
An Overview of Biosimilars—Development, Quality, Regulatory Issues, and Management in Healthcare

 ,
,  , , , ,
, , , ,  and
and 
Abstract

1. Introduction
Key Points
- −
- The use of biosimilar medicines contributes to greater patient access to biological therapies. However, for this to happen, healthcare professionals and patients must understand the concept of biosimilarity.
- −
- The development of a biosimilar involves conducting a comparability assessment between the intended biosimilar and the reference biological. The primary objective is not to establish their efficacy and safety independently but rather to validate the similarity between the two products.
- −
- Despite their recognized value, biosimilars are still subject to some controversies, such as immunogenicity, interchangeability and substitution, extrapolation, and nomenclature.
2. Biological Medicines
2.1. Biosimilars
2.2. Intended Copies, Biobetters, and Standalone Biologics
3. Development and Regulatory Approval of Biosimilars
3.1. Development of a Biosimilar
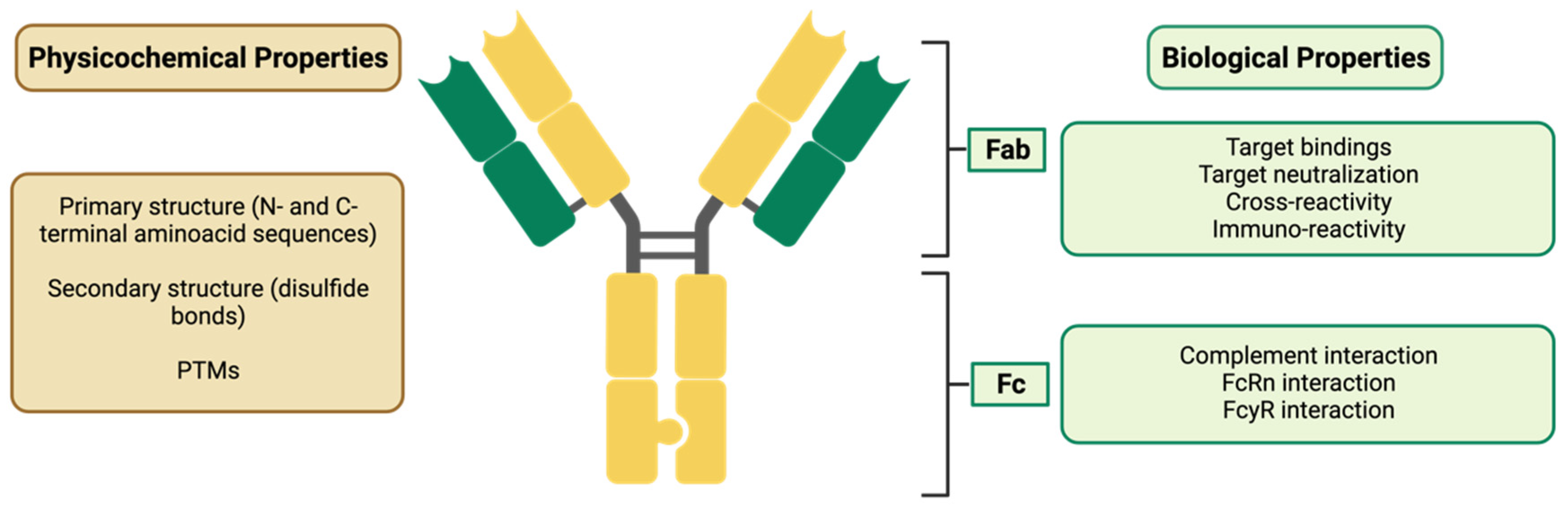
Post-Translational Modifications (PTMs)
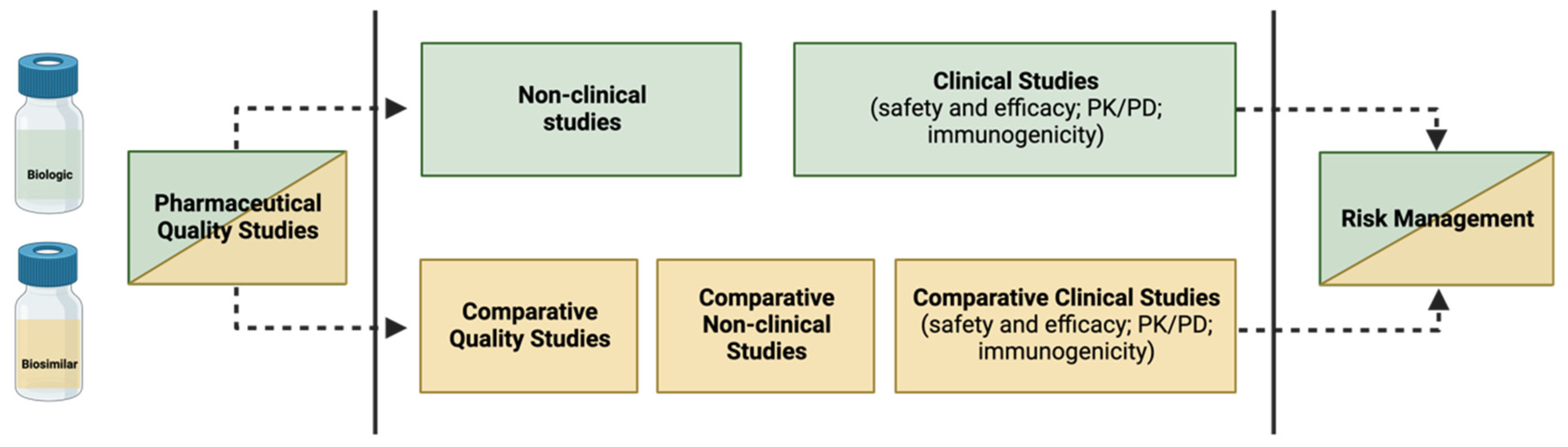
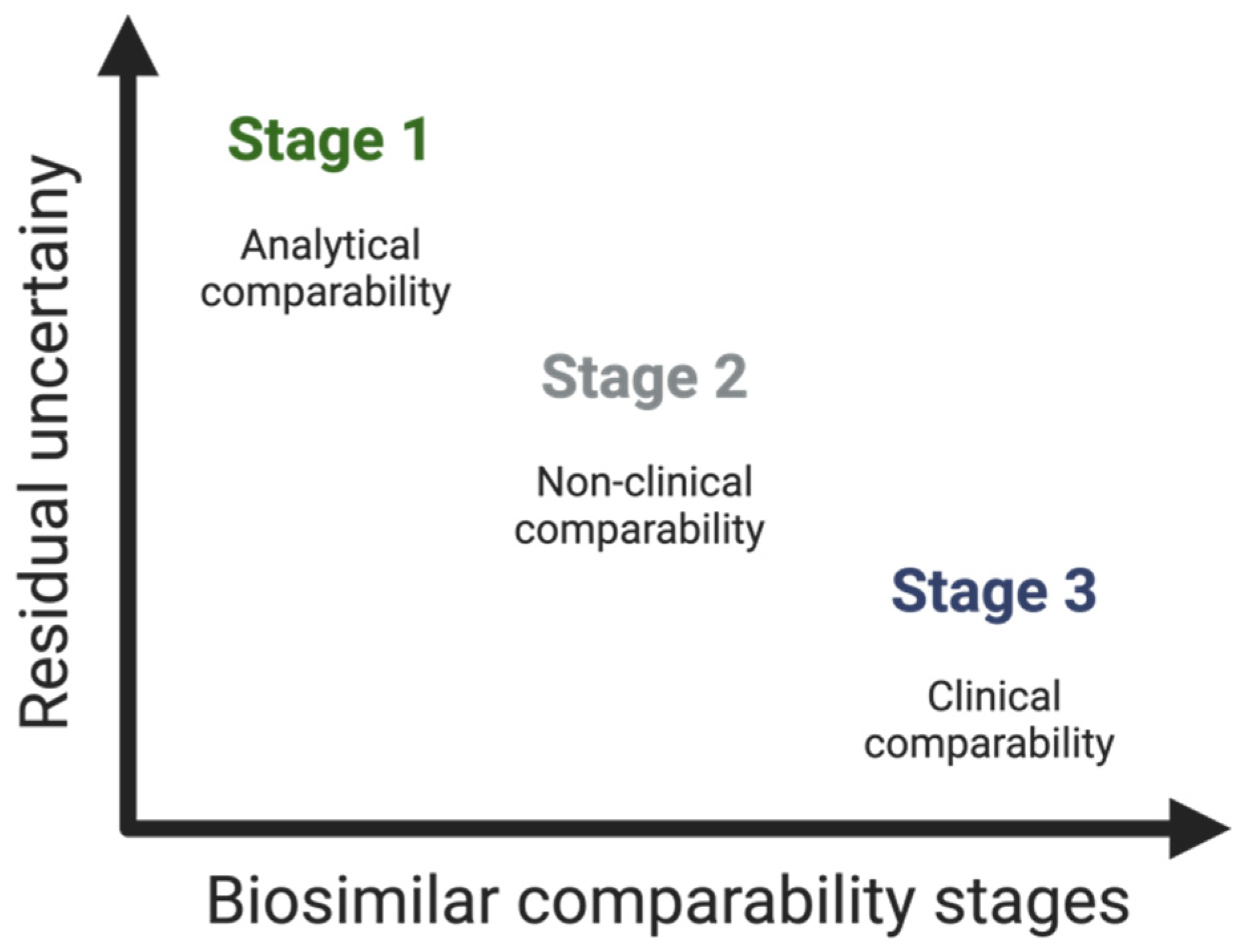
3.2. How to Build the Evidence for Biosimilarity
3.2.1. Demonstration of Analytical Similarity—Comparative Quality Studies
