
Hyperlipidemia Increases Nalbuphine Brain Accumulation with Multiple Dosing without Affecting Its Analgesic Response—Its Respiratory Depression Potential Should Be Investigated in Future Studies


Abstract
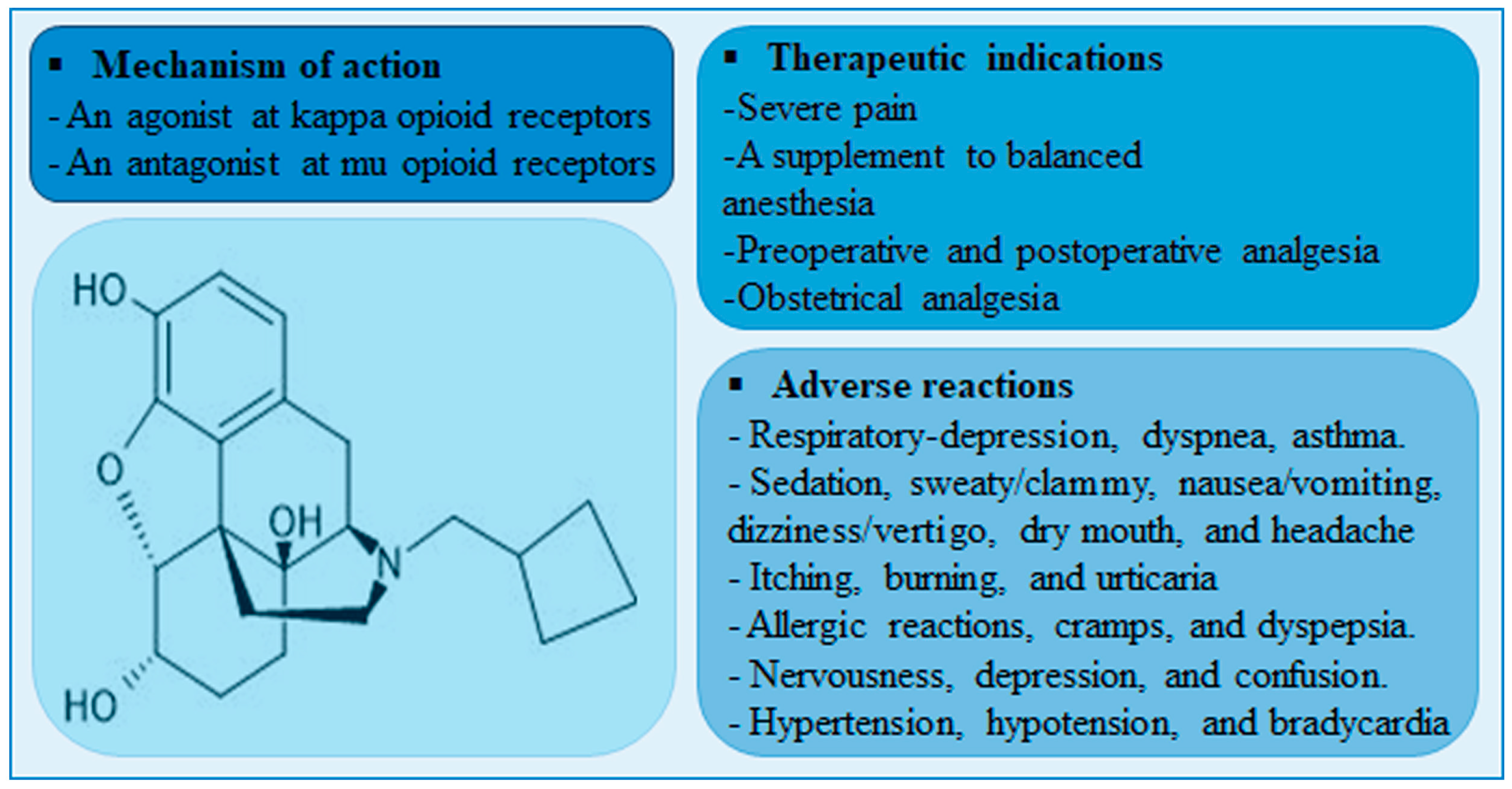
1. Introduction
2. Results
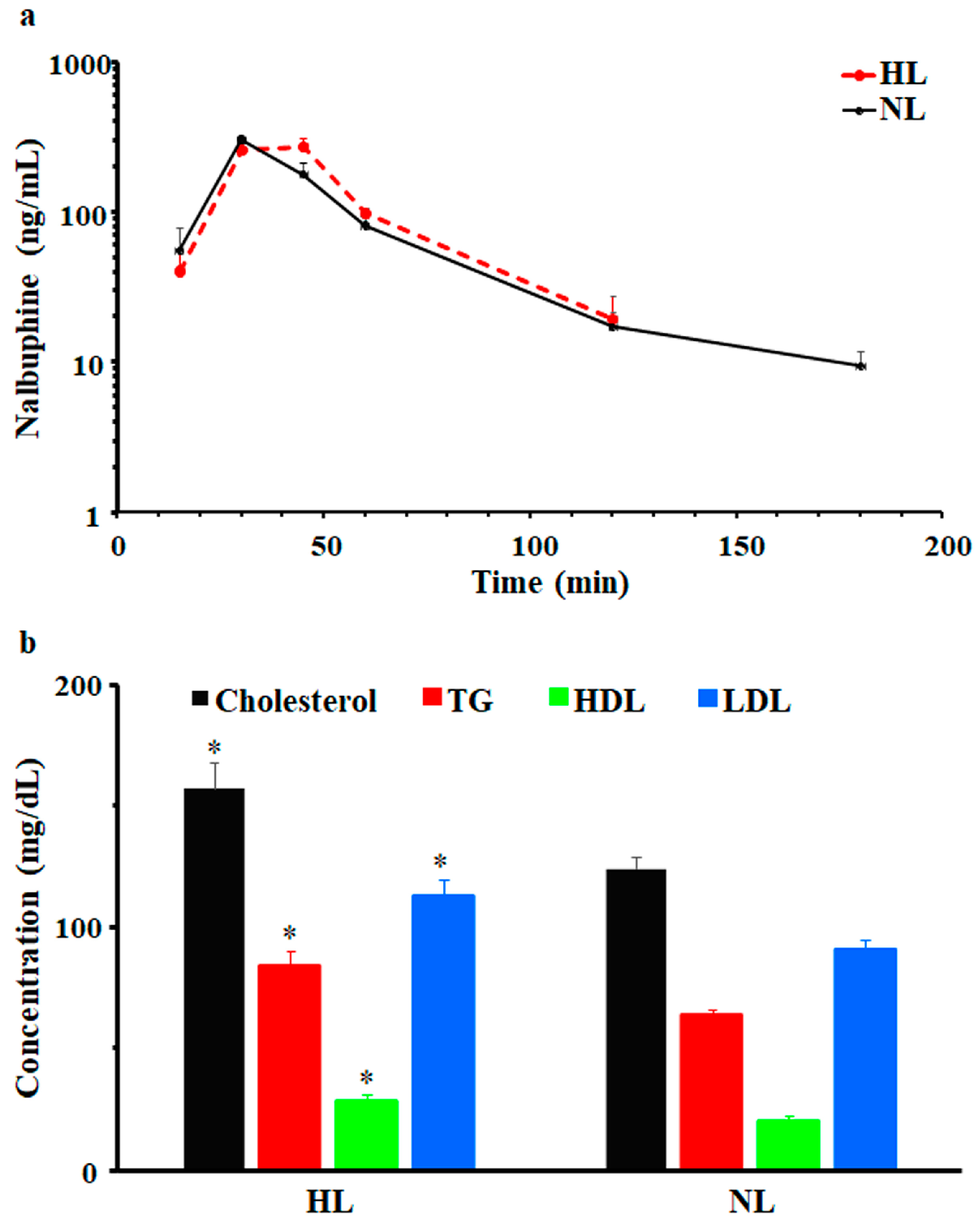
2.1. Single-Dose Pharmacokinetic Study
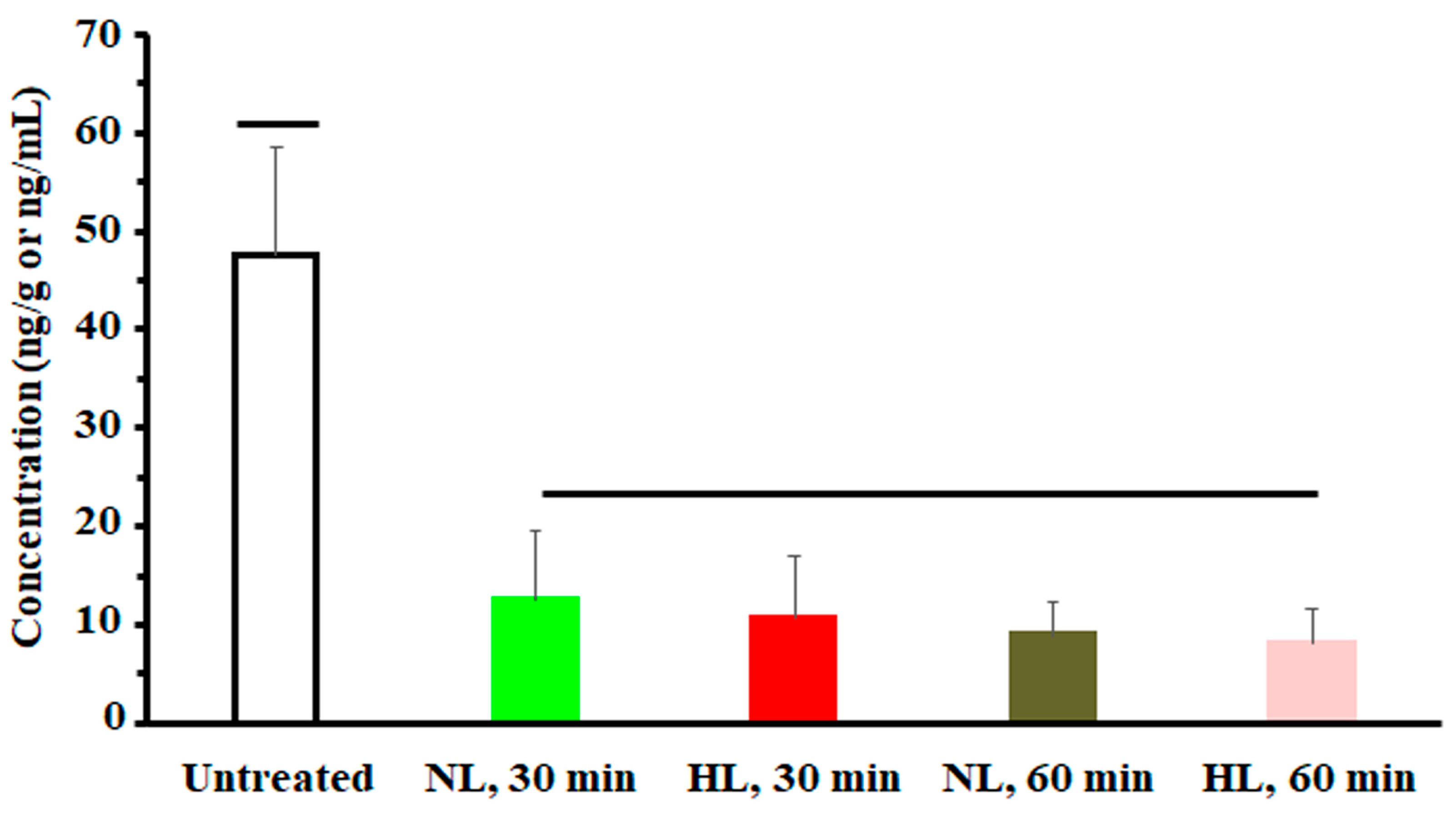
2.2. Multiple-Dose Tissue Distribution Study
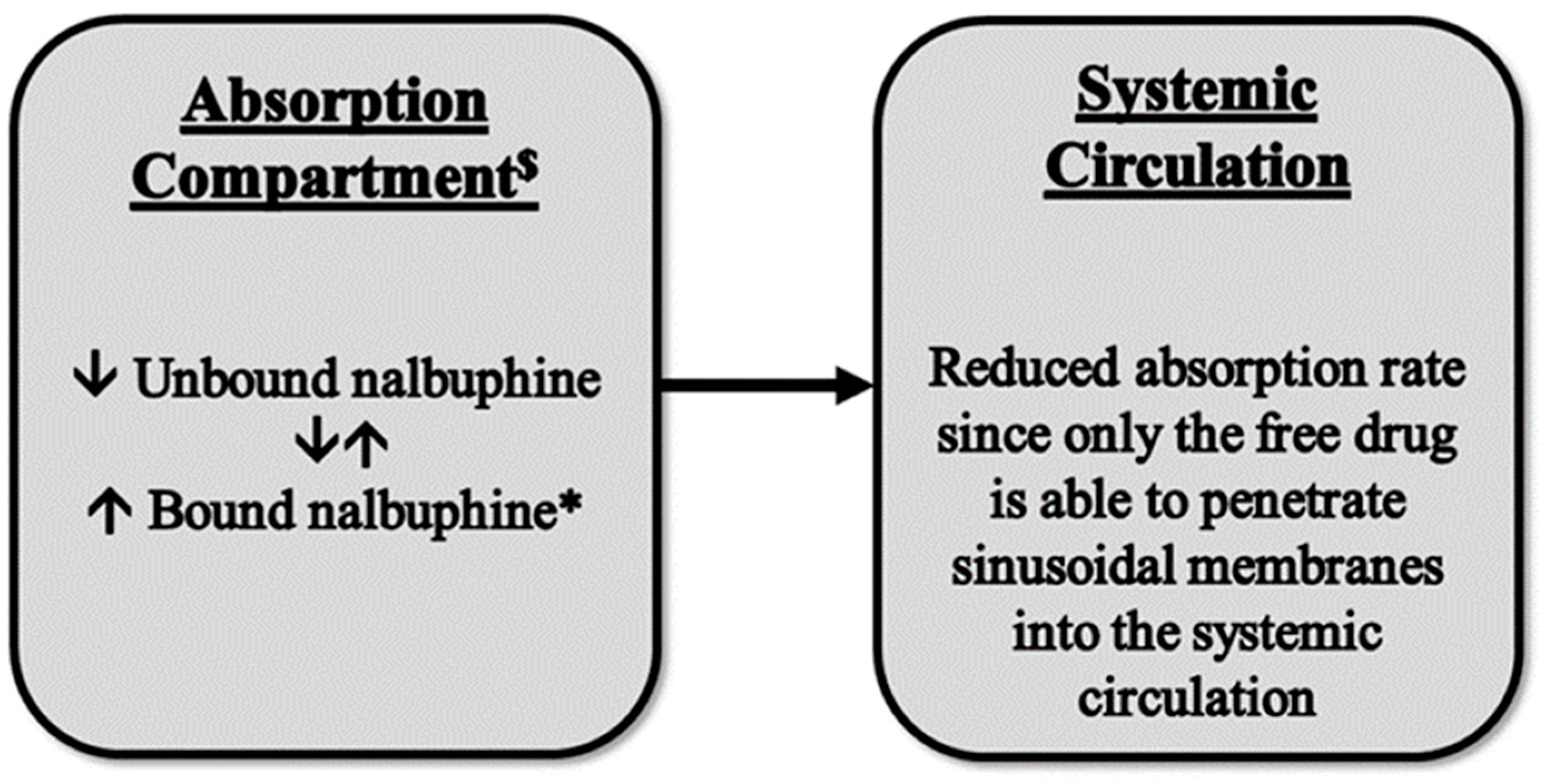
