
Structure–Activity Relationship Studies of Chalcones and Diarylpentanoids with Antitumor Activity: Potency and Selectivity Optimization

 , ,
, ,  ,
,  and
and 
Abstract
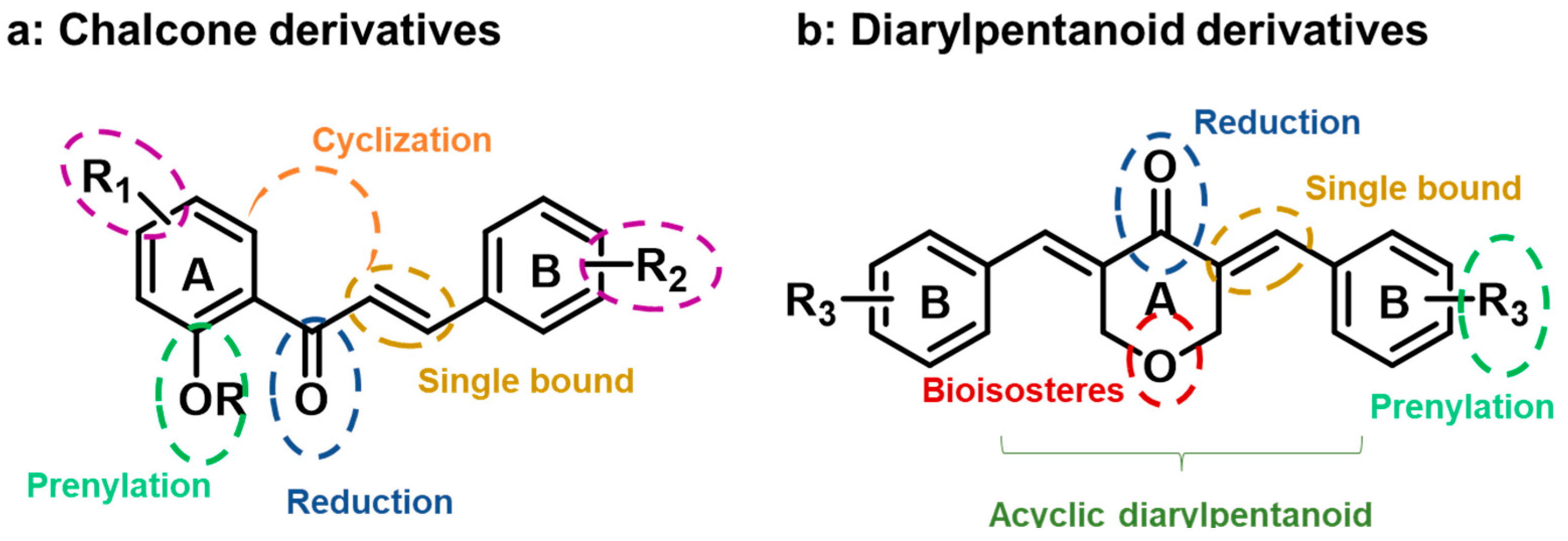
:1. Introduction
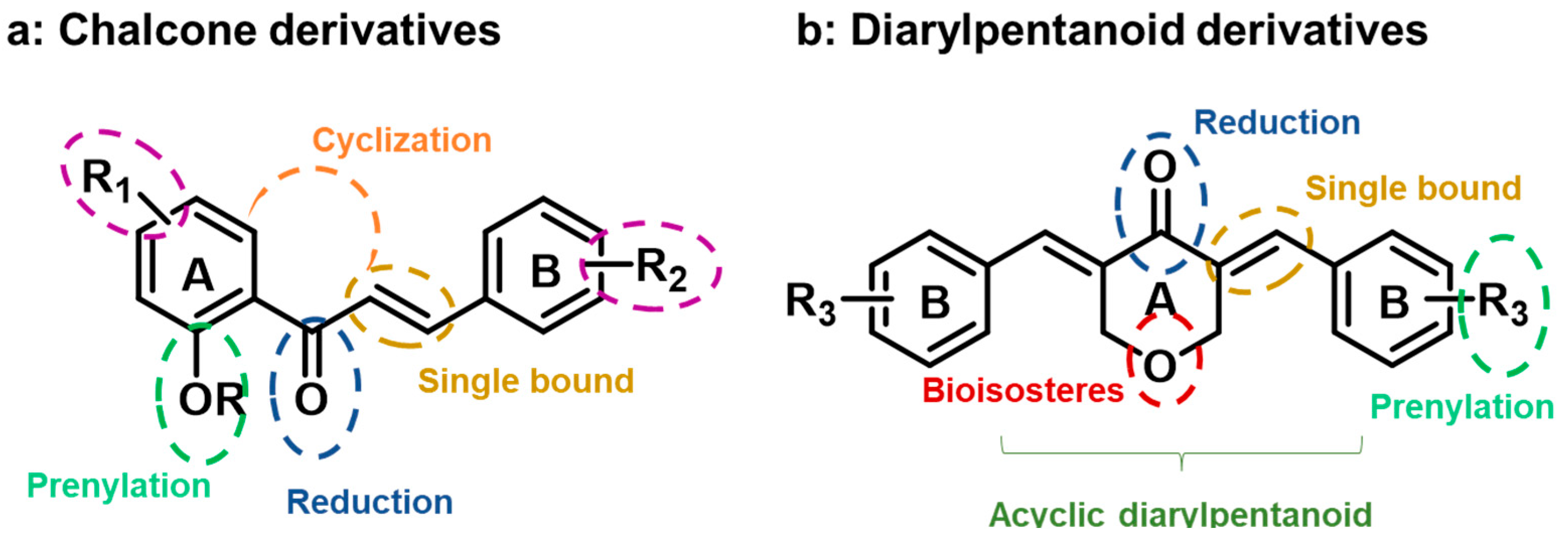
2. Results and Discussion
