
Synergistic Autophagy-Related Mechanisms of Protection Against Brain Aging and AD: Cellular Pathways and Therapeutic Strategies

 ,
, 
Abstract
1. Introduction
2. Mismatch Between Senescence and Autophagy, a Critical Switch Initiating and Propagating Neurovascular Dysfunction and Blood–Brain Barrier Integrity
3. Mechanisms of Impaired Autophagy, Associated Neurodegeneration, and Novel Therapeutic Strategies for Protection of the Neurovascular Unit (NVU) and BBB
3.1. Senolytic Mechanisms and Pharmacological Intervention for Neurovascular Protection
3.2. Re-Normalization of Autophagy to Balance Senolysis, and Restoration of the NVU/BBB Microenvironment
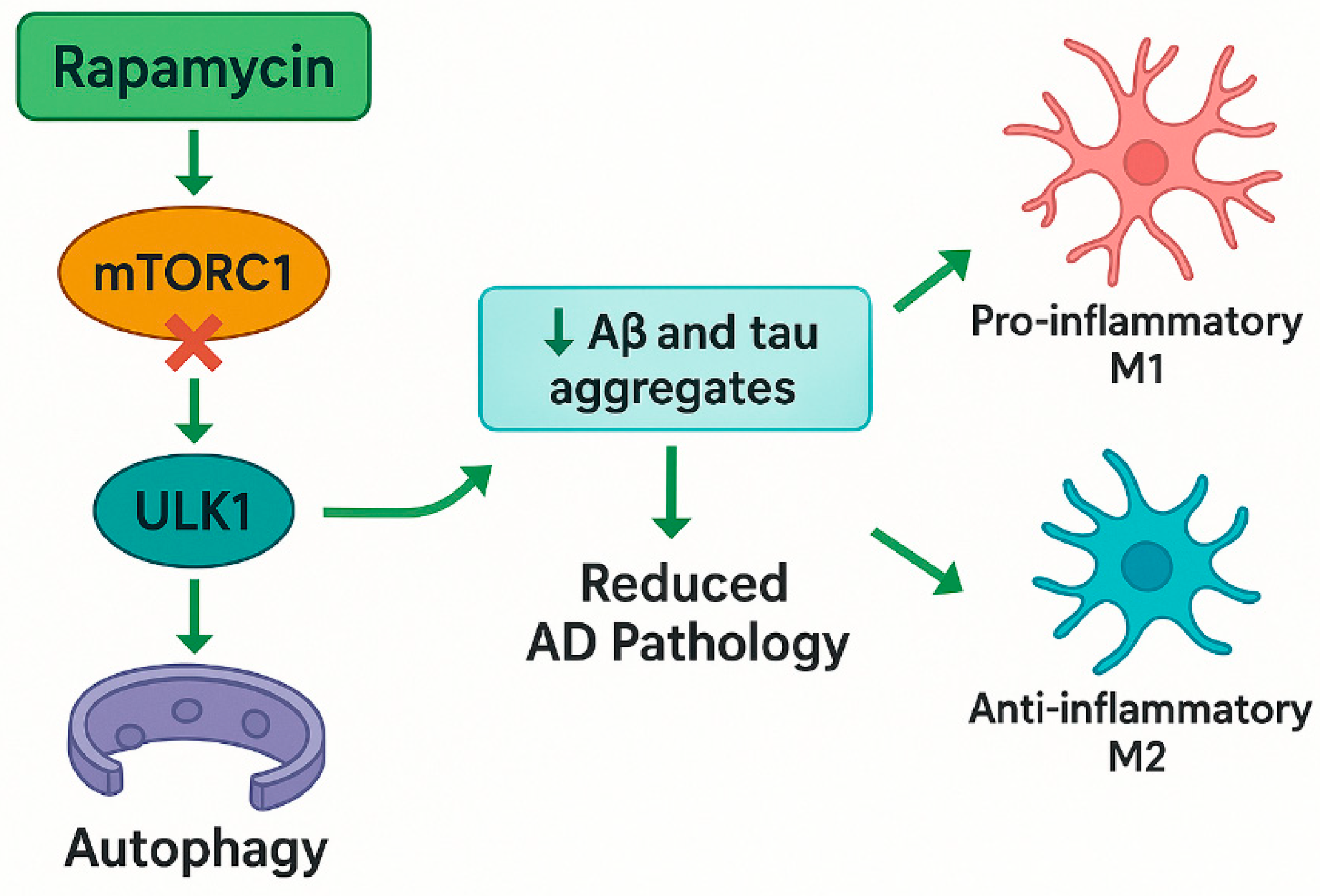
3.2.1. Rapamycin and mTOR Inhibition, a Cornerstone of Autophagic Modulation
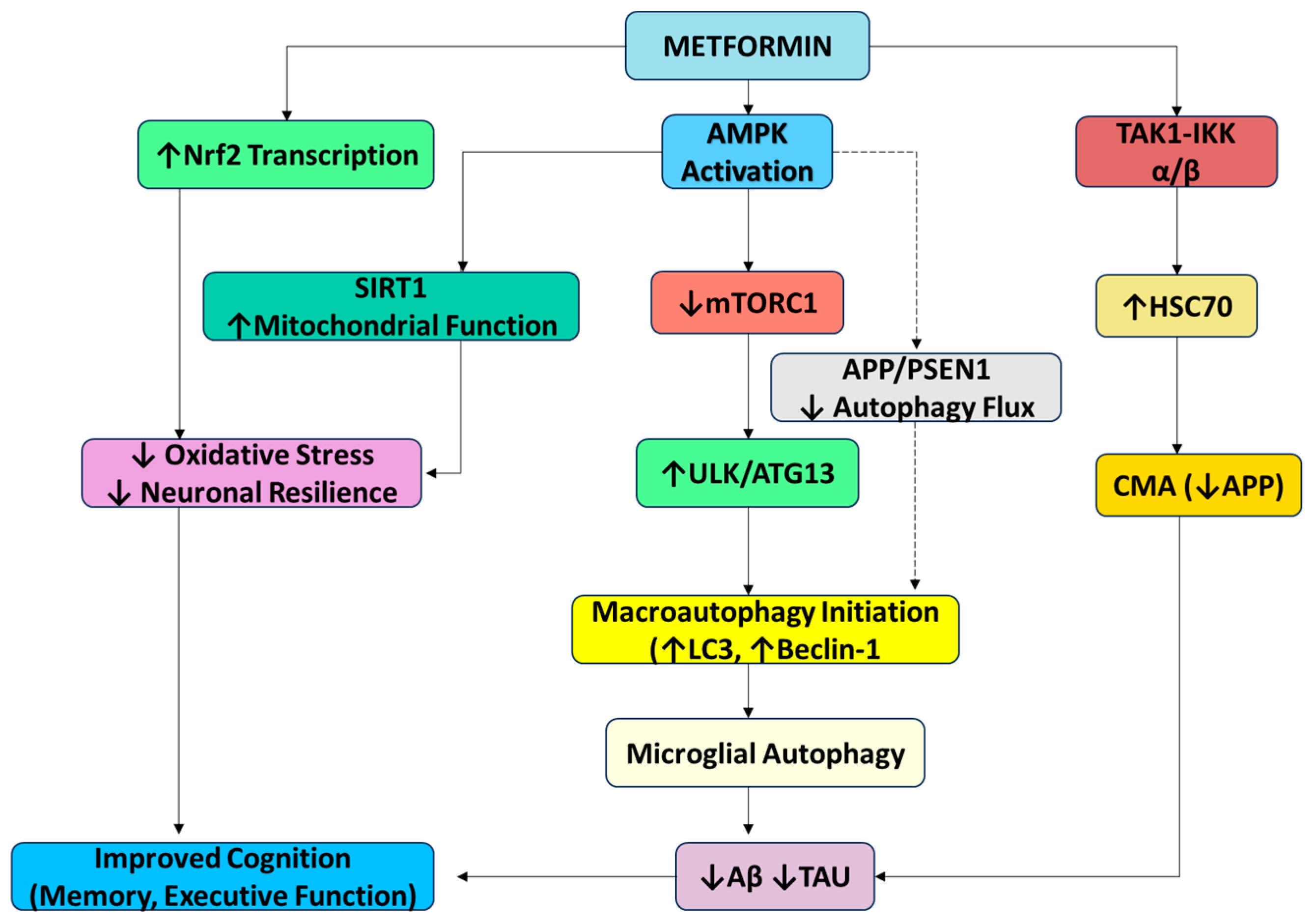
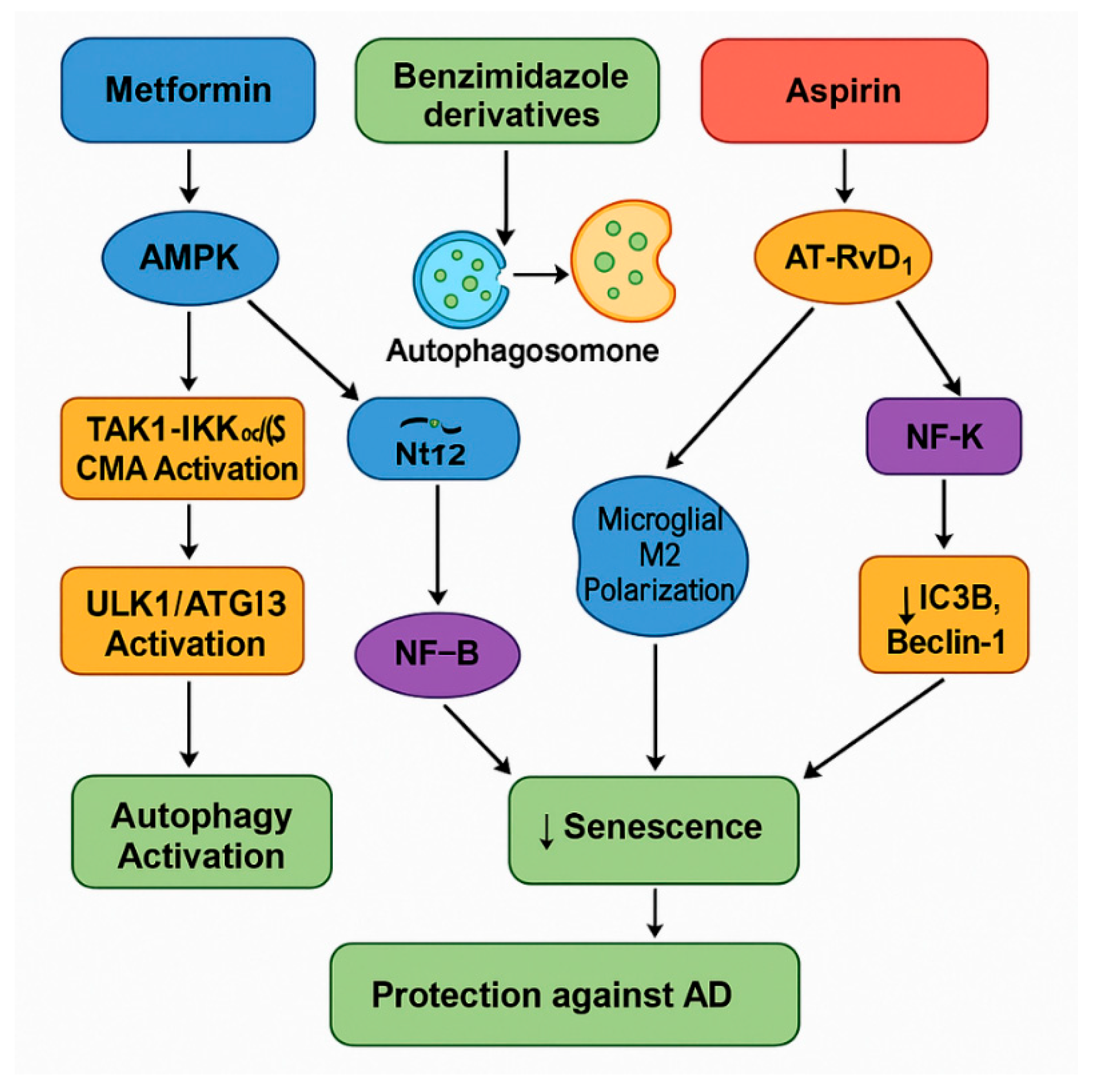
3.2.2. Metformin (AMPK Activator) Could Be a Multifaceted Therapeutic Agent in Autophagy, AD, and Anti-Aging
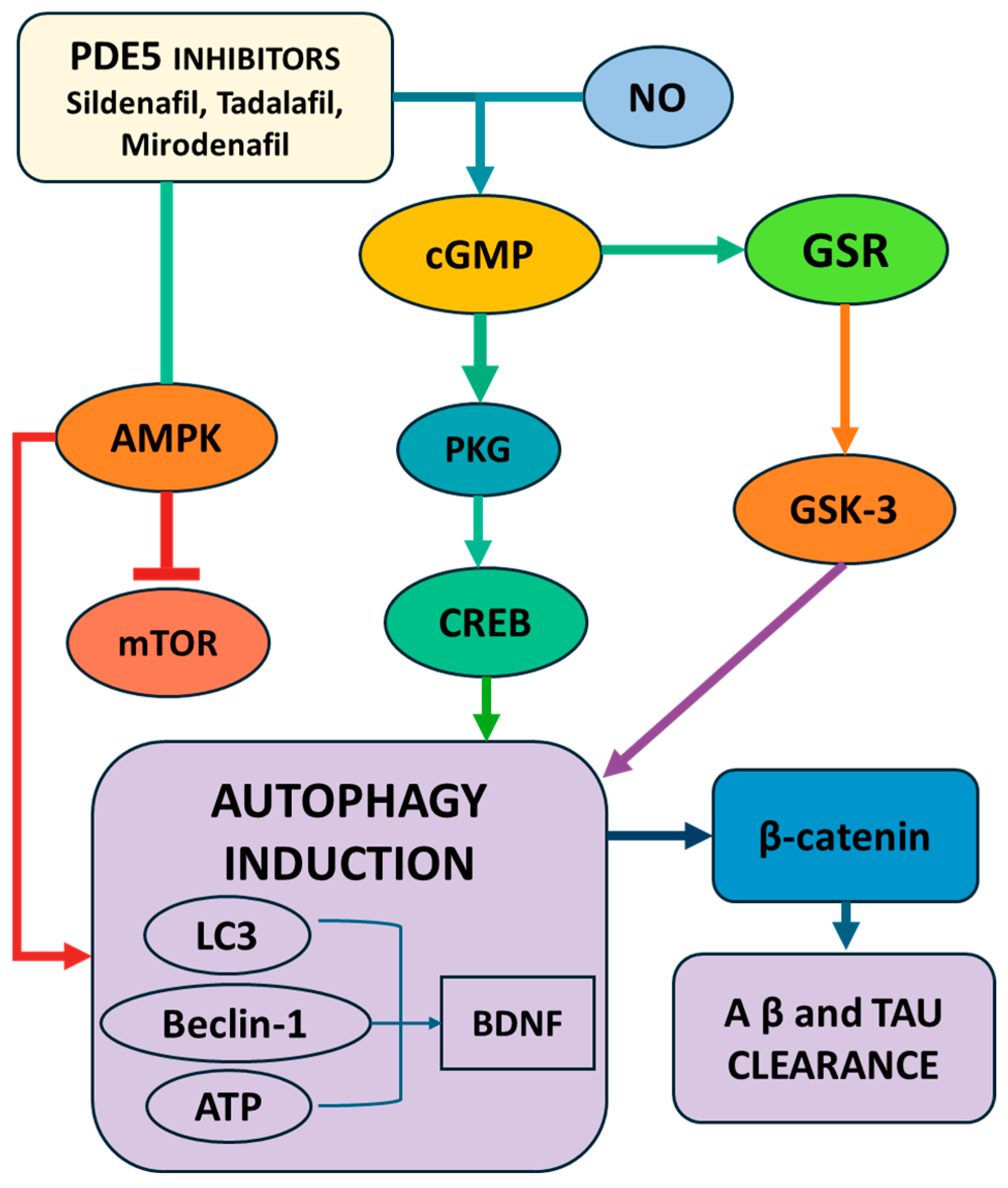
3.2.3. PDE5 Inhibitors as Novel Therapeutics in AD; Mechanisms and Evidence
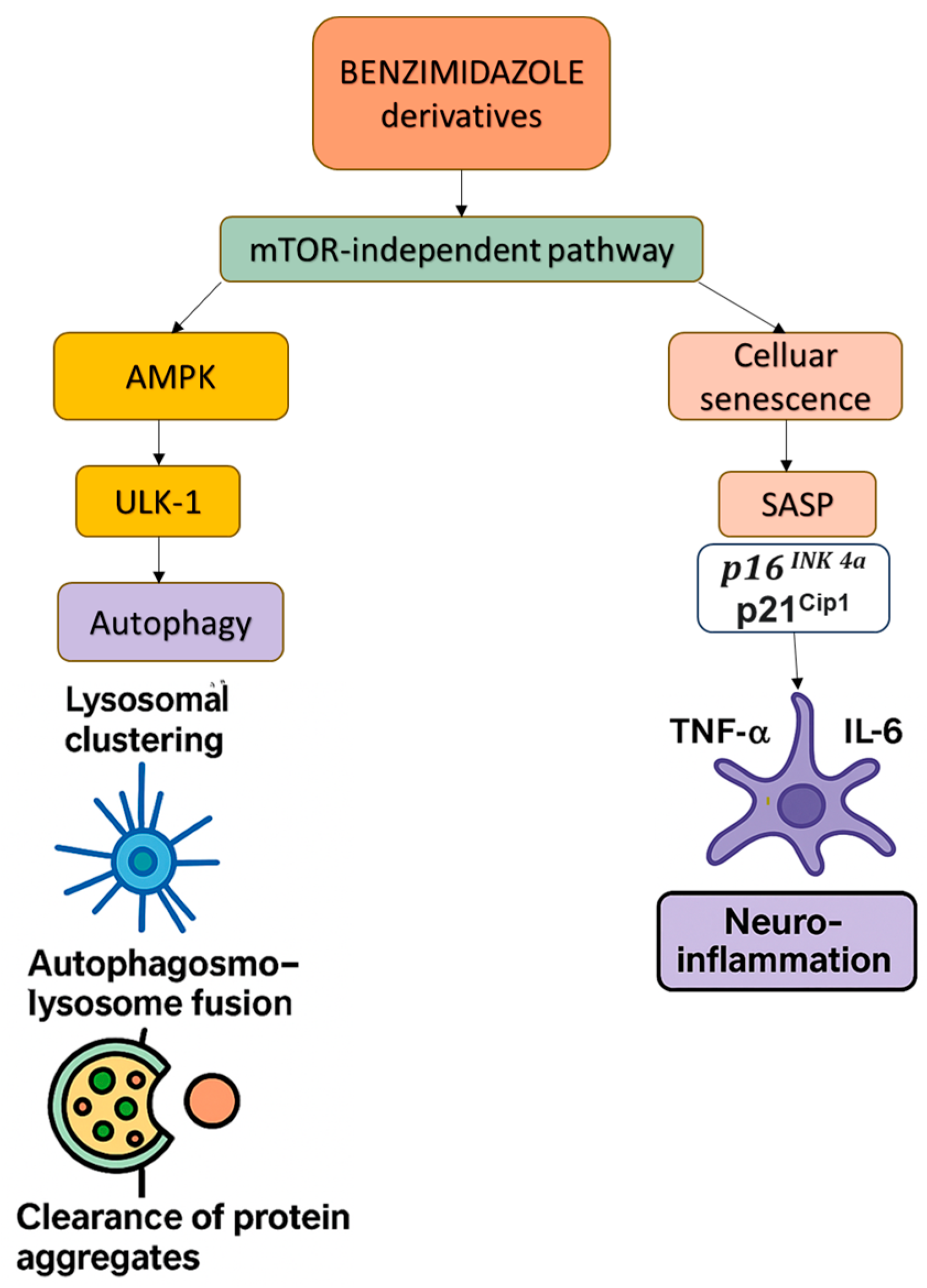
3.2.4. Benzimidazole Derivatives: Repurposed for Autophagy and Senescence Modulation in AD
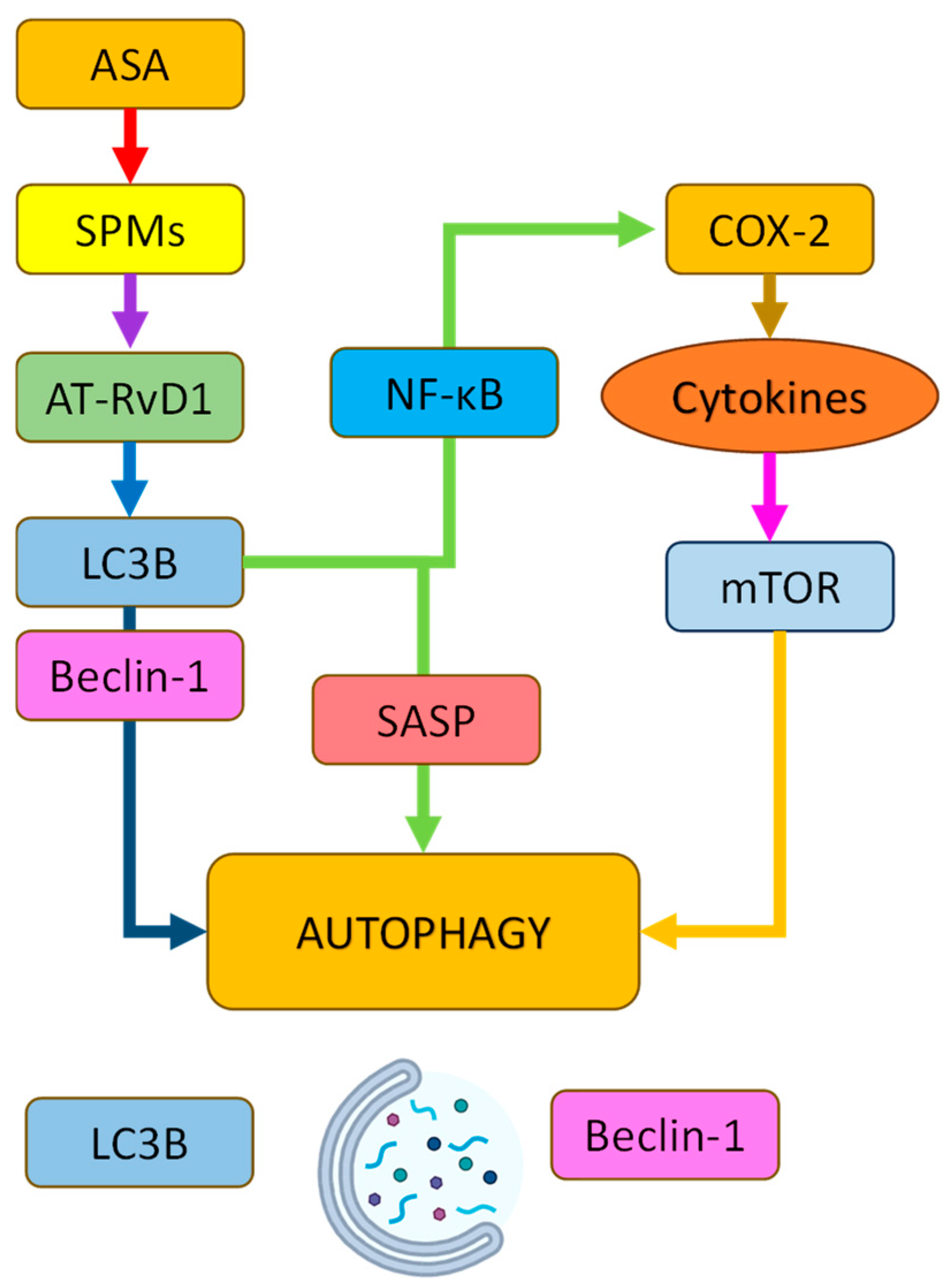
3.2.5. Acetylsalicylic Acid (ASA) in Autophagy, Senescence, and Neurodegeneration: Mechanisms of Action and Therapeutic Potential
3.2.6. A Note About Statins
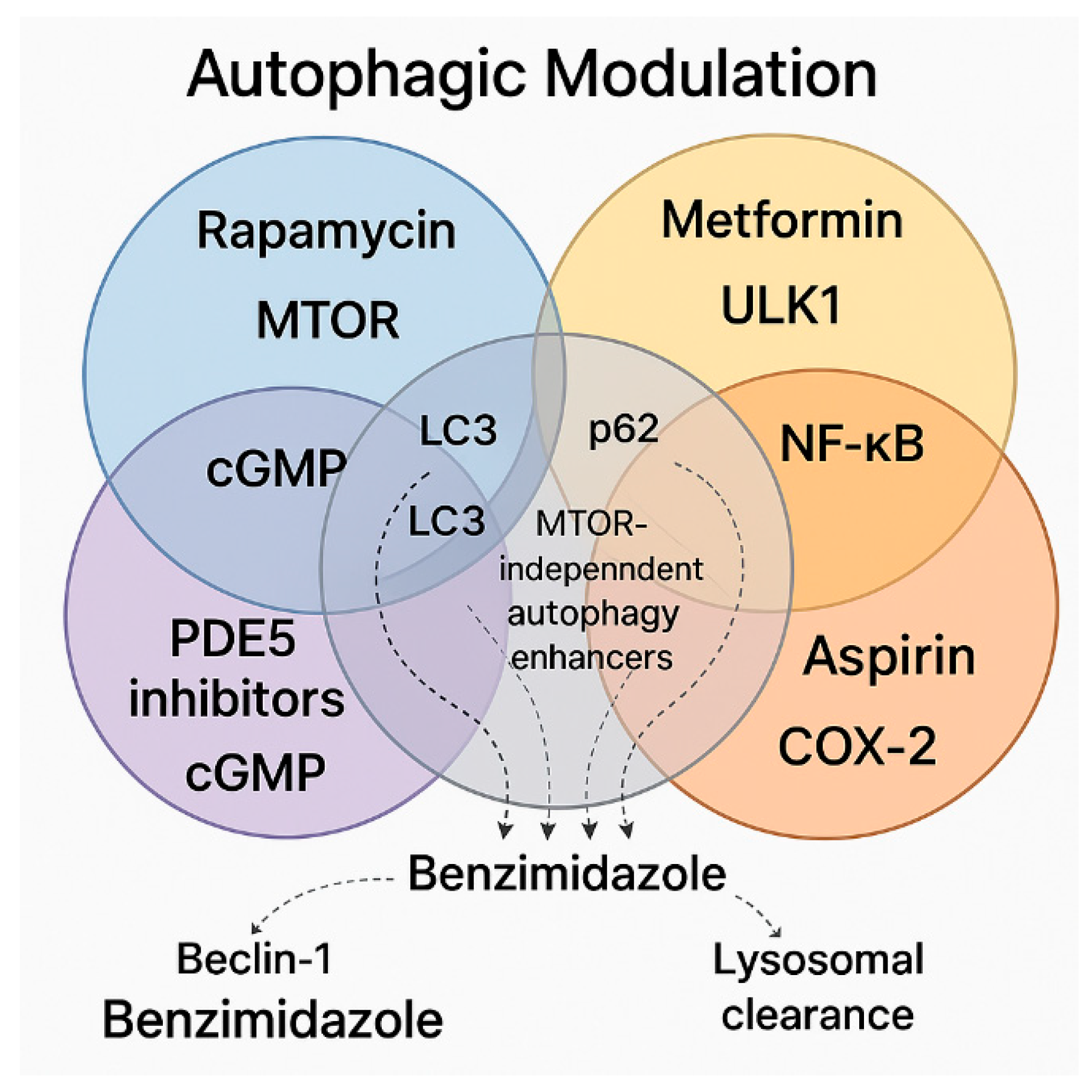
4. Synergistic and Overlapping Signaling Molecules in Autophagic Modulation: A Case for Therapeutic Combination
4.1. Key Pathways and Unique Synergies
4.2. Therapeutic–Protective Composite
4.3. Further Hypothesized Combinational Strategies That Could Support AD Protection
4.4. A Targeted and Personalized/Stratified Approach
4.5. Limitations and Considerations for This Approach
4.6. Alternative Autophagy-Modulating Compounds for AD
5. Conclusions
6. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 8-OHdG | 8-hydroxy-2′-deoxyguanosine |
| AChE | Acetylcholinesterase |
| AD | Alzheimer’s disease |
| Akt | Protein Kinase B |
| ALS | Amyotrophic Lateral Sclerosis |
| AMPK | AMP-activated protein kinase |
| APOE4 | Apolipoprotein E4 |
| APP | Amyloid Precursor Protein |
| ASA | Acetylsalicylic Acid |
| AT-RvD1 | Aspirin-Triggered Resolvin D1 |
| Aβ | β-amyloid |
| BBB | Blood–Brain Barrier |
| BChE | Butyrylcholinesterase |
| BDNF | Brain-Derived Neurotrophic Factor |
| BPH | Benign Prostatic Hyperplasia |
| cGMP | Cyclic Guanosine Monophosphate |
| CMA | Chaperone-Mediated Autophagy |
| COX | Cyclooxygenase |
| CREB | cAMP Response Element-Binding Protein |
| ED | Erectile Dysfunction |
| FLBZ | Flubendazole |
| GGPP | Geranylgeranyl Pyrophosphate |
| GSK3β | Glycogen Synthase Kinase 3 Beta |
| HMGB1 | High mobility group box 1 protein |
| HOMA-IR | Homeostasis Model Assessment of Insulin Resistance |
| IL-6 | Interleukin-6 |
| LC3 | Microtubule-associated protein 1 light chain 3 |
| LTP | Long-Term Potentiation |
| MAPK | Mitogen-Activated Protein Kinase |
| MMP | Matrix Metalloproteinase |
| mTOR | Mammalian Target of Rapamycin |
| mTORC1 | Mammalian Target of Rapamycin Complex 1 |
| NF-κB | Nuclear factor kappa B |
| NO | Nitric Oxide |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| OXPHOS | Oxidative Phosphorylation |
| PAH | Pulmonary Arterial Hypertension |
| PD | Parkinson’s disease |
| PDE5 | Phosphodiesterase-5 |
| PGC-1α | Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-alpha |
| PPAR-γ | Peroxisome proliferator-activated receptor gamma |
| ROS | Reactive Oxygen Species |
| RPE | Retinal Pigment Epithelial Cells |
| SASP | Senescence-Associated Secretory Phenotype |
| SIRT1 | Sirtuin 1 |
| SPMs | Specialized Pro-resolving Mediators |
| TNF-α | Tumor Necrosis Factor alpha |
| ULK-1 | Unc-51-Like Autophagy Activating Kinase 1 |
References
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A New Immune–Metabolic Viewpoint for Age-Related Diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cué, C.; Rueda, N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef]
