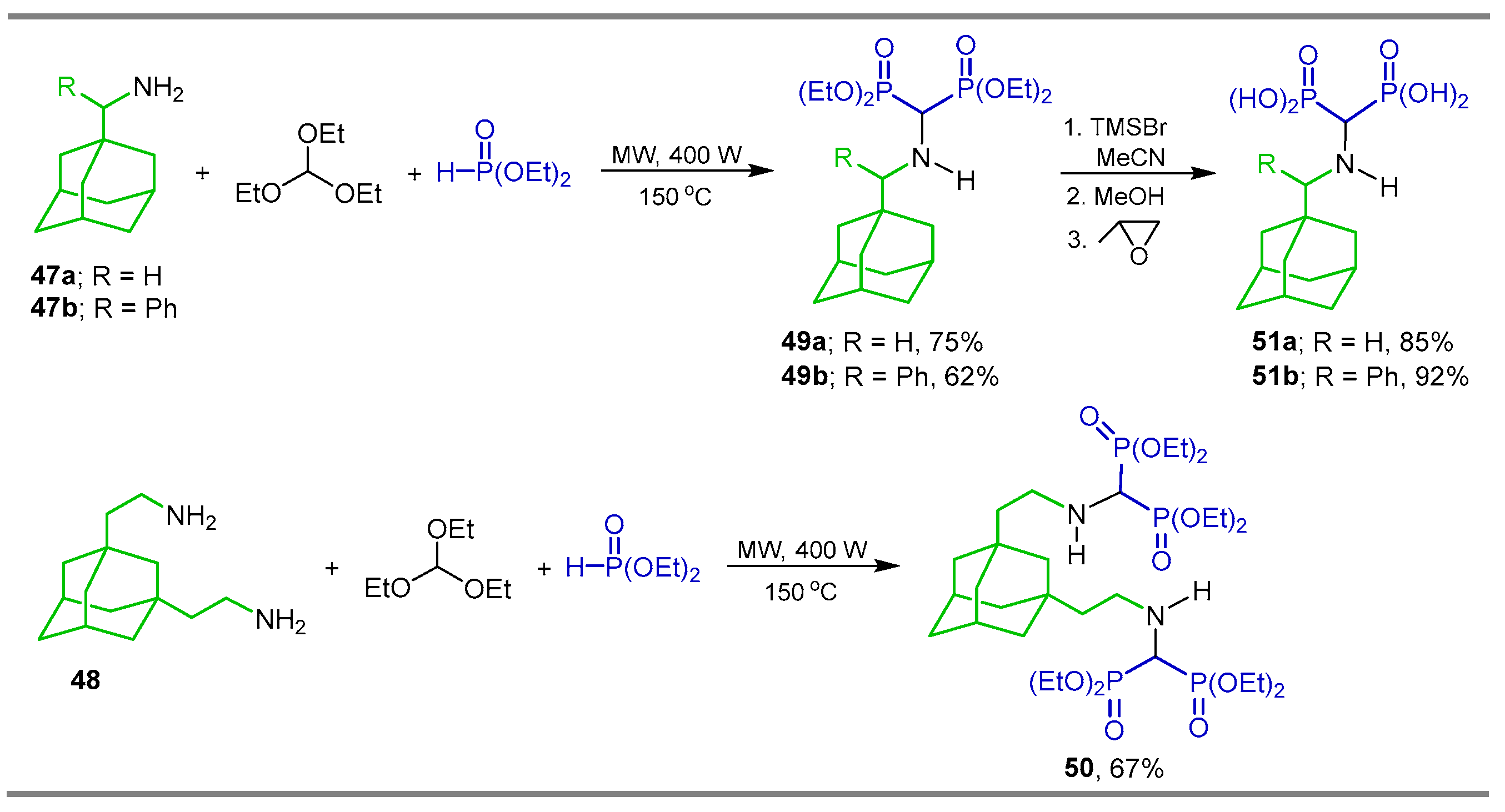
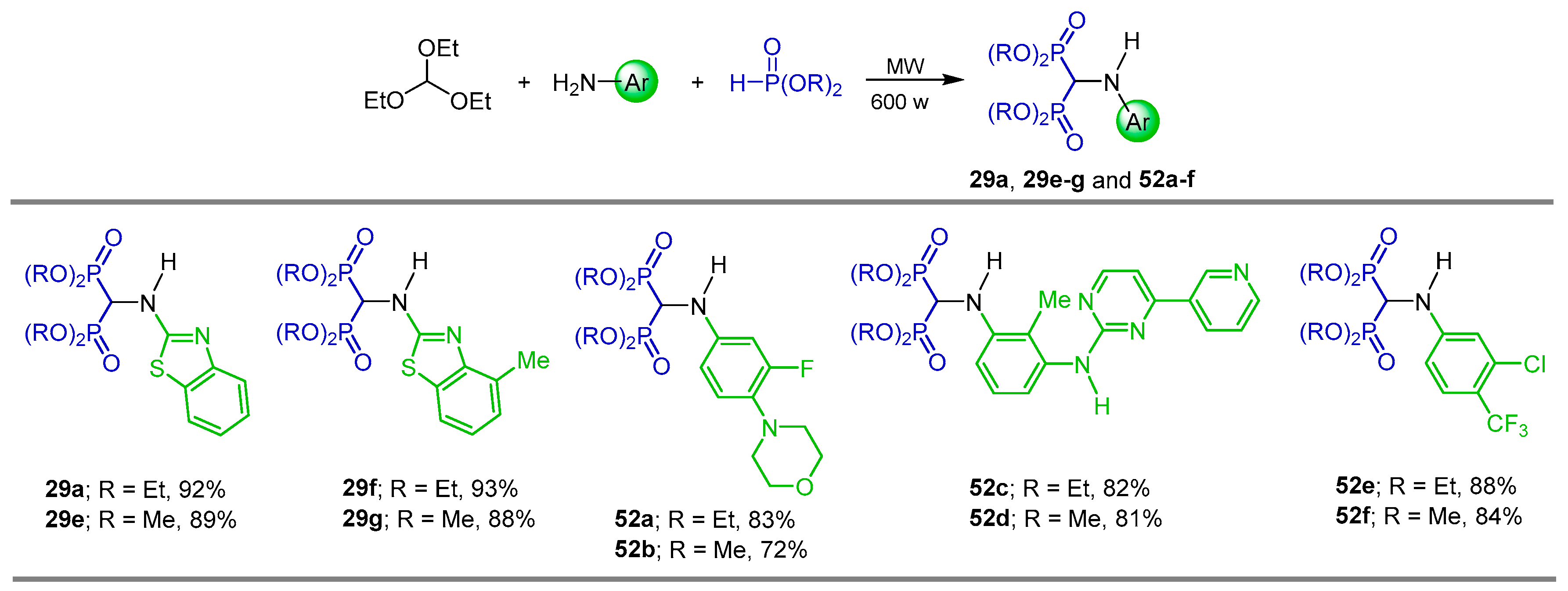

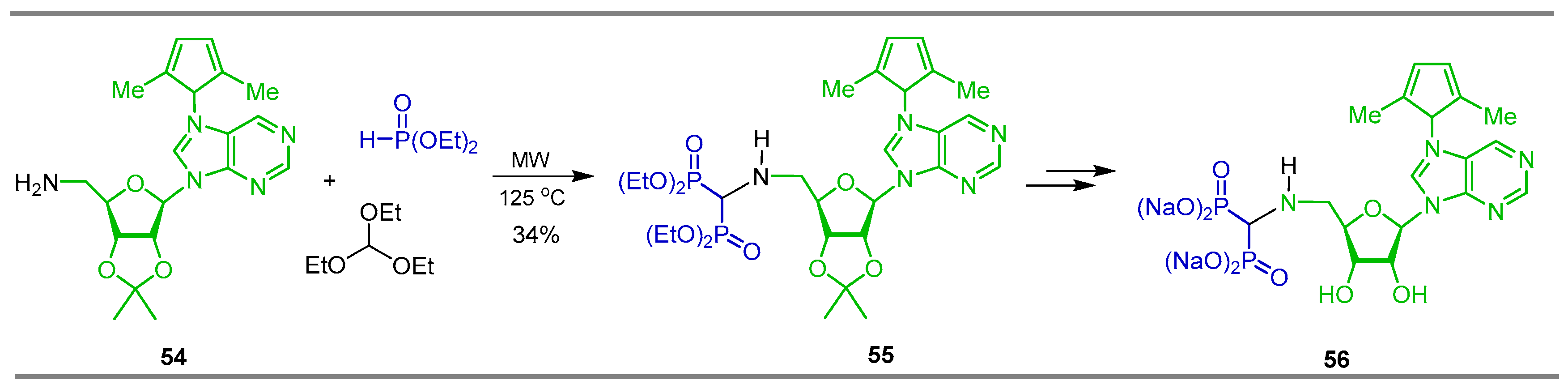
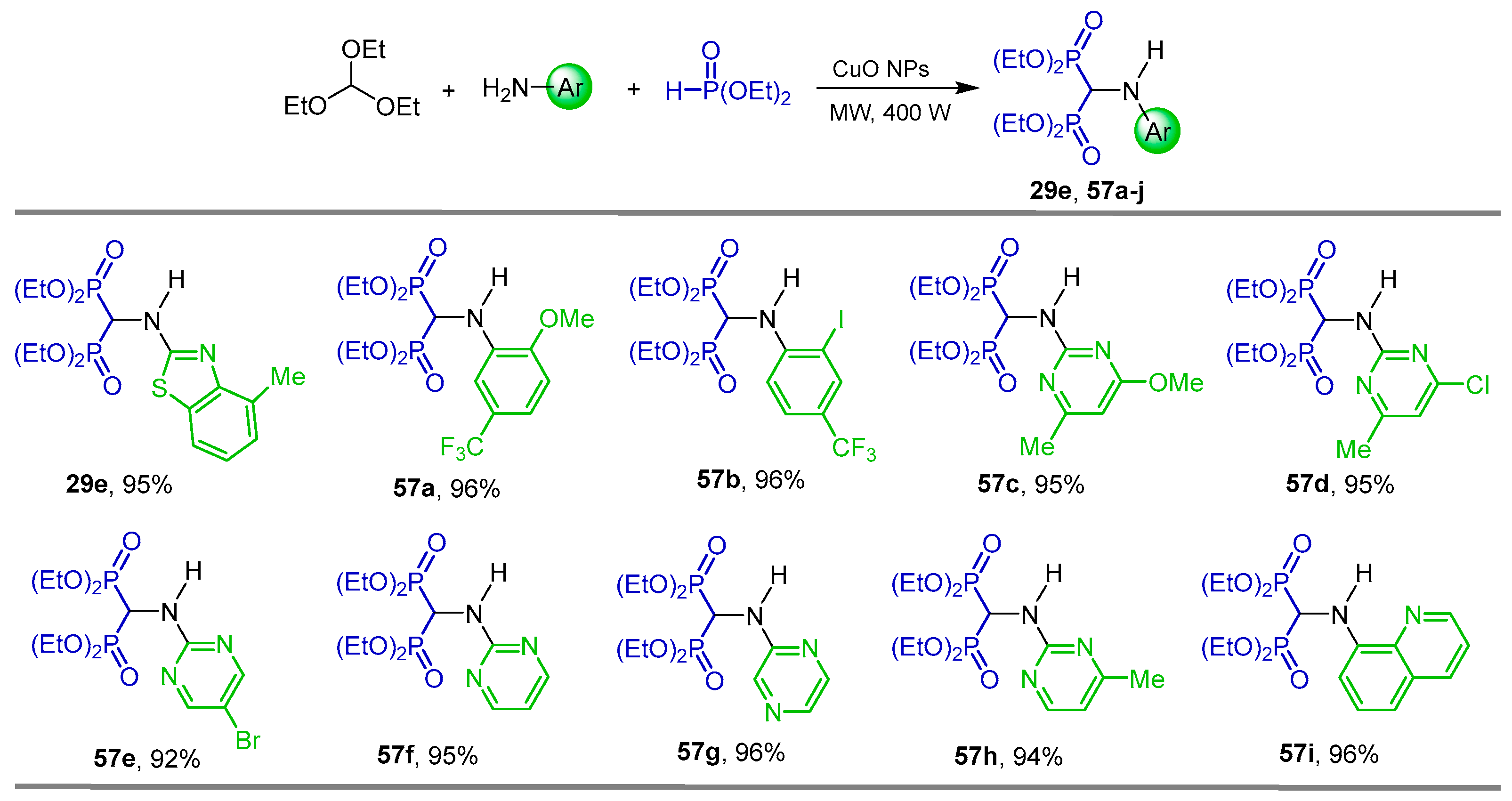
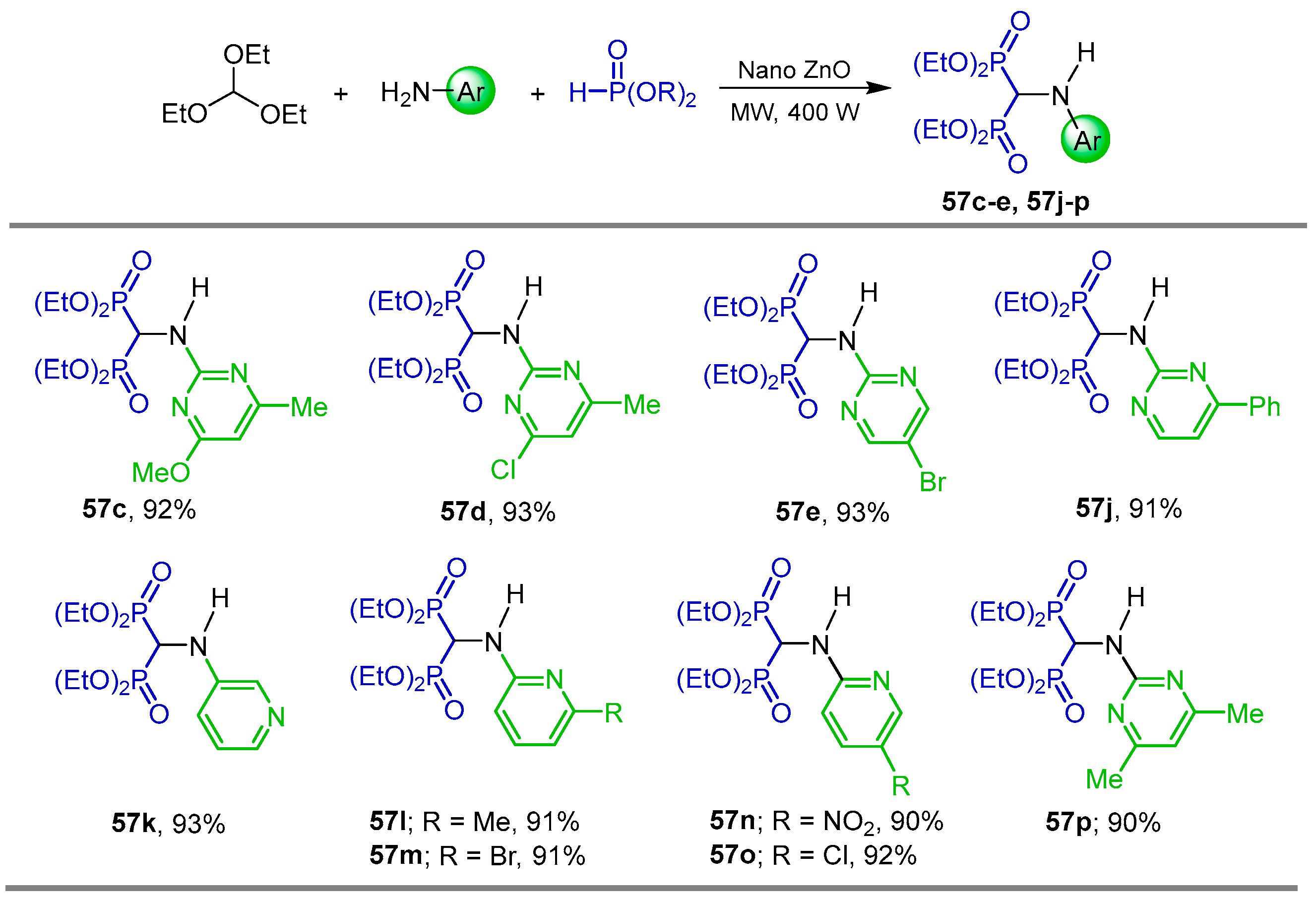
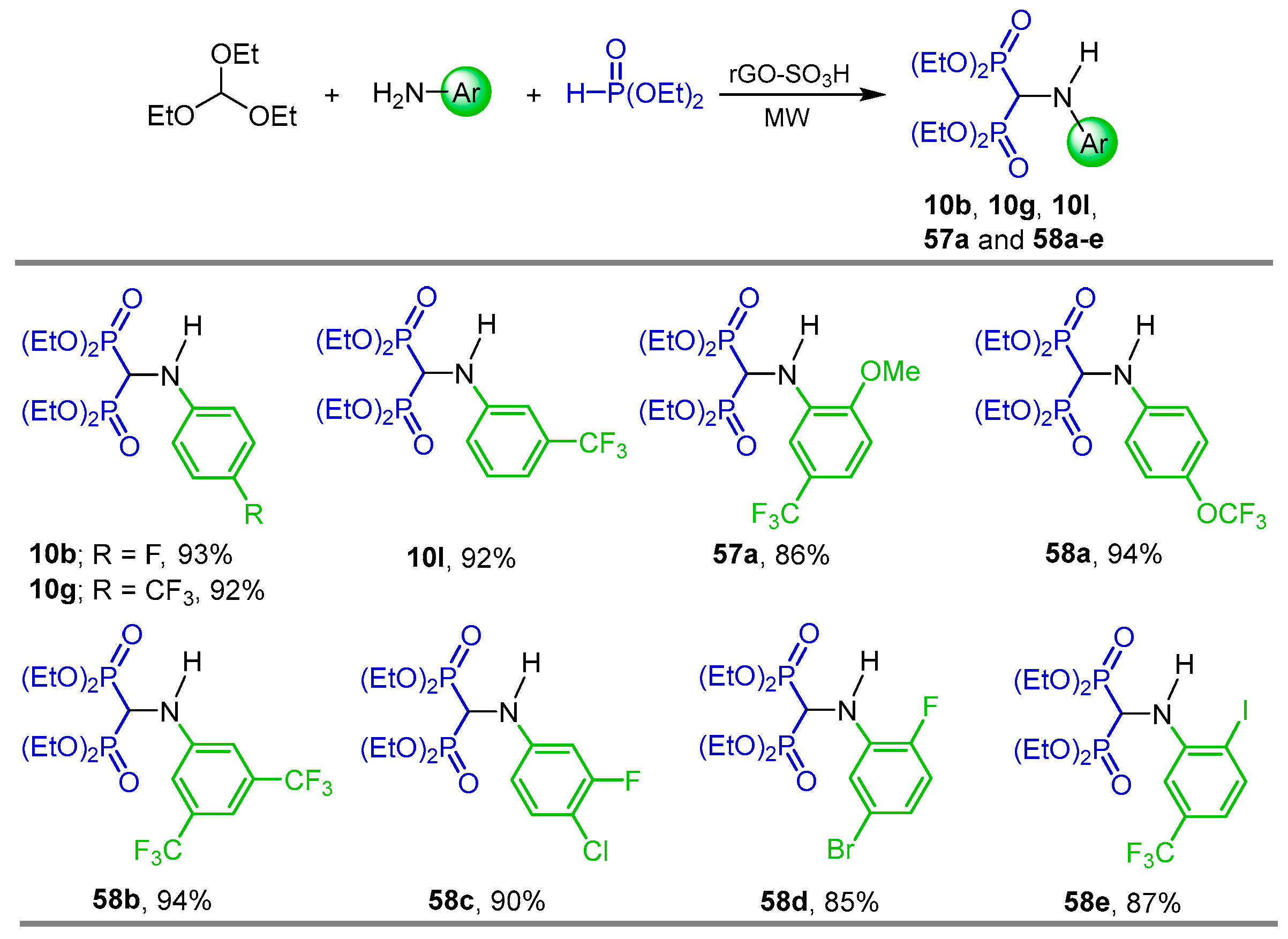

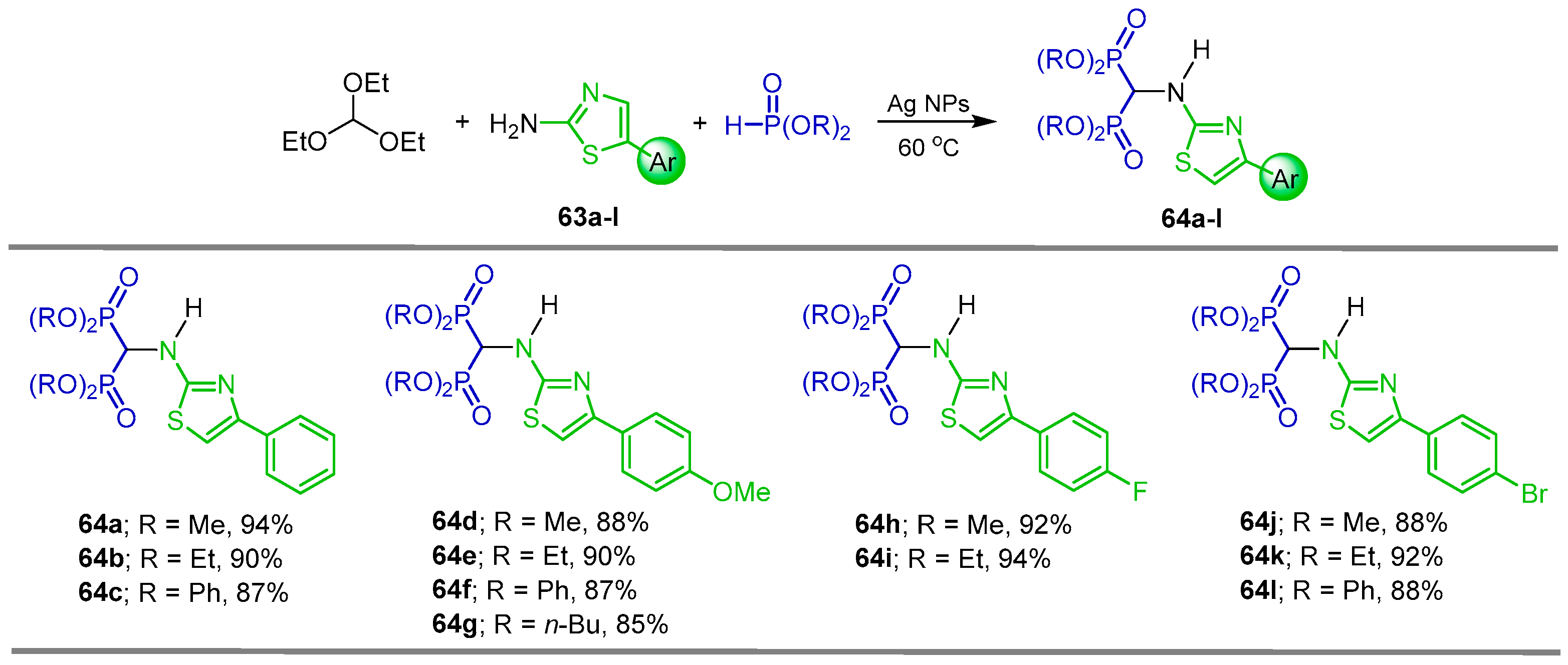
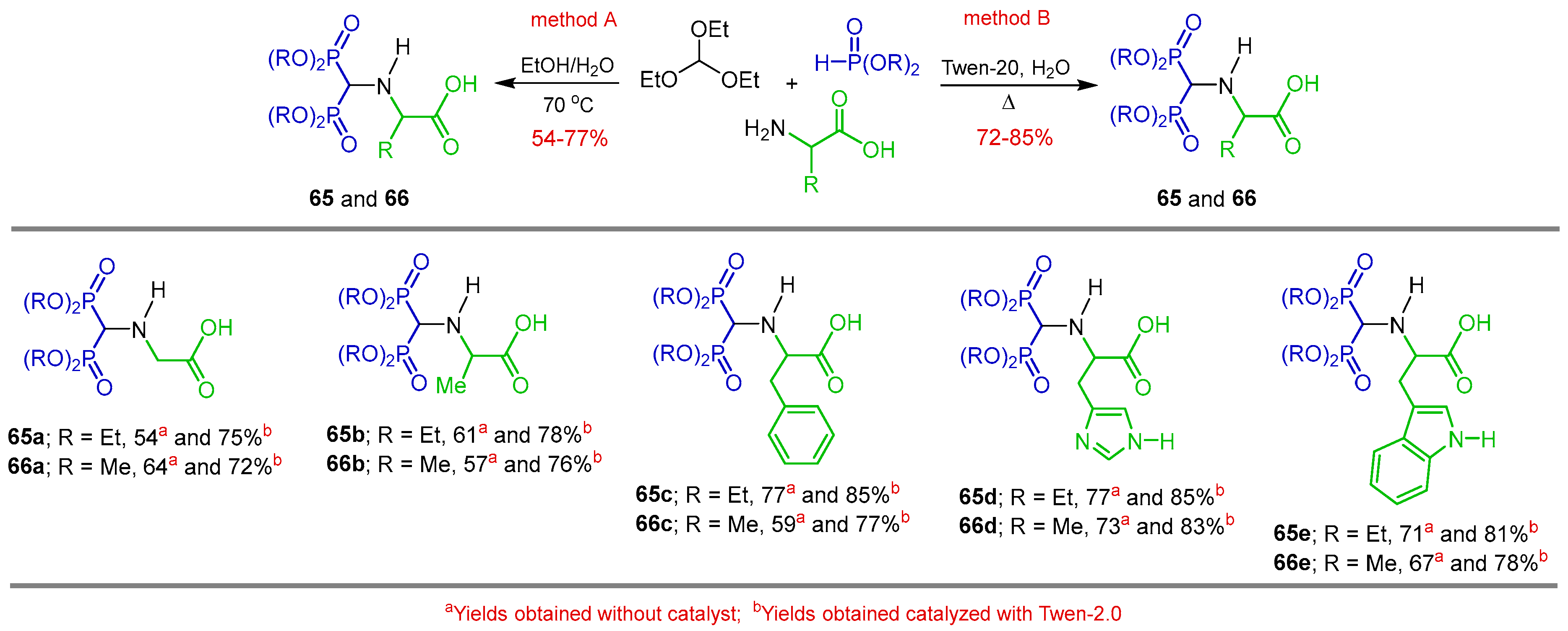
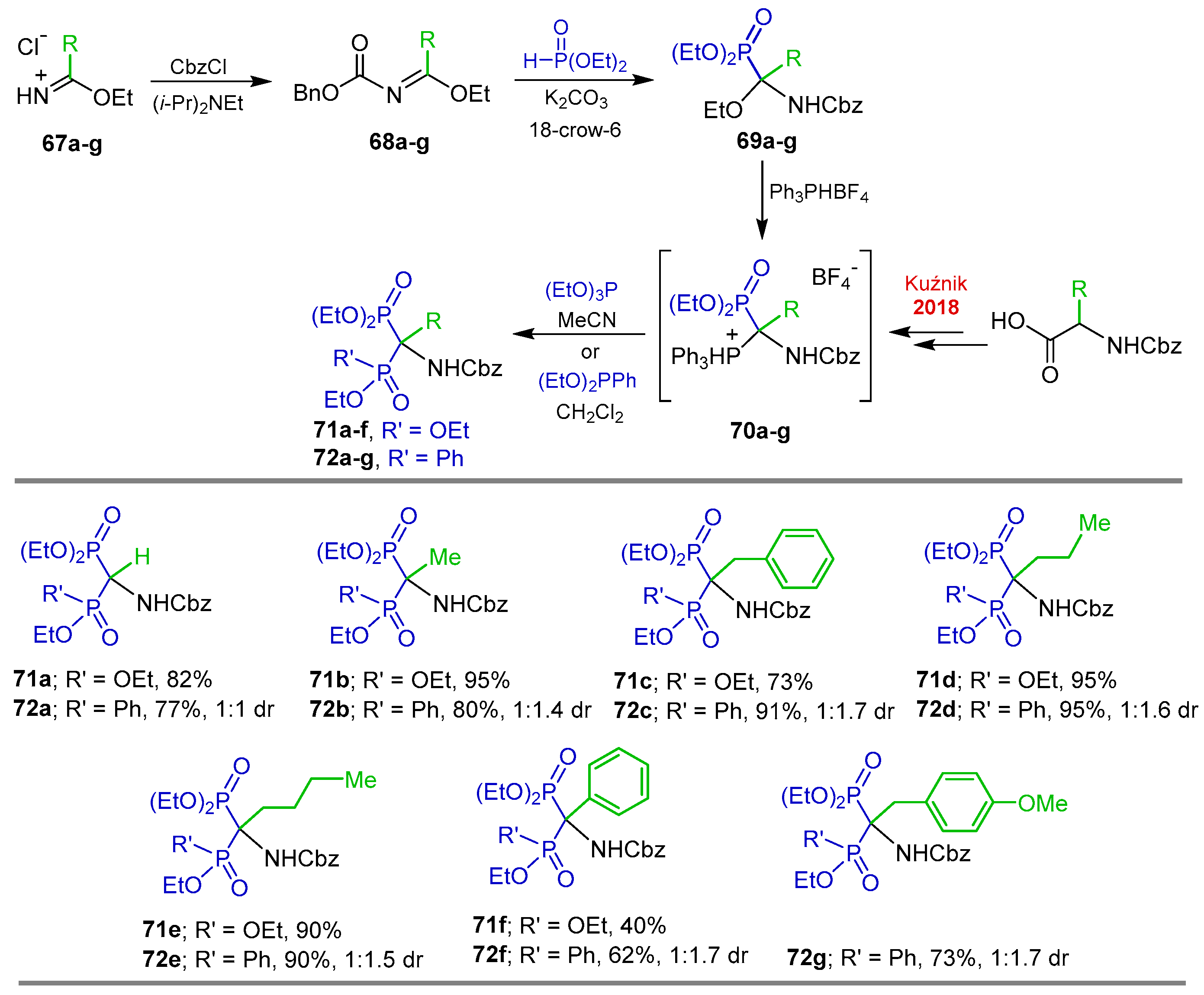
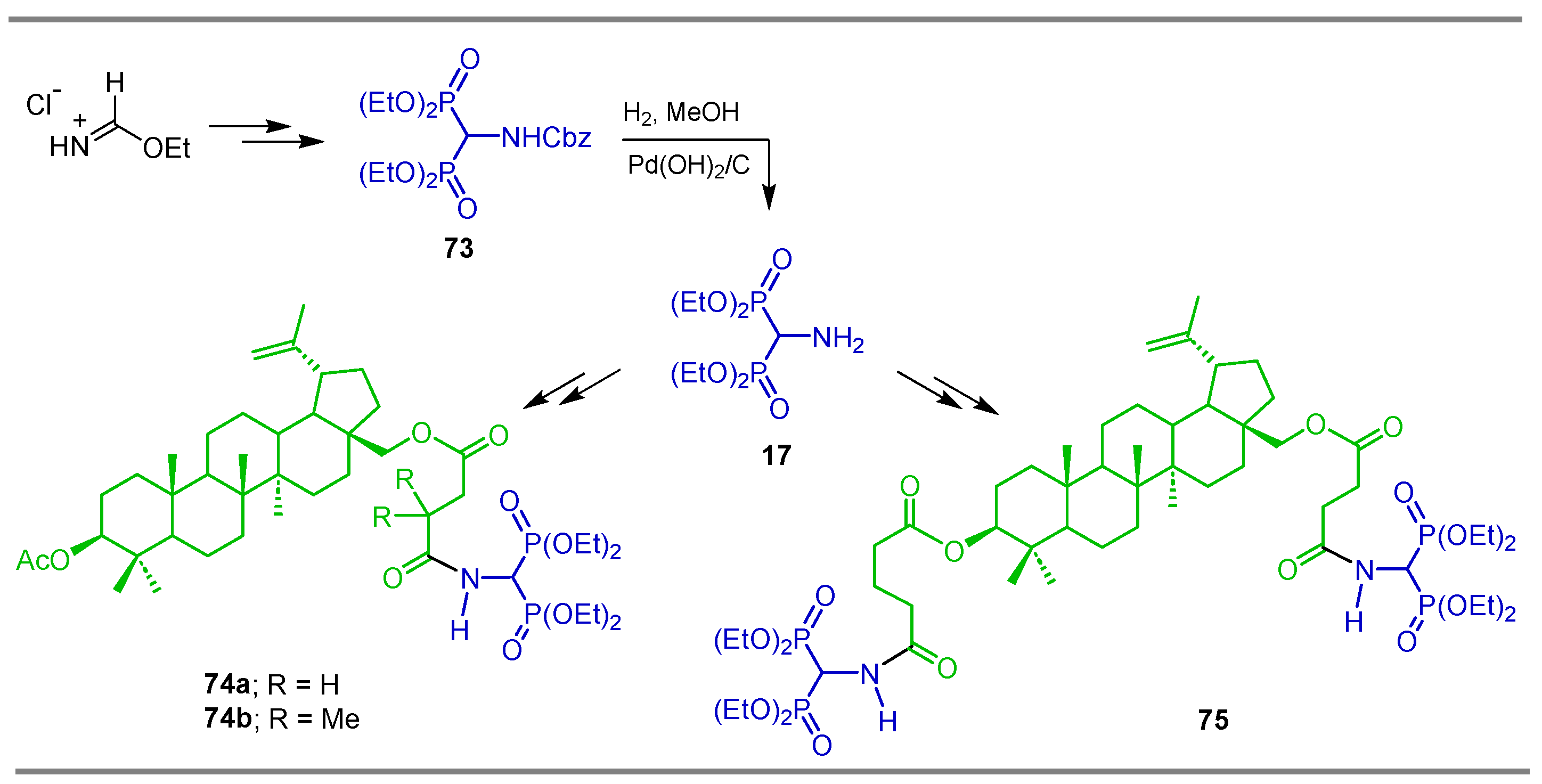
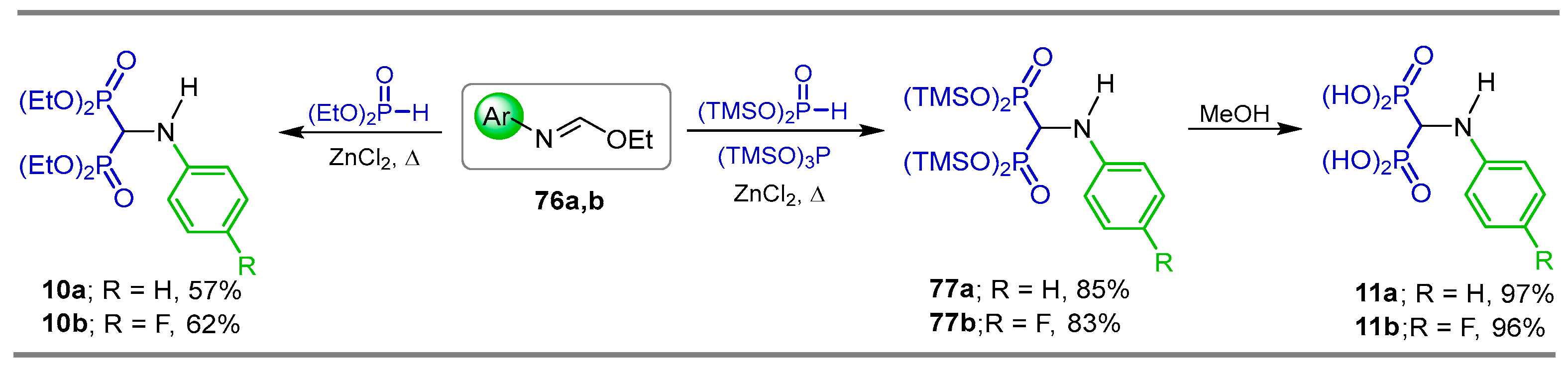
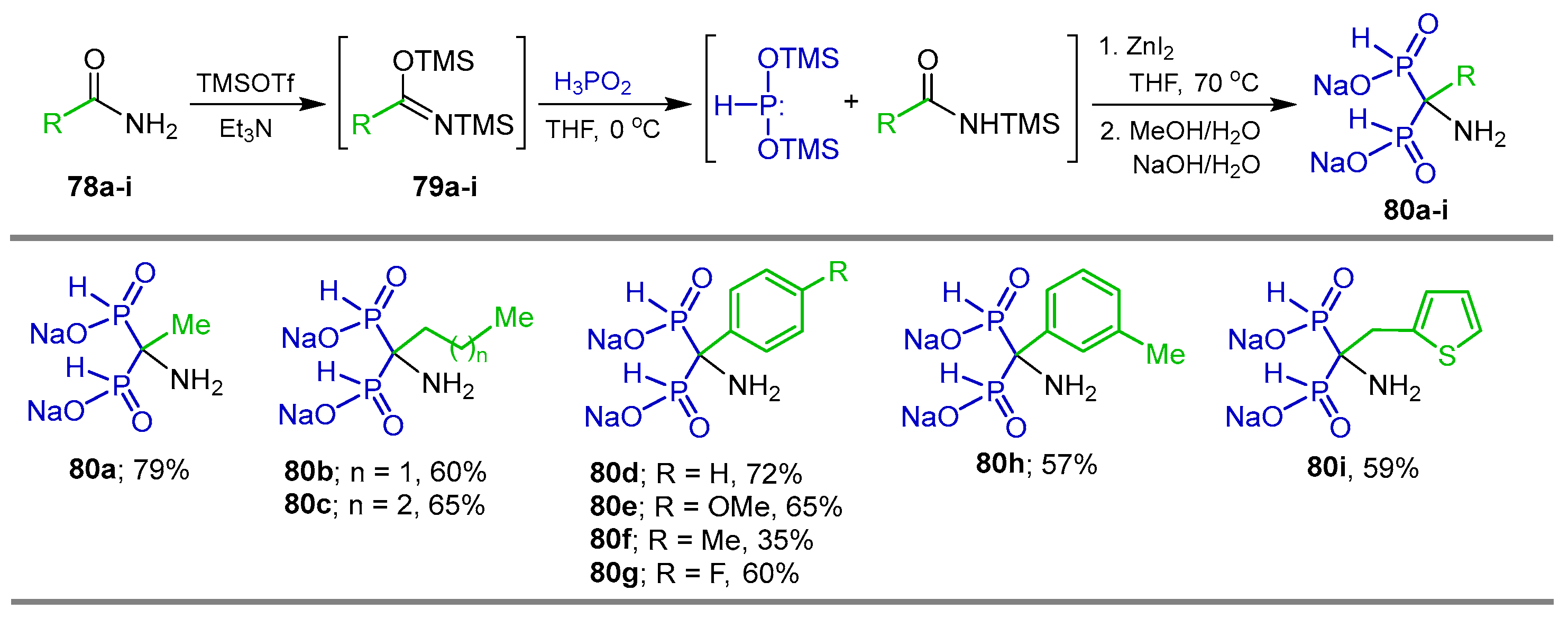
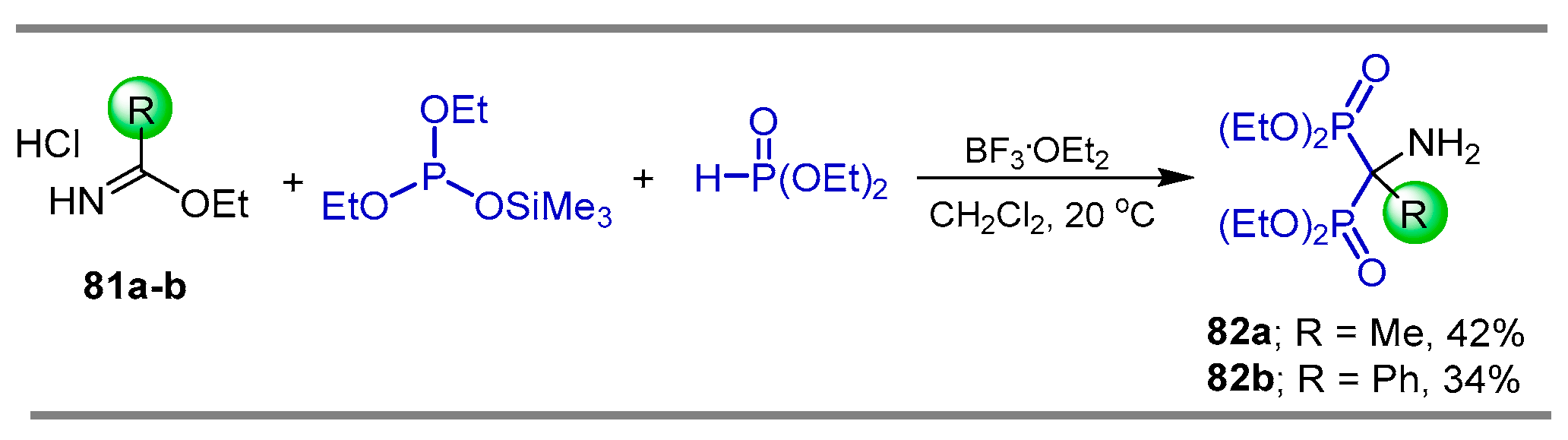
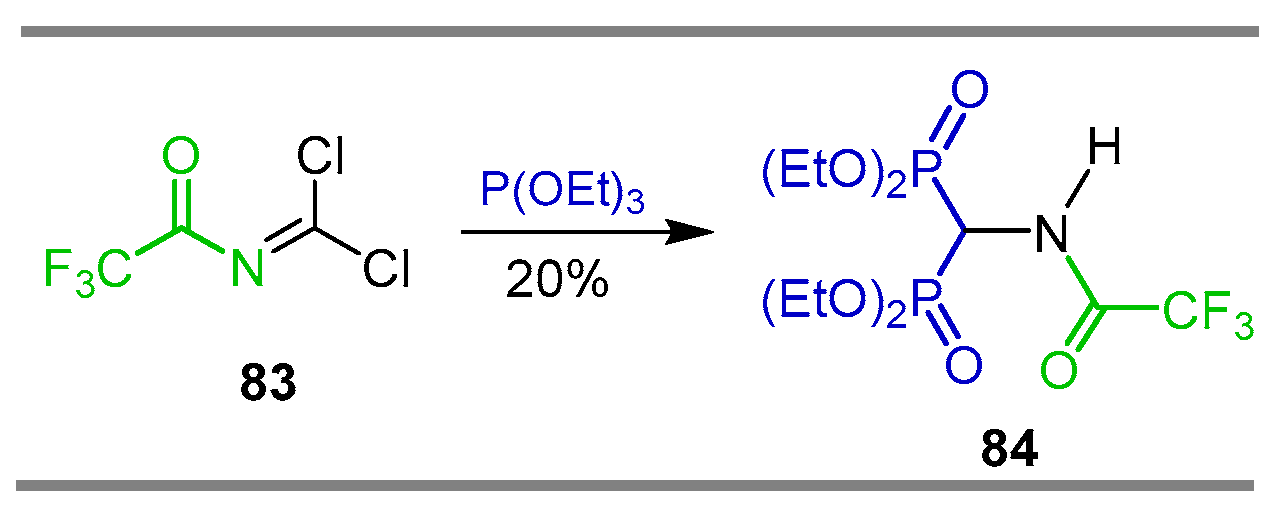
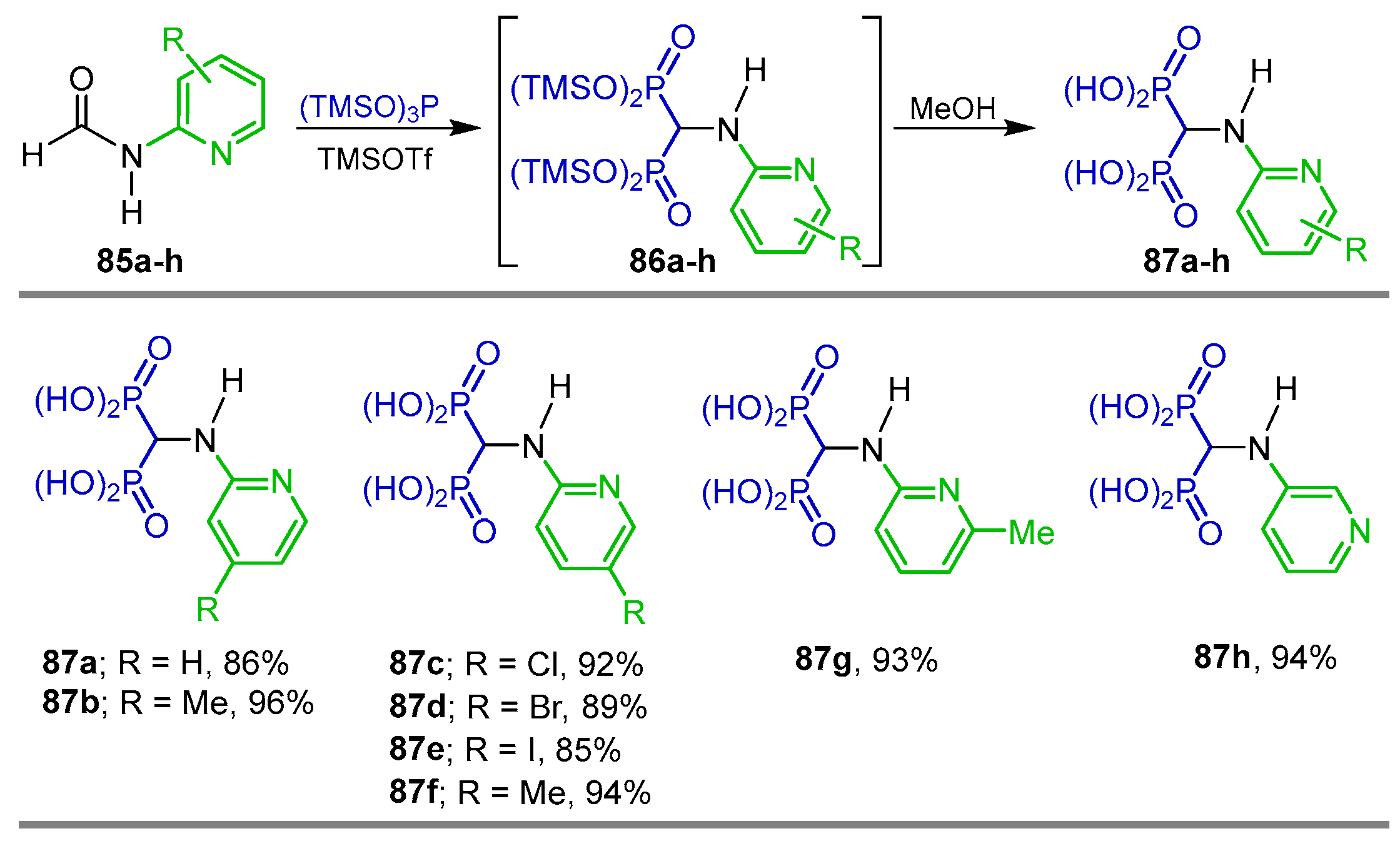
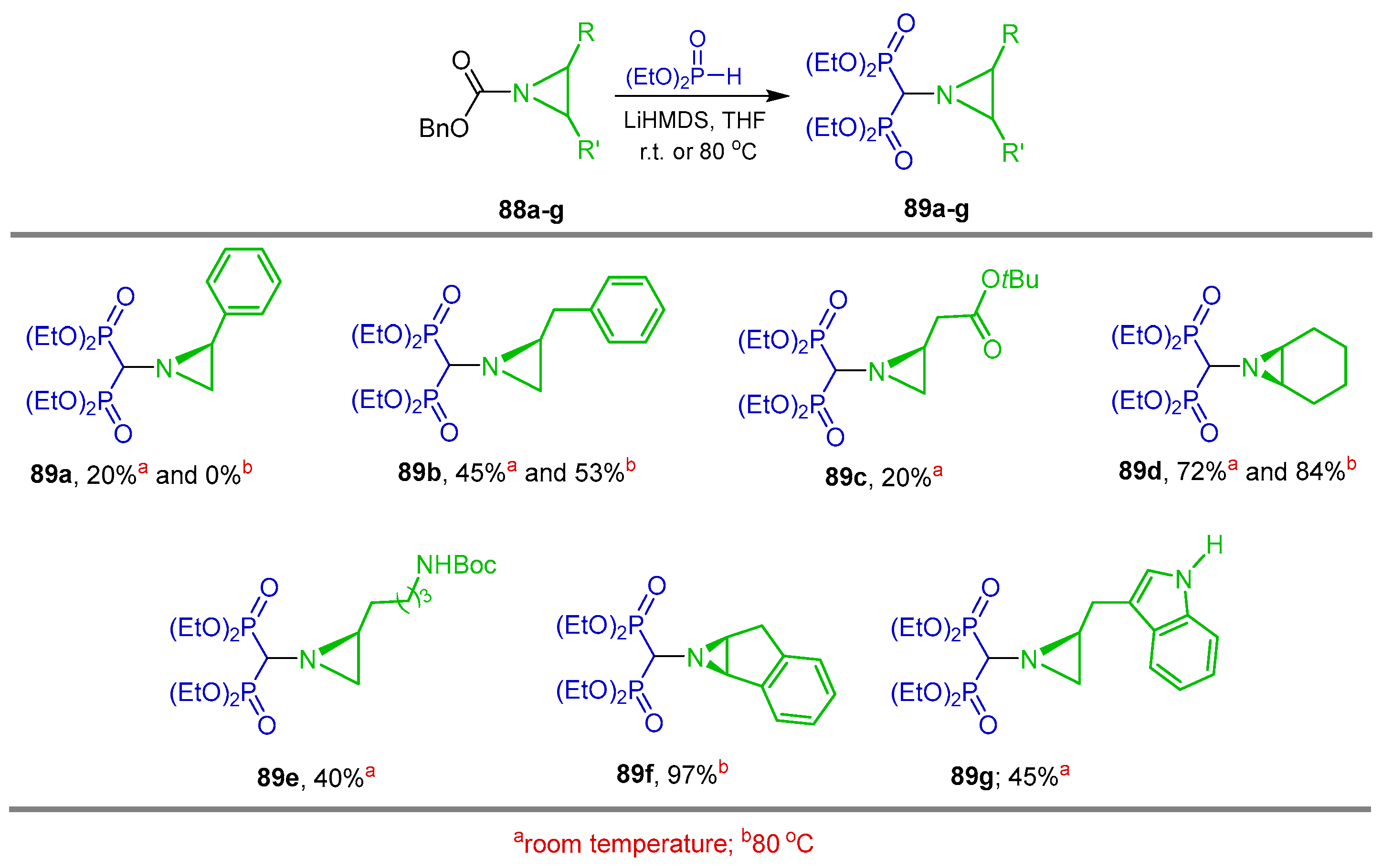
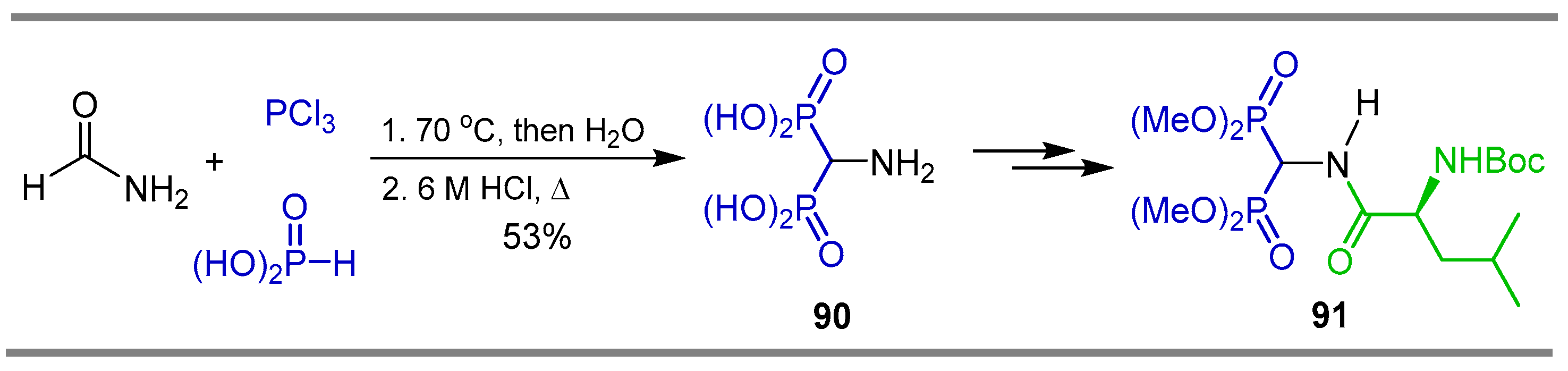
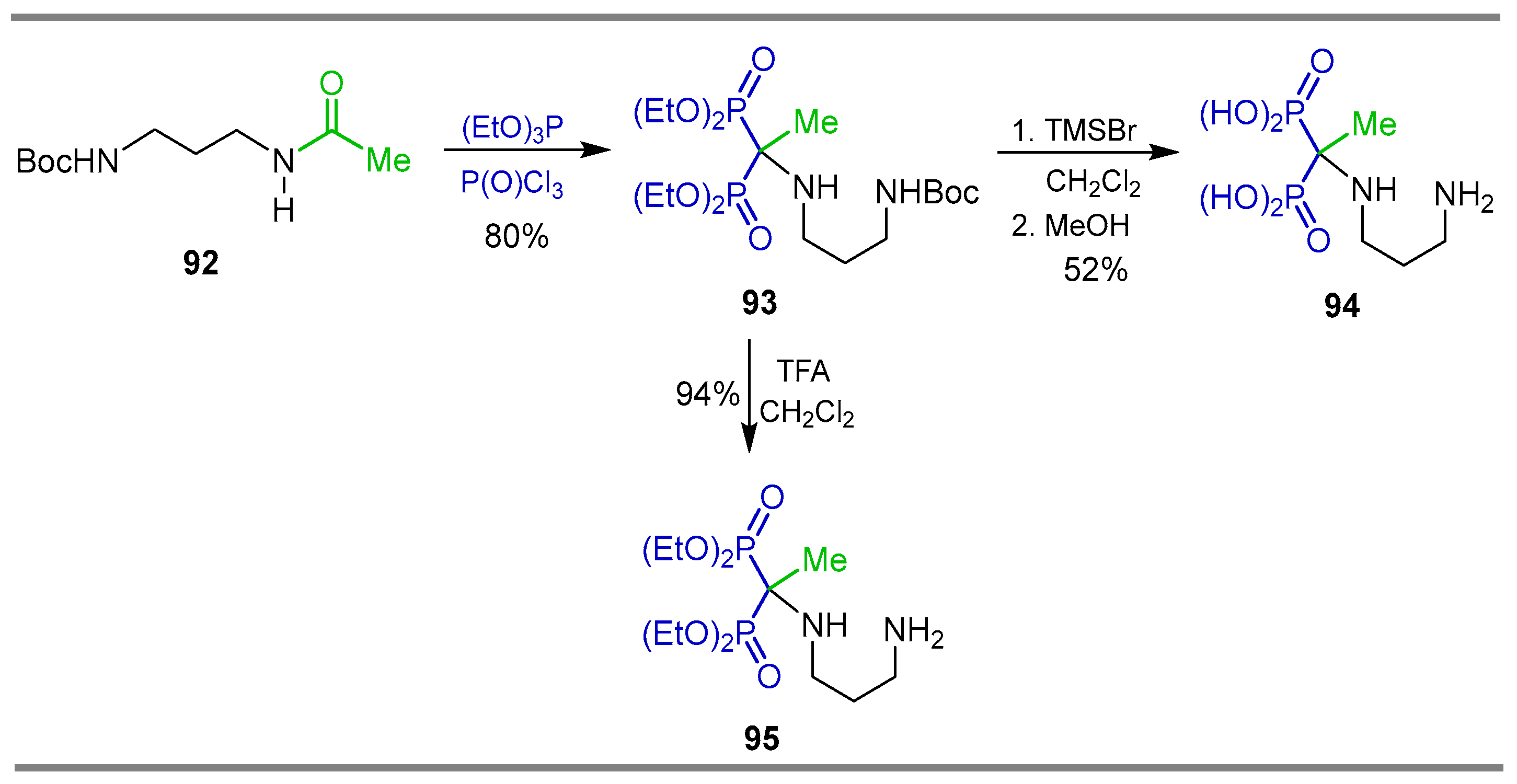
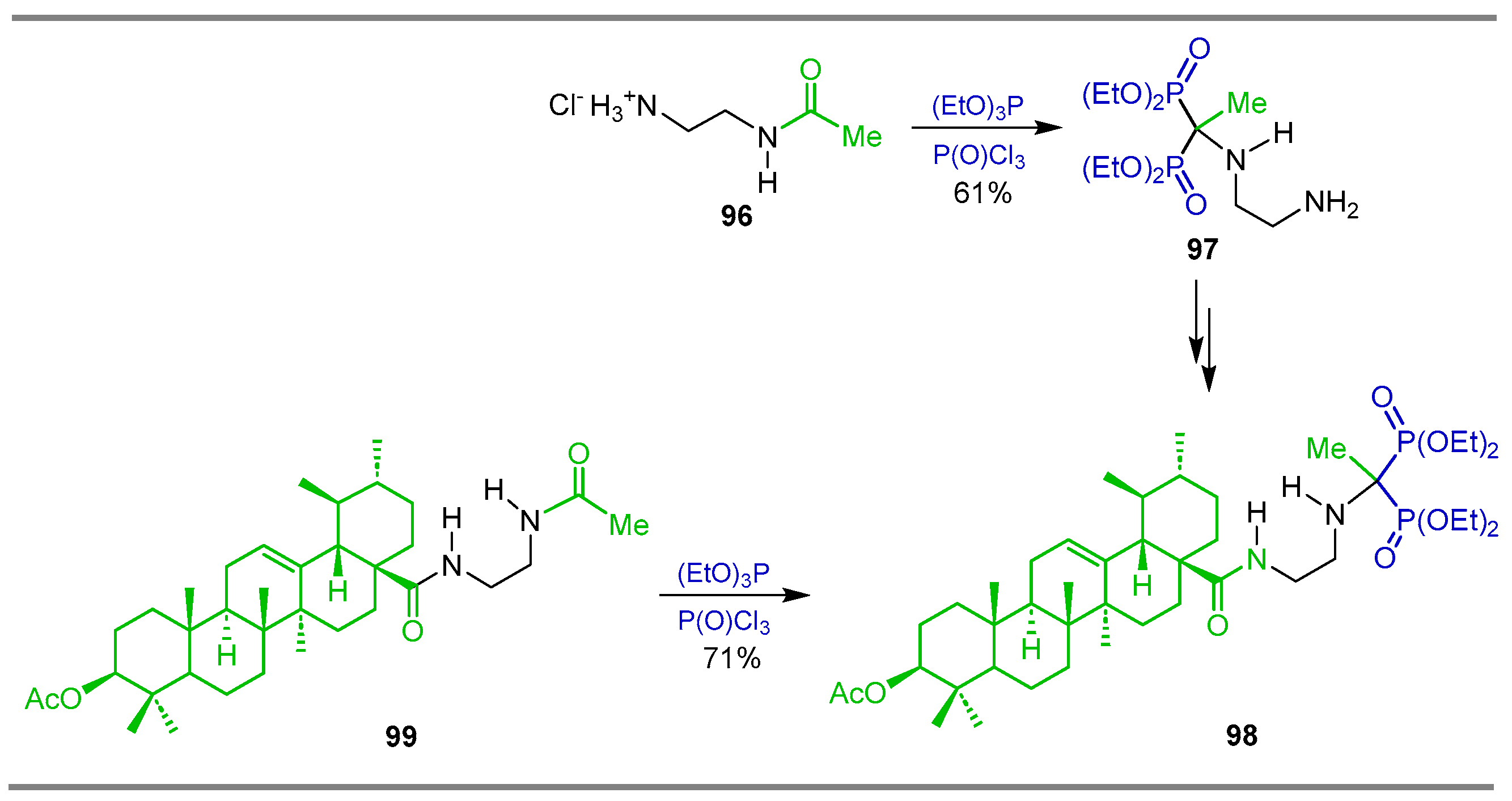
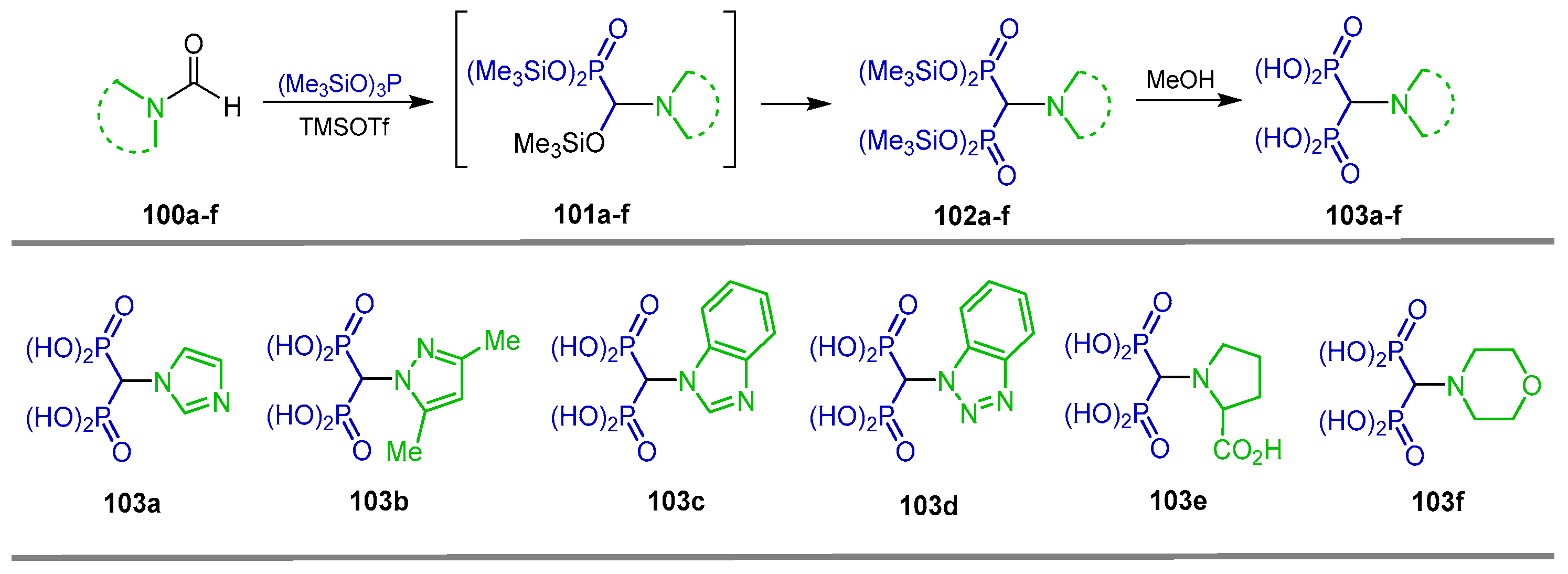