3.2.2. Establishing Non-Clinical Biosimilarity
3.2.3. Clinical Considerations—The Supporting Role of Phase I and Phase III Clinical Studies
Pharmacokinetic and Pharmacodynamic Studies
Efficacy Studies
Safety Evaluation
3.3. Regulatory Concerns
4. Post-Marketing Monitoring of Safety of Biosimilars—Pharmacovigilance
5. Controversies in the Use of Biosimilars
5.1. Immunogenicity
5.2. Extrapolation
5.3. Interchangeability and Substitution
5.4. Nomenclature
6. Implementation Model of Biosimilars in Hospital Settings
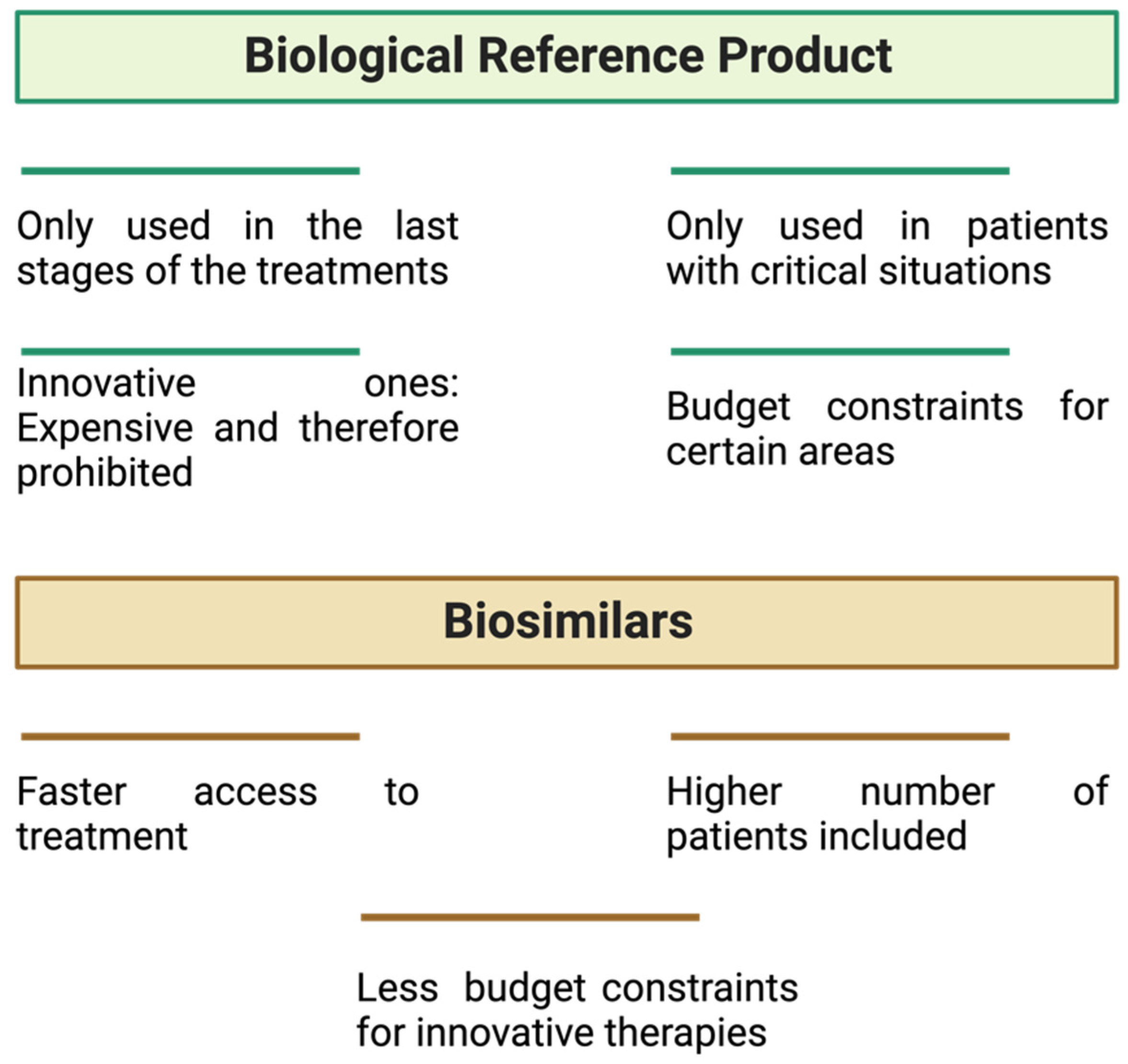
6.1. Economic Aspects of the Use of Biosimilars
6.2. What a Prescriber Needs to Know
6.3. Patient Needs
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AEs | Adverse Events |
| ADAs | Anti-Drug Antibodies |
| BD | Biodisponibility |
| CHMP | Committee for Medicinal Products for Human Use |
| CQAs | Critical Quality Attributes |
| EMA | European Medicine Administration |
| EEE | European Economic Area |
| EU | European Union |
| Fab | Fragment Antigen-Binding |
| Fc | Fragment Crystallizable |
| FcRn | Neonatal Fragment Crystallizable Receptor |
| FcγR | Fragment Crystallizable-Gamma Receptor |
| FDA | Food and Drug Administration |
| FIMEA | Finnish Medicines Agency |
| GCSF | Granulocyte Colony-Stimulating Factor |
| GH | Growth Hormone |
| ICH | International Conference of Harmonization |