- Xue, L.; He, W.; Zhang, Y.; Wang, Z.; Chen, H.; Chen, Z.; Zhu, W.; Liu, D.; Jia, H.; Jiang, Y.; et al. Origins of Biallelic Inactivation of NF2 in Neurofibromatosis Type 2. Neuro-Oncol. 2022, 24, 903–913. [Google Scholar] [CrossRef]
- Ting, K.K.; Coleman, P.; Kim, H.J.; Zhao, Y.; Mulangala, J.; Cheng, N.C.; Li, W.; Gunatilake, D.; Johnstone, D.M.; Loo, L.; et al. Vascular Senescence and Leak Are Features of the Early Breakdown of the Blood–Brain Barrier in Alzheimer’s Disease Models. GeroScience 2023, 45, 3307–3331. [Google Scholar] [CrossRef]
- Herdy, J.R.; Traxler, L.; Agarwal, R.K.; Karbacher, L.; Schlachetzki, J.C.M.; Boehnke, L.; Zangwill, D.; Galasko, D.; Glass, C.K.; Mertens, J.; et al. Increased Post-Mitotic Senescence in Aged Human Neurons Is a Pathological Feature of Alzheimer’s Disease. Cell Stem Cell 2022, 29, 1637–1652.e6. [Google Scholar] [CrossRef]
- Han, X.; Zhang, T.; Liu, H.; Mi, Y.; Gou, X. Astrocyte Senescence and Alzheimer’s Disease: A Review. Front. Aging Neurosci. 2020, 12, 148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Q.; Li, S.; Li, X.-J.; Yang, W.; He, D. Microglial Autophagy in Alzheimer’s Disease and Parkinson’s Disease. Front. Aging Neurosci. 2023, 14, 1065183. [Google Scholar] [CrossRef]
- Nixon, R.A. Amyloid Precursor Protein and Endosomal-lysosomal Dysfunction in Alzheimer’s Disease: Inseparable Partners in a Multifactorial Disease. FASEB J. 2017, 31, 2729–2743. [Google Scholar] [CrossRef]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in Neurodegenerative Diseases: Pathogenesis and Therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a Risk Factor for Neurodegenerative Disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-A. Neuronal Autophagy: A Housekeeper or a Fighter in Neuronal Cell Survival? Exp. Neurobiol. 2012, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, X.; Song, Y.-Q.; Tu, J. Autophagy in Alzheimer’s Disease Pathogenesis: Therapeutic Potential and Future Perspectives. Ageing Res. Rev. 2021, 72, 101464. [Google Scholar] [CrossRef]
- Lambert, M.; Miquel, G.; Villeneuve, L.; Thorin-Trescases, N.; Thorin, E. The Senolytic ABT-263 Improves Cognitive Functions in Middle-Aged Male, but Not Female, Atherosclerotic LDLr−/−;hApoB100+/+ Mice. GeroScience 2025, 1–24. [Google Scholar] [CrossRef]
- Lelarge, V.; Capelle, R.; Oger, F.; Mathieu, T.; Le Calvé, B. Senolytics: From Pharmacological Inhibitors to Immunotherapies, a Promising Future for Patients’ Treatment. npj Aging 2024, 10, 12. [Google Scholar] [CrossRef]
- Mannarino, M.; Cherif, H.; Ghazizadeh, S.; Martinez, O.W.; Sheng, K.; Cousineau, E.; Lee, S.; Millecamps, M.; Gao, C.; Gilbert, A.; et al. Senolytic Treatment for Low Back Pain. Sci. Adv. 2025, 11, eadr1719. [Google Scholar] [CrossRef] [PubMed]
- Aguado, J.; Amarilla, A.A.; Taherian Fard, A.; Albornoz, E.A.; Tyshkovskiy, A.; Schwabenland, M.; Chaggar, H.K.; Modhiran, N.; Gómez-Inclán, C.; Javed, I.; et al. Senolytic Therapy Alleviates Physiological Human Brain Aging and COVID-19 Neuropathology. Nat. Aging 2023, 3, 1561–1575. [Google Scholar] [CrossRef]
- Mansfield, L.; Ramponi, V.; Gupta, K.; Stevenson, T.; Mathew, A.B.; Barinda, A.J.; Herbstein, F.; Morsli, S. Emerging Insights in Senescence: Pathways from Preclinical Models to Therapeutic Innovations. npj Aging 2024, 10, 53. [Google Scholar] [CrossRef]
- Faakye, J.; Nyúl-Tóth, Á.; Muranyi, M.; Gulej, R.; Csik, B.; Shanmugarama, S.; Tarantini, S.; Negri, S.; Prodan, C.; Mukli, P.; et al. Preventing Spontaneous Cerebral Microhemorrhages in Aging Mice: A Novel Approach Targeting Cellular Senescence with ABT263/Navitoclax. GeroScience 2023, 46, 21–37. [Google Scholar] [CrossRef]
- Fatt, M.P.; Tran, L.M.; Vetere, G.; Storer, M.A.; Simonetta, J.V.; Miller, F.D.; Frankland, P.W.; Kaplan, D.R. Restoration of Hippocampal Neural Precursor Function by Ablation of Senescent Cells in the Aging Stem Cell Niche. Stem Cell Rep. 2022, 17, 259–275. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of Senescent Cells by ABT263 Rejuvenates Aged Hematopoietic Stem Cells in Mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Millar, C.L.; Iloputaife, I.; Baldyga, K.; Norling, A.M.; Boulougoura, A.; Vichos, T.; Tchkonia, T.; Deisinger, A.; Pirtskhalava, T.; Kirkland, J.L.; et al. A Pilot Study of Senolytics to Improve Cognition and Mobility in Older Adults at Risk for Alzheimer’s Disease. eBioMedicine 2025, 113, 105612. [Google Scholar] [CrossRef] [PubMed]