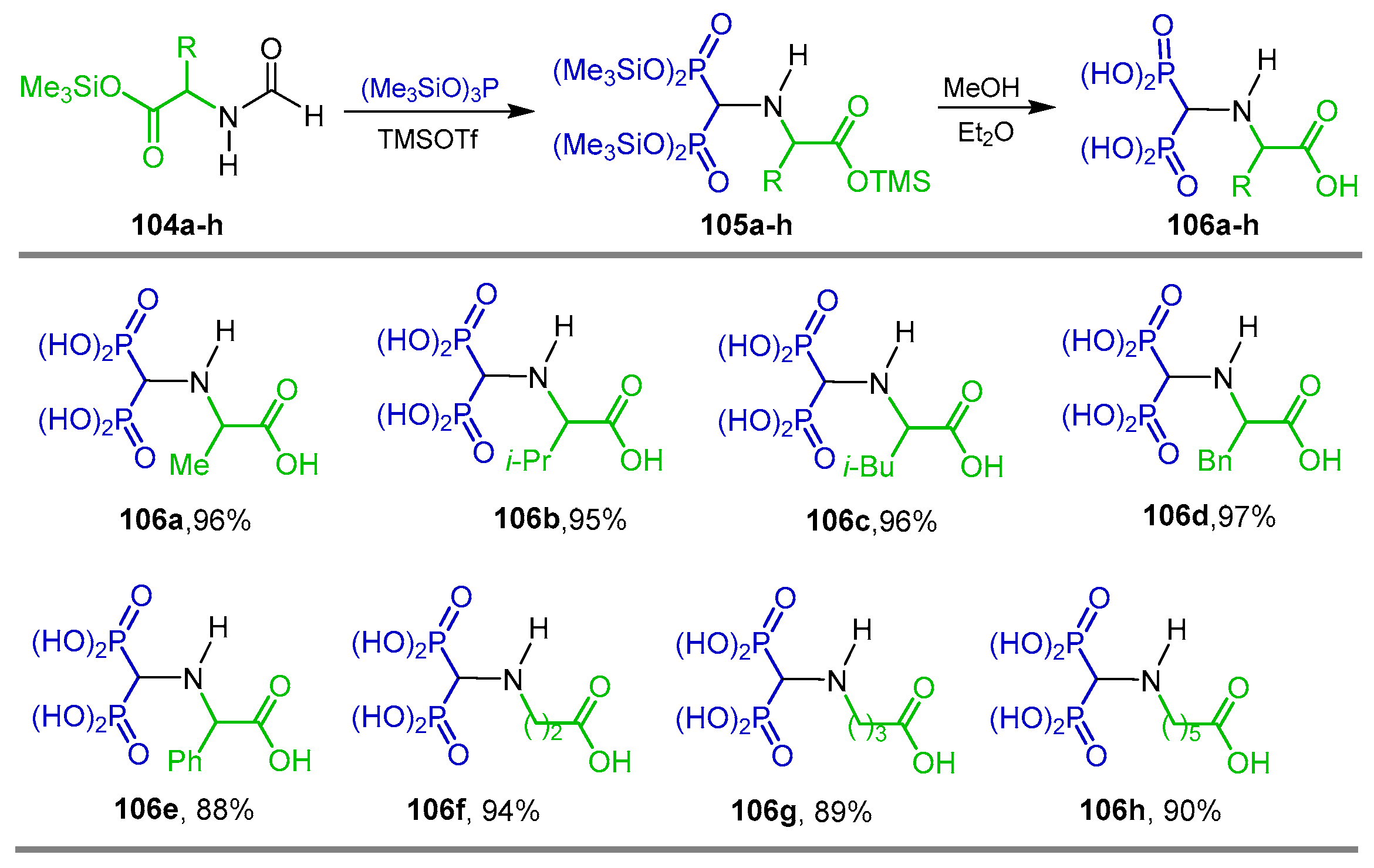
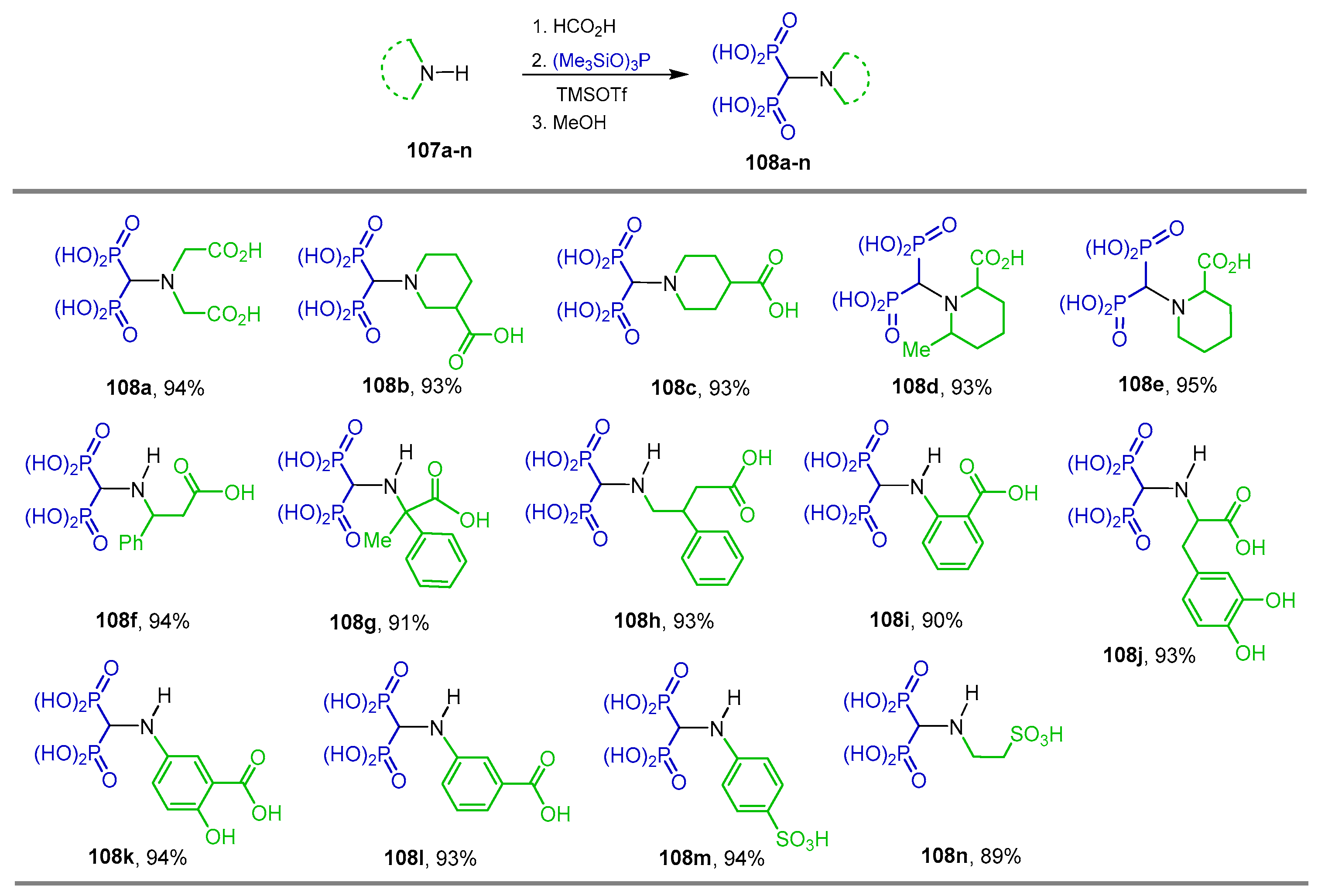
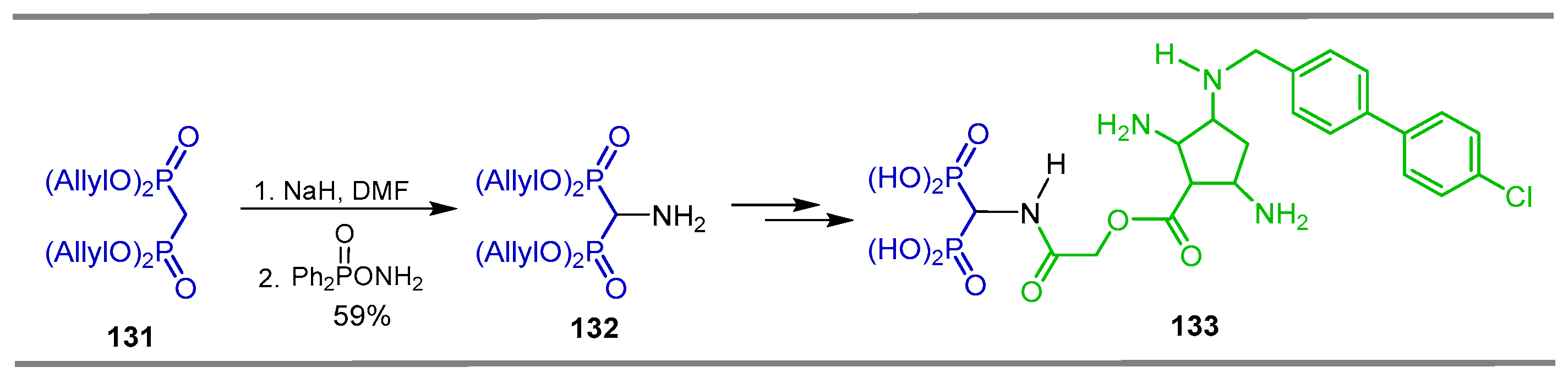
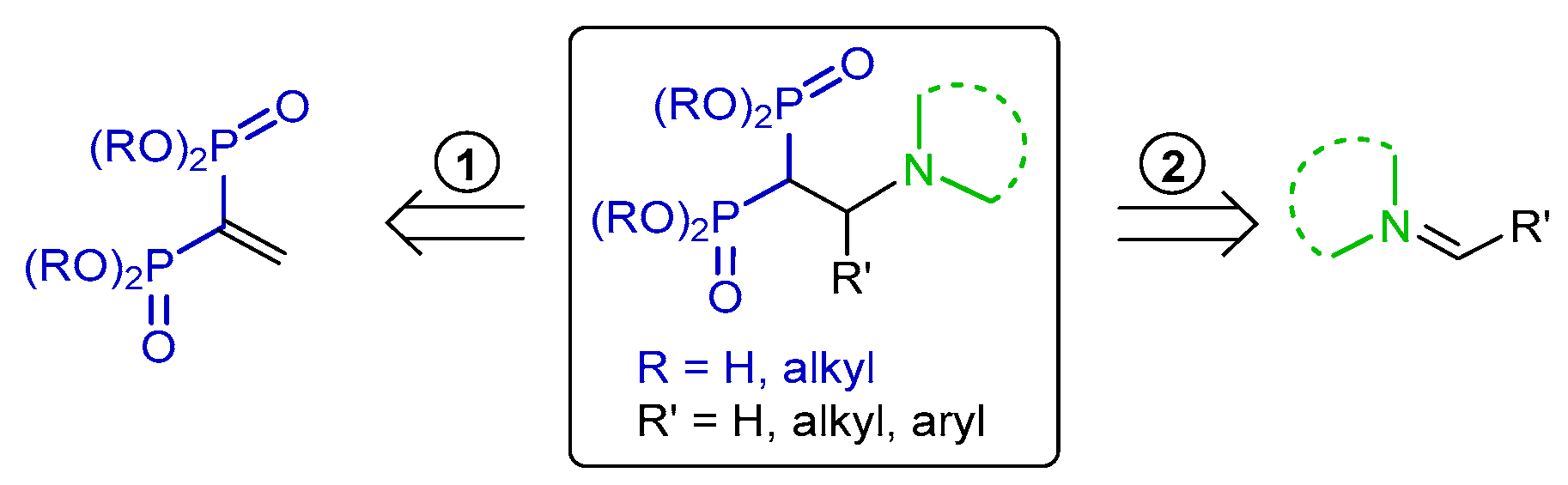
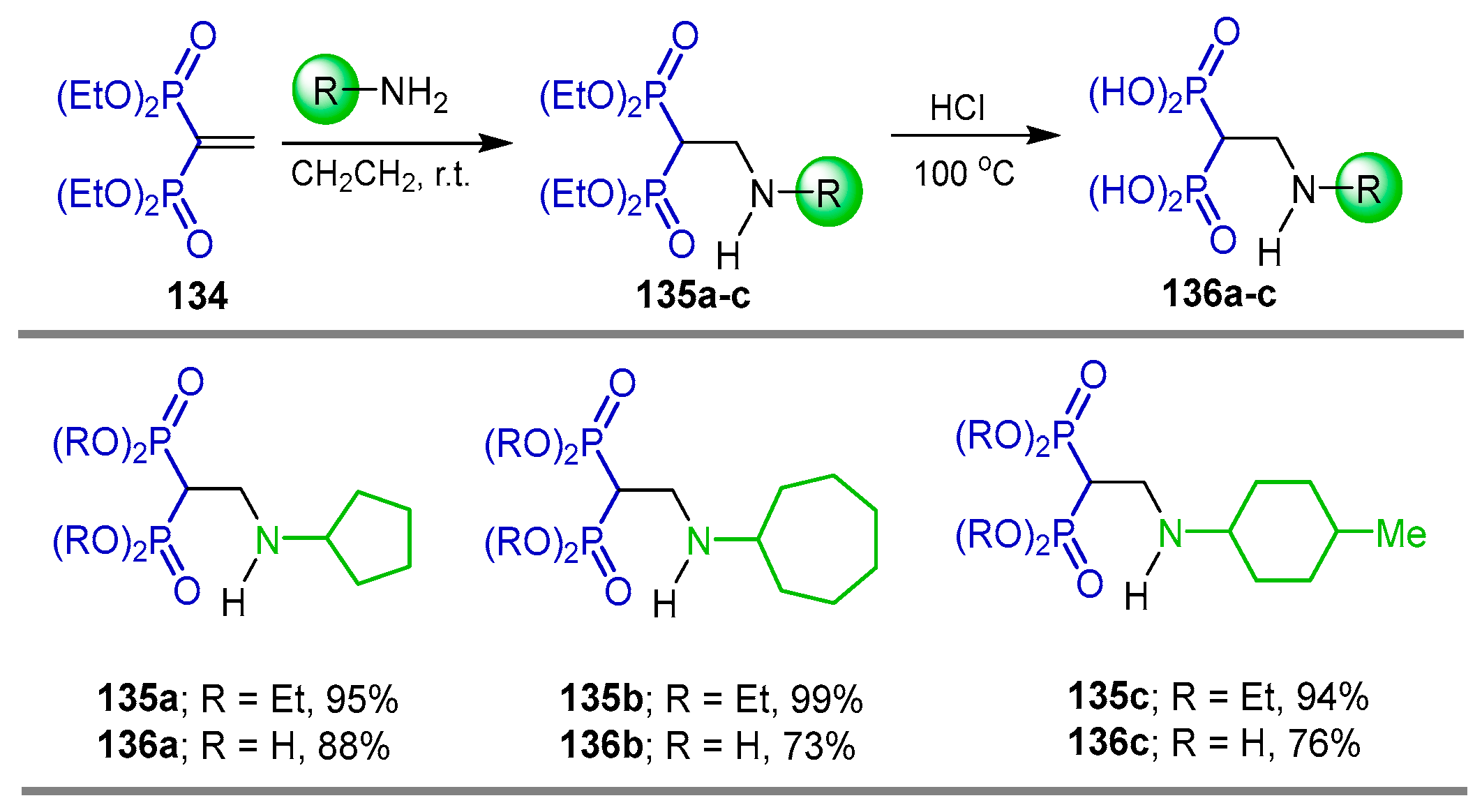
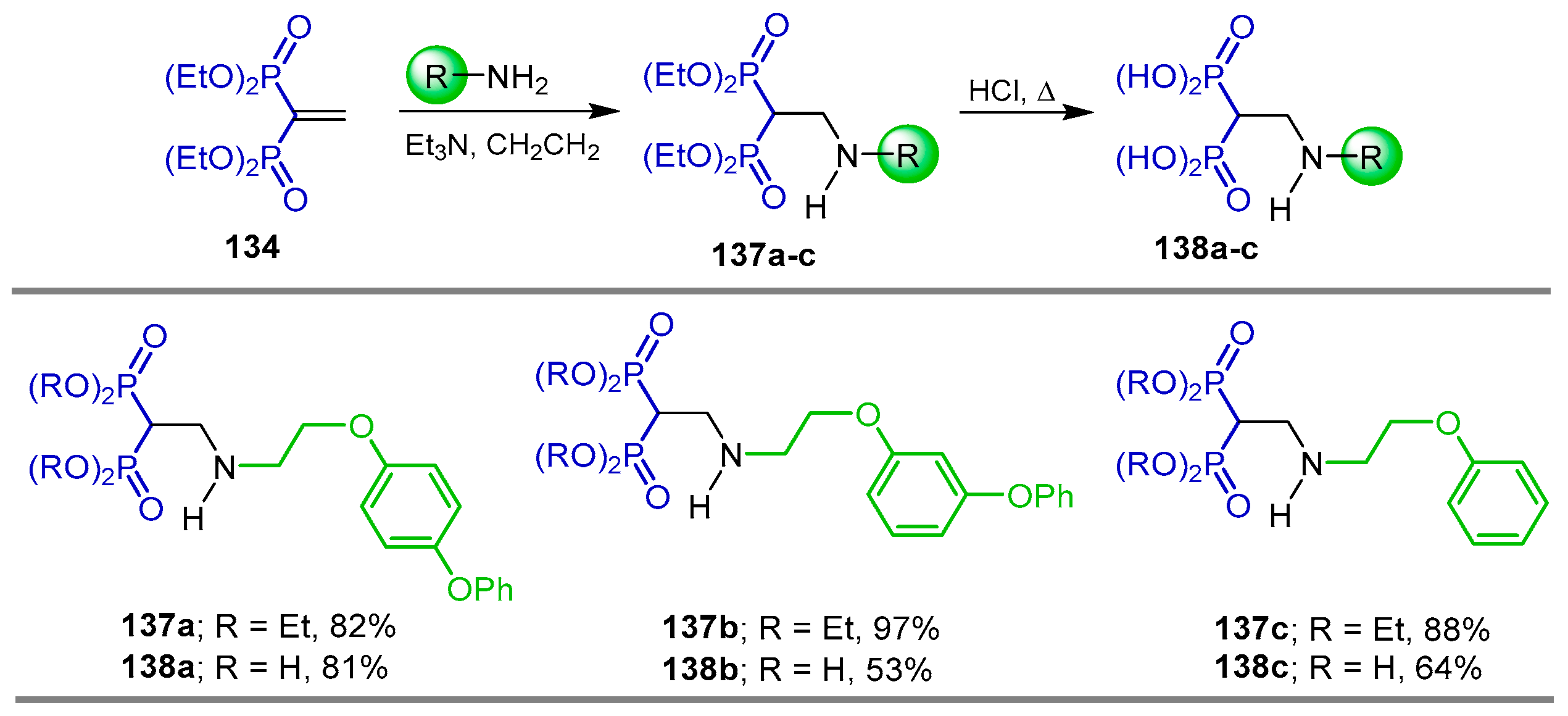
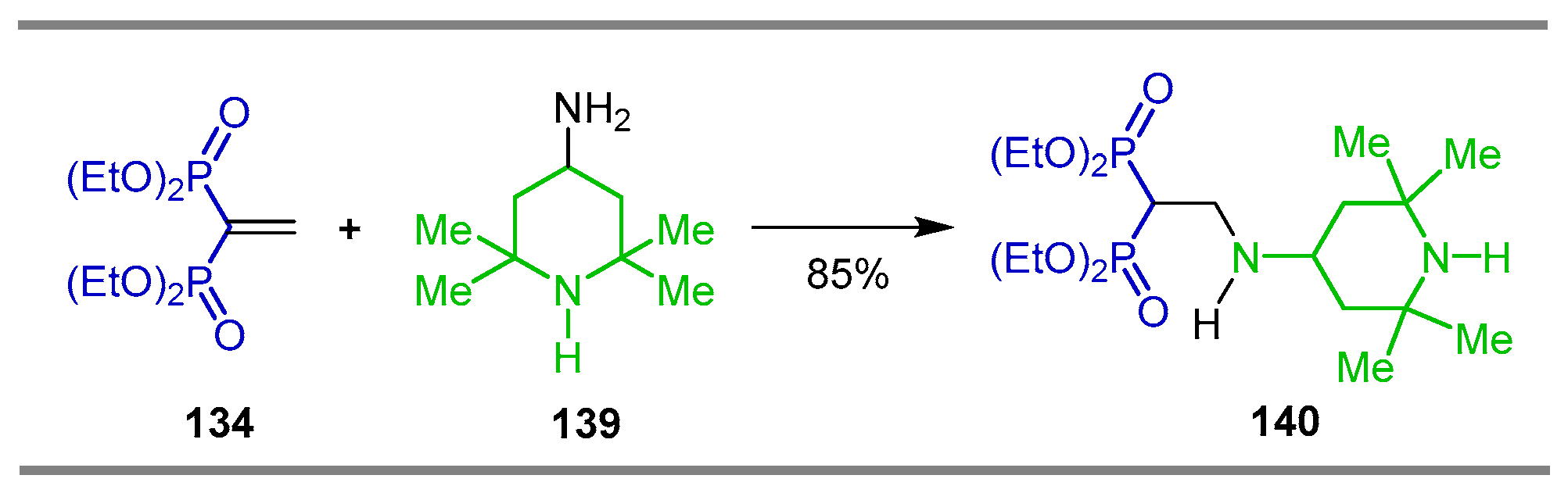
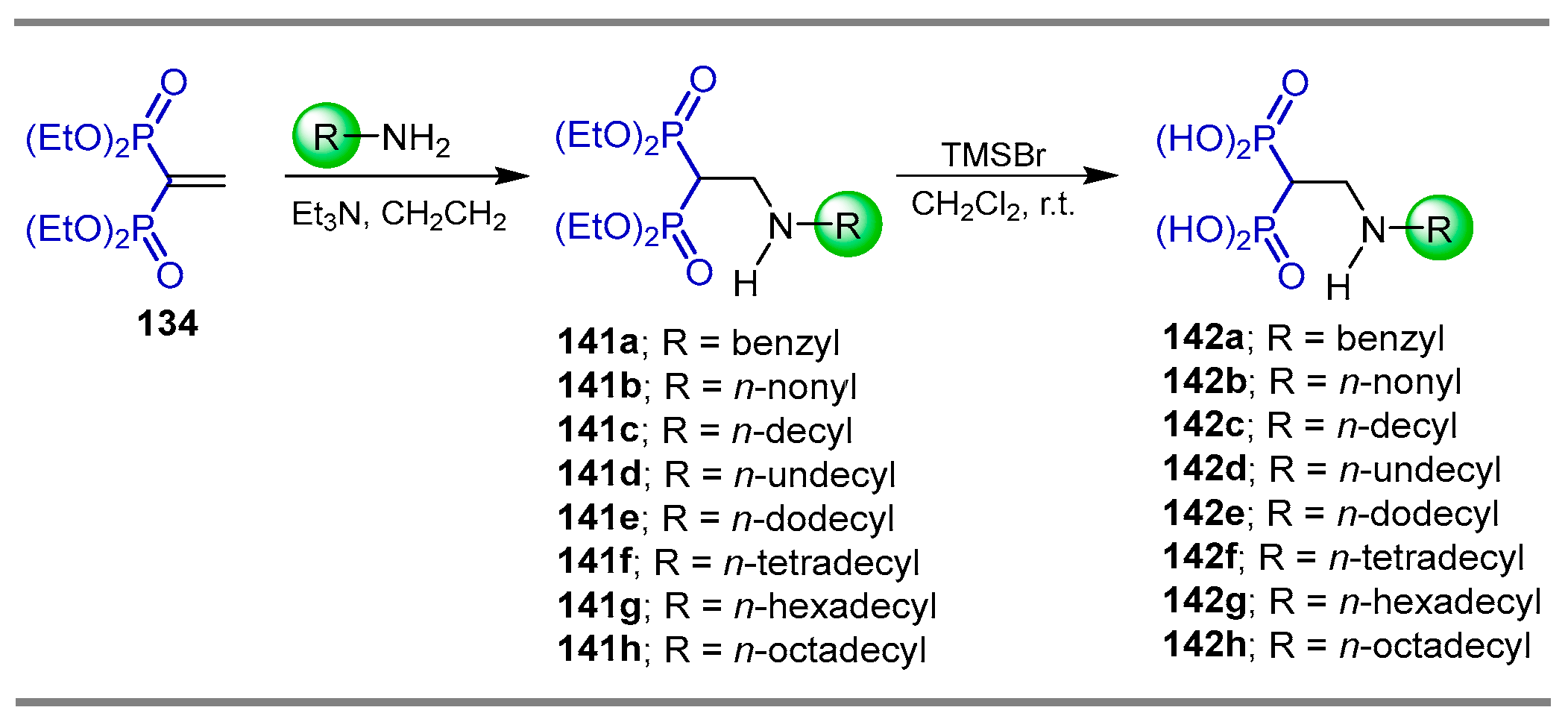
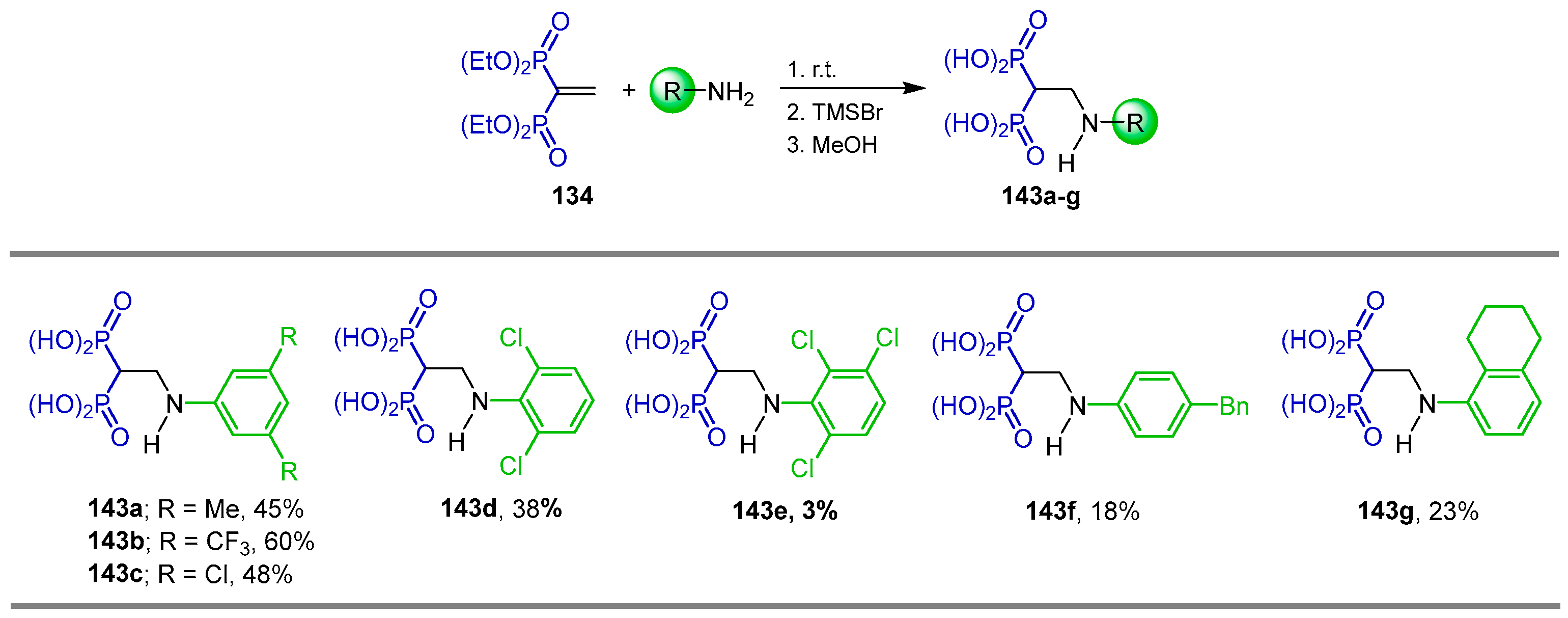
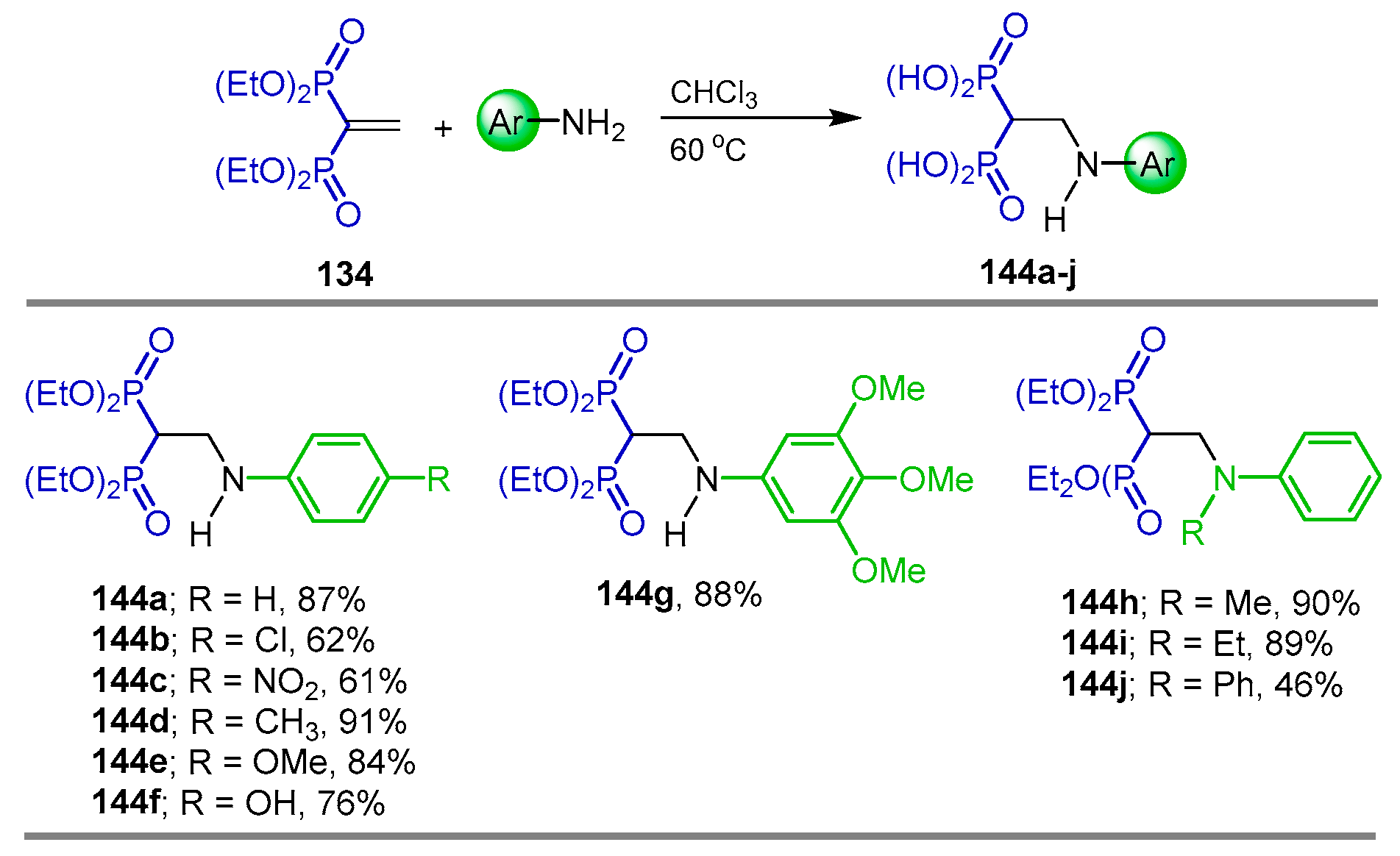
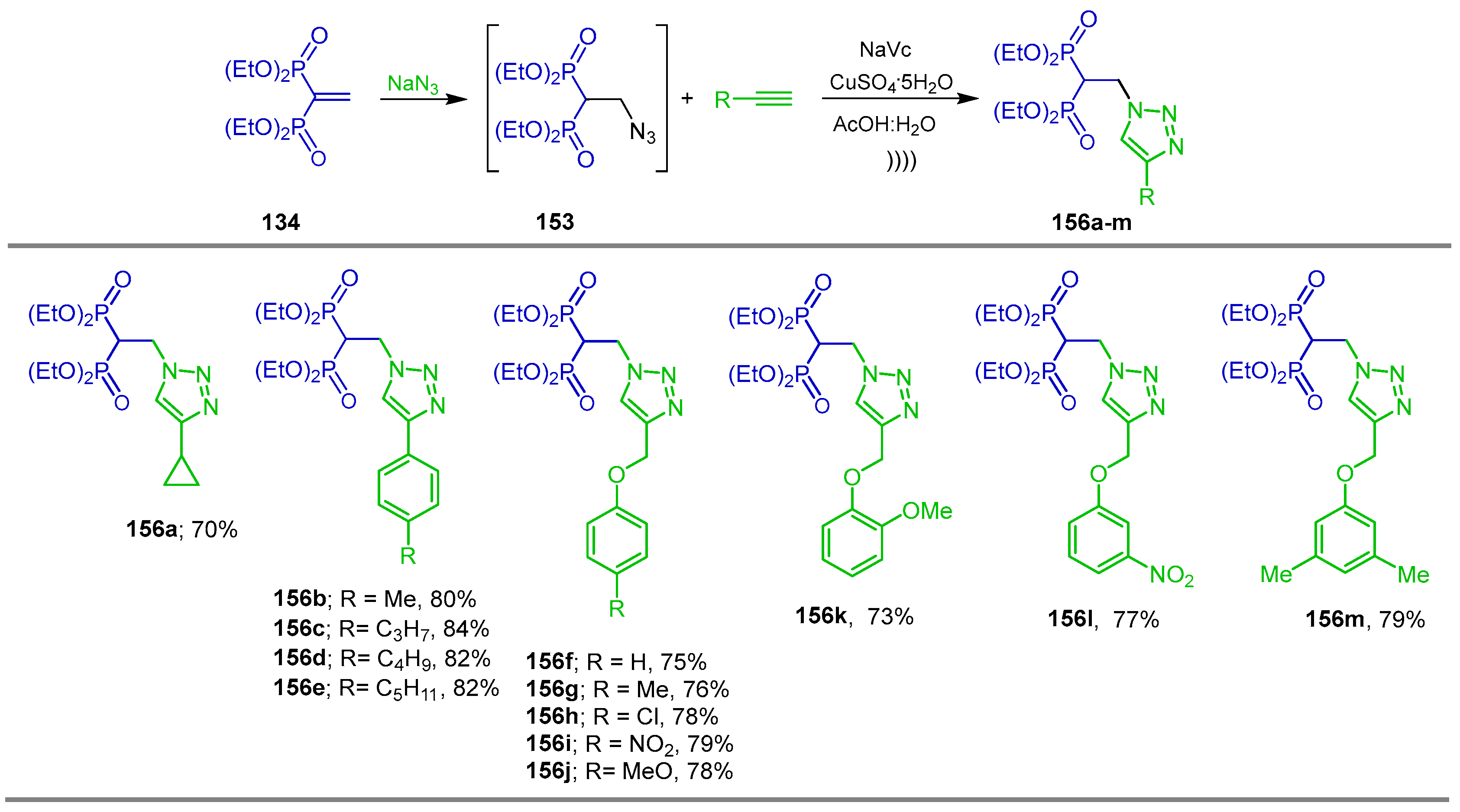
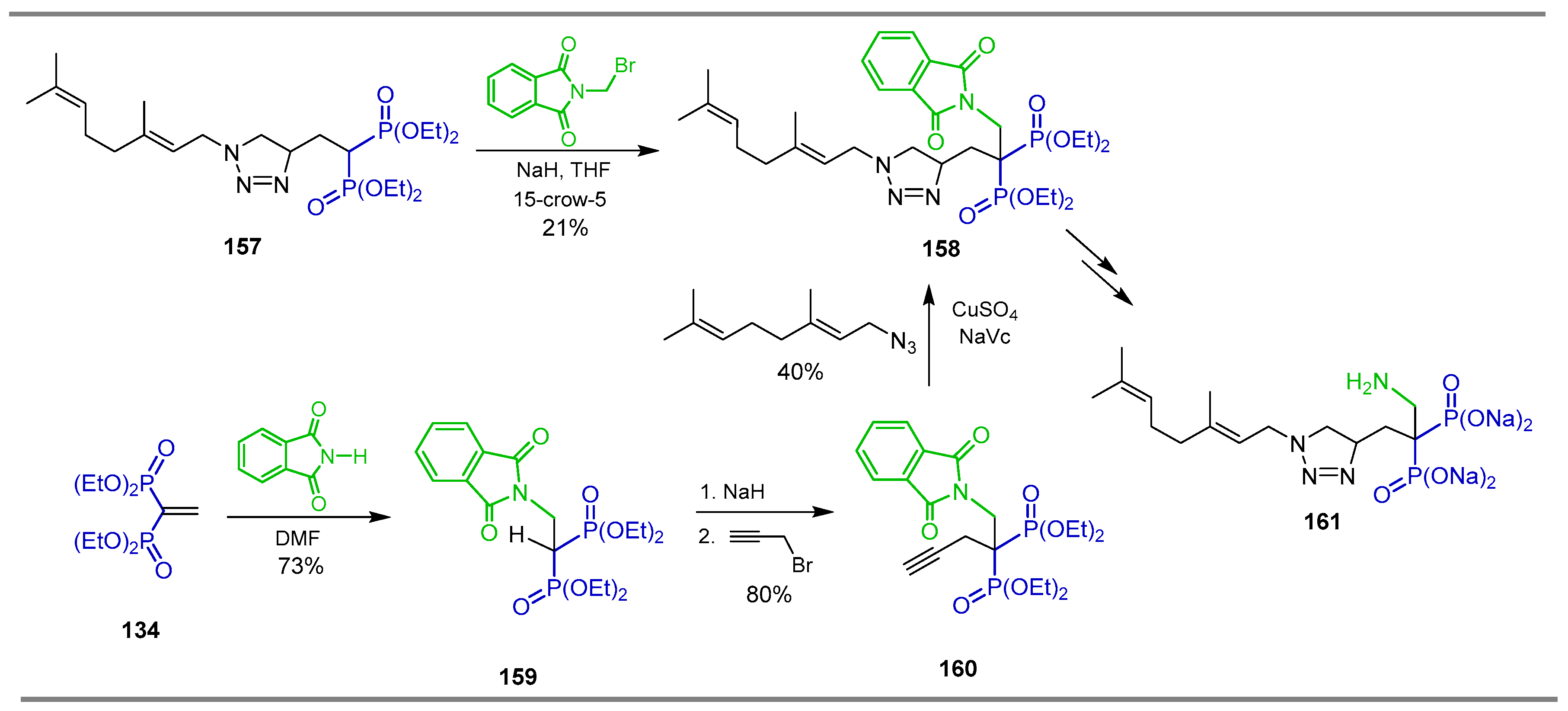
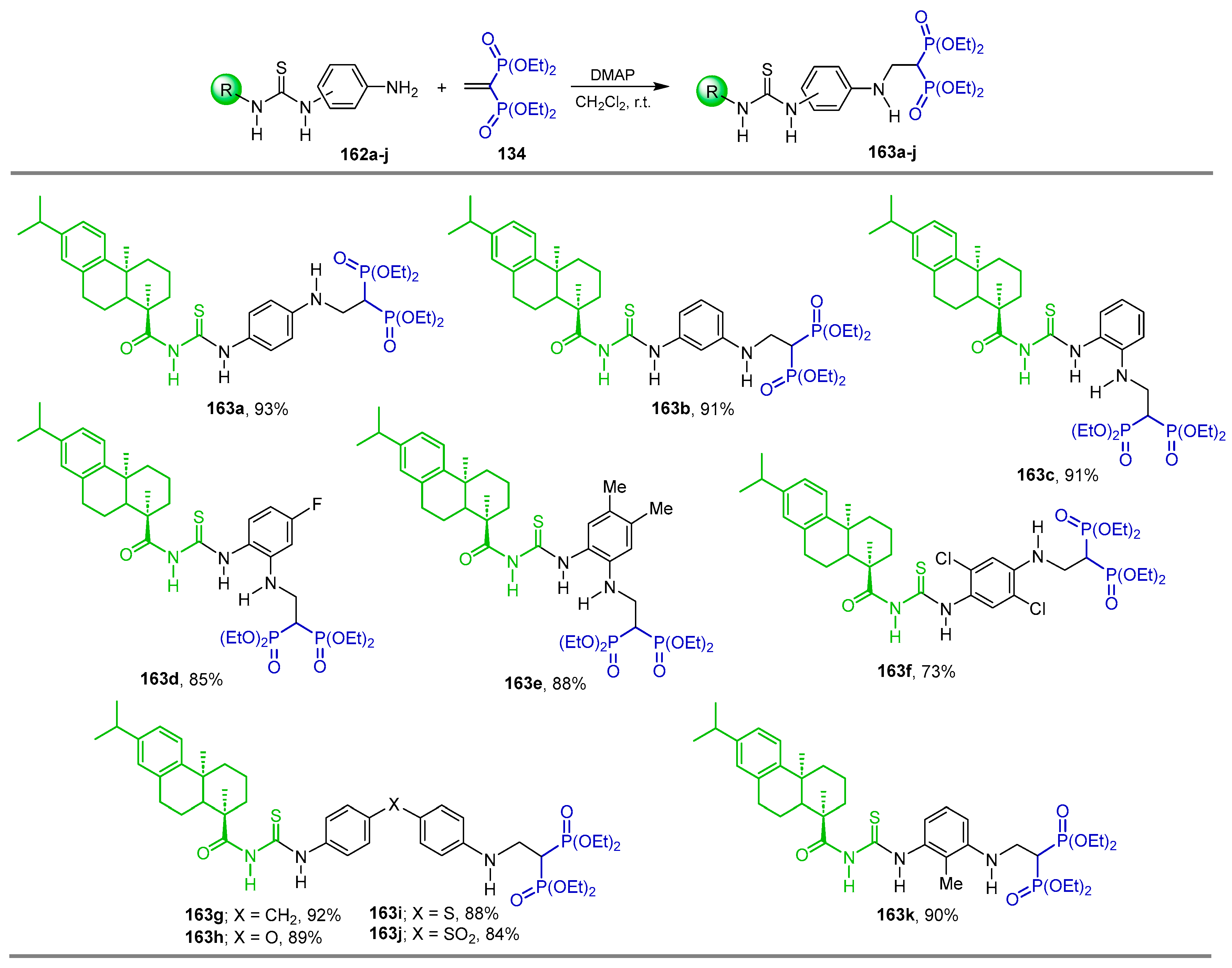
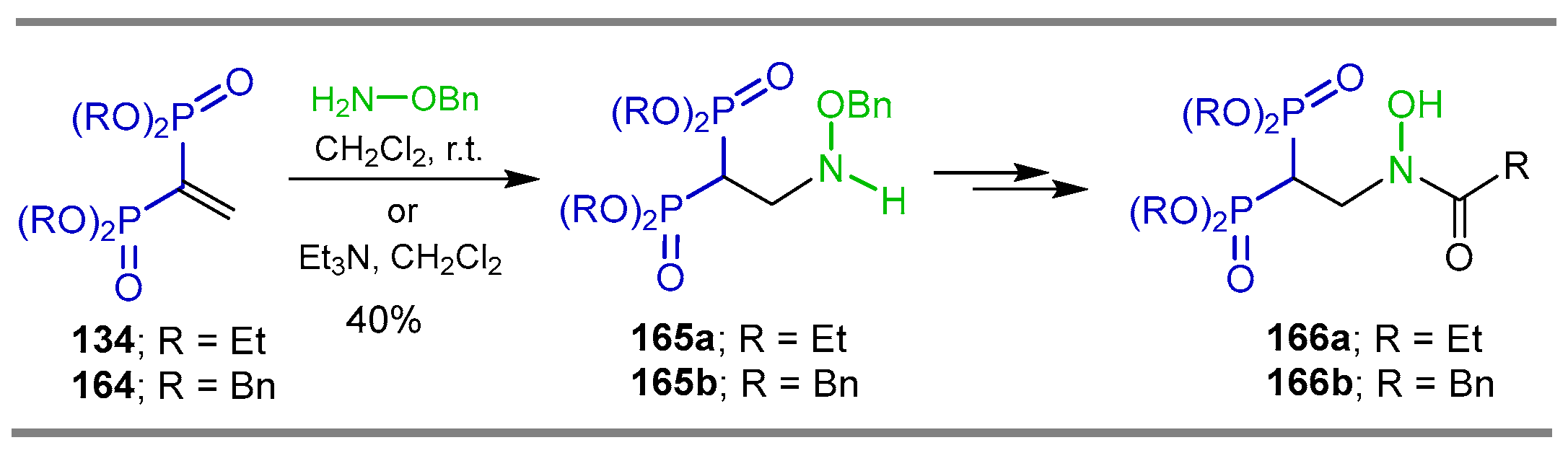
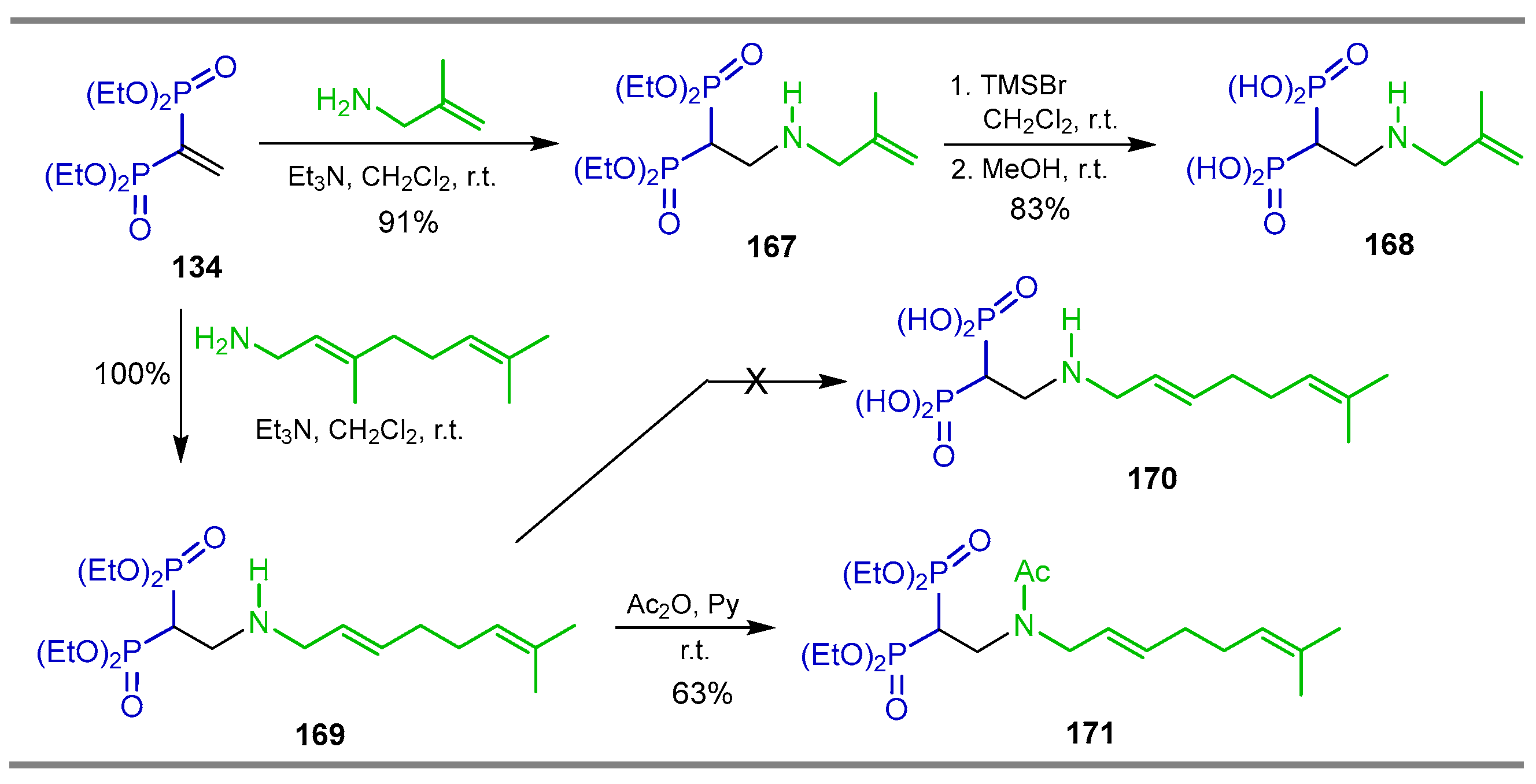
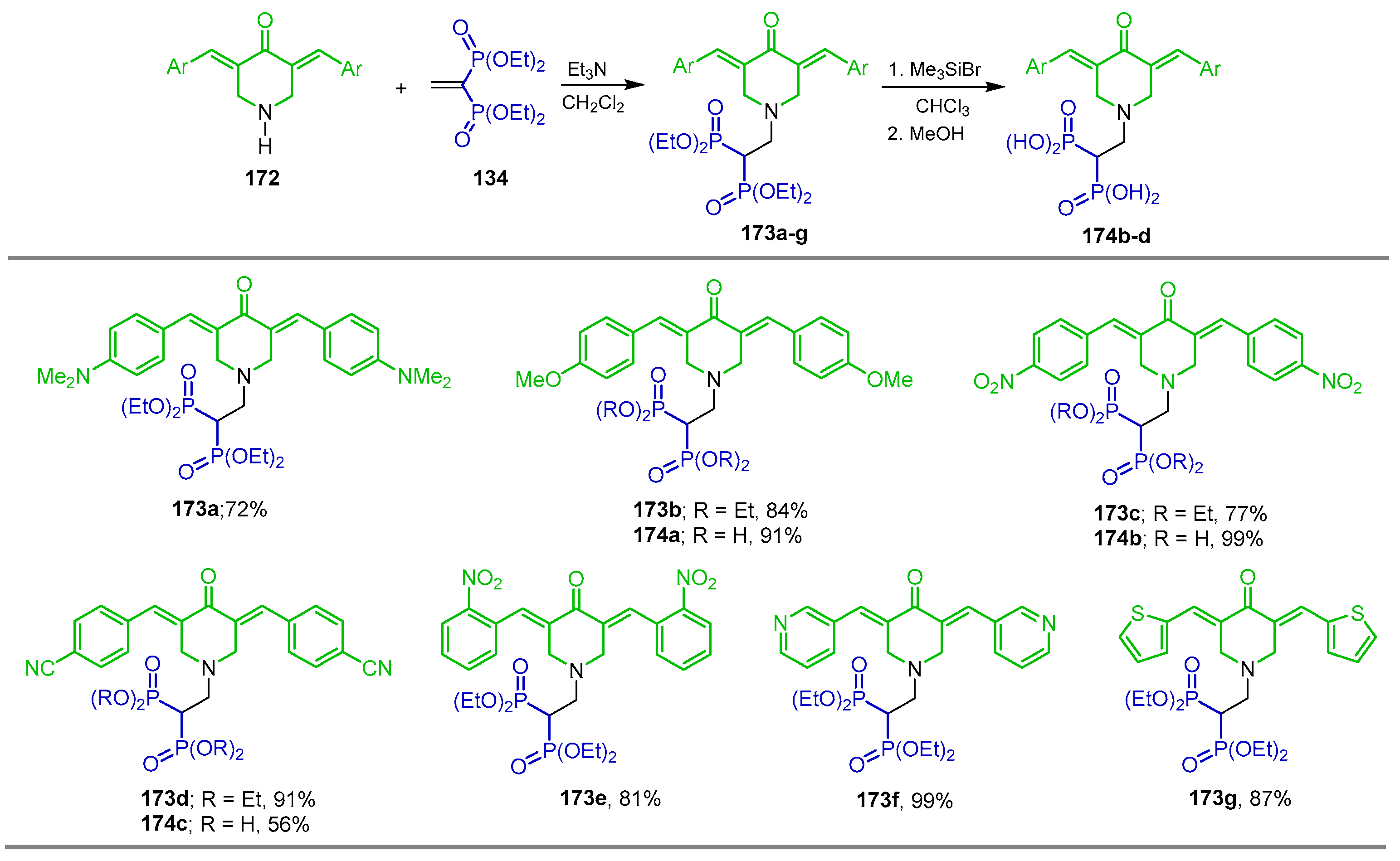
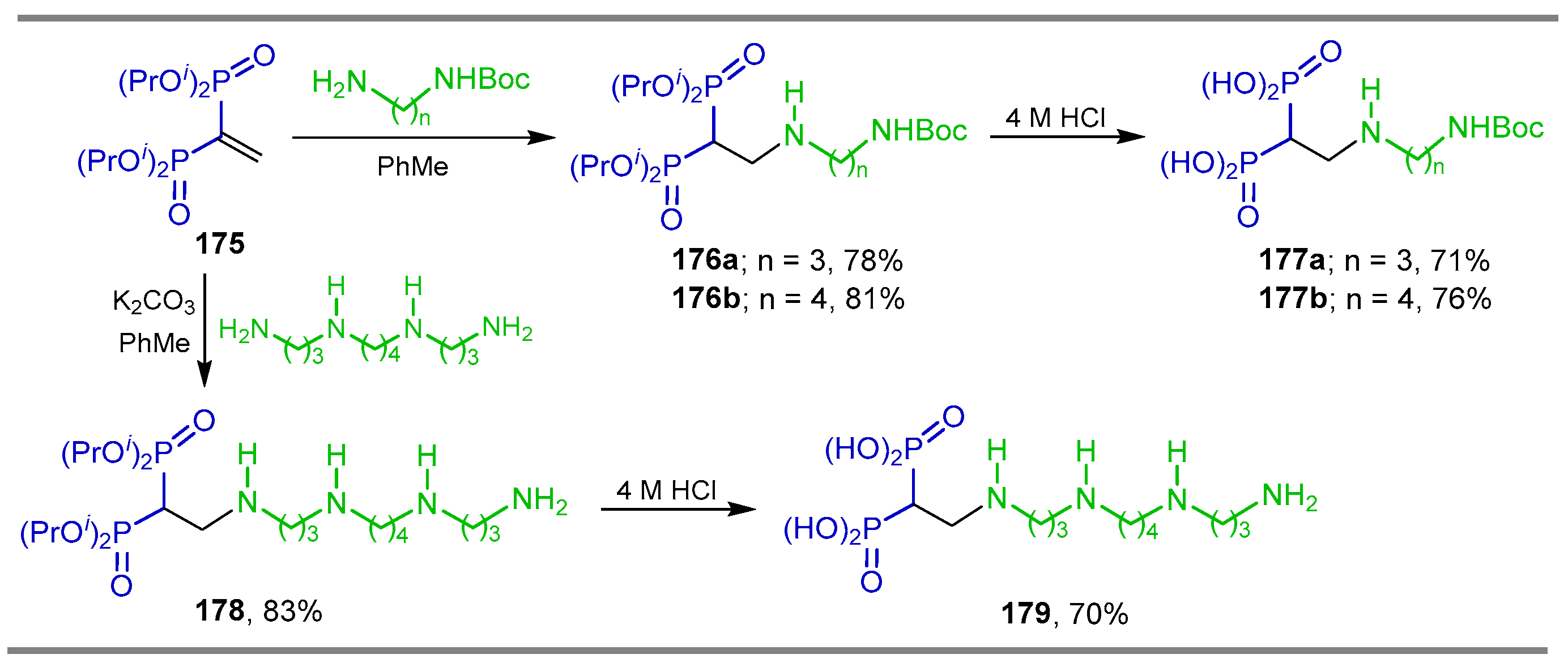
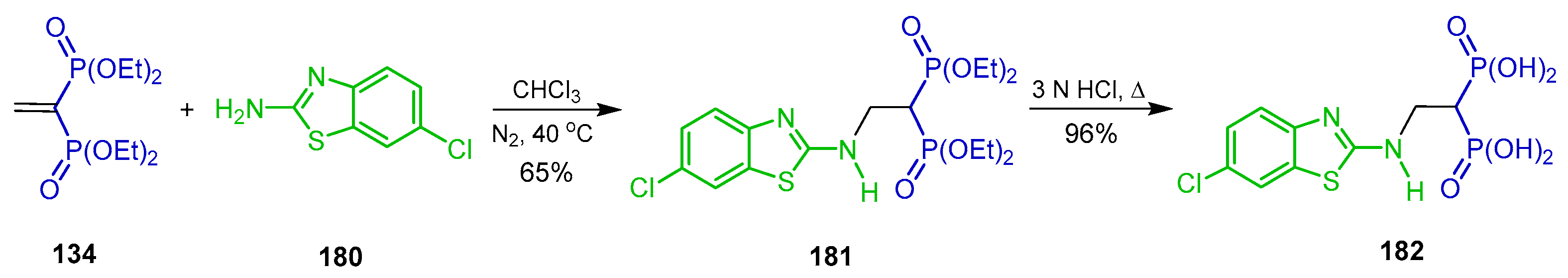
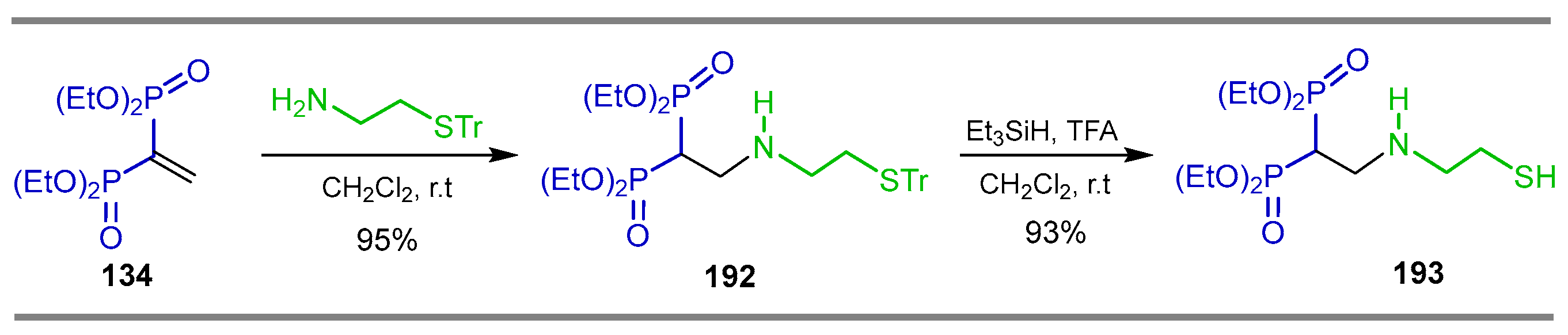
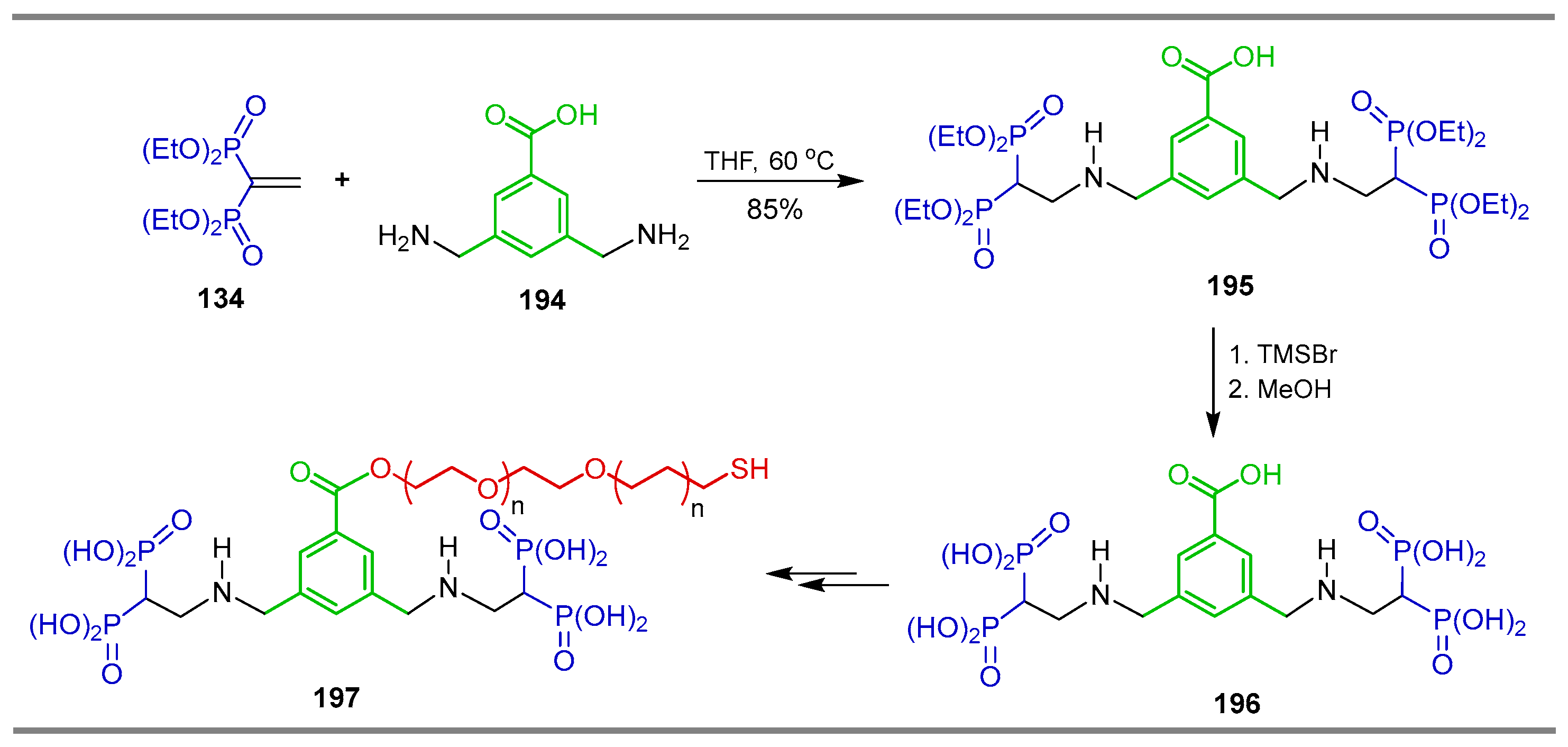
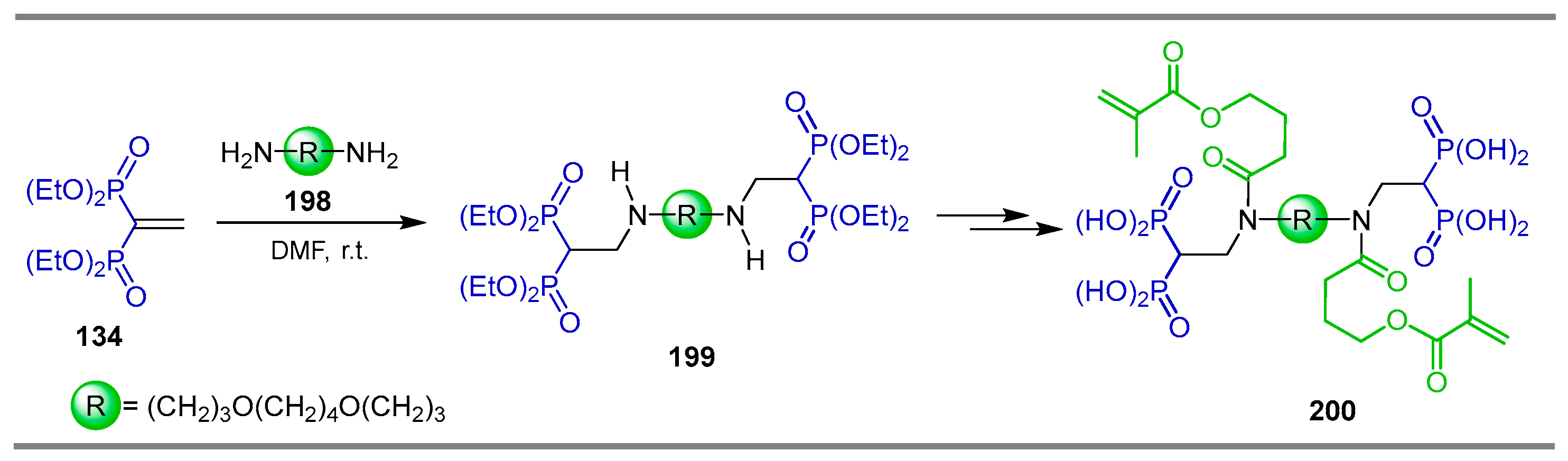
