
Resveratrol Enhances Cytotoxic Effects of Cisplatin by Inducing Cell Cycle Arrest and Apoptosis in Ovarian Adenocarcinoma SKOV-3 Cells through Activating the p38 MAPK and Suppressing AKT

 , , ,
, , ,  , , and
, , and 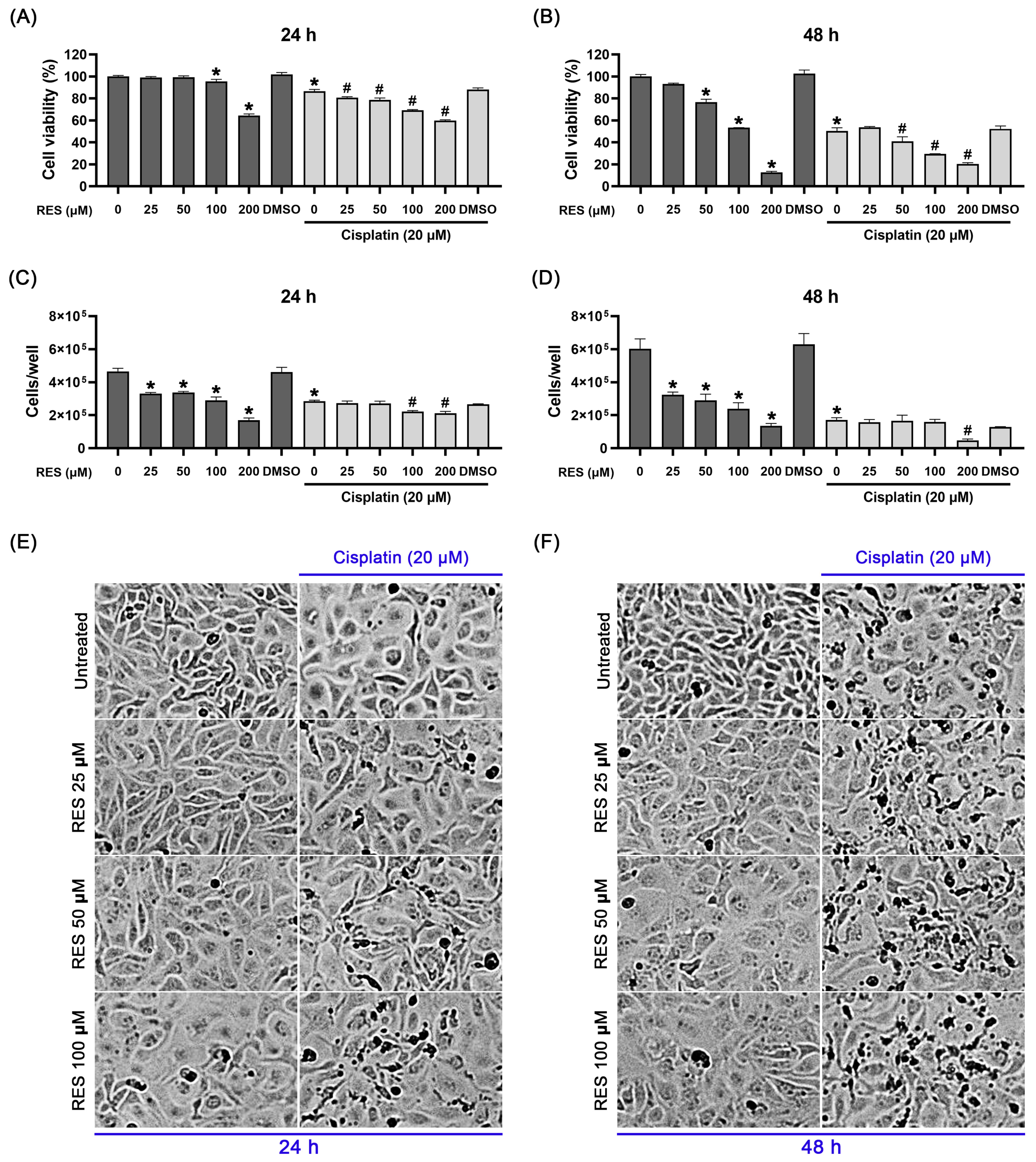{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. RES in Combination with Cisplatin Effectively Suppresses SKOV-3 Ovarian Cancer Cell Growth and Viability
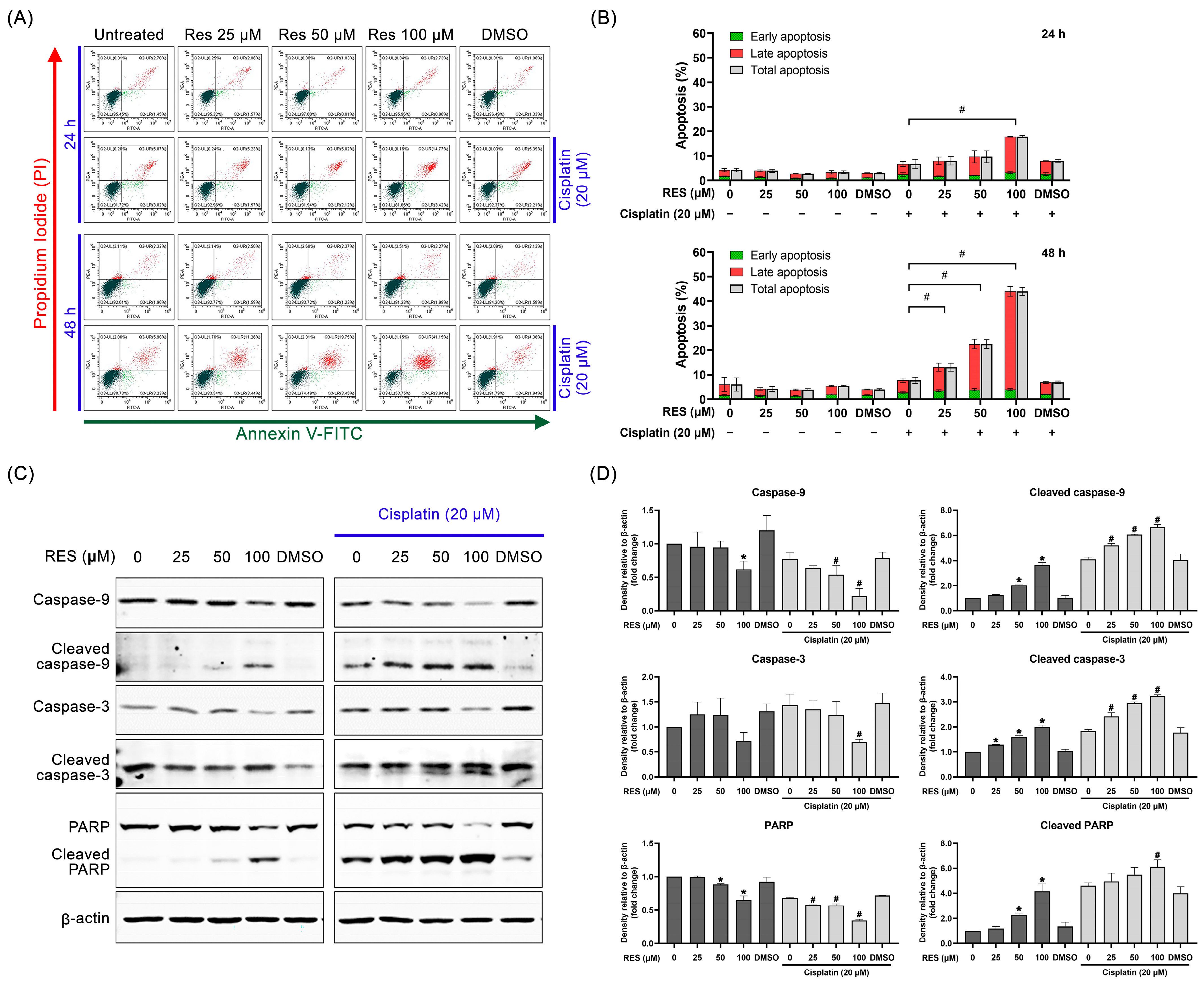
2.2. RES Amplifies SKOV-3 Cell Death through Enhancing Caspase-Dependent Apoptosis
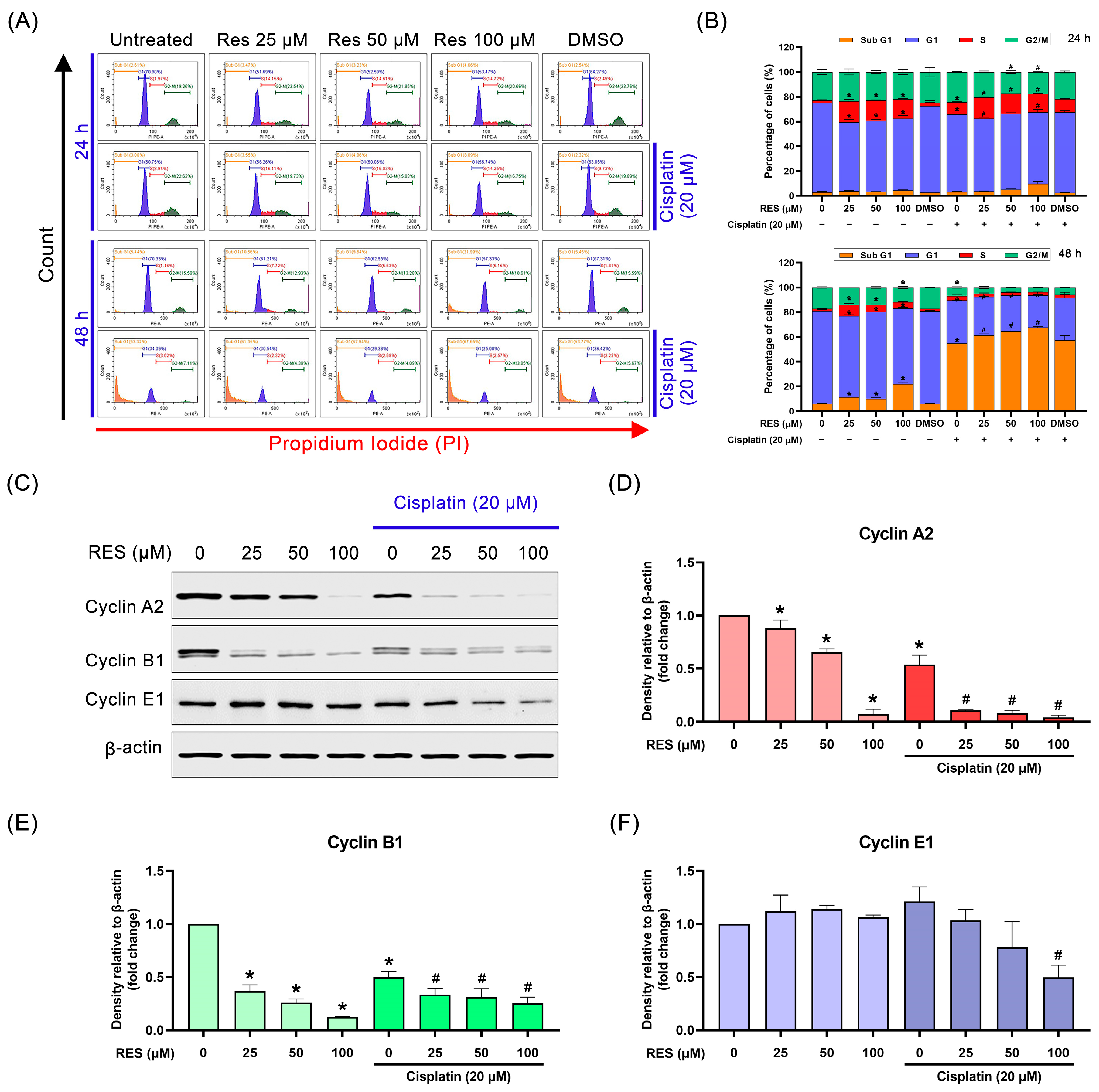
2.3. RES Arrests SKOV-3 Cell Cycle in the S-Phase
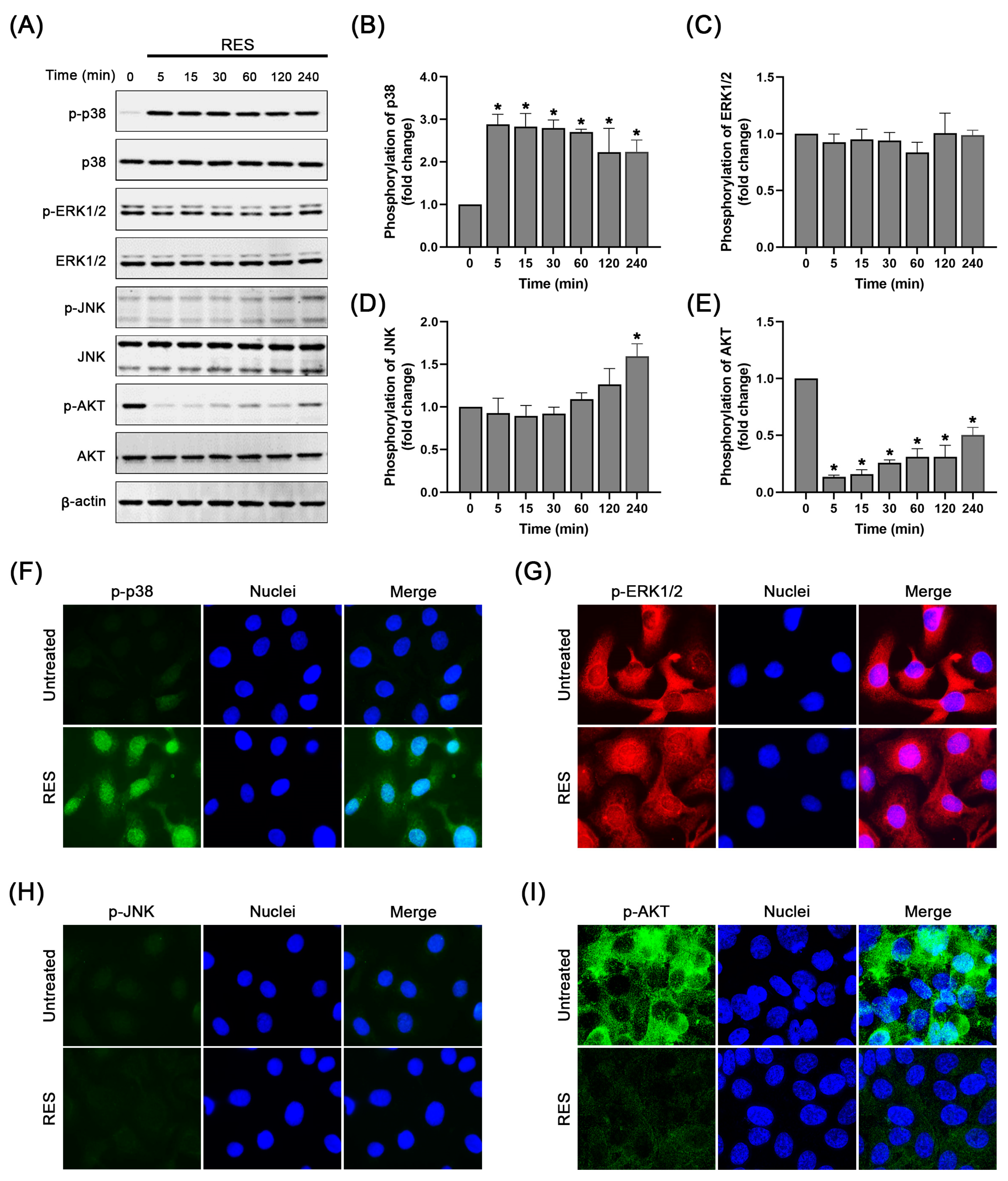
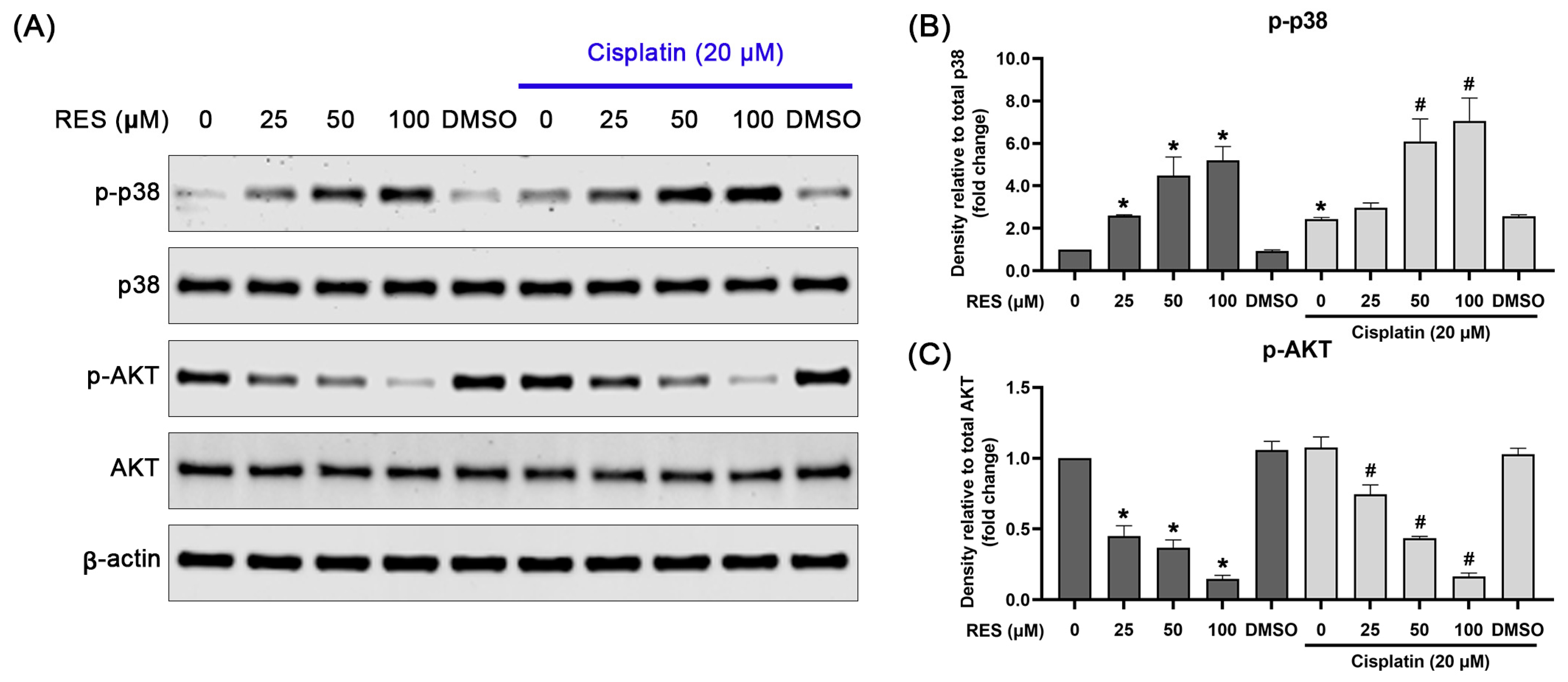
2.4. RES Specifically Induces p38 MAPK Activation and Suppresses AKT Activation
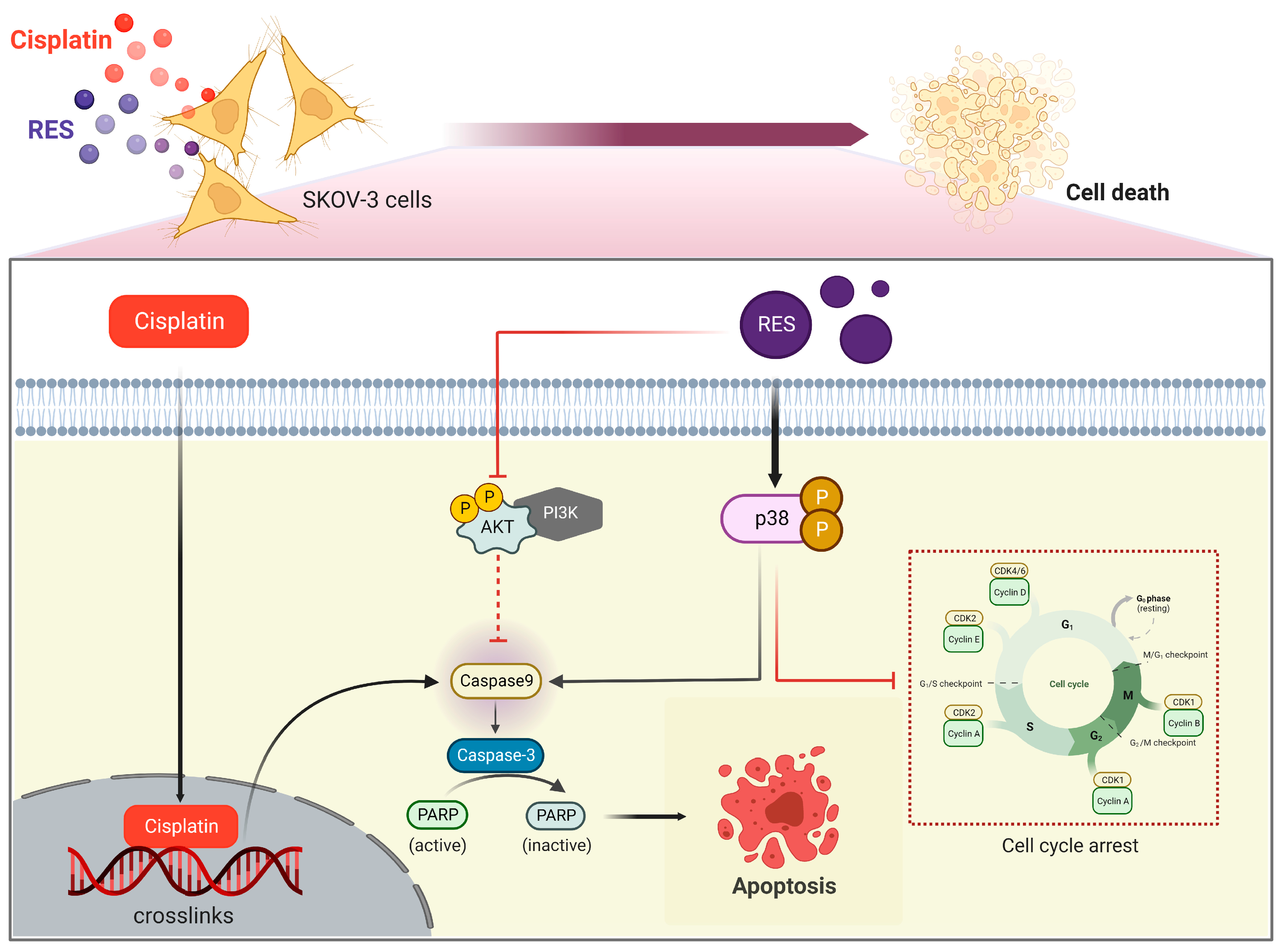
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. Cell Culture and RES Treatment
4.3. Cell Viability Assay
4.4. Cell Counting
4.5. Cell Apoptosis Analysis by Annexin V/PI Staining
4.6. Cell Cycle Analysis via Flow Cytometry
4.7. Western Blot Analysis
4.8. Immunofluorescence Study
4.9. Statistical Analysis
4.10. Synergy Quotient Calculation for Synergism
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian cancer in the world: Epidemiology and risk factors. Int. J. Womens Health 2019, 11, 287–299. [Google Scholar] [CrossRef]
- Sankaranarayanan, R.; Ferlay, J. Worldwide burden of gynaecological cancer: The size of the problem. Best Pract. Res. Clin. Obstet. Gynaecol. 2006, 20, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. The origin and pathogenesis of epithelial ovarian cancer: A proposed unifying theory. Am. J. Surg. Pathol. 2010, 34, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Levanon, K.; Crum, C.; Drapkin, R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J. Clin. Oncol. 