2.1. Synthesis
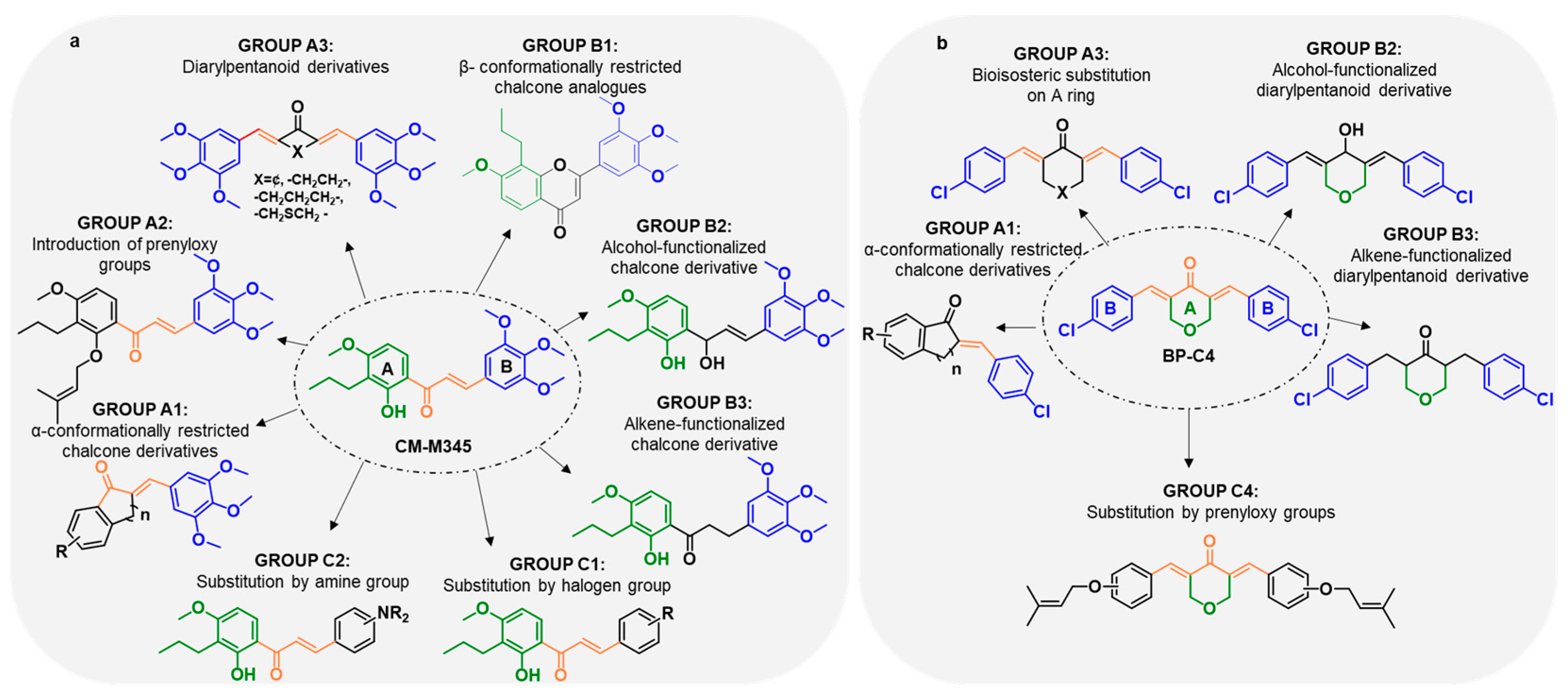
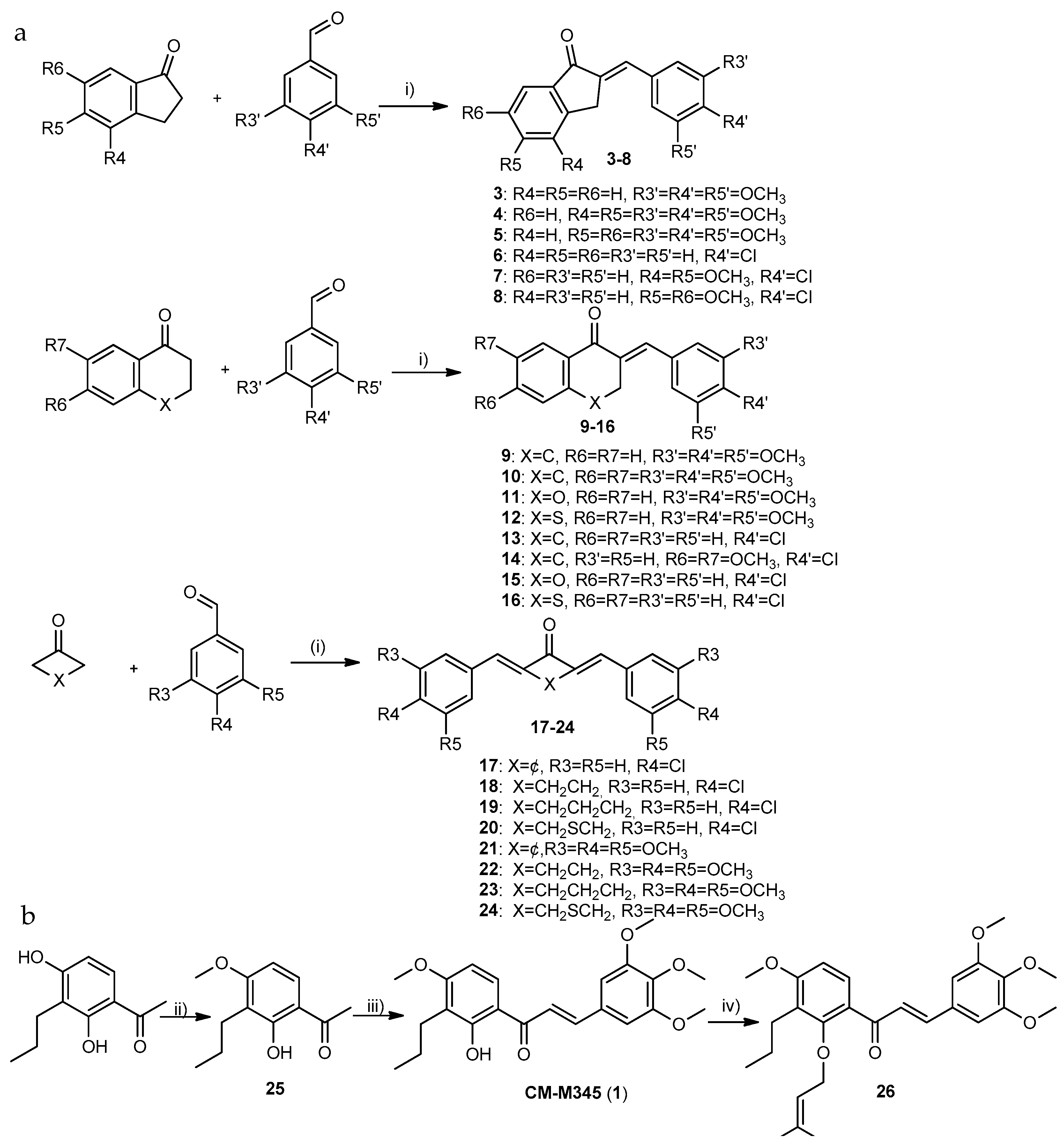
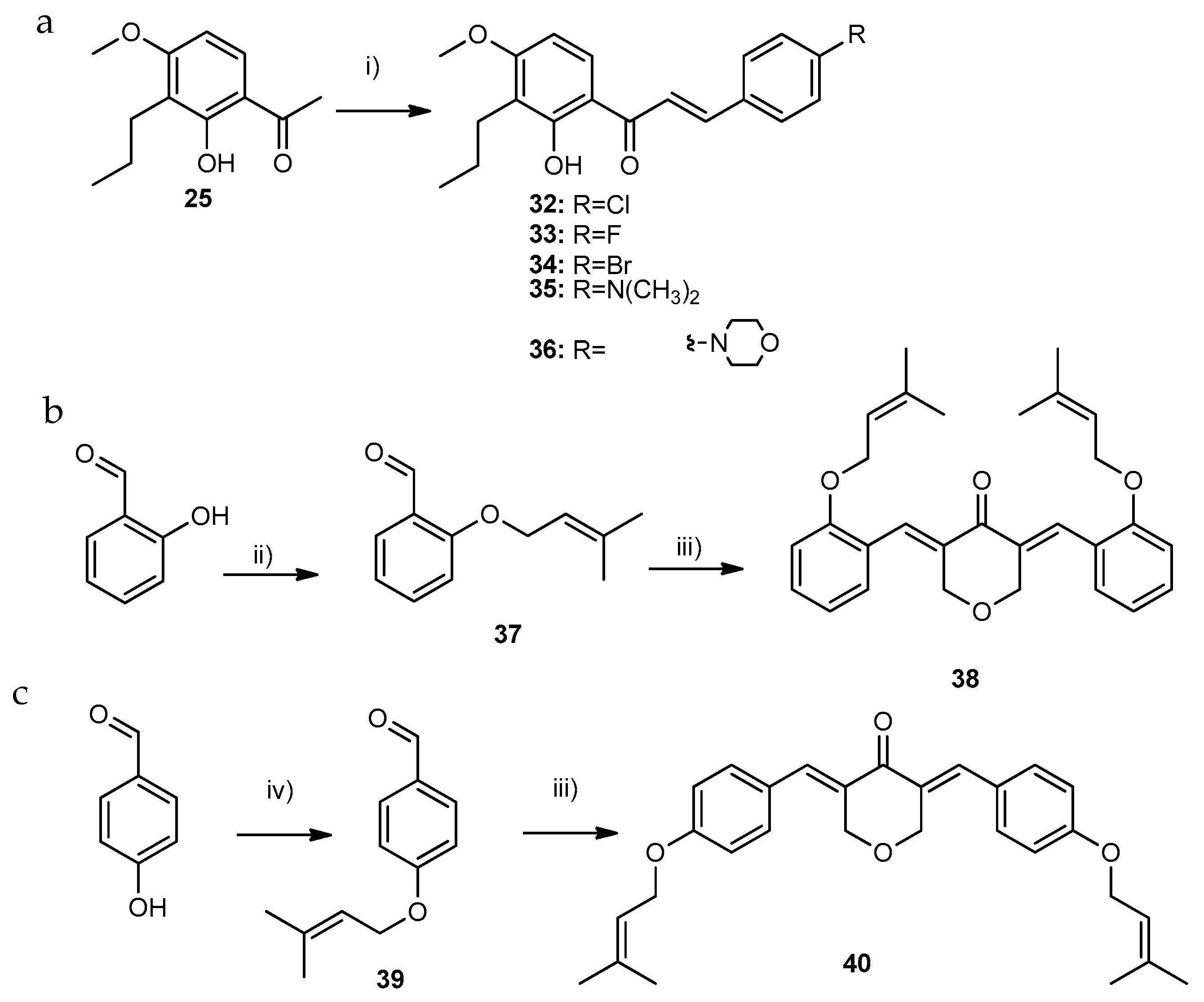
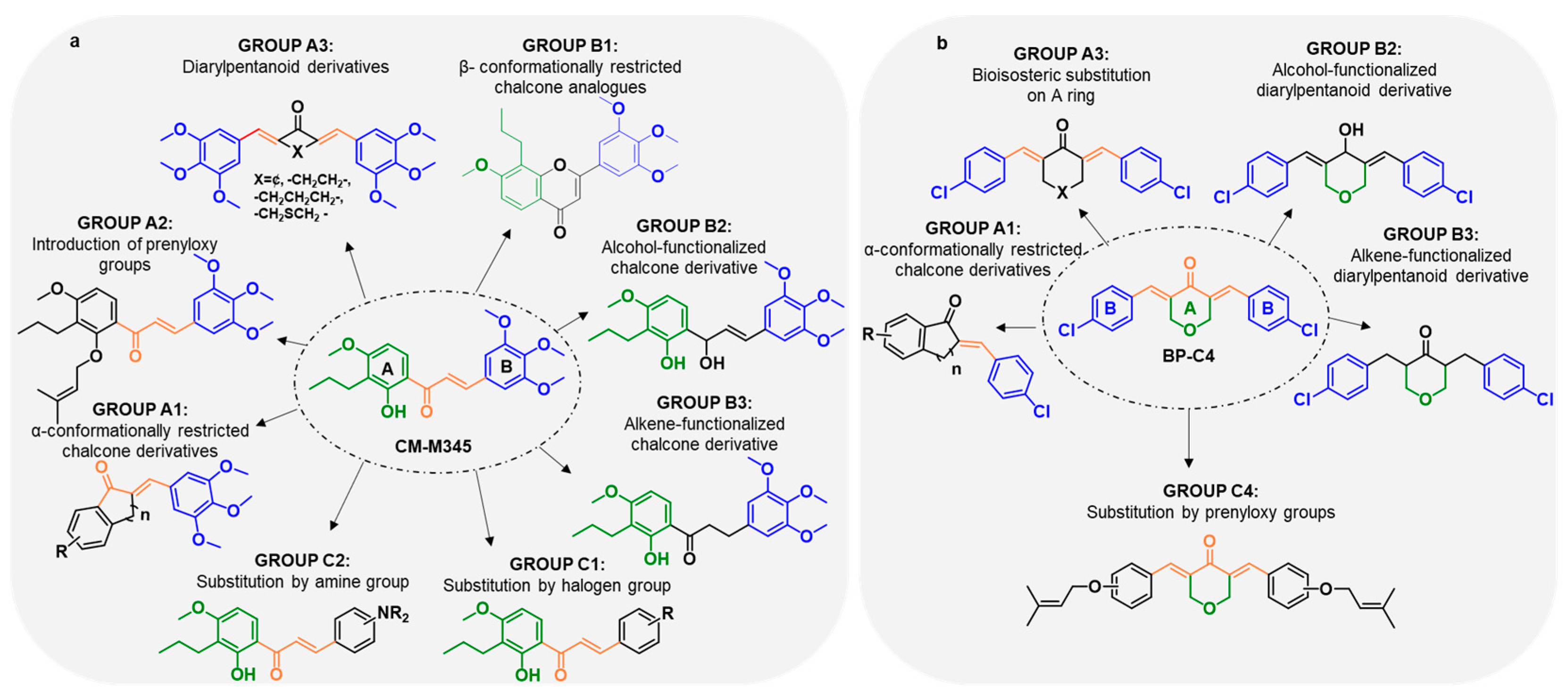
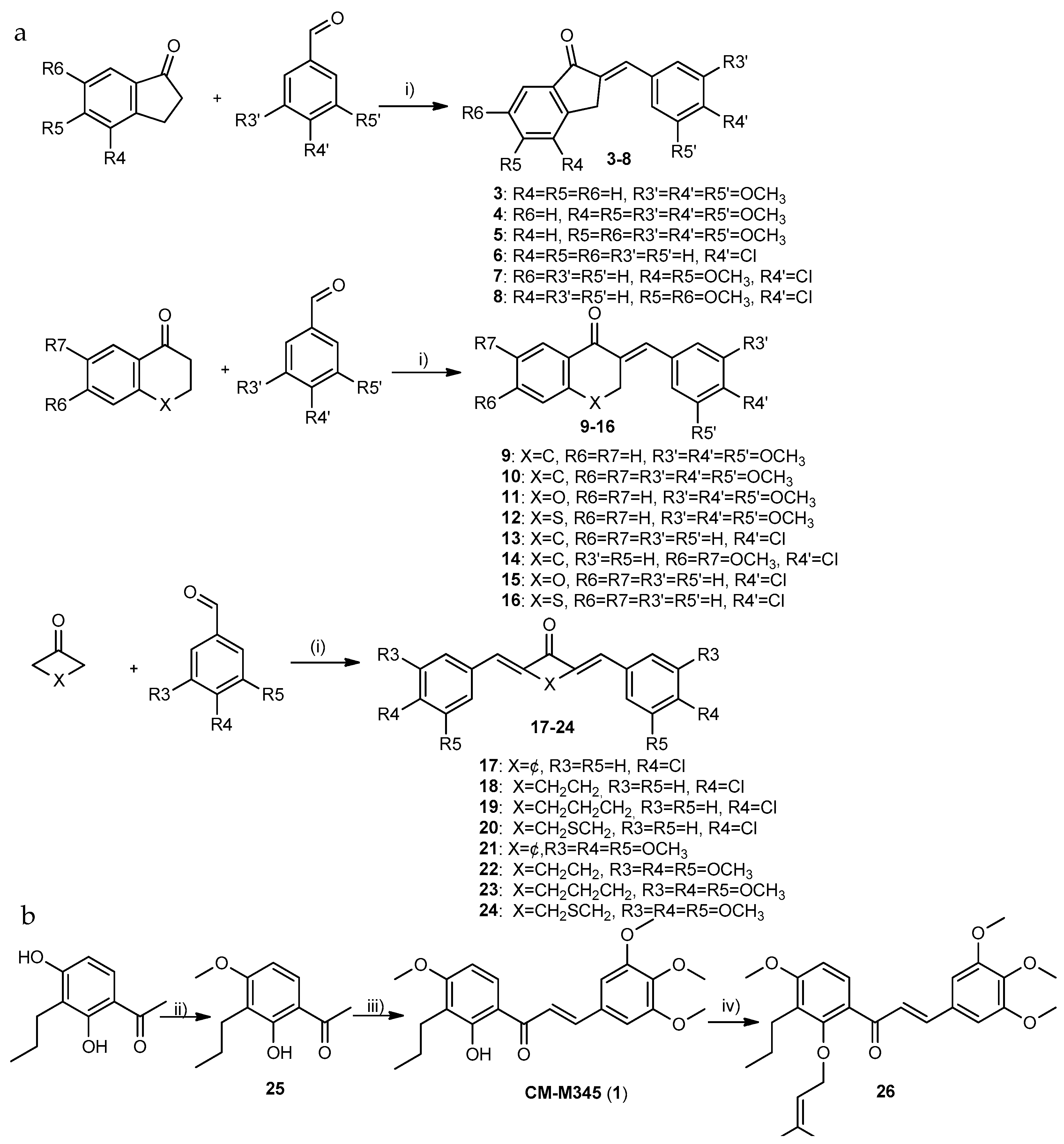
2.1.1. Synthesis of Compounds of Group A
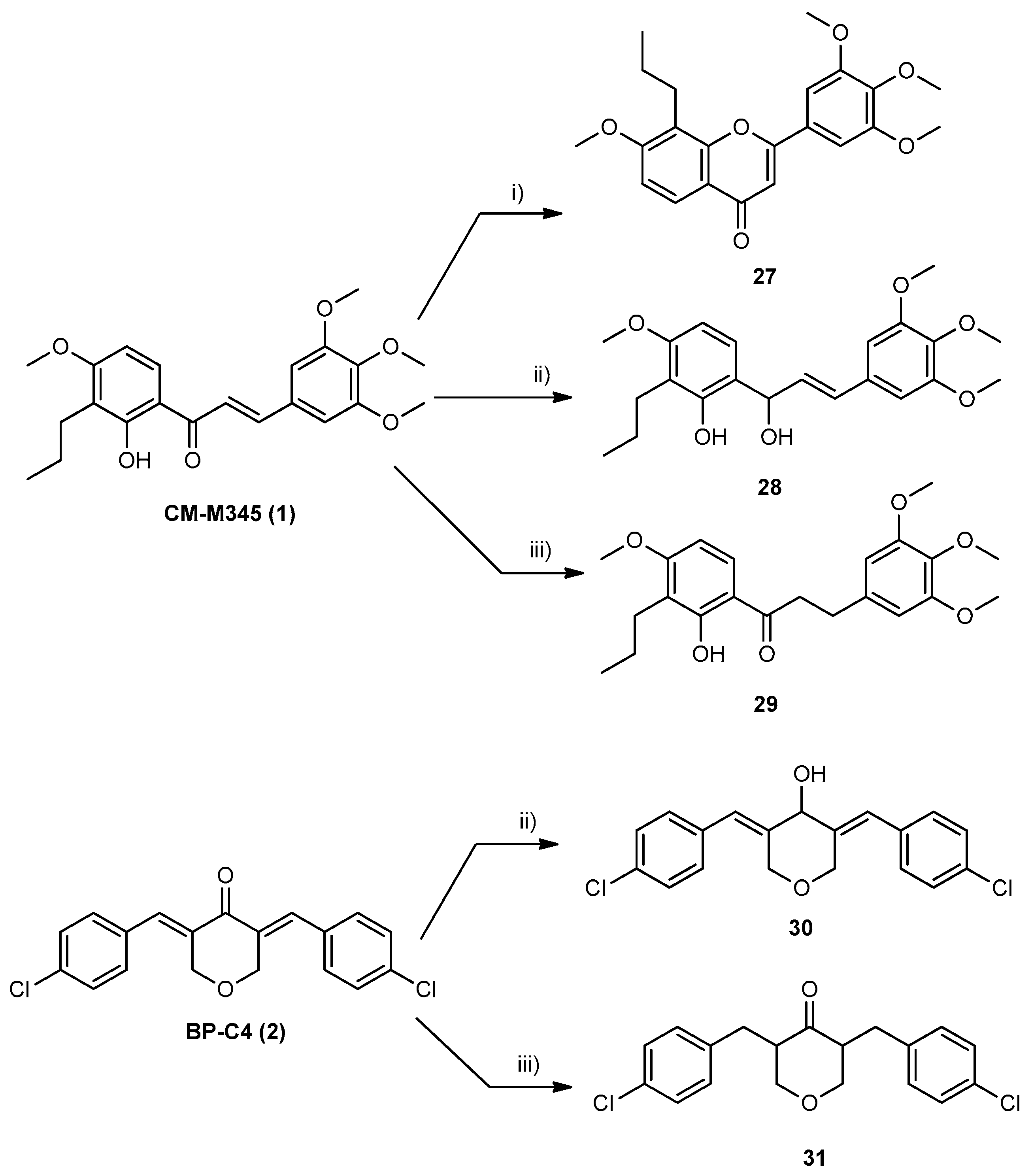