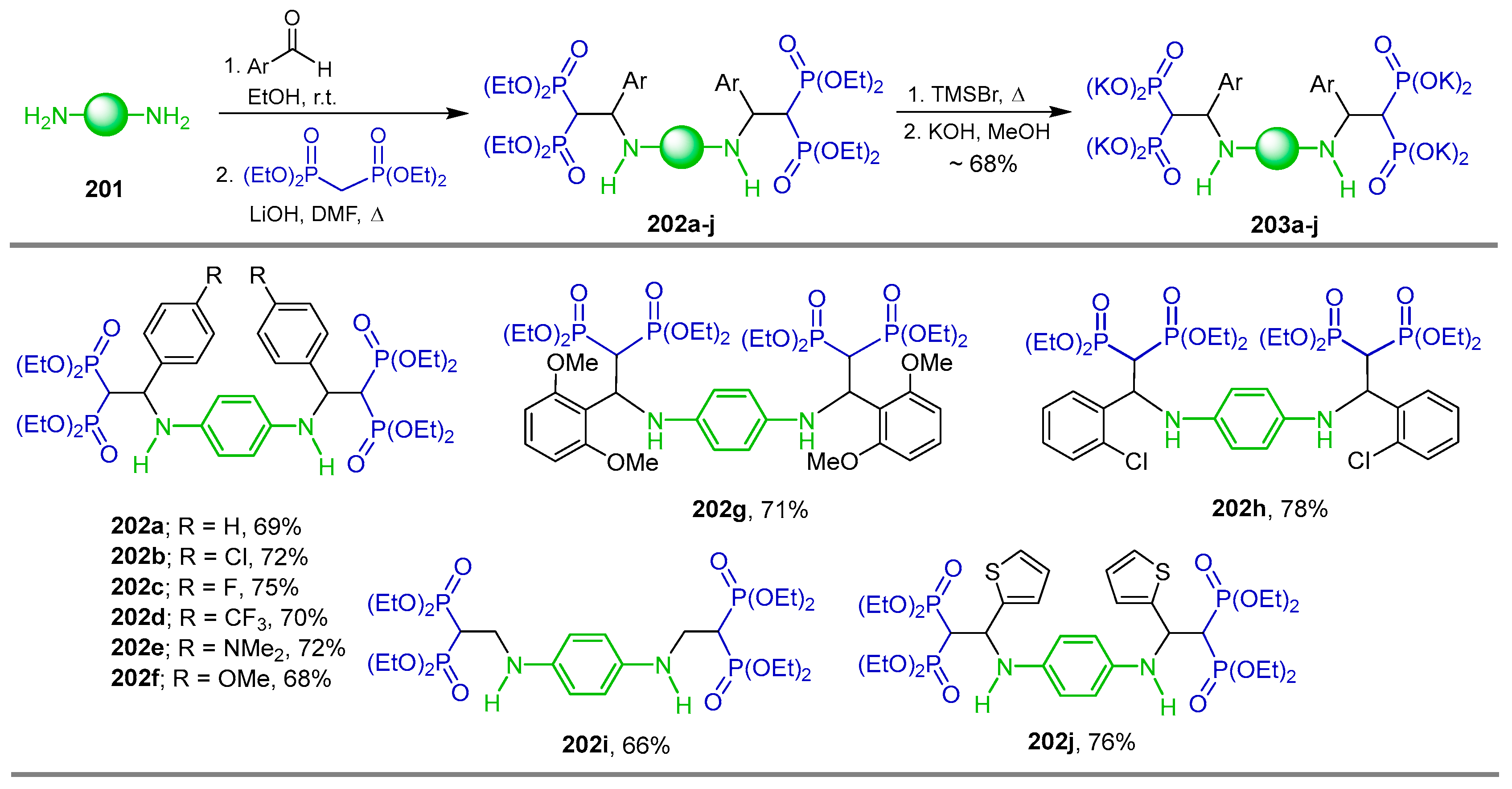


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Biological Activity | Ref. | Compound | Biological Activity | Ref. |
|---|---|---|---|---|---|
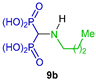 | Cytoprotection against cholesterol-dependent cy-tolysin | [28] | 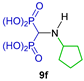 | Antiparasitary against Trypanosoma cruzi and Toxoplasma gondii | [29] |
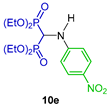 | Antibacterial (Gram positive and Gram negative) and fungi activity and antioxidant activity | [30] |  | Antimycobacterial against Mycobacterium tuberculosis | [31] |
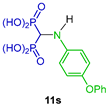 | Antimetastatic agents for bone malignancies | [32] | 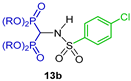 | Antiresorptive | [33] |