2008, 26, 5284–5293. [Google Scholar] [CrossRef]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Li, S.S.; Ma, J.; Wong, A.S.T. Chemoresistance in ovarian cancer: Exploiting cancer stem cell metabolism. J. Gynecol. Oncol. 2018, 29, e32. [Google Scholar] [CrossRef]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark. Cancer 2019, 11, 1179299x19860815. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.K.; Lin, Y.H.; Chang, H.A.; Lai, Y.S.; Chen, Y.C.; Huang, S.C.; Chou, C.Y.; Chiu, W.T. Chemoresistant ovarian cancer enhances its migration abilities by increasing store-operated Ca(2+) entry-mediated turnover of focal adhesions. J. Biomed. Sci. 2020, 27, 36. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Wiegand, K.C.; Melnyk, N.; Chow, C.; Salamanca, C.; Prentice, L.M.; Senz, J.; Yang, W.; Spillman, M.A.; Cochrane, D.R.; et al. Type-specific cell line models for type-specific ovarian cancer research. PLoS ONE 2013, 8, e72162. [Google Scholar] [CrossRef]
- Marks, J.R.; Davidoff, A.M.; Kerns, B.J.; Humphrey, P.A.; Pence, J.C.; Dodge, R.K.; Clarke-Pearson, D.L.; Iglehart, J.D.; Bast, R.C., Jr.; Berchuck, A. Overexpression and mutation of p53 in epithelial ovarian cancer. Cancer Res. 1991, 51, 2979–2984. [Google Scholar]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Banno, K.; Okawa, R.; Yanokura, M.; Iijima, M.; Irie-Kunitomi, H.; Nakamura, K.; Iida, M.; Adachi, M.; Umene, K.; et al. ARID1A gene mutation in ovarian and endometrial cancers (Review). Oncol. Rep. 2016, 35, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Gasparri, M.L.; Bardhi, E.; Ruscito, I.; Papadia, A.; Farooqi, A.A.; Marchetti, C.; Bogani, G.; Ceccacci, I.; Mueller, M.D.; Benedetti Panici, P. PI3K/AKT/mTOR Pathway in Ovarian Cancer Treatment: Are We on the Right Track? Geburtshilfe Frauenheilkd. 2017, 77, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.G.; Pak, J.H.; Choi, W.H.; Park, J.Y.; Nam, J.H.; Kim, J.H. The relationship between cisplatin resistance and histone deacetylase isoform overexpression in epithelial ovarian cancer cell lines. J. Gynecol. Oncol. 