2.1.2. Synthesis of Compounds of Group B
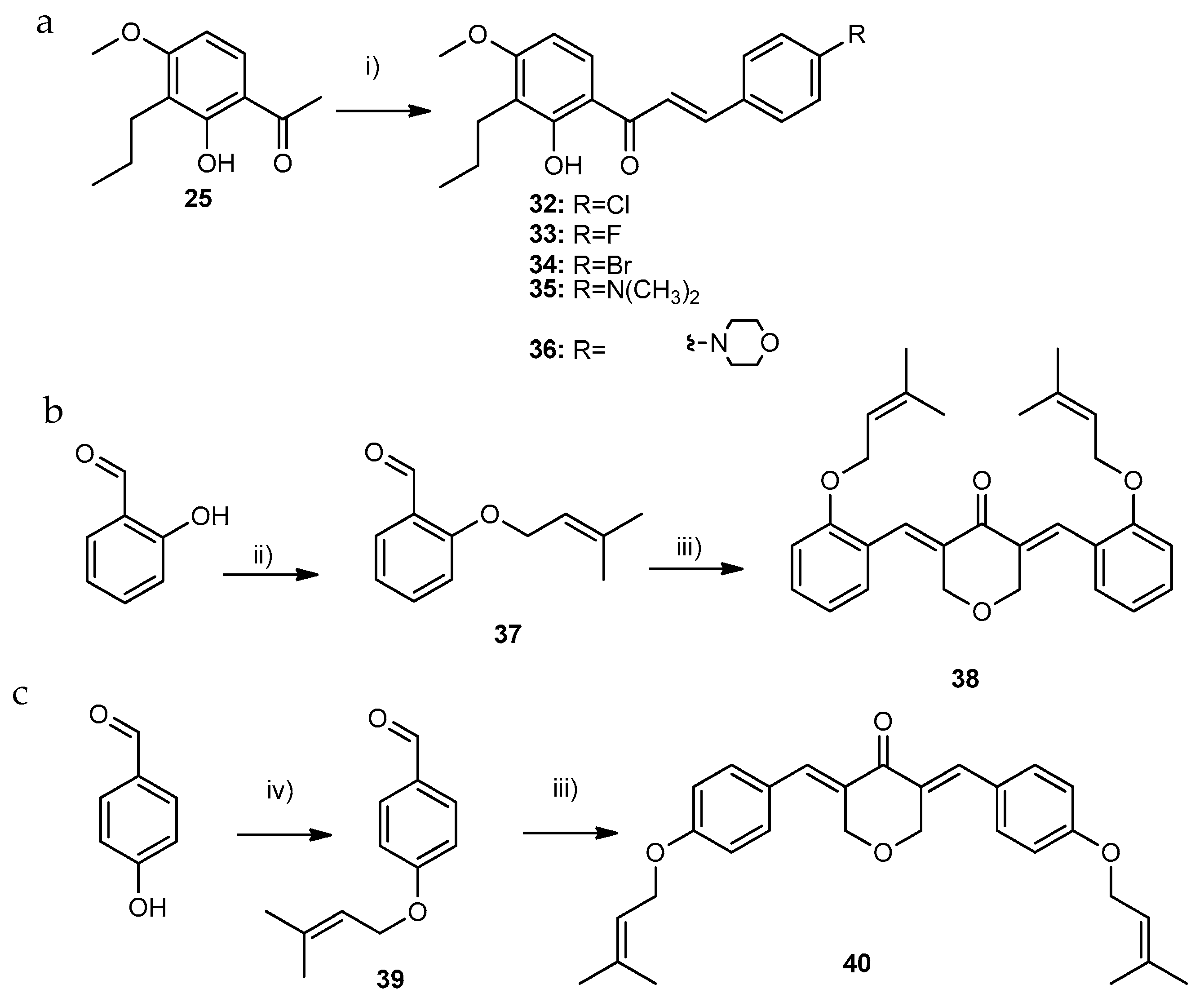
2.1.3. Synthesis of Compounds of Group C
2.2. Biological Activity Evaluation
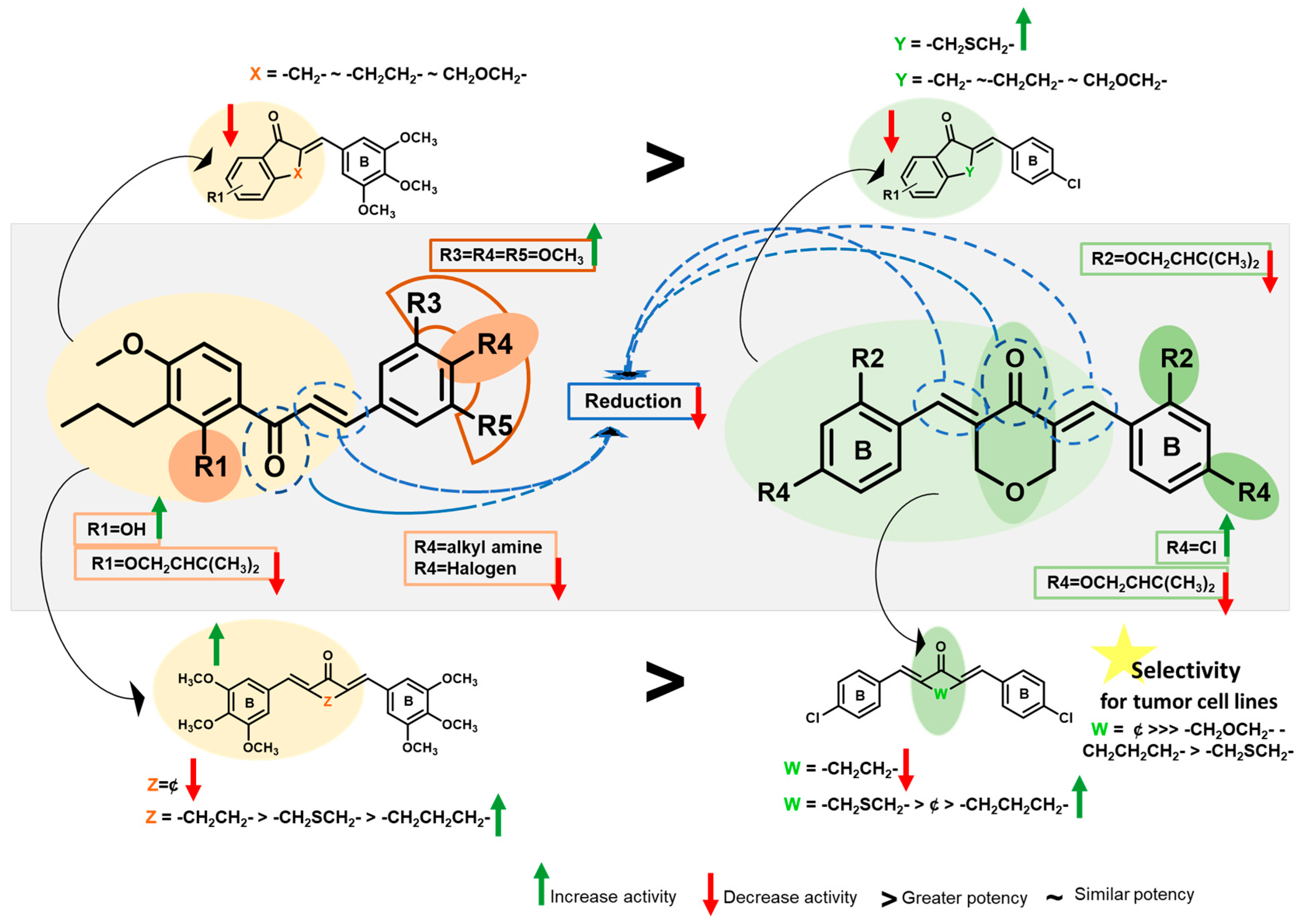
2.2.1. Growth Inhibitory Activity of 3–40 in Human Cancer Cells
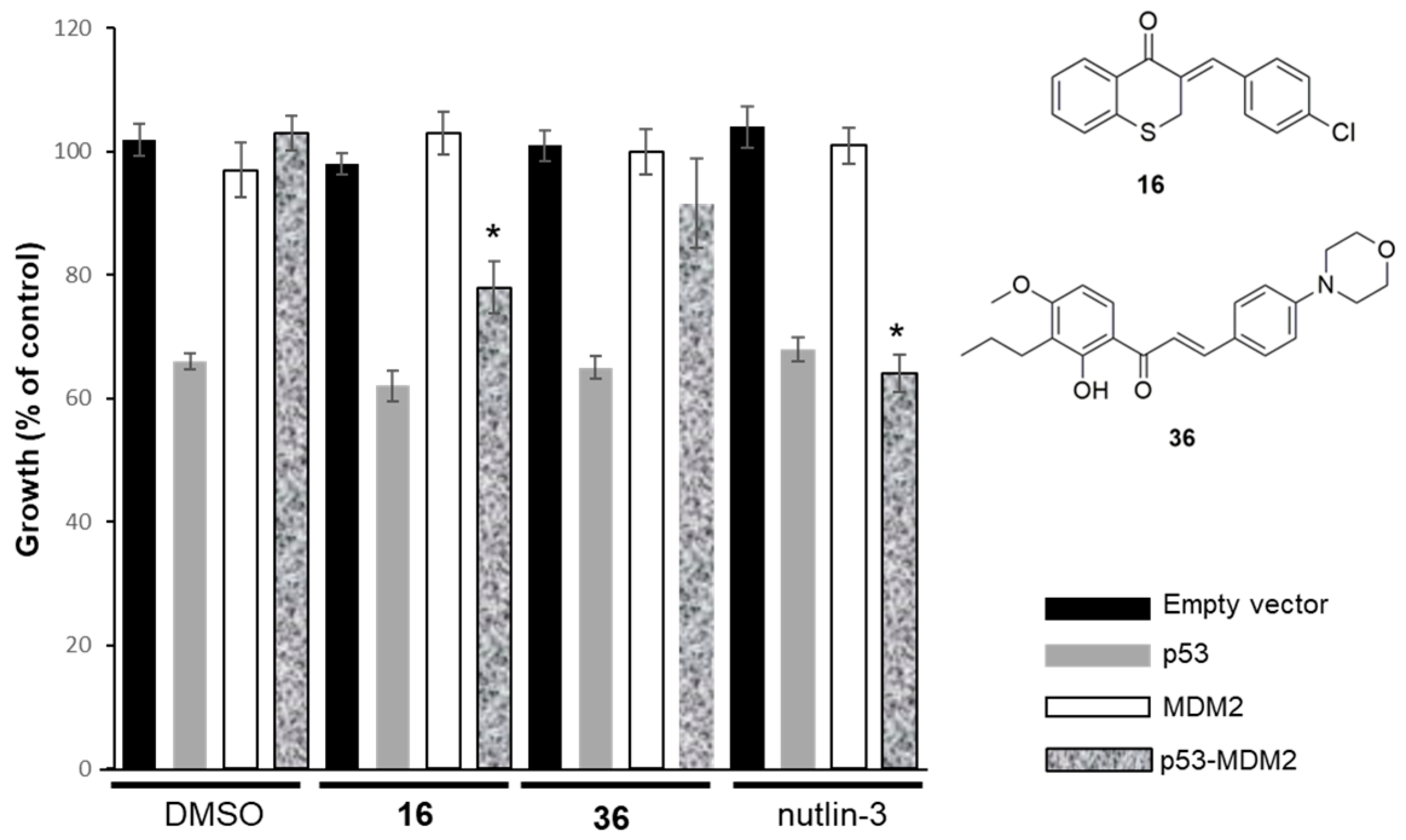