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Single-Dose Pharmacokinetic Study
4.3. Multiple-Dose Tissue Distribution Study
4.4. Serum Lipid Measurement
4.5. Serum and Tissue Sample Preparation
4.6. Nalbuphine Analysis
4.7. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Cmax | The maximum serum concentration |
| Tmax | Time to reach Cmax |
| AUC0-Tlast | Area under serum concentration vs. time curve from 0 to the last time point |
| AUC0-inf | Area under serum concentration vs. time curve from 0 to infinity |
| t1/2 | Serum half-life |
| CL/F | Apparent intraperitoneal clearance |
| Vdss/F | Apparent intraperitoneal volume of distribution at steady state |
| NL | Normolipidemic group |
| HL | Hyperlipidemic group |
| TG | Triglycerides |
| HDL | High density lipoprotein |
| LDL | Low density lipoprotein |
| VLDL | Very low-density lipoprotein |
| i.p. | Intraperitoneal injection |
| r | Pearson correlation coefficient |
| HBF | Hepatic blood flow |
| HPF | Hepatic plasms flow |
| min | Minutes |
References
- Gunion, M.W.; Marchionne, A.M.; Anderson, C.T. Use of the mixed agonist–antagonist nalbuphine in opioid based analgesia. Acute Pain 2004, 6, 29–39. [Google Scholar] [CrossRef]
- FDA. NUBAIN—(Nalbuphine Hydrochloride) Injection, for Intramuscular, Subcutaneous, or Intravenous Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/018024s042lbl.pdf (accessed on 1 July 2022).