2012, 23, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Han, J. The p38 signal transduction pathway: Activation and function. Cell. Signal. 2000, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Huth, H.W.; Santos, D.M.; Gravina, H.D.; Resende, J.M.; Goes, A.M.; de Lima, M.E.; Ropert, C. Upregulation of p38 pathway accelerates proliferation and migration of MDA-MB-231 breast cancer cells. Oncol. Rep. 2017, 37, 2497–2505. [Google Scholar] [CrossRef]
- Chen, L.; Mayer, J.A.; Krisko, T.I.; Speers, C.W.; Wang, T.; Hilsenbeck, S.G.; Brown, P.H. Inhibition of the p38 kinase suppresses the proliferation of human ER-negative breast cancer cells. Cancer Res. 2009, 69, 8853–8861. [Google Scholar] [CrossRef]
- Olson, J.M.; Hallahan, A.R. p38 MAP kinase: A convergence point in cancer therapy. Trends Mol. Med. 2004, 10, 125–129. [Google Scholar] [CrossRef]
- Bradham, C.; McClay, D.R. p38 MAPK in development and cancer. Cell Cycle 2006, 5, 824–828. [Google Scholar] [CrossRef]
- Han, X.; Chen, H.; Zhou, J.; Steed, H.; Postovit, L.M.; Fu, Y. Pharmacological Inhibition of p38 MAPK by SB203580 Increases Resistance to Carboplatin in A2780cp Cells and Promotes Growth in Primary Ovarian Cancer Cells. Int. J. Mol. Sci. 2018, 19, 2184. [Google Scholar] [CrossRef] [PubMed]
- Koushki, M.; Amiri-Dashatan, N.; Ahmadi, N.; Abbaszadeh, H.A.; Rezaei-Tavirani, M. Resveratrol: A miraculous natural compound for diseases treatment. Food Sci. Nutr. 2018, 6, 2473–2490. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.; Xiao, D.; Muhammed, A.; Deng, J.; Chen, L.; He, J. Anti-Inflammatory Action and Mechanisms of Resveratrol. Molecules 2021, 26, 229. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, M.; Ingmer, H. Antibacterial and antifungal properties of resveratrol. Int. J. Antimicrob. Agents 2019, 53, 716–723. [Google Scholar] [CrossRef]