| INFα | Interferon Alpha |
| INFβ | Interferon Beta |
| INN | International Naming System |
| RP | Reference Product |
| WHO | World Health Organization |
| mAb | Monoclonal Antibody |
| MA | Marketing Authorization |
| MS | Member State |
| PK | Pharmacokinetic |
| PD | Pharmacodynamics |
| PTMs | Post-Translational Modifications |
| PV | Pharmacovigilance |
| QbD | Quality-by-Design |
| QTPP | Quality Target Product Profile |
| SmPC | Summary of Product Characteristics |
| tRNA | Transfer RNA |
References
- Wolff-Holz, E.; Tiitso, K.; Vleminckx, C.; Weise, M. Evolution of the EU Biosimilar Framework: Past and Future. BioDrugs 2019, 33, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Afzali, A.; Furtner, D.; Melsheimer, R.; Molloy, P.J. The Automatic Substitution of Biosimilars: Definitions of Interchangeability are not Interchangeable. Adv. Ther. 2021, 38, 2077–2093. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.; Gonçalves, J.; Strohal, R.; Castañeda-Hernández, G.; Azevedo, V.; Dörner, T.; McInnes, I. The biosimilar approval process: How different is it. Consid. Med. 2017, 1, 3–6. [Google Scholar] [CrossRef]