- Zeng, Z.; Lu, J.; Shu, C.; Chen, Y.; Guo, T.; Wu, Q.-P.; Yao, S.-L.; Yin, P. A Comparision of Nalbuphine with Morphine for Analgesic Effects and Safety: Meta-Analysis of Randomized Controlled Trials. Sci. Rep. 2015, 5, 10927. [Google Scholar] [CrossRef] [PubMed]
- Larsen, D.; Maani, C.V. Nalbuphine, 2023: Treasure Island (FL) Ineligible Companies. In Disclosure: Christopher Maani Declares No Relevant Financial Relationships with Ineligible Companies; StatPearls: Tampa, FL, USA, 2023. [Google Scholar]
- de Cazanove, F.; Kinowski, J.M.; Audran, M.; Rochette, A.; Bressolle, F. Determination of nalbuphine in human plasma by high-performance liquid chromatography with electrochemical detection. Application to a pharmacokinetic study. J. Chromatogr. B Biomed. Sci. Appl. 1997, 690, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, P.; E Gardin, M.; Lecocq, B.; O Richard, M.; Meignan, S.; Blondel, Y.; Grippat, J.C.; Bergnieres, J.; Vergnoux, O. Pharmacokinetics of nalbuphine in infants, young healthy volunteers, and elderly patients. Clin. Pharmacol. Ther. 1989, 46, 226–233. [Google Scholar] [CrossRef]
- Bressolle, F.; Khier, S.; Rochette, A.; Kinowski, J.M.; Dadure, C.; Capdevila, X. Population pharmacokinetics of nalbuphine after surgery in children. Br. J. Anaesth. 2011, 106, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.-J.; Shih, Y.-N.; Chen, Y.-L.; Liu, W.-Y.; Yang, W.-L.; Lee, S.-Y.; Wang, H.-J. A dual system platform for drug metabolism: Nalbuphine as a model compound. Eur. J. Pharm. Sci. 2019, 141, 105093. [Google Scholar] [CrossRef]
- Koyyalagunta, D.; Waldman, S.D. Opioid Analgesics. In Pain Management, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2011; pp. 890–912. [Google Scholar]
- Doan, K.M.M.; Humphreys, J.E.; Webster, L.O.; Wring, S.A.; Shampine, L.J.; Serabjit-Singh, C.J.; Adkison, K.K.; Polli, J.W. Passive Permeability and P-Glycoprotein-Mediated Efflux Differentiate Central Nervous System (CNS) and Non-CNS Marketed Drugs. J. Pharmacol. Exp. Ther. 2002, 303, 1029–1037. [Google Scholar] [CrossRef]
- Tournier, N.; Declèves, X.; Saubaméa, B.; Scherrmann, J.M.; Cisternino, S. Opioid transport by ATP-binding cassette transporters at the blood-brain barrier: Implications for neuropsychopharmacology. Curr. Pharm. Des. 2011, 17, 2829–2842. [Google Scholar] [CrossRef]
- Coluzzi, F.; Scerpa, M.S.; Rocco, M.; Fornasari, D. The Impact of P-Glycoprotein on Opioid Analgesics: What’s the Real Meaning in Pain Management and Palliative Care? Int. J. Mol. Sci. 2022, 23, 14125. [Google Scholar] [CrossRef]
- Viscusi, E.R.; Viscusi, A.R. Blood-brain barrier: Mechanisms governing permeability and interaction with peripherally acting mu-opioid receptor antagonists. Reg. Anesth. Pain Med. 2020, 45, 688–695. [Google Scholar] [CrossRef]
- Mégarbane, B.; Alhaddad, H. P-glycoprotein should be considered as an additional factor contributing to opioid-induced respiratory depression in paediatrics: The buprenorphine example. Br. J. Anaesth. 2013, 110, 842. [Google Scholar] [CrossRef]
- Brocks, D.R.; Chaudhary, H.R.; Ben-Eltriki, M.; Elsherbiny, M.E.; El-Kadi, A.O. Effects of serum lipoproteins on cyclosporine A cellular uptake and renal toxicity in vitro. Can. J. Physiol. Pharmacol. 2014, 92, 140–148. [Google Scholar] [CrossRef]
- Siti, Z.; Seoparjoo, A.; Shahrul, H. Lipoproteins modulate growth and P-glycoprotein expression in drug-resistant HER2-overexpressed breast cancer cells. Heliyon 2019, 5, e01573. [Google Scholar] [CrossRef] [PubMed]
- Shayeganpour, A.; Jun, A.S.; Brocks, D.R. Pharmacokinetics of Amiodarone in hyperlipidemic and simulated high fat-meal rat models. Biopharm. Drug Dispos. 2005, 26, 249–257. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Raised Cholesterol. Available online: https://www.who.int/data/gho/indicator-metadata-registry/imr-details/3236 (accessed on 18 January 2024).
- World Health Organization. Noncommunicable Diseases: Risk Factors. Available online: https://www.who.int/data/gho/data/themes/topics/topic-details/GHO/ncd-risk-factors (accessed on 15 July 2023).