- Zhou, D.D.; Luo, M.; Huang, S.Y.; Saimaiti, A.; Shang, A.; Gan, R.Y.; Li, H.B. Effects and Mechanisms of Resveratrol on Aging and Age-Related Diseases. Oxidative Med. Cell. Longev. 2021, 2021, 9932218. [Google Scholar] [CrossRef]
- Nanjan, M.J.; Betz, J. Resveratrol for the Management of Diabetes and its Downstream Pathologies. Eur. Endocrinol. 2014, 10, 31–35. [Google Scholar] [CrossRef]
- Dyck, G.J.B.; Raj, P.; Zieroth, S.; Dyck, J.R.B.; Ezekowitz, J.A. The Effects of Resveratrol in Patients with Cardiovascular Disease and Heart Failure: A Narrative Review. Int. J. Mol. Sci. 2019, 20, 904. [Google Scholar] [CrossRef]
- Gülçin, İ. Antioxidant properties of resveratrol: A structure–activity insight. Innov. Food Sci. Emerg. Technol. 2010, 11, 210–218. [Google Scholar] [CrossRef]
- Jang, J.Y.; Im, E.; Kim, N.D. Mechanism of Resveratrol-Induced Programmed Cell Death and New Drug Discovery against Cancer: A Review. Int. J. Mol. Sci. 2022, 23, 13689. [Google Scholar] [CrossRef]
- She, Q.B.; Bode, A.M.; Ma, W.Y.; Chen, N.Y.; Dong, Z. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinases and p38 kinase. Cancer Res. 2001, 61, 1604–1610. [Google Scholar]
- Chang, W.S.; Tsai, C.W.; Yang, J.S.; Hsu, Y.M.; Shih, L.C.; Chiu, H.Y.; Bau, D.T.; Tsai, F.J. Resveratrol inhibited the metastatic behaviors of cisplatin-resistant human oral cancer cells via phosphorylation of ERK/p-38 and suppression of MMP-2/9. J. Food Biochem. 2021, 45, e13666. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Cao, N.; Li, Z.; Han, J.; Li, L. Resveratrol, an activator of SIRT1, induces protective autophagy in non-small-cell lung cancer via inhibiting Akt/mTOR and activating p38-MAPK. Onco Targets Ther. 2018, 11, 7777–7786. [Google Scholar] [CrossRef] [PubMed]