- Yabluchanskiy, A.; Tarantini, S.; Balasubramanian, P.; Kiss, T.; Csipo, T.; Fülöp, G.A.; Lipecz, A.; Ahire, C.; DelFavero, J.; Nyul-Toth, A.; et al. Pharmacological or Genetic Depletion of Senescent Astrocytes Prevents Whole Brain Irradiation–Induced Impairment of Neurovascular Coupling Responses Protecting Cognitive Function in Mice. GeroScience 2020, 42, 409–428. [Google Scholar] [CrossRef] [PubMed]
- Mançano, A.S.F.; Pina, J.G.; Froes, B.R.; Sciani, J.M. Autophagy-Lysosomal Pathway Impairment and Cathepsin Dysregulation in Alzheimer’s Disease. Front. Mol. Biosci. 2024, 11, 1490275. [Google Scholar] [CrossRef]
- Davoody, S.; Asgari Taei, A.; Khodabakhsh, P.; Dargahi, L. mTOR Signaling and Alzheimer’s Disease: What We Know and Where We Are? CNS Neurosci. Ther. 2024, 30, e14463. [Google Scholar] [CrossRef]
- Ríos, J.A.; Bórquez, J.C.; Godoy, J.A.; Zolezzi, J.M.; Furrianca, M.C.; Inestrosa, N.C. Emerging Role of Metformin in Alzheimer’s Disease: A Translational View. Ageing Res. Rev. 2024, 100, 102439. [Google Scholar] [CrossRef]
- Cai, J.; Xie, D.; Kong, F.; Zhai, Z.; Zhu, Z.; Zhao, Y.; Xu, Y.; Sun, T. Effect and Mechanism of Rapamycin on Cognitive Deficits in Animal Models of Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Preclinical Studies. J. Alzheimer’s Dis. 2024, 99, 53–84. [Google Scholar] [CrossRef]
- Hwang, J.-Y.; Gertner, M.; Pontarelli, F.; Court-Vazquez, B.; Bennett, M.V.L.; Ofengeim, D.; Zukin, R.S. Global Ischemia Induces Lysosomal-Mediated Degradation of mTOR and Activation of Autophagy in Hippocampal Neurons Destined to Die. Cell Death Differ. 2017, 24, 317–329. [Google Scholar] [CrossRef]
- Carosi, J.M.; Sargeant, T.J. Rapamycin and Alzheimer Disease: A Hypothesis for the Effective Use of Rapamycin for Treatment of Neurodegenerative Disease. Autophagy 2023, 19, 2386–2390. [Google Scholar] [CrossRef]
- Yun, Q.; Ma, S.-F.; Zhang, W.-N.; Gu, M.; Wang, J. FoxG1 as a Potential Therapeutic Target for Alzheimer’s Disease: Modulating NLRP3 Inflammasome via AMPK/mTOR Autophagy Pathway. Cell Mol. Neurobiol. 2024, 44, 35. [Google Scholar] [CrossRef]
- Rodríguez-Muela, N.; Germain, F.; Mariño, G.; Fitze, P.S.; Boya, P. Autophagy Promotes Survival of Retinal Ganglion Cells after Optic Nerve Axotomy in Mice. Cell Death Differ. 2012, 19, 162–169. [Google Scholar] [CrossRef]
- Tarantini, S.; Balasubramanian, P.; Delfavero, J.; Csipo, T.; Yabluchanskiy, A.; Kiss, T.; Nyúl-Tóth, Á.; Mukli, P.; Toth, P.; Ahire, C.; et al. Treatment with the BCL-2/BCL-xL Inhibitor Senolytic Drug ABT263/Navitoclax Improves Functional Hyperemia in Aged Mice. GeroScience 2021, 43, 2427–2440. [Google Scholar] [CrossRef]
- Baghdadi, M.; Nespital, T.; Monzó, C.; Deelen, J.; Grönke, S.; Partridge, L. Intermittent Rapamycin Feeding Recapitulates Some Effects of Continuous Treatment While Maintaining Lifespan Extension. Mol. Metab. 2024, 81, 101902. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, Y.; Cen, X.; Shan, B.; Zhao, Q.; Xie, T.; Wang, Z.; Hou, T.; Xue, Y.; Zhang, M.; et al. Metformin Activates Chaperone-Mediated Autophagy and Improves Disease Pathologies in an Alzheimer Disease Mouse Model. Protein Cell 2021, 12, 769–787. [Google Scholar] [CrossRef] [PubMed]
- Isop, L.M.; Neculau, A.E.; Necula, R.D.; Kakucs, C.; Moga, M.A.; Dima, L. Metformin: The Winding Path from Understanding Its Molecular Mechanisms to Proving Therapeutic Benefits in Neurodegenerative Disorders. Pharmaceuticals 2023, 16, 1714. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lu, X.; Liu, N.; Ma, S.; Zhang, H.; Zhang, Z.; Yang, K.; Jiang, M.; Zheng, Z.; Qiao, Y.; et al. Metformin Decelerates Aging Clock in Male Monkeys. Cell 2024, 187, 6358–6378.e29. [Google Scholar] [CrossRef]
- Chung, M.-M.; Chen, Y.-L.; Pei, D.; Cheng, Y.-C.; Sun, B.; Nicol, C.J.; Yen, C.-H.; Chen, H.-M.; Liang, Y.-J.; Chiang, M.-C. The Neuroprotective Role of Metformin in Advanced Glycation End Product Treated Human Neural Stem Cells Is AMPK-Dependent. Biochim. Et Biophys. Acta (BBA)—Mol. Basis Dis. 2015, 1852, 720–731. [Google Scholar] [CrossRef]