- Blandizzi, C.; Meroni, P.L.; Lapadula, G. Comparing Originator Biologics and Biosimilars: A Review of the Relevant Issues. Clin. Ther. 2017, 39, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Declerck, P.; Danesi, R.; Petersel, D.; Jacobs, I. The Language of Biosimilars: Clarification, Definitions, and Regulatory Aspects. Drugs 2017, 77, 671–677. [Google Scholar] [CrossRef]
- Mirjalili, S.Z.; Sabourian, R.; Sadeghalvad, M.; Rezaei, N. Therapeutic applications of biosimilar monoclonal antibodies: Systematic review of the efficacy, safety, and immunogenicity in autoimmune disorders. Int. Immunopharmacol. 2021, 101, 108305. [Google Scholar] [CrossRef]
- Ramanan, S.; Grampp, G. Drift, evolution, and divergence in biologics and biosimilars manufacturing. BioDrugs 2014, 28, 363–372. [Google Scholar] [CrossRef]
- Ingrasciotta, Y.; Cutroneo, P.M.; Marciano, I.; Giezen, T.; Atzeni, F.; Trifiro, G. Safety of Biologics, Including Biosimilars: Perspectives on Current Status and Future Direction. Drug Saf. 2018, 41, 1013–1022. [Google Scholar] [CrossRef]
- Halimi, V.; Daci, A.; Ancevska Netkovska, K.; Suturkova, L.; Babar, Z.U.; Grozdanova, A. Clinical and Regulatory Concerns of Biosimilars: A Review of Literature. Int. J. Environ. Res. Public Health 2020, 17, 5800. [Google Scholar] [CrossRef]
- European Commission. What You Need to Know about Biosimilar Medicinal Products. Consensus Information Paper [Internet]. 2013. Available online: https://ec.europa.eu/docsroom/documents/8242 (accessed on 7 February 2024).