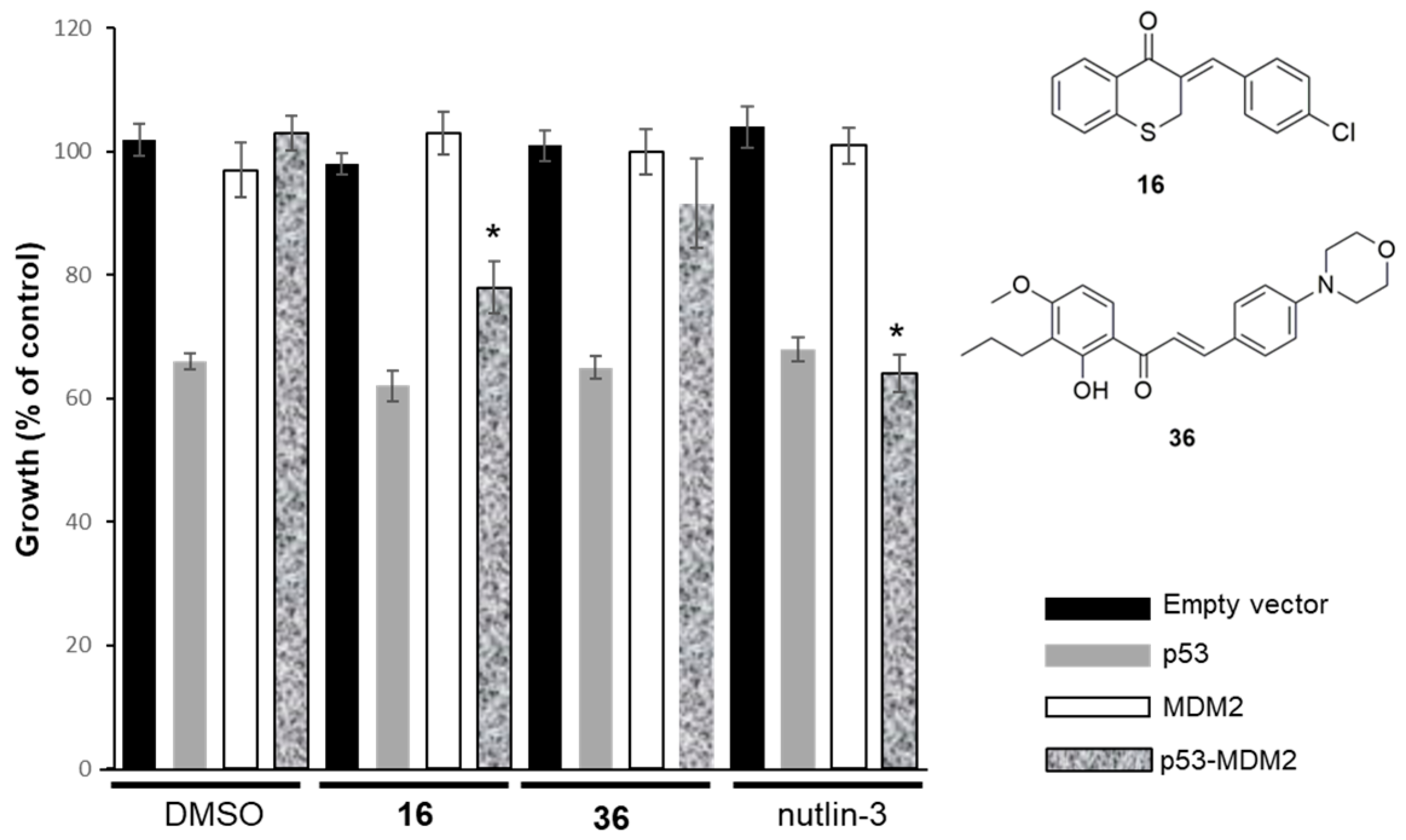
2.2.2. Effect of Compounds 16 and 36 on the p53–MDM2 Interaction Analyzed Using Yeast-Based Assays
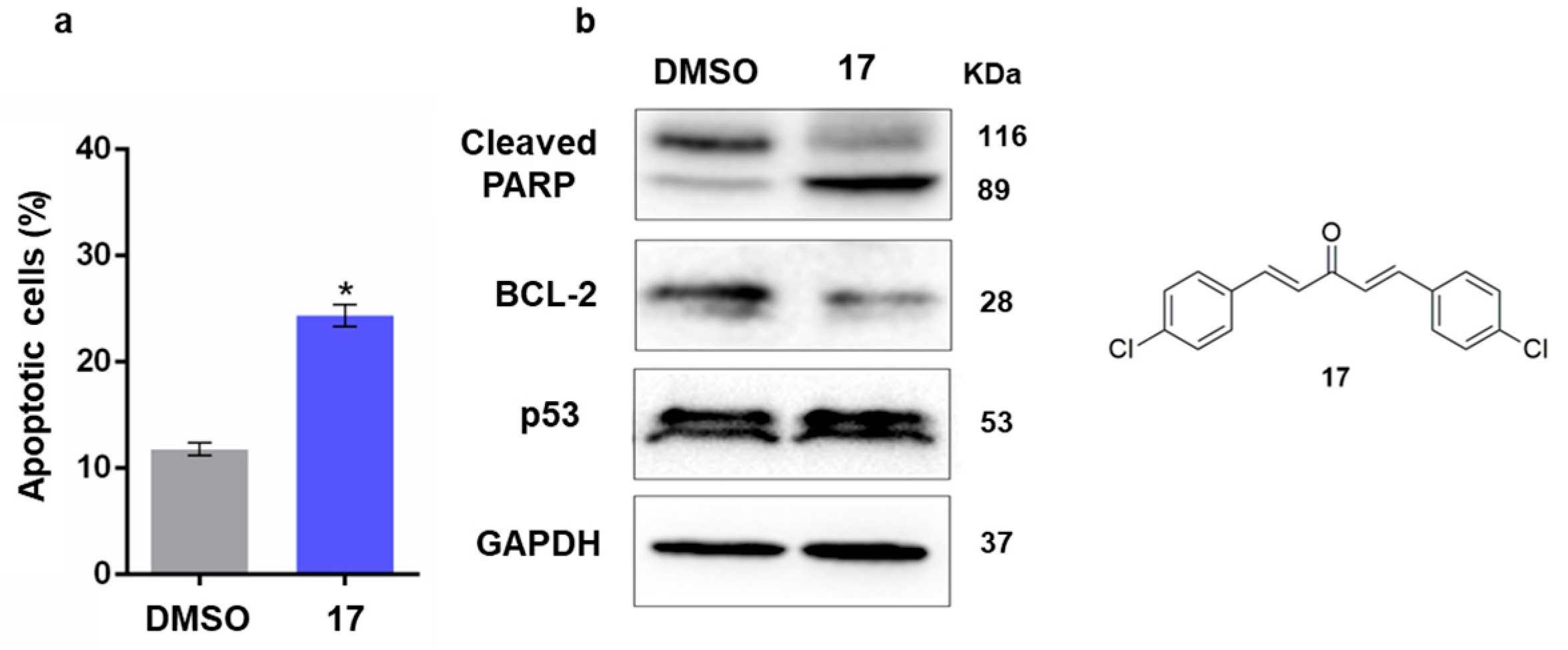
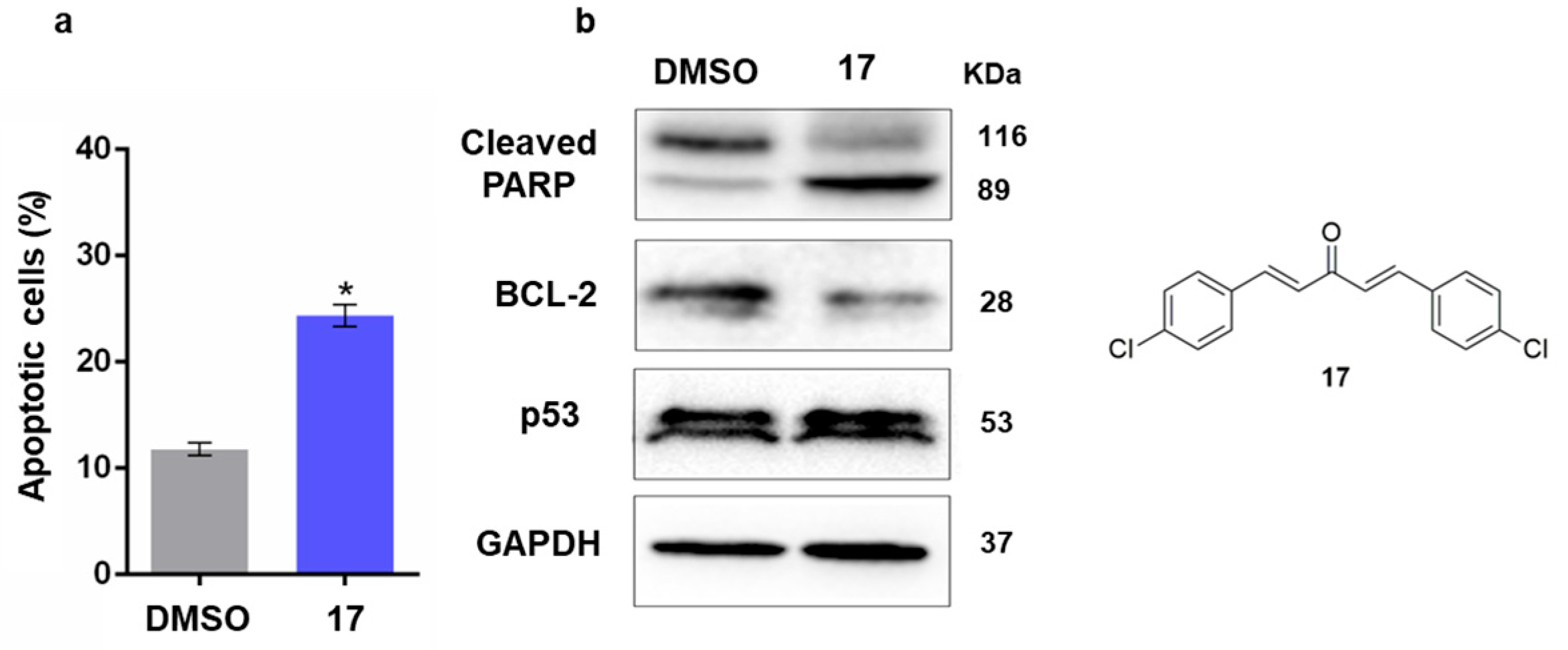
2.2.3. Study of the Mechanism of Action of Compound 17 in Human Cancer Cells