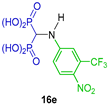 | Antiosteoporotic | [34] | 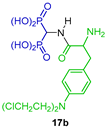 | Affinity to hydroxyapatite | [35] |
 | Anti-osteolytic agent | [36] | 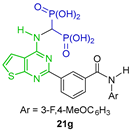 | Inhibit proliferation of human multiple myeloma cells (hGGPPS) | [37] |
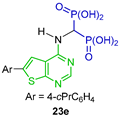 | Inhibit proliferation of human multiple myeloma cells (hFPPS) | [38] | 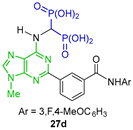 | Inhibit proliferation of human multiple myeloma cells (hGGPPS) | [39] |
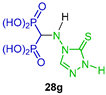 | Anti-inflammatory | [40] | 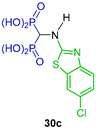 | Bone-targeted MMP-13 inhibitors | [41] |
 | Human farnesyl pyrophosphate synthase inhibitor | [42] | 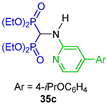 | Human farnesyl pyrophosphate synthase inhibitor | [43] |
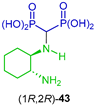 | Cytotoxic activity against human melanoma A375 and human colorectal adenocarcinoma HT29 cells | [44] | 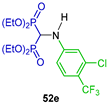 | Antibacterial (E. coli, Klebsiellapneumonia and Pseudomonasaerogenos) and antifungal activity (Canadida albicans and Aspergillus fumigates) | [45] |
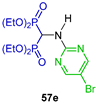 | Antibacterial (Gram positive and Gram negative) and fungi activity and antioxidant activity | [46] | 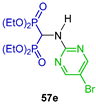 | Cytotoxic activity against human breast (MCF-7), prostate (DU-145), osteosarcoma (MG-63), fibrosarcoma (HT-1080) and multiple myeloma (RPMI-8226) cancer cell lines | [47] |
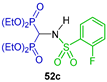 | Anti-inflammatory | [48] | 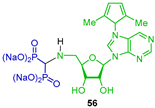 | Recombinant CD73 and cellular CD73 inhibition | [30] |