- Mitra, S.; Murthy, G.S. Bioreactor control systems in the biopharmaceutical industry: A critical perspective. Syst. Microbiol. Biomanuf. 2022, 2, 91–112. [Google Scholar] [CrossRef]
- Declerck, P.; Farouk Rezk, M. The road from development to approval: Evaluating the body of evidence to confirm biosimilarity. Rheumatology 2017, 56, iv4–iv13. [Google Scholar] [CrossRef]
- Millán-Martín, S.; Zaborowska, I.; Jakes, C.; Carillo, S.; Bones, J. Comparability Study for the Determination of Post-Translational Modifications of Biotherapeutic Drug Products and Biosimilars by Automated Peptide Mapping Analysis. Available online: https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/an-21850-lc-ms-comparability-biosimilars-an21850-en.pdf (accessed on 7 February 2024).
- Ismail, S.; Abu Esba, L.; Khan, M.; Al-Abdulkarim, H.; Modimagh, H.; Yousef, C. An Institutional Guide for Formulary Decisions of Biosimilars. Hosp. Pharm. 2023, 58, 38–48. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use. Guideline on Similar Biological Medicinal Products Containing Biotechnology-derived Proteins as Active Substance: Non-Clinical and Clinical Issues; European Medicines Agency: London, UK, 2006.
- Ishii-Watabe, A.; Kuwabara, T. Biosimilarity assessment of biosimilar therapeutic monoclonal antibodies. Drug Metab. Pharmacokinet. 2019, 34, 64–70. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Bissig, M.; Curigliano, G.; Coppola, J.; Latymer, M. Talking to patients about biosimilars. Future Oncol. 2018, 14, 2403–2414. [Google Scholar] [CrossRef] [PubMed]
- Gamez-Belmonte, R.; Hernandez-Chirlaque, C.; Arredondo-Amador, M.; Aranda, C.J.; Gonzalez, R.; Martinez-Augustin, O.; Sanchez de Medina, F. Biosimilars: Concepts and controversies. Pharmacol. Res. 2018, 133, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Agbogbo, F.K.; Ecker, D.M.; Farrand, A.; Han, K.; Khoury, A.; Martin, A.; McCool, J.; Rasche, U.; Rau, T.D.; Schmidt, D.; et al. Current perspectives on biosimilars. J. Ind. Microbiol. Biotechnol. 2019, 46, 1297–1311. [Google Scholar] [CrossRef] [PubMed]
- Alsamil, A.M.; Giezen, T.J.; Egberts, T.C.; Leufkens, H.G.; Vulto, A.G.; van der Plas, M.R.; Gardarsdottir, H. Reporting of quality attributes in scientific publications presenting biosimilarity assessments of (intended) biosimilars: A systematic literature review. Eur. J. Pharm. Sci. 2020, 154, 105501. [Google Scholar] [CrossRef] [PubMed]
- Markus, R.; Liu, J.; Ramchandani, M.; Landa, D.; Born, T.; Kaur, P. Developing the totality of evidence for biosimilars: Regulatory considerations and building confidence for the healthcare community. BioDrugs 2017, 31, 175–187. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Guideline on Similar Biological Medicinal Products. CHMP/437/04 Rev 1. 2014. Available online: https://www.ema.europa.eu/system/files/documents/scientific-guideline/wc500176768_en.pdf (accessed on 7 February 2024).