- Lamboy-Caraballo, R.; Ortiz-Sanchez, C.; Acevedo-Santiago, A.; Matta, J.; Monteiro, A.N.; Armaiz-Pena, G.N. Norepinephrine-Induced DNA Damage in Ovarian Cancer Cells. Int. J. Mol. Sci. 2020, 21, 2250. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Bahar, E.; Lee, J.Y.; Chang, S.; Kim, S.H.; Park, E.Y.; Do, S.I.; Yoon, H.; Kim, H.S. ARL6IP5 reduces cisplatin-resistance by suppressing DNA repair and promoting apoptosis pathways in ovarian carcinoma. Cell Death Dis. 2022, 13, 239. [Google Scholar] [CrossRef]
- He, M.; Wang, D.; Zou, D.; Wang, C.; Lopes-Bastos, B.; Jiang, W.G.; Chester, J.; Zhou, Q.; Cai, J. Re-purposing of curcumin as an anti-metastatic agent for the treatment of epithelial ovarian cancer: In vitro model using cancer stem cell enriched ovarian cancer spheroids. Oncotarget 2016, 7, 86374–86387. [Google Scholar] [CrossRef]
- Goh, J.C.H.; Gourley, C.; Tan, D.S.P.; Nogueira-Rodrigues, A.; Elghazaly, H.; Edy Pierre, M.; Giornelli, G.; Kim, B.G.; Morales-Vasquez, F.; Tyulyandina, A. Optimizing treatment selection and sequencing decisions for first-line maintenance therapy of newly diagnosed advanced ovarian cancer. Gynecol. Oncol. Rep. 2022, 42, 101028. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Jessmon, P.; Boulanger, T.; Zhou, W.; Patwardhan, P. Epidemiology and treatment patterns of epithelial ovarian cancer. Expert. Rev. Anticancer. Ther. 2017, 17, 427–437. [Google Scholar] [CrossRef]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef]
- Buys, S.S.; Partridge, E.; Black, A.; Johnson, C.C.; Lamerato, L.; Isaacs, C.; Reding, D.J.; Greenlee, R.T.; Yokochi, L.A.; Kessel, B.; et al. Effect of screening on ovarian cancer mortality: The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA 2011, 305, 2295–2303. [Google Scholar] [CrossRef]