- Benito-Cuesta, I.; Ordóñez-Gutiérrez, L.; Wandosell, F. AMPK Activation Does Not Enhance Autophagy in Neurons in Contrast to MTORC1 Inhibition: Different Impact on β-Amyloid Clearance. Autophagy 2021, 17, 656–671. [Google Scholar] [CrossRef]
- García-Juan, M.; Villa, M.; Benito-Cuesta, I.; Ordóñez-Gutiérrez, L.; Wandosell, F. Reassessing the AMPK-MTORC1 Balance in Autophagy in the Central Nervous System. Neural Regen. Res. 2025, 20, 3209–3210. [Google Scholar] [CrossRef]
- Liu, S.; Yue, C.; Chen, H.; Chen, Y.; Li, G. Metformin Promotes Beclin1-Dependent Autophagy to Inhibit the Progression of Gastric Cancer. OncoTargets Ther. 2020, 13, 4445–4455. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, S.; Fan, Z.; Li, Z.; Zhu, Y.; Shen, T.; Li, K.; Yan, Y.; Tian, J.; Liu, Z.; et al. Metformin Attenuates Plaque-Associated Tau Pathology and Reduces Amyloid-β Burden in APP/PS1 Mice. Alzheimer’s Res. Ther. 2021, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Du, M.-R.; Gao, Q.-Y.; Liu, C.-L.; Bai, L.-Y.; Li, T.; Wei, F.-L. Exploring the Pharmacological Potential of Metformin for Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 838173. [Google Scholar] [CrossRef]
- Tahmi, M.; Luchsinger, J.A. Metformin in the Prevention of Alzheimer’s Disease and Alzheimer’s Disease Related Dementias. J. Prev. Alzheimer’s Dis. 2023, 10, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Luo, A.; Ning, P.; Lu, H.; Huang, H.; Shen, Q.; Zhang, D.; Xu, F.; Yang, L.; Xu, Y. Association Between Metformin and Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Clinical Observational Studies. J. Alzheimer’s Dis. 2022, 88, 1311–1323. [Google Scholar] [CrossRef]
- Ha, J.; Choi, D.-W.; Kim, K.J.; Cho, S.Y.; Kim, H.; Kim, K.Y.; Koh, Y.; Nam, C.M.; Kim, E. Association of Metformin Use with Alzheimer’s Disease in Patients with Newly Diagnosed Type 2 Diabetes: A Population-Based Nested Case–Control Study. Sci. Rep. 2021, 11, 24069. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Zhang, X.-Y.; Sun, Y.-Q.; Lv, R.-H.; Chen, M.; Li, M. Metformin Use Is Associated with a Reduced Risk of Cognitive Impairment in Adults with Diabetes Mellitus: A Systematic Review and Meta-Analysis. Front. Neurosci. 2022, 16, 984559. [Google Scholar] [CrossRef]
- Samaras, K.; Makkar, S.; Crawford, J.D.; Kochan, N.A.; Wen, W.; Draper, B.; Trollor, J.N.; Brodaty, H.; Sachdev, P.S. Metformin Use Is Associated With Slowed Cognitive Decline and Reduced Incident Dementia in Older Adults With Type 2 Diabetes: The Sydney Memory and Ageing Study. Diabetes Care 2020, 43, 2691–2701. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, K.; Ma, C.; Wang, X.; Gong, Z.; Zhang, R.; Zang, D.; Cheng, Y. Evaluation of Metformin on Cognitive Improvement in Patients With Non-Dementia Vascular Cognitive Impairment and Abnormal Glucose Metabolism. Front. Aging Neurosci. 2018, 10, 227. [Google Scholar] [CrossRef]
- Xue, Y.; Xie, X. The Association between Metformin Use and Risk of Developing Severe Dementia among AD Patients with Type 2 Diabetes. Biomedicines 2023, 11, 2935. [Google Scholar] [CrossRef]
- ElHady, A.K.; El-Gamil, D.S.; Abdel-Halim, M.; Abadi, A.H. Advancements in Phosphodiesterase 5 Inhibitors: Unveiling Present and Future Perspectives. Pharmaceuticals 2023, 16, 1266. [Google Scholar] [CrossRef]
- Kang, B.W.; Kim, F.; Cho, J.-Y.; Kim, S.; Rhee, J.; Choung, J.J. Phosphodiesterase 5 Inhibitor Mirodenafil Ameliorates Alzheimer-like Pathology and Symptoms by Multimodal Actions. Alzheimer’s Res. Ther. 2022, 14, 92. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Silva, E.; De Melo, M.G.; Maes, M.; Filho, A.J.M.C.; Macedo, D.; Peixoto, C.A. Shared Metabolic and Neuroimmune Mechanisms Underlying Type 2 Diabetes Mellitus and Major Depressive Disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 111, 110351. [Google Scholar] [CrossRef] [PubMed]
- Koka, S.; Aluri, H.S.; Xi, L.; Lesnefsky, E.J.; Kukreja, R.C. Chronic Inhibition of Phosphodiesterase 5 with Tadalafil Attenuates Mitochondrial Dysfunction in Type 2 Diabetic Hearts: Potential Role of NO/SIRT1/PGC-1α Signaling. Am. J. Physiol. -Heart Circ. Physiol. 2014, 306, H1558–H1568. [Google Scholar] [CrossRef]