- Andrade, C. Bioequivalence of generic drugs. J. Clin. Psychiatry 2015, 76, 1905. [Google Scholar] [CrossRef]
- Beninger, P. Pharmacovigilance: An Overview. Clin. Ther. 2018, 40, 1991–2004. [Google Scholar] [CrossRef] [PubMed]
- Schreitmuller, T.; Barton, B.; Zharkov, A.; Bakalos, G. Comparative immunogenicity assessment of biosimilars. Future Oncol. 2019, 15, 319–329. [Google Scholar] [CrossRef]
- de Mora, F. Biosimilars: A Value Proposition. BioDrugs 2019, 33, 353–356. [Google Scholar] [CrossRef]
- Scheckel, C.J.; Rajkumar, S.V. Generics and biosimilars: Barriers and opportunities. Mayo Clin. Proc. 2021, 96, 2947–2957. [Google Scholar] [CrossRef]
- Kurki, P.; van Aerts, L.; Wolff-Holz, E.; Giezen, T.; Skibeli, V.; Weise, M. Interchangeability of Biosimilars: A European Perspective. BioDrugs 2017, 31, 83–91. [Google Scholar] [CrossRef]
- De Mora, F. Biosimilar: What it is not. Br. J. Clin. Pharmacol. 2015, 80, 949–956. [Google Scholar] [CrossRef]
- Ghia, C.; Shah, D.; Rambhad, G.; Mubashir, A.; Upadhyaya, S. Biologics, biosimilars, intended copies and the era of competitive medicine. Apollo Med. 2015, 12, 103–111. [Google Scholar] [CrossRef]
- Elgundi, Z.; Reslan, M.; Cruz, E.; Sifniotis, V.; Kayser, V. The state-of-play and future of antibody therapeutics. Adv. Drug Deliv. Rev. 2017, 122, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Kapur, M.; Nirula, S.; Naik, M.P. Future of anti-VEGF: Biosimilars and biobetters. Int. J. Retin. Vitr. 2022, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kumar, N.; Kuppermann, B.D.; Bandello, F.; Loewenstein, A. Biologics, biosilimars, and biobetters: Different terms or different drugs? Eye 2019, 33, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, E.A.; Joseph, P.F. The economics of biosimilars. Am. Health Drug Benefits 2013, 6, 469–478. [Google Scholar] [PubMed]
- Socinski, M.A.; Curigliano, G.; Jacobs, I.; Gumbiner, B.; MacDonald, J.; Thomas, D. Clinical considerations for the development of biosimilars in oncology. mAbs 2015, 7, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.B.; Taraban, M.B.; Wang, W.; Briggs, K.T. Improving biopharmaceutical safety through verification-based quality control. Trends Biotechnol. 2017, 35, 1140–1155. [Google Scholar] [CrossRef]
- Mellstedt, H.; Niederwieser, D.; Ludwig, H. The challenge of biosimilars. Ann. Oncol. 2008, 19, 411–419. [Google Scholar] [CrossRef]
- Vulto, A.G.; Jaquez, O.A. The process defines the product: What really matters in biosimilar design and production? Rheumatology 2017, 56, iv14–iv29. [Google Scholar] [CrossRef]
- Lee, S.J.; Chow, S.-C. Biosimilar Product Development. In Methodologies in Biosimilar Product Development; CRC Press: Boca Raton, FL, USA, 2021; pp. 1–22. [Google Scholar]
- Shrivastava, A.; Joshi, S.; Guttman, A.; Rathore, A.S. N-Glycosylation of monoclonal antibody therapeutics: A comprehensive review on significance and characterization. Anal. Chim. Acta 2022, 1209, 339828. [Google Scholar] [CrossRef]
- Xie, H.; Chakraborty, A.; Ahn, J.; Yu, Y.Q.; Dakshinamoorthy, D.P.; Gilar, M.; Chen, W.; Skilton, S.J.; Mazzeo, J.R. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. MAbs 2010, 2, 379–394. [Google Scholar] [CrossRef]
- Wang, T.; Chu, L.; Li, W.; Lawson, K.; Apostol, I.; Eris, T. Application of a Quantitative LC-MS Multiattribute Method for Monitoring Site-Specific Glycan Heterogeneity on a Monoclonal Antibody Containing Two N-Linked Glycosylation Sites. Anal. Chem. 2017, 89, 3562–3567. [Google Scholar] [CrossRef] [PubMed]
- Largy, E.; Cantais, F.; Van Vyncht, G.; Beck, A.; Delobel, A. Orthogonal liquid chromatography-mass spectrometry methods for the comprehensive characterization of therapeutic glycoproteins, from released glycans to intact protein level. J. Chromatogr. A 2017, 1498, 128–146. [Google Scholar] [CrossRef] [PubMed]
- Mouchahoir, T.; Schiel, J.E. Development of an LC-MS/MS peptide mapping protocol for the NISTmAb. Anal. Bioanal. Chem. 2018, 410, 2111–2126. [Google Scholar] [CrossRef]
- Gajadhar, A.; Fisher, T.; Scientific, S.J. SureQuant Intelligence-Driven MS: A New Paradigm for Targeted Quantitation; Thermo Fischer Scientific: Waltham, MA, USA, 2020. [Google Scholar]
- European Medicines Agency; European Commission. Biosimilars in the EU—Information Guide for Healthcare Professionals; European Medicines Agency: London, UK, 2019.