- Stordal, B.; Davey, M. Understanding cisplatin resistance using cellular models. IUBMB Life 2007, 59, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G. Clinical perspectives on platinum resistance. Drugs 2000, 59 (Suppl. 4), 9–17. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Calderon Iglesias, R.; Ruiz, R.; Aparicio, S.; Crespo, J.; Dzul Lopez, L.; Giampieri, F.; Battino, M. The use of natural compounds for the targeting and chemoprevention of ovarian cancer. Cancer Lett. 2017, 411, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Athar, M.; Back, J.H.; Tang, X.; Kim, K.H.; Kopelovich, L.; Bickers, D.R.; Kim, A.L. Resveratrol: A review of preclinical studies for human cancer prevention. Toxicol. Appl. Pharmacol. 2007, 224, 274–283. [Google Scholar] [CrossRef]
- Li, F.; Gong, Q.; Dong, H.; Shi, J. Resveratrol, a neuroprotective supplement for Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 27–33. [Google Scholar] [CrossRef]
- Kim, M.K.; Kim, K.; Han, J.Y.; Lim, J.M.; Song, Y.S. Modulation of inflammatory signaling pathways by phytochemicals in ovarian cancer. Genes. Nutr. 2011, 6, 109–115. [Google Scholar] [CrossRef]
- Opipari, A.W., Jr.; Tan, L.; Boitano, A.E.; Sorenson, D.R.; Aurora, A.; Liu, J.R. Resveratrol-induced autophagocytosis in ovarian cancer cells. Cancer Res. 2004, 64, 696–703. [Google Scholar] [CrossRef]
- Vergara, D.; Simeone, P.; Toraldo, D.; Del Boccio, P.; Vergaro, V.; Leporatti, S.; Pieragostino, D.; Tinelli, A.; De Domenico, S.; Alberti, S.; et al. Resveratrol downregulates Akt/GSK and ERK signalling pathways in OVCAR-3 ovarian cancer cells. Mol. Biosyst. 2012, 8, 1078–1087. [Google Scholar] [CrossRef]
- Engelke, L.H.; Hamacher, A.; Proksch, P.; Kassack, M.U. Ellagic Acid and Resveratrol Prevent the Development of Cisplatin Resistance in the Epithelial Ovarian Cancer Cell Line A2780. J. Cancer 2016, 7, 353–363. [Google Scholar] [CrossRef]
- Park, S.Y.; Jeong, K.J.; Lee, J.; Yoon, D.S.; Choi, W.S.; Kim, Y.K.; Han, J.W.; Kim, Y.M.; Kim, B.K.; Lee, H.Y. Hypoxia enhances LPA-induced HIF-1alpha and VEGF expression: Their inhibition by resveratrol. Cancer Lett. 2007, 258, 63–69. [Google Scholar] [CrossRef]