- Puzzo, D.; Staniszewski, A.; Deng, S.X.; Privitera, L.; Leznik, E.; Liu, S.; Zhang, H.; Feng, Y.; Palmeri, A.; Landry, D.W.; et al. Phosphodiesterase 5 Inhibition Improves Synaptic Function, Memory, and Amyloid- Load in an Alzheimer’s Disease Mouse Model. J. Neurosci. 2009, 29, 8075–8086. [Google Scholar] [CrossRef]
- Abouelmagd, M.E.; Abdelmeseh, M.; Elrosasy, A.; Saad, Y.H.; Alnajjar, A.Z.; Eid, M.; Hassan, A.; Abbas, A. Phosphodiesterase-5 Inhibitors Use and the Risk of Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Neurol. Sci. 2024, 45, 5261–5270. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Zhang, P.; Zhou, Y.; Chiang, C.-W.; Tan, J.; Hou, Y.; Stauffer, S.; Li, L.; Pieper, A.A.; Cummings, J.; et al. Endophenotype-Based in Silico Network Medicine Discovery Combined with Insurance Record Data Mining Identifies Sildenafil as a Candidate Drug for Alzheimer’s Disease. Nat. Aging 2021, 1, 1175–1188. [Google Scholar] [CrossRef]
- Zhang, Q.; Presswalla, F.; Ali, R.R.; Zacks, D.N.; Thompson, D.A.; Miller, J.M. Pharmacologic Activation of Autophagy without Direct mTOR Inhibition as a Therapeutic Strategy for Treating Dry Macular Degeneration. Aging 2021, 13, 10866–10890. [Google Scholar] [CrossRef]
- Date, Y.; Sasazawa, Y.; Kitagawa, M.; Gejima, K.; Suzuki, A.; Saya, H.; Kida, Y.; Imoto, M.; Itakura, E.; Hattori, N.; et al. Novel Autophagy Inducers by Accelerating Lysosomal Clustering against Parkinson’s Disease. eLife 2024, 13, e98649. [Google Scholar] [CrossRef]
- Ullah, A.; Al Kury, L.T.; Althobaiti, Y.S.; Ali, T.; Shah, F.A. Benzimidazole Derivatives as New Potential NLRP3 Inflammasome Inhibitors That Provide Neuroprotection in a Rodent Model of Neurodegeneration and Memory Impairment. J. Inflamm. Res. 2022, 15, 3873–3890. [Google Scholar] [CrossRef]
- Wildenberg, M.E.; Levin, A.D.; Ceroni, A.; Guo, Z.; Koelink, P.J.; Hakvoort, T.B.M.; Westera, L.; Bloemendaal, F.M.; Brandse, J.F.; Simmons, A.; et al. Benzimidazoles Promote Anti-TNF Mediated Induction of Regulatory Macrophages and Enhance Therapeutic Efficacy in a Murine Model. J. Crohn’s Colitis 2017, 11, 1480–1490. [Google Scholar] [CrossRef]
- Al-Nasser, S.; Abdulla, M.H.; Alhassan, N.; Vaali-Mohammed, M.-A.; Al-Omar, S.; Hamdi, N.; Elnakady, Y.; Matou-Nasri, S.; Mansour, L. A Benzimidazole-Based N-Heterocyclic Carbene Derivative Exhibits Potent Antiproliferative and Apoptotic Effects against Colorectal Cancer. Medicina 2024, 60, 1379. [Google Scholar] [CrossRef] [PubMed]
- Acar Cevik, U.; Saglik, B.N.; Levent, S.; Osmaniye, D.; Kaya Cavuşoglu, B.; Ozkay, Y.; Kaplancikli, Z.A. Synthesis and AChE-Inhibitory Activity of New Benzimidazole Derivatives. Molecules 2019, 24, 861. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.; Dagar, A.; Lee, S.; Cho, I.; Lee, D.; Kim, K.; Park, I.; Yoon, S.; Kim, H.Y.; Kim, I.; et al. Benzo[d]Imidazole-Pyrrolo[1,2-a]Pyrazine Hybrids Ameliorate Amyloid Aggregates in the Brain of Alzheimer Transgenic Mice. ACS Chem. Neurosci. 2023, 14, 3025–3034. [Google Scholar] [CrossRef]
- Rosales Hernández, M.C.; Olvera-Valdez, M.; Velazquez Toledano, J.; Mendieta Wejebe, J.E.; Fragoso Morales, L.G.; Cruz, A. Exploring the Benzazoles Derivatives as Pharmacophores for AChE, BACE1, and as Anti-Aβ Aggregation to Find Multitarget Compounds against Alzheimer’s Disease. Molecules 2024, 29, 4780. [Google Scholar] [CrossRef]
- Zhang, H.; Su, Y.; Sun, Z.; Chen, M.; Han, Y.; Li, Y.; Dong, X.; Ding, S.; Fang, Z.; Li, W.; et al. Ginsenoside Rg1 Alleviates Aβ Deposition by Inhibiting NADPH Oxidase 2 Activation in APP/PS1 Mice. J. Ginseng Res. 2021, 45, 665–675. [Google Scholar] [CrossRef]
- Chen, Q.; Qu, M.; Chen, Q.; Meng, X.; Fan, H. Phosphoproteomics Analysis of the Effect of Target of Rapamycin Kinase Inhibition on Cucumis Sativus in Response to Podosphaera Xanthii. Plant Physiol. Biochem. 2023, 197, 107641. [Google Scholar] [CrossRef]