- The Food and Drug Administration. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Guidance for Industry; 2015. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/scientific-considerations-demonstrating-biosimilarity-reference-product (accessed on 7 February 2024).
- Rugo, H.S.; Linton, K.M.; Cervi, P.; Rosenberg, J.A.; Jacobs, I. A clinician’s guide to biosimilars in oncology. Cancer Treat. Rev. 2016, 46, 73–79. [Google Scholar] [CrossRef]
- Kim, H.; Alten, R.; Avedano, L.; Dignass, A.; Gomollón, F.; Greveson, K.; Halfvarson, J.; Irving, P.M.; Jahnsen, J.; Lakatos, P.L. The future of biosimilars: Maximizing benefits across immune-mediated inflammatory diseases. Drugs 2020, 80, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, B.; Wiendl, H.; Roth, K.; Wessels, H.; Hofler, J.; Hornuss, C.; Liedert, B.; Selmaj, K. Efficacy and Safety of Proposed Biosimilar Natalizumab (PB006) in Patients with Relapsing-Remitting Multiple Sclerosis: The Antelope Phase 3 Randomized Clinical Trial. JAMA Neurol. 2023, 80, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Kesik-Brodacka, M. Progress in biopharmaceutical development. Biotechnol. Appl. Biochem. 2018, 65, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Cazap, E.; Jacobs, I.; McBride, A.; Popovian, R.; Sikora, K. Global Acceptance of Biosimilars: Importance of Regulatory Consistency, Education, and Trust. Oncologist 2018, 23, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Christl, L. FDA’s Overview of the Regulatory Guidance for the Development and Approval of Biosimilar Products in the US; US Food and Drug Administration: Silver Spring, MD, USA, 2016.
- Singh, A.; Kalaivani, M.; Srivastava, S.; Goyal, R.K.; Gupta, S.K. Postmarketing Safety of Biosimilars: Current Status, Challenges, and Opportunities in the Spontaneous Reporting System. Ther. Innov. Regul. Sci. 2020, 54, 667–680. [Google Scholar] [CrossRef]
- Kabir, E.R.; Moreino, S.S.; Sharif Siam, M.K. The Breakthrough of Biosimilars: A Twist in the Narrative of Biological Therapy. Biomolecules 2019, 9, 410. [Google Scholar] [CrossRef]
- Ascef, B.O.; Lopes, A.C.F.; de Soarez, P.C. Health technology assessment of biosimilars worldwide: A scoping review. Health Res. Policy Syst. 2020, 18, 95. [Google Scholar] [CrossRef]
- Konstantinidou, S.; Papaspiliou, A.; Kokkotou, E. Current and future roles of biosimilars in oncology practice. Oncol. Lett. 2020, 19, 45–51. [Google Scholar] [CrossRef]
- Tkaczuk, K.H.R.; Jacobs, I.A. Biosimilars in oncology: From development to clinical practice. Semin. Oncol. 2014, 41, S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Lyman, G.H.; Balaban, E.; Diaz, M.; Ferris, A.; Tsao, A.; Voest, E.; Zon, R.; Francisco, M.; Green, S.; Sherwood, S.; et al. American Society of Clinical Oncology Statement: Biosimilars in Oncology. J. Clin. Oncol. 2018, 36, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Lonnfors, S.; Roblin, X.; Danese, S.; Avedano, L. Patient Perspectives on Biosimilars: A Survey by the European Federation of Crohn’s and Ulcerative Colitis Associations. J. Crohns Colitis 2017, 11, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Planes, S.; Villier, C.; Mallaret, M. The nocebo effect of drugs. Pharmacol. Res. Perspect. 2016, 4, e00208. [Google Scholar] [CrossRef] [PubMed]
- Rezk, M.F.; Pieper, B. Treatment Outcomes with Biosimilars: Be Aware of the Nocebo Effect. Rheumatol. Ther. 2017, 4, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Ghil, J.; Niebrzydowski, J.; Zielińska, A.; Lee, Y. FRI0198 Usability and safety of SB5 (an adalimumab biosimilar) pre-filled syringe and pre-filled pen in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 556. [Google Scholar]
- Kijanka, M.; Dorresteijn, B.; Oliveira, S.; van Bergen en Henegouwen, P.M. Nanobody-based cancer therapy of solid tumors. Nanomedicine 2015, 10, 161–174. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biosimilars | Generics | Ref. | |
|---|---|---|---|
| Product characteristics | - Large complex molecules (up to 270,000 Da) | - Small and simple molecules (up to 300 Da) | [5] |