- Seino, M.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, S.; Takeda, H.; Ohta, T.; Kurachi, H.; Kitanaka, C. Differential contribution of ROS to resveratrol-induced cell death and loss of self-renewal capacity of ovarian cancer stem cells. Anticancer. Res. 2015, 35, 85–96. [Google Scholar] [PubMed]
- Paramee, S.; Sookkhee, S.; Sakonwasun, C.; Na Takuathung, M.; Mungkornasawakul, P.; Nimlamool, W.; Potikanond, S. Anti-cancer effects of Kaempferia parviflora on ovarian cancer SKOV3 cells. BMC Complement. Altern. Med. 2018, 18, 178. [Google Scholar] [CrossRef] [PubMed]
- Potikanond, S.; Sookkhee, S.; Na Takuathung, M.; Mungkornasawakul, P.; Wikan, N.; Smith, D.R.; Nimlamool, W. Kaempferia parviflora Extract Exhibits Anti-cancer Activity against HeLa Cervical Cancer Cells. Front. Pharmacol. 2017, 8, 630. [Google Scholar] [CrossRef] [PubMed]
- Suradej, B.; Sookkhee, S.; Panyakaew, J.; Mungkornasawakul, P.; Wikan, N.; Smith, D.R.; Potikanond, S.; Nimlamool, W. Kaempferia parviflora Extract Inhibits STAT3 Activation and Interleukin-6 Production in HeLa Cervical Cancer Cells. Int. J. Mol. Sci. 2019, 20, 4226. [Google Scholar] [CrossRef] [PubMed]
- Thaklaewphan, P.; Ruttanapattanakul, J.; Monkaew, S.; Buatoom, M.; Sookkhee, S.; Nimlamool, W.; Potikanond, S. Kaempferia parviflora extract inhibits TNF-alpha-induced release of MCP-1 in ovarian cancer cells through the suppression of NF-kappaB signaling. Biomed. Pharmacother. 2021, 141, 111911. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, H.; Safrit, J.T.; Bonavida, B. Synergistic effect of tumor necrosis factor-alpha- and diphtheria toxin-mediated cytotoxicity in sensitive and resistant human ovarian tumor cell lines. J. Immunol. 1991, 147, 2609–2616. [Google Scholar] [CrossRef] [PubMed]
- Muhanmode, Y.; Wen, M.K.; Maitinuri, A.; Shen, G. Curcumin and resveratrol inhibit chemoresistance in cisplatin-resistant epithelial ovarian cancer cells via targeting P13K pathway. Hum. Exp. Toxicol. 2022, 41, 9603271221095929. [Google Scholar] [CrossRef]