2.3. In Silico Studies
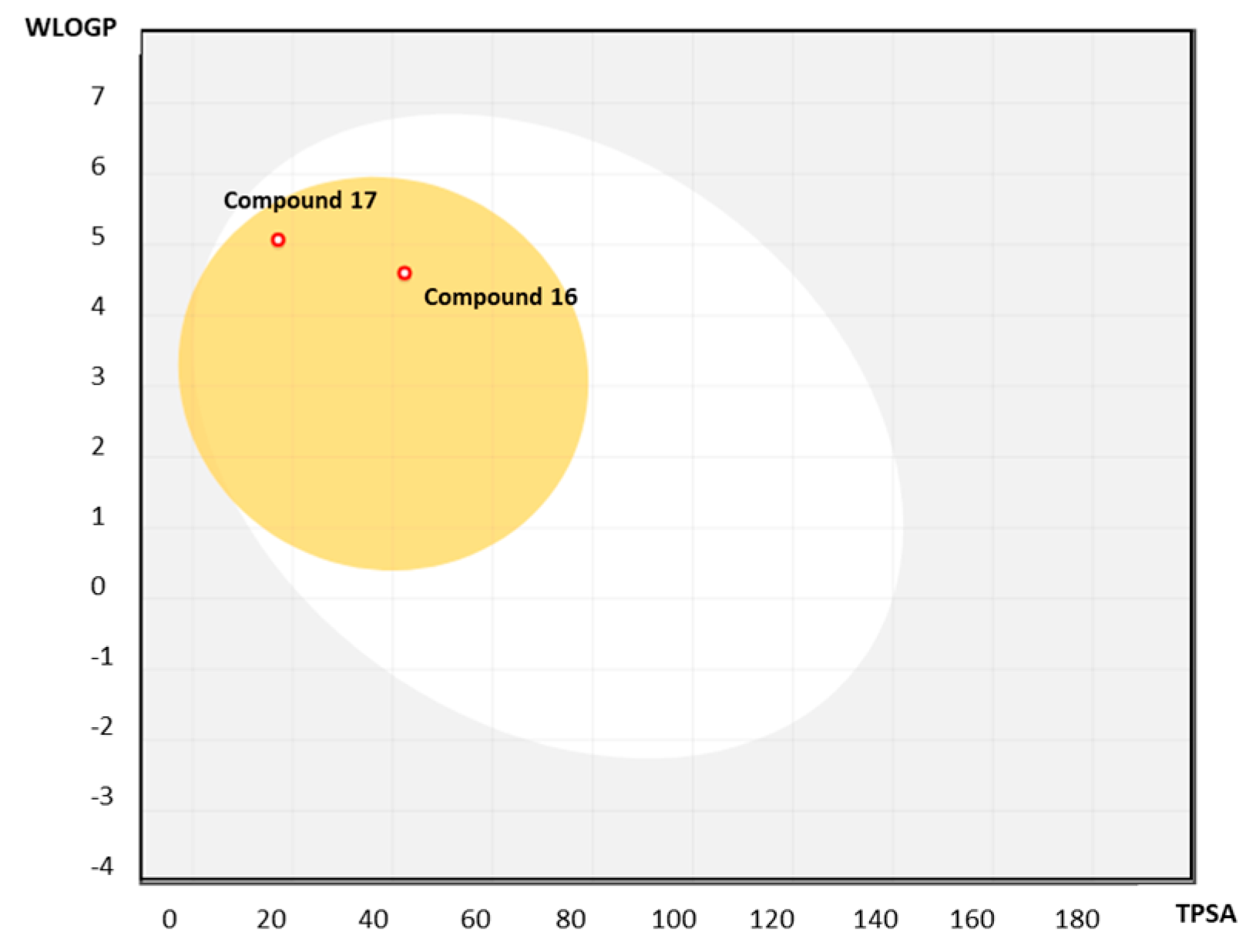
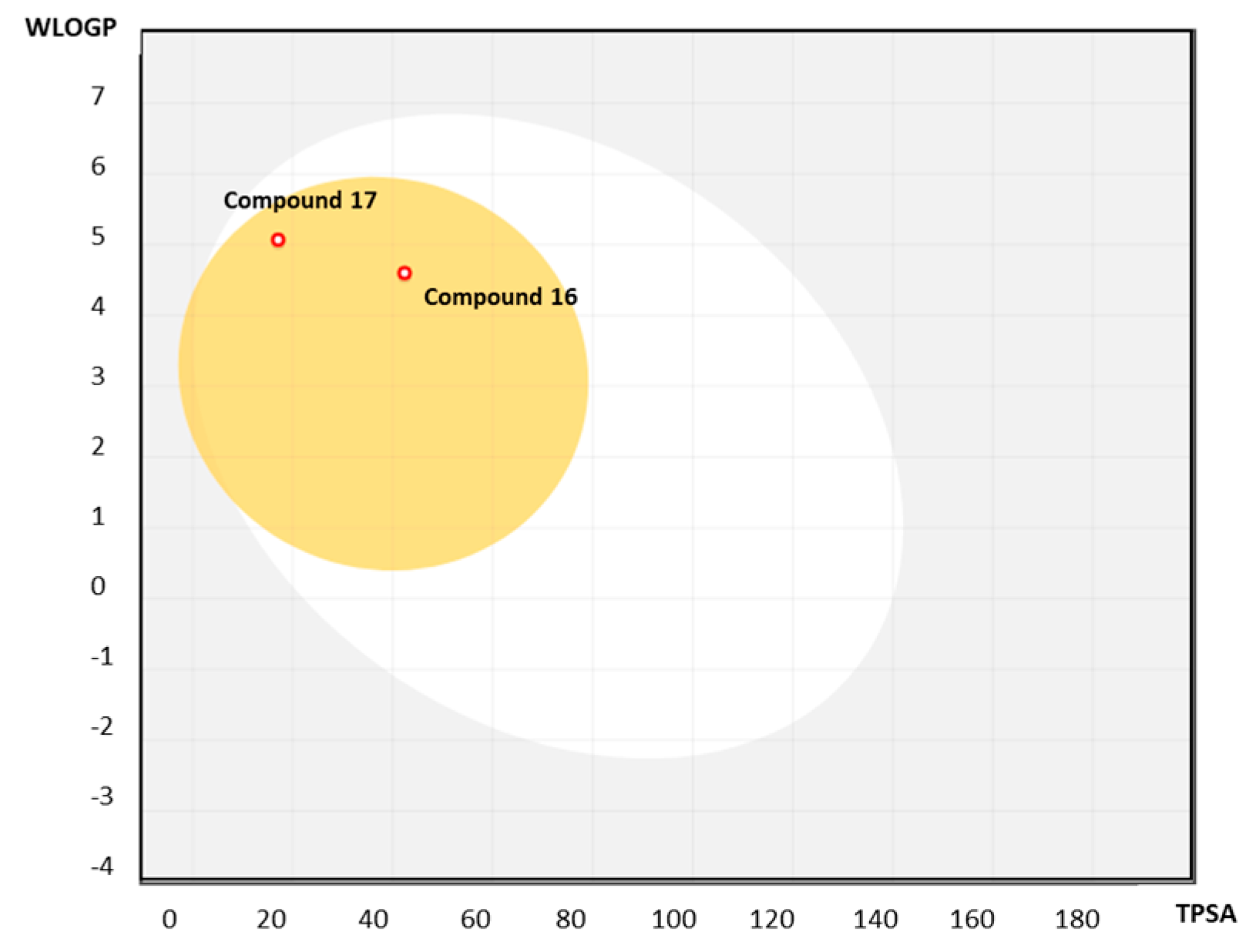
2.3.1. Predicting Druglikeness
2.3.2. ADMET Properties Prediction
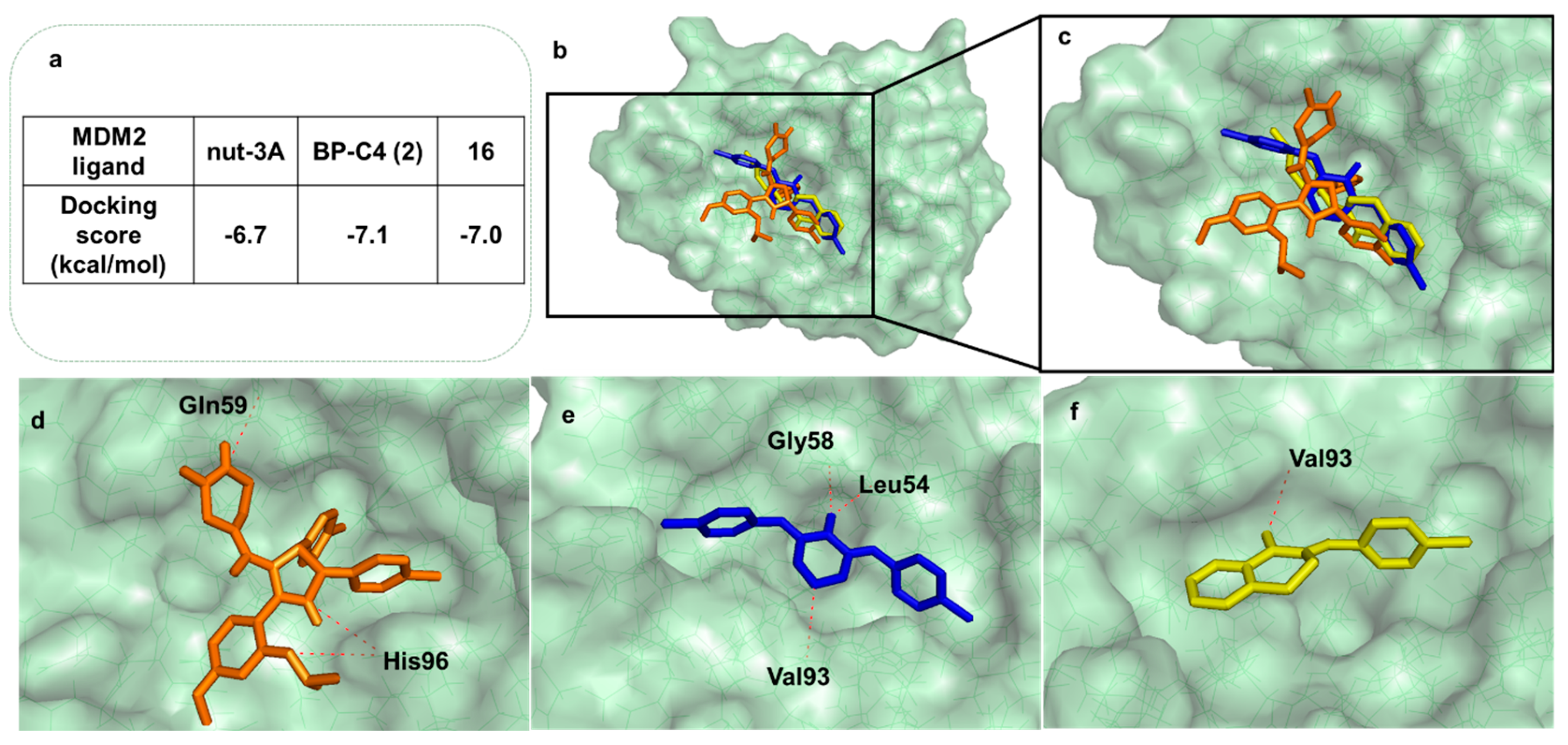
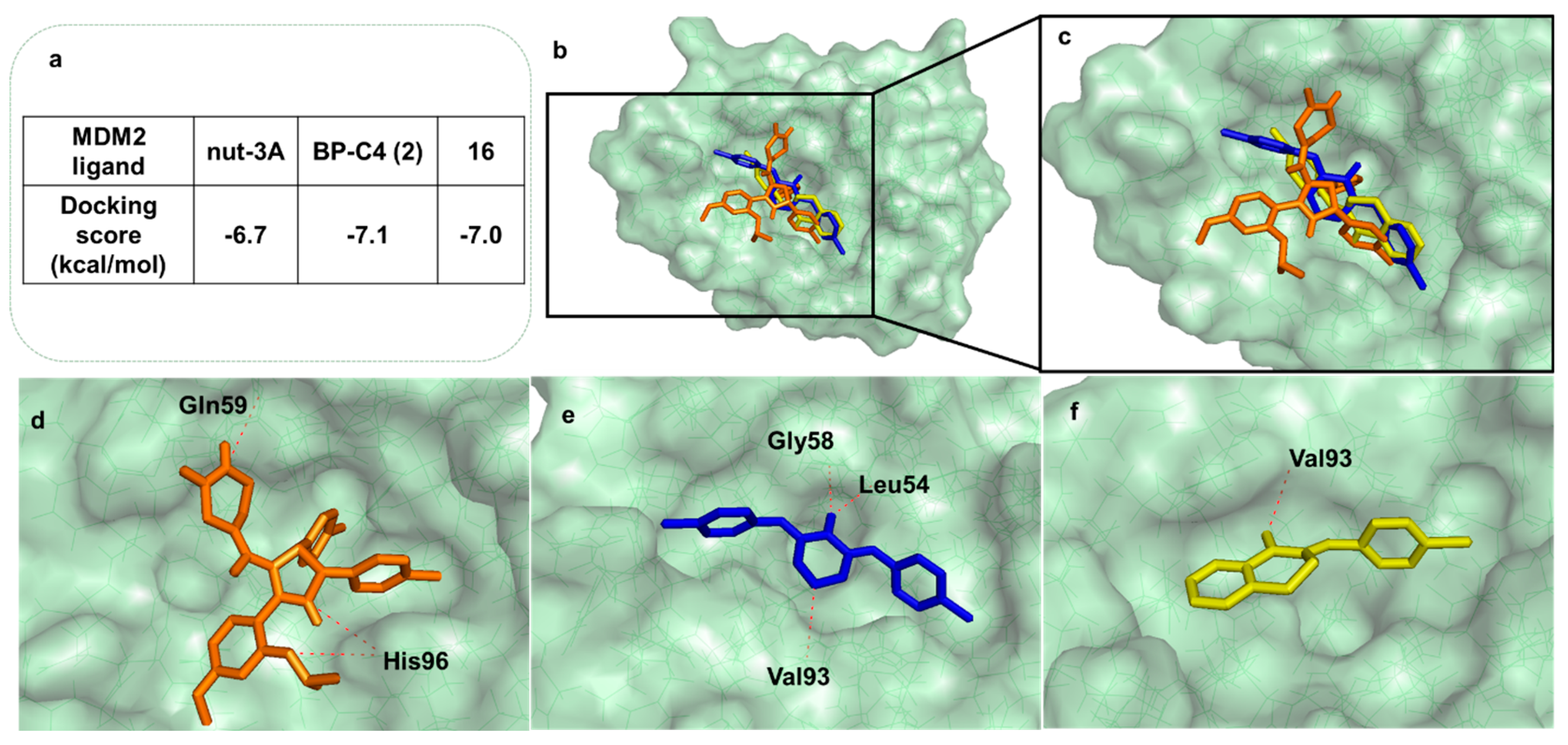