| Production | - Produced using live organisms (highly sensitive to manufacturing changes) - 5–9 years - High production costs | - Produced by chemical synthesis - 2–3 year - Lower production costs | [5,21] |
| Structural comparison to reference medication | - Highly similar to the RP: same amino acid sequence; - There may be differences in minor parts of the structure | - Structurally identical to the reference medicine | [4,5,12] |
| Development | - Comparability studies between the biosimilar and the RP | - Bioequivalence between the generic and the RP is evaluated | [4,22] |
| Nomenclature | - Rules vary from country to country | - Same chemical name (active ingredient) as the reference medicine | [12,16,18,23] |
| Requirements for approval | - Animal and clinical studies (toxicity, PK, PD, and immunogenicity) | - No animal or clinical studies (only bioequivalence studies) - The active ingredient must be identical in strength, dosage form, and route of administration | [3,5,12,23] |
| Post-authorization activities | - Pharmacovigilance (PV) | - Phase IV, risk management plan including PV | [9,18,24,25,26] |
| Immunogenicity | - Immunogenic | - Mostly nonimmunogenic | [23,24,25,27] |
| Equivalence | - Data must demonstrate, in each indication, that clinically significant differences, related to safety and efficacy, are not verified - Conclusive clinical studies may not be necessary for all indications | - Demonstration of bioequivalence is enough to grant all approved indications for the RP, without requiring any additional clinical studies | [5,12,18,23,26] |
| Interchangeability and substitution | - EMA see biosimilars to be scientifically interchangeable (2022), but any decision on the use of biosimilars is the mandate of the EU member states - FDA can designate a biosimilar interchangeable if a sponsor applies for this (but it is up to US State Law to permit substitution at pharmacies) - Automatic substitution is generally the decision of each country | - If permitted by state law, pharmacists may automatically substitute the generic for the reference medicine | [2,18,23,27,28] |
| Topic | Title | Application |
|---|---|---|
| Overarching | - Guideline on similar biological medicinal products | General—applies to all biosimilars |
| Quality | - Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues | |
| Nonclinical and clinical | - Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues | |
| Annexes | - Recombinant human erythropoietin - Recombinant GCSF - Recombinant human insulin - Recombinant human GH - INF-α and INF-β - Low-molecular-weight heparins - Monoclonal antibodies - Recombinant follicle-stimulating hormone | Specific—product data requirements |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mascarenhas-Melo, F.; Diaz, M.; Gonçalves, M.B.S.; Vieira, P.; Bell, V.; Viana, S.; Nunes, S.; Paiva-Santos, A.C.; Veiga, F. An Overview of Biosimilars—Development, Quality, Regulatory Issues, and Management in Healthcare. Pharmaceuticals 2024, 17, 235. https://doi.org/10.3390/ph17020235
Mascarenhas-Melo F, Diaz M, Gonçalves MBS, Vieira P, Bell V, Viana S, Nunes S, Paiva-Santos AC, Veiga F. An Overview of Biosimilars—Development, Quality, Regulatory Issues, and Management in Healthcare. Pharmaceuticals. 2024; 17(2):235. https://doi.org/10.3390/ph17020235
Chicago/Turabian StyleMascarenhas-Melo, Filipa, Mariana Diaz, Maria Beatriz S. Gonçalves, Pedro Vieira, Victoria Bell, Sofia Viana, Sara Nunes, Ana Cláudia Paiva-Santos, and Francisco Veiga. 2024. "An Overview of Biosimilars—Development, Quality, Regulatory Issues, and Management in Healthcare" Pharmaceuticals 17, no. 2: 235. https://doi.org/10.3390/ph17020235
APA StyleMascarenhas-Melo, F., Diaz, M., Gonçalves, M. B. S., Vieira, P., Bell, V., Viana, S., Nunes, S., Paiva-Santos, A. C., & Veiga, F. (2024). An Overview of Biosimilars—Development, Quality, Regulatory Issues, and Management in Healthcare. Pharmaceuticals, 17(2), 235. https://doi.org/10.3390/ph17020235