- Chen, D.; Chen, Q.; Qin, X.; Tong, P.; Peng, L.; Zhang, T.; Xia, C. Development and Evolution of Human Glutaminyl Cyclase Inhibitors (QCIs): An Alternative Promising Approach for Disease-Modifying Treatment of Alzheimer’s Disease. Front. Aging Neurosci. 2023, 15, 1209863. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Zhao, G.; Weng, L.; Guan, J.; for the Alzheimer’s Disease Neuroimaging Initiative. Aspirin Using Was Associated with Slower Cognitive Decline in Patients with Alzheimer’s Disease. PLoS ONE 2021, 16, e0252969. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Tang, Y.-R.; Gao, X.; Zhang, N.-N.; Lv, Q.-Q.; Liu, J.; Li, Y. Aspirin-Triggered Resolvin D1 Ameliorates Activation of the NLRP3 Inflammasome via Induction of Autophagy in a Rat Model of Neuropathic Pain. Front. Pharmacol. 2023, 14, 971136. [Google Scholar] [CrossRef]
- Vergil Andrews, J.F.; Selvaraj, D.B.; Kumar, A.; Roshan, S.A.; Anusuyadevi, M.; Kandasamy, M. A Mild Dose of Aspirin Promotes Hippocampal Neurogenesis and Working Memory in Experimental Ageing Mice. Brain Sci. 2023, 13, 1108. [Google Scholar] [CrossRef]
- De Cristóbal, J.; Madrigal, J.L.M.; Lizasoain, I.; Lorenzo, P.; Leza, J.C.; Moro, M.A. Aspirin Inhibits Stress-Induced Increase in Plasma Glutamate, Brain Oxidative Damage and ATP Fall in Rats. Neuroreport 2002, 13, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Yu, Q.; Ou, J.; Lou, J.; Zhu, J.; Lin, Z. The Neuroprotective Mechanisms of PPAR-γ: Inhibition of Microglia-Mediated Neuroinflammation and Oxidative Stress in a Neonatal Mouse Model of Hypoxic-Ischemic White Matter Injury. CNS Neurosci. Ther. 2024, 30, e70081. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cao, J.; Hao, Z.; Liu, A.; Li, X.; Li, H.; Xia, N.; Wang, Z.; Zhang, Z.; Bai, J.; et al. Aspirin Ameliorates the Cognition Impairment in Mice Following Benzo[a]Pyrene Treatment via down-Regulating BDNF IV Methylation. Neurotoxicology 2022, 89, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Gholami, M.; Sadegh, M.; Koroush-arami, M.; Norouzi, S.; Arismani, R.J.; Asadi, E.; Amini, M.; Khodayari, N. Targeting Memory Loss with Aspirin, a Molecular Mechanism Perspective for Future Therapeutic Approaches. Inflammopharmacol 2023, 31, 2827–2842. [Google Scholar] [CrossRef]
- Ryan, J.; Storey, E.; Murray, A.M.; Woods, R.L.; Wolfe, R.; Reid, C.M.; Nelson, M.R.; Chong, T.T.J.; Williamson, J.D.; Ward, S.A.; et al. Randomized Placebo-Controlled Trial of the Effects of Aspirin on Dementia and Cognitive Decline. Neurology 2020, 95, e320–e331. [Google Scholar] [CrossRef]
- Doost Mohammadpour, J.; Hosseinmardi, N.; Janahmadi, M.; Fathollahi, Y.; Motamedi, F.; Rohampour, K. Non-Selective NSAIDs Improve the Amyloid-β-Mediated Suppression of Memory and Synaptic Plasticity. Pharmacol. Biochem. Behav. 2015, 132, 33–41. [Google Scholar] [CrossRef]
- Chandra, S.; Roy, A.; Patel, D.R.; Pahan, K. PPARα Between Aspirin and Plaque Clearance. J. Alzheimer’s Dis. 2019, 71, 389–397. [Google Scholar] [CrossRef]
- Ghosh, B.; Datta, A.; Gupta, V.; Sodnar, B.; Sarkar, A.; Singh, U.; Raut, S.; Suthar, P.; Thongire, V.; Sarmah, D.; et al. Simvastatin Exerts Neuroprotective Effects Post-Stroke by Ameliorating Endoplasmic Reticulum Stress and Regulating Autophagy/Apoptosis Balance through pAMPK/LC3B/ LAMP2 Axis. Exp. Neurol. 2024, 381, 114940. [Google Scholar] [CrossRef]
- Qi, W.; Yan, L.; Liu, Y.; Zhou, X.; Li, R.; Wang, Y.; Bai, L.; Chen, J.; Nie, X. Simvastatin Aggravates Impaired Autophagic Flux in NSC34-hSOD1G93A Cells through Inhibition of Geranylgeranyl Pyrophosphate Synthesis. Neuroscience 2019, 409, 130–141. [Google Scholar] [CrossRef]
- Olmastroni, E.; Molari, G.; De Beni, N.; Colpani, O.; Galimberti, F.; Gazzotti, M.; Zambon, A.; Catapano, A.L.; Casula, M. Statin Use and Risk of Dementia or Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Observational Studies. Eur. J. Prev. Cardiol. 2022, 29, 804–814. [Google Scholar] [CrossRef]
- VanFossen, B.T.; Watson, G.S.; Baker, L.D.; Rhoads, K.W.; Cholerton, B.A.; Reger, M.A.; Plymate, S.R.; Schellenberg, G.; Craft, S. Statin Users Without an APOE-Ε4 Allele Have Increased Insulin Resistance. J. Alzheimer’s Dis. 2010, 19, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.-C.; Chuang, Y.-S.; Hsieh, H.-M.; Lee, T.-C.; Chiu, K.-F.; Liu, C.-K.; Wu, M.-T. Early Statin Use and the Progression of Alzheimer Disease: A Total Population-Based Case-Control Study. Medicine 2015, 94, e2143. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Wang, Y.; Huo, J.; Li, S.; Wen, Y.; Liu, Q.; Yang, J.; Liu, Y.; Li, R. Simvastatin Accelerated Motoneurons Death in SOD1G93A Mice through Inhibiting Rab7-Mediated Maturation of Late Autophagic Vacuoles. Cell Death Dis. 2021, 12, 392. [Google Scholar] [CrossRef] [PubMed]