- Chaudhary, H.R.; Brocks, D.R. The Single Dose Poloxamer 407 Model of Hyperlipidemia; Systemic Effects on Lipids Assessed Using Pharmacokinetic Methods, and its Effects on Adipokines. J. Pharm. Pharm. Sci. 2013, 16, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Chávez, J.J.; López-Cervantes, M.; Naïk, A.; Kalia, Y.N.; Quintanar-Guerrero, D.; Ganem-Quintanar, A. Applications of thermo-reversible pluronic F-127 gels in pharmaceutical formulations. J. Pharm. Pharm. Sci. 2006, 9, 339–358. [Google Scholar] [PubMed]
- Ma, Y.; Zhang, C.; Chen, X.; Jiang, H.; Pan, S.; Easteal, A.J.; Sun, X. The Influence of Modified Pluronic F127 Copolymers with Higher Phase Transition Temperature on Arsenic Trioxide-Releasing Properties and Toxicity in a Subcutaneous Model of Rats. AAPS PharmSciTech 2012, 13, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Dziwenka, M.; Li, X.; Li, W.; Zong, J.; Zong, L.; Wang, S.; Li, L.; Liu, Z.; Zhang, Z.; Wang, M. Safety evaluation of CuminUP60® –A novel curcumin complex. Toxicol. Rep. 2022, 9, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Zuberu, J.; Saleh, M.I.A.; Alhassan, A.W.; Adamu, B.Y.; Aliyu, M.; Iliya, B.T. Hepatoprotective Effect of Camel Milk on Poloxamer 407 Induced Hyperlipidaemic Wistar Rats. Open Access Maced. J. Med. Sci. 2017, 5, 852–858. [Google Scholar] [CrossRef]
- Korolenko, T.; Johnston, T.P.; Dubrovina, N.I.; Kisarova, Y.A.; Zhanaeva, S.Y.; Cherkanova, M.S.; Filjushina, E.E.; Alexeenko, T.V.; Machova, E.; Zhukova, N.A. Effect of poloxamer 407 administration on the serum lipids profile, anxiety level and protease activity in the heart and liver of mice. Interdiscip. Toxicol. 2013, 6, 18–25. [Google Scholar] [CrossRef]
- Korolenko, T.A.; Johnston, T.P.; Tuzikov, F.V.; Tuzikova, N.A.; Pupyshev, A.B.; Spiridonov, V.K.; Goncharova, N.V.; Maiborodin, I.V.; Zhukova, N.A. Early-stage atherosclerosis in poloxamer 407-induced hyperlipidemic mice: Pathological features and changes in the lipid composition of serum lipoprotein fractions and subfractions. Lipids Health Dis. 2016, 15, 16. [Google Scholar] [CrossRef]
- Akl, M.A.; Ryad, S.; Ibrahim, M.F.; Kassem, A.A. Formulation, and optimization of transdermal Atorvastatin Calcium-Loaded Ultra-flexible vesicles; ameliorates poloxamer 407-caused dyslipidemia. Int. J. Pharm. 2023, 638, 122917. [Google Scholar] [CrossRef]
- US Department of Justice, D.E.A. Diversion Control Division, Drug & Chemical Evaluation Section. 2023. Available online: https://www.deadiversion.usdoj.gov/drug_chem_info/nalbuphine.pdf (accessed on 15 July 2023).