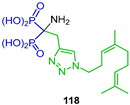 | Inhibitory activity against GGDPS | [49] | 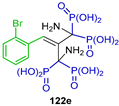 | Anti-osteoclastogenic | [50] |
 | Anti-osteolytic | [51] | 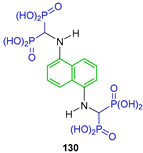 | Activity toward osteoclast precursors in vitro/antiproliferative and proapoptotic | [52] |
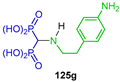
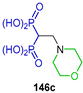
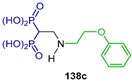 | Action against Toxoplasma gondii proliferation | [53] | 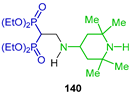 | Antitumor activity against carcinoma CRL1642 LLC | [54] |
 | Inhibition of human osteoclast activity | [55] | 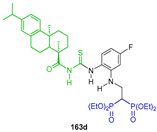 | Anticancer activity against the SK-OV-3, BEL-7404, A549, HCT-116 and NCI-H460 tumor cell lines in vitro | [56] |
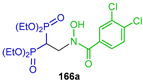 | Proherbicide | [57] | 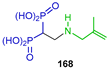 | Activity against the target enzymes TcFPPS and TgFPPS | [58] |
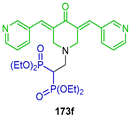 | Inhibitory properties towards human carcinoma cell lines (Caov3, A549, PC3 and KB 3-1) | [59] |  | Theranostic applications | [60] |
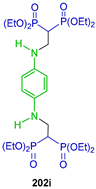 | Antitumor activity against SK-MEL-2 and SK-MEL-5 (I/II-stage), UACC-257, UACC-62 and LOX-IMVI cell lines | [61] |  | Cytogenetic activity in normal human lymphocyte cultures | [62] |
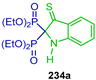 | Anti-inflamatory | [63] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ordoñez, M.; Argüello Velasco, R.O. Synthesis of Amino-gem-Bisphosphonate Derivatives and Their Application as Synthons for the Preparation of Biorelevant Compounds. Pharmaceuticals 2025, 18, 1063. https://doi.org/10.3390/ph18071063
Ordoñez M, Argüello Velasco RO. Synthesis of Amino-gem-Bisphosphonate Derivatives and Their Application as Synthons for the Preparation of Biorelevant Compounds. Pharmaceuticals. 2025; 18(7):1063. https://doi.org/10.3390/ph18071063
Chicago/Turabian StyleOrdoñez, Mario, and Rubén Oswaldo Argüello Velasco. 2025. "Synthesis of Amino-gem-Bisphosphonate Derivatives and Their Application as Synthons for the Preparation of Biorelevant Compounds" Pharmaceuticals 18, no. 7: 1063. https://doi.org/10.3390/ph18071063
APA StyleOrdoñez, M., & Argüello Velasco, R. O. (2025). Synthesis of Amino-gem-Bisphosphonate Derivatives and Their Application as Synthons for the Preparation of Biorelevant Compounds. Pharmaceuticals, 18(7), 1063. https://doi.org/10.3390/ph18071063