- Lipid and Blood Pressure Meta-analysis Collaboration (LBPMC) Group; Banach, M.; Serban, C.; Sahebkar, A.; Mikhailidis, D.P.; Ursoniu, S.; Ray, K.K.; Rysz, J.; Toth, P.P.; Muntner, P.; et al. Impact of Statin Therapy on Coronary Plaque Composition: A Systematic Review and Meta-Analysis of Virtual Histology Intravascular Ultrasound Studies. BMC Med. 2015, 13, 229. [Google Scholar] [CrossRef]
- Din, F.V.N.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin Inhibits mTOR Signaling, Activates AMP-Activated Protein Kinase, and Induces Autophagy in Colorectal Cancer Cells. Gastroenterology 2012, 142, 1504–1515.e3. [Google Scholar] [CrossRef]
- Parnetti, L.; Chipi, E.; Salvadori, N.; D’Andrea, K.; Eusebi, P. Prevalence and Risk of Progression of Preclinical Alzheimer’s Disease Stages: A Systematic Review and Meta-Analysis. Alzheimer’s Res. Ther. 2019, 11, 1–13. [Google Scholar] [CrossRef]
- Tropea, T.F.; Waligorska, T.; Xie, S.X.; Nasrallah, I.M.; Cousins, K.A.Q.; Trojanowski, J.Q.; Grossman, M.; Irwin, D.J.; Weintraub, D.; Lee, E.B.; et al. Plasma Phosphorylated Tau181 Predicts Cognitive and Functional Decline. Ann. Clin. Transl. Neurol. 2023, 10, 18–31. [Google Scholar] [CrossRef]
- Palmqvist, S.; Stomrud, E.; Cullen, N.; Janelidze, S.; Manuilova, E.; Jethwa, A.; Bittner, T.; Eichenlaub, U.; Suridjan, I.; Kollmorgen, G.; et al. An Accurate Fully Automated Panel of Plasma Biomarkers for Alzheimer’s Disease. Alzheimer’s Dement. 2023, 19, 1204–1215. [Google Scholar] [CrossRef]
- Dakterzada, F.; Benítez, I.D.; Targa, A.; Carnes, A.; Pujol, M.; Jové, M.; Mínguez, O.; Vaca, R.; Sánchez-de-la-Torre, M.; Barbé, F.; et al. Cerebrospinal Fluid Lipidomic Fingerprint of Obstructive Sleep Apnoea in Alzheimer’s Disease. Alzheimer’s Res. Ther. 2023, 15, 134. [Google Scholar] [CrossRef]
- Sasazawa, Y.; Date, Y.; Hattori, N.; Saiki, S. Clustering Lysosomes around the MTOC: A Promising Strategy for SNCA/Alpha-Synuclein Breakdown Leading to Parkinson Disease Treatment. Autophagy 2024, 20, 2839–2840. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-Activated Protein Kinase in Mechanism of Metformin Action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Khalifeh, M.; Read, M.I.; Barreto, G.E.; Sahebkar, A. Trehalose against Alzheimer’s Disease: Insights into a Potential Therapy. BioEssays 2020, 42, 1900195. [Google Scholar] [CrossRef] [PubMed]
- Motoi, Y.; Shimada, K.; Ishiguro, K.; Hattori, N. Lithium and Autophagy. ACS Chem. Neurosci. 2014, 5, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.W.; Gandy, S. Latrepirdine (Dimebon®), a Potential Alzheimer Therapeutic, Regulates Autophagy and Neuropathology in an Alzheimer Mouse Model. Autophagy 2013, 9, 617–618. [Google Scholar] [CrossRef]
- Bourdenx, M.; Martín-Segura, A.; Scrivo, A.; Rodriguez-Navarro, J.A.; Kaushik, S.; Tasset, I.; Diaz, A.; Storm, N.J.; Xin, Q.; Juste, Y.R.; et al. Chaperone-Mediated Autophagy Prevents Collapse of the Neuronal Metastable Proteome. Cell 2021, 184, 2696–2714.e25. [Google Scholar] [CrossRef]
- Billes, V.; Kovács, T.; Hotzi, B.; Manzéger, A.; Tagscherer, K.; Komlós, M.; Tarnóci, A.; Pádár, Z.; Erdős, A.; Bjelik, A.; et al. AUTEN-67 (Autophagy Enhancer-67) Hampers the Progression of Neurodegenerative Symptoms in a Drosophila Model of Huntington’s Disease. J. Huntington’s Dis. 2016, 5, 133–147. [Google Scholar] [CrossRef]
- Niikura, T. Humanin and Alzheimer’s Disease: The Beginning of a New Field. Biochim. Et Biophys. Acta (BBA)—Gen. Subj. 2022, 1866, 130024. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Pharmacological Class | Mechanisms of Action | Cellular Targets/Effects |
|---|---|---|---|
| Rapamycin | mTOR inhibitor (macrolide antibiotic) | Inhibits mTORC1 → activates ULK1/ATG13 → promotes autophagy; upregulates Beclin-1, reduces p62, and modulates MEK/ERK pathway | Enhances autophagy in neurons; reduces Aβ and tau; improves synaptic plasticity; modulates microglial polarization |