- Kick, B.L.; Shu, P.; Wen, B.; Sun, D.; Taylor, D.K. Pharmacokinetic Profiles of Nalbuphine after Intraperitoneal and Subcutaneous Administration to C57BL/6 Mice. J. Am. Assoc. Lab. Anim. Sci. 2017, 56, 534–538. [Google Scholar]
- Hussain, M.A.; Aungst, B.J.; Shefter, E. Buccal and Oral Bioavailability of Nalbuphine in Rats. J. Pharm. Sci. 1986, 75, 218–219. [Google Scholar] [CrossRef]
- Groenendaal, D.; Blom-Roosemalen, M.C.; Danhof, M.; de Lange, E.C. High-performance liquid chromatography of nalbuphine, butorphanol and morphine in blood and brain microdialysate samples: Application to pharmacokinetic/pharmacodynamic studies in rats. J. Chromatogr. B 2005, 822, 230–237. [Google Scholar] [CrossRef]
- Aungst, B.J.; Myers, M.J.; Shefter, E.; Shami, E.G. Prodrugs for improved oral nalbuphine bioavailability: Inter-species differences in the disposition of nalbuphine and its acetylsalicylate and anthranilate esters. Int. J. Pharm. 1987, 38, 199–209. [Google Scholar] [CrossRef]
- Gibson, C.R.; Gleason, A.; Messina, E. Measurement of total liver blood flow in intact anesthetized rats using ultrasound imaging. Pharmacol. Res. Perspect. 2021, 9, e00731. [Google Scholar] [CrossRef]
- Xie, C.; Wei, W.; Zhang, T.; Dirsch, O.; Dahmen, U. Monitoring of systemic and hepatic hemodynamic parameters in mice. J. Vis. Exp. 2014, 92, e51955. [Google Scholar]
- Lemaire, M.; Urien, S.; Albengres, E.; Riant, P.; Zini, R.; Tillement, J.P. Lipoprotein binding of drugs. Drug-Protein Bind. 1986, 93–108. [Google Scholar]
- Yamamoto, H.; Takada, T.; Yamanashi, Y.; Ogura, M.; Masuo, Y.; Harada-Shiba, M.; Suzuki, H. VLDL/LDL acts as a drug carrier and regulates the transport and metabolism of drugs in the body. Sci. Rep. 2017, 7, 633. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.R.; Ala, S.; Aliabadi, H.M. The effect of increased lipoprotein levels on the pharmacokinetics of cyclosporine A in the laboratory rat. Biopharm. Drug Dispos. 2005, 27, 7–16. [Google Scholar] [CrossRef]
- Hawi, A.; Alcorn, H.; Berg, J.; Hines, C.; Hait, H.; Sciascia, T. Pharmacokinetics of nalbuphine hydrochloride extended release tablets in hemodialysis patients with ex-ploratory effect on pruritus. BMC Nephrol. 2015, 16, 47. [Google Scholar] [CrossRef]
- Han, Y.-H.; Onufer, E.J.; Huang, L.-H.; Sprung, R.W.; Davidson, W.S.; Czepielewski, R.S.; Wohltmann, M.; Sorci-Thomas, M.G.; Warner, B.W.; Randolph, G.J. Enterically derived high-density lipoprotein restrains liver injury through the portal vein. Science 2021, 373, 410. [Google Scholar] [CrossRef]
- Ganesan, L.P.; Mates, J.M.; Cheplowitz, A.M.; Avila, C.L.; Zimmerer, J.M.; Yao, Z.; Maiseyeu, A.; Rajaram, M.V.S.; Robinson, J.M.; Anderson, C.L. Scavenger receptor B1, the HDL receptor, is expressed abundantly in liver sinusoidal endothelial cells. Sci. Rep. 2016, 6, 20646. [Google Scholar] [CrossRef]
- Kayden, H.; Traber, M. Absorption, lipoprotein transport, and regulation of plasma concentrations of vitamin E in humans. J. Lipid Res. 1993, 34, 343–358. [Google Scholar] [CrossRef]
- Bock, H.H.; May, P.; Herz, J. Lipoprotein Transport, in Transgenic Models in Pharmacology. In Handbook of Experimental Pharmacology; Offermanns, S., Hein, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 397–421. [Google Scholar]
- Gawade, S. Acetic acid induced painful endogenous infliction in writhing test on mice. J. Pharmacol. Pharmacother. 2012, 3, 348. [Google Scholar] [CrossRef]
- Ismail, N.I.; Ming-Tatt, L.; Lajis, N.; Akhtar, M.N.; Akira, A.; Perimal, E.K.; Israf, D.A.; Sulaiman, M.R. Antinociceptive Effect of 3-(2,3-Dimethoxyphenyl)-1-(5-methylfuran-2-yl)prop-2-en-1-one in Mice Models of Induced Nociception. Molecules 2016, 21, 1077. [Google Scholar] [CrossRef]