- Bian, Y.; Wang, X.; Zheng, Z.; Ren, G.; Zhu, H.; Qiao, M.; Li, G. Resveratrol drives cancer cell senescence via enhancing p38MAPK and DLC1 expressions. Food Funct. 2022, 13, 3283–3293. [Google Scholar] [CrossRef]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hankittichai, P.; Thaklaewphan, P.; Wikan, N.; Ruttanapattanakul, J.; Potikanond, S.; Smith, D.R.; Nimlamool, W. Resveratrol Enhances Cytotoxic Effects of Cisplatin by Inducing Cell Cycle Arrest and Apoptosis in Ovarian Adenocarcinoma SKOV-3 Cells through Activating the p38 MAPK and Suppressing AKT. Pharmaceuticals 2023, 16, 755. https://doi.org/10.3390/ph16050755
Hankittichai P, Thaklaewphan P, Wikan N, Ruttanapattanakul J, Potikanond S, Smith DR, Nimlamool W. Resveratrol Enhances Cytotoxic Effects of Cisplatin by Inducing Cell Cycle Arrest and Apoptosis in Ovarian Adenocarcinoma SKOV-3 Cells through Activating the p38 MAPK and Suppressing AKT. Pharmaceuticals. 2023; 16(5):755. https://doi.org/10.3390/ph16050755
Chicago/Turabian StyleHankittichai, Phateep, Phatarawat Thaklaewphan, Nitwara Wikan, Jirapak Ruttanapattanakul, Saranyapin Potikanond, Duncan R. Smith, and Wutigri Nimlamool. 2023. "Resveratrol Enhances Cytotoxic Effects of Cisplatin by Inducing Cell Cycle Arrest and Apoptosis in Ovarian Adenocarcinoma SKOV-3 Cells through Activating the p38 MAPK and Suppressing AKT" Pharmaceuticals 16, no. 5: 755. https://doi.org/10.3390/ph16050755
APA StyleHankittichai, P., Thaklaewphan, P., Wikan, N., Ruttanapattanakul, J., Potikanond, S., Smith, D. R., & Nimlamool, W. (2023). Resveratrol Enhances Cytotoxic Effects of Cisplatin by Inducing Cell Cycle Arrest and Apoptosis in Ovarian Adenocarcinoma SKOV-3 Cells through Activating the p38 MAPK and Suppressing AKT. Pharmaceuticals, 16(5), 755. https://doi.org/10.3390/ph16050755