| Metformin | AMPK activator (antidiabetic biguanide) | Activates AMPK → inhibits mTORC1; induces CMA via TAK1–IKK pathway; stimulates SIRT1 and Nrf2 for antioxidant effects | Enhances autophagy (neurons and microglia); improves mitochondrial function; effects are cell-type and age dependent |
| Navitoclax | Senolytic (Bcl-2/Bcl-xL inhibitor) | Induces apoptosis in senescent cells by inhibiting anti-apoptotic Bcl-2 family proteins | Eliminates senescent astrocytes and endothelial cells; restores neurovascular function and cognitive performance |
| Sildenafil | PDE5 inhibitor | ↑ NO/cGMP → activates AMPK and eNOS → inhibits mTOR; activates CREB/BDNF; modulates NF-κB; increases autophagy markers (LC3, Beclin-1, ATG5) | Promotes autophagy in neurons and glia; improves blood flow, synaptic plasticity, and cognitive performance |
| Tadalafil | PDE5 inhibitor | ↑ NO → activates SIRT1–PGC-1α axis → improves mitochondrial function and reduces ROS | Improves mitochondrial biogenesis and function in diabetic models; potential neuroprotective effects |
| Mirodenafil | BBB-penetrant PDE5 inhibitor | Activates AMPK and autophagy; modulates cGMP/PKG/CREB, Wnt/β-catenin, GSK-3β, and glucocorticoid signaling | Enhances autophagy; reduces Aβ and tau; improves cognitive function and synaptic signaling |
| FLBZ/Albendazole | Benzimidazole derivatives | Promote lysosomal clustering, JIP4–TRPML1 activation, enhance autophagosome–lysosome fusion; modulate NF-κB and NLRP3 pathways | Induce autophagy without mTOR inhibition; reduce SASP and neuroinflammation; clear protein aggregates |
| Acetylsalicylic acid (ASA) | NSAID/COX-1/2 inhibitor | Induces autophagy via COX-2 inhibition and AT-RvD1 generation; inhibits NF-κB and NLRP3; activates TFEB via PPARα | Promotes clearance of Aβ/tau; reduces senescence and inflammation; preserves synaptic function |
| Biomarker | Pathway Dysfunction Indicated | Relevant Drug | Therapeutic Action |
|---|---|---|---|
| ↓ LAMP1, ↓ Cathepsin D | Impaired autophagosome–lysosome fusion | Benzimidazole derivatives | Enhance autophagic degradation via JIP4–TRPML1 pathway |
| ↑ IL-6, ↑ TNF-α, ↑ MMPs | Chronic inflammation and SASP activation | ASA and benzimidazoles | Suppress NF-κB signaling and SASP factors |
| ↑ NF-κB activation (p65 subunit) | Inflammatory and senescence pathways | ASA and benzimidazoles | NF-κB inhibition and anti-inflammatory action |
| ↓ AMPK activation, ↑ mTORC1 activity | Impaired autophagy initiation and metabolic stress | Metformin | AMPK activation and mTORC1 inhibition to restore autophagy |
| ↑ 8-OHdG, mitochondrial DNA damage | Oxidative stress and mitochondrial dysfunction | Metformin | Nrf2-mediated antioxidant defense and mitochondrial support |
| ↑ HOMA-IR score, insulin resistance | Metabolic dysfunction | Metformin | Improvement of insulin sensitivity and energy homeostasis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordos, B.; Tero-Vescan, A.; Hampson, I.N.; Oliver, A.W.; Slevin, M. Synergistic Autophagy-Related Mechanisms of Protection Against Brain Aging and AD: Cellular Pathways and Therapeutic Strategies. Pharmaceuticals 2025, 18, 829. https://doi.org/10.3390/ph18060829
Cordos B, Tero-Vescan A, Hampson IN, Oliver AW, Slevin M. Synergistic Autophagy-Related Mechanisms of Protection Against Brain Aging and AD: Cellular Pathways and Therapeutic Strategies. Pharmaceuticals. 2025; 18(6):829. https://doi.org/10.3390/ph18060829
Chicago/Turabian StyleCordos, Bogdan, Amelia Tero-Vescan, Ian N. Hampson, Anthony W. Oliver, and Mark Slevin. 2025. "Synergistic Autophagy-Related Mechanisms of Protection Against Brain Aging and AD: Cellular Pathways and Therapeutic Strategies" Pharmaceuticals 18, no. 6: 829. https://doi.org/10.3390/ph18060829
APA StyleCordos, B., Tero-Vescan, A., Hampson, I. N., Oliver, A. W., & Slevin, M. (2025). Synergistic Autophagy-Related Mechanisms of Protection Against Brain Aging and AD: Cellular Pathways and Therapeutic Strategies. Pharmaceuticals, 18(6), 829. https://doi.org/10.3390/ph18060829