- Ruan, D.; Wang, Y.; Li, S.; Zhang, C.; Zheng, W.; Yu, C. Nalbuphine alleviates inflammation by down-regulating NF-kappaB in an acute inflammatory visceral pain rat model. BMC Pharmacol. Toxicol. 2022, 23, 34. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Chang, H.Y.; Rappsilber, J.; Ishihama, Y. Isolation of Acetylated and Unmodified Protein N-Terminal Peptides by Strong Cation Exchange Chromatographic Separation of TrypN-Digested Peptides. Mol. Cell. Proteom. 2021, 20, 100003. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T.; Nafady, A.; Talpur, F.N.; Agheem, M.H.; Shah, M.R.; Sherazi, S.T.H.; Soomro, R.A.; Siddiqui, S. Tranexamic acid derived gold nanoparticles modified glassy carbon electrode as sensitive sensor for de-termination of nalbuphine. Sens. Actuators B Chem. 2015, 211, 359–369. [Google Scholar] [CrossRef]
- Elsherbiny, M.E.; Brocks, D.R. The effect of CYP1A induction on amiodarone disposition in the rat. J. Pharm. Sci. 2010, 99, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Scheerans, C.; Heinig, R.; Mueck, W. Proposal for defining the relevance of drug accumulation derived from single dose study data for modified release dosage forms. Biopharm. Drug Dispos. 2014, 36, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, C.W.; Wiggins, B.S.; Spinler, S.A. Role of P-glycoprotein in Statin Drug Interactions. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2006, 26, 1601–1607. [Google Scholar] [CrossRef] [PubMed]
- Wessler, J.D.; Grip, L.T.; Mendell, J.; Giugliano, R.P. The P-Glycoprotein Transport System and Cardio-vascular Drugs. J. Am. Coll. Cardiol. 2013, 61, 2495–2502. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmax (ng/mL) | Tmax (min) | AUC0-Tlast (ng.min/mL) | AUC0-inf (ng.min/mL) | t1/2 (min) | CL/F (mL/min/kg) | Vdss/F (mL/kg) |
|---|---|---|---|---|---|---|
| NL rats | ||||||
| 303 ± 6.7 | 30 | 13,307 ± 1850 | 13,869 ± 1836 | 26 ± 3.0 | 195 ± 16.7 | 2165 ± 315 |
| HL rats | ||||||
| 256 ± 26 | 30 | 12,860 ± 1269 | 13,988 ± 1267 | 27 ± 3.4 | 188 ± 15.4 | 3193 ± 820 |
| Correlation of Nalbuphine Cmax with Different Serum Lipids | |
|---|---|
| Total cholesterol | r = −0.606→(0.0129) n = 16 |
| Triglycerides | r = −0.455→(0.0767) n = 16 |
| High-density lipoproteins | r = −0.712→(0.00196) n = 16 |
| Low-density lipoproteins | r = −0.568→(0.0217) n = 16 |
| Correlation of Brain Nalbuphine Concentration with Different Serum Lipids | |
| Total cholesterol | r = −0.513→(0.298) n = 6 |
| Triglycerides | r = −0.842→(0.0353) n = 6 |
| High-density lipoproteins | r = 0.385→(0.451) n = 6 |
| Low-density lipoproteins | r = −0.467→(0.350) n = 6 |
| Correlation of Liver Nalbuphine Concentration with Different Serum Lipids | |
| Total cholesterol | r = 0.450→(0.371) n = 6 |
| Triglycerides | r = −0.146→(0.782) n = 6 |
| High-density lipoproteins | r = 0.764→(0.0767) n = 6 |
| Low-density lipoproteins | r = 0.290→(0.578) n = 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsherbiny, M.E.; Almukainzi, M.; Amer, E.; Emara, M. Hyperlipidemia Increases Nalbuphine Brain Accumulation with Multiple Dosing without Affecting Its Analgesic Response—Its Respiratory Depression Potential Should Be Investigated in Future Studies. Pharmaceuticals 2024, 17, 282. https://doi.org/10.3390/ph17030282
Elsherbiny ME, Almukainzi M, Amer E, Emara M. Hyperlipidemia Increases Nalbuphine Brain Accumulation with Multiple Dosing without Affecting Its Analgesic Response—Its Respiratory Depression Potential Should Be Investigated in Future Studies. Pharmaceuticals. 2024; 17(3):282. https://doi.org/10.3390/ph17030282
Chicago/Turabian StyleElsherbiny, Marwa E., May Almukainzi, Eman Amer, and Marwan Emara. 2024. "Hyperlipidemia Increases Nalbuphine Brain Accumulation with Multiple Dosing without Affecting Its Analgesic Response—Its Respiratory Depression Potential Should Be Investigated in Future Studies" Pharmaceuticals 17, no. 3: 282. https://doi.org/10.3390/ph17030282
APA StyleElsherbiny, M. E., Almukainzi, M., Amer, E., & Emara, M. (2024). Hyperlipidemia Increases Nalbuphine Brain Accumulation with Multiple Dosing without Affecting Its Analgesic Response—Its Respiratory Depression Potential Should Be Investigated in Future Studies. Pharmaceuticals, 17(3), 282. https://doi.org/10.3390/ph17030282