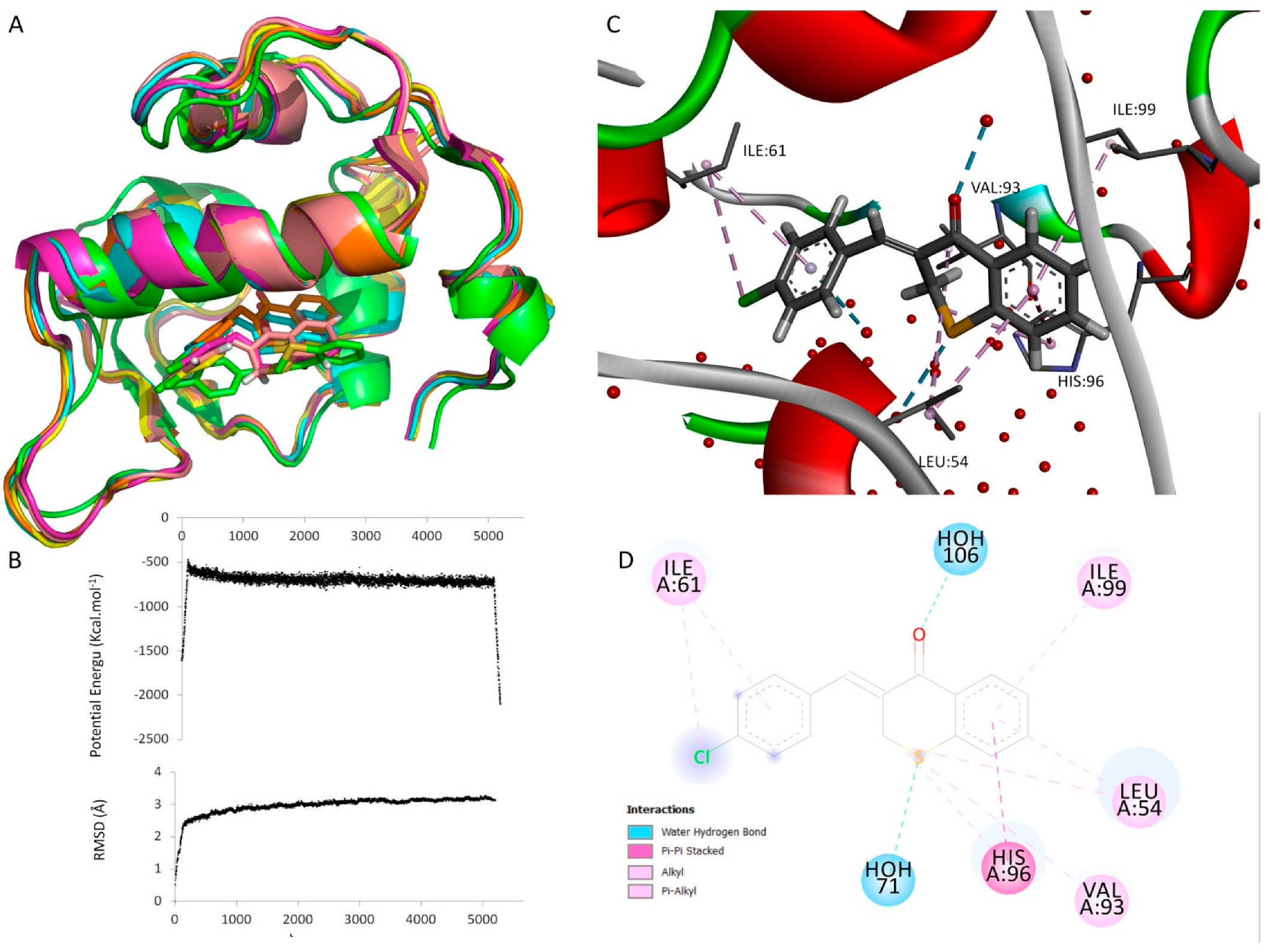
2.3.3. Docking and Molecular Dynamics Studies
2.3.4. Quantitative Structure–Activity Relationship (QSAR)
(n = 25, R2 = 0.7217, F = 9.85, S2 = 0.1029, Q2 = 0.6650)
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of Compounds of Group A1 (3–16)
- (E)-2-(3,4,5-trimethoxybenzylidene)-4,5-dimethoxy-2,3-dihydro-1H-inden-1-one (4): Purified by crystallization from methanol. Yield: 86% as a white solid; 99.8% purity; mp 165–168 °C (methanol); 1H NMR (CDCl3, 300.13 MHz) δ: 7.68 (d, J = 8.3 Hz, 1H, H-7), 7.54 (t, J = 2.2 Hz, 1H, H-1″), 7.02 (d, J = 8.4 Hz, 1H, H-6), 6.91 (s, 2H, H-2′,-6′), 3.99 (d, J = 2.0 Hz, 2H, H-3), 3.99 (s, 3H, 5-OCH3), 3.98 (s, 3H, 4-OCH3), 3.94 (s, 6H, 3′,5′-OCH3), 3.91 (s, 3H, 4′-OCH3) ppm; 13C NMR (CDCl3, 75.47 MHz) δ: 192.9 (C=O), 157.7 (C-5), 153.5 (C-3′,-5′), 145.3 (C-4), 142.3 (C-9), 139.8 (C-4′), 134.3 (C-2), 133.6 (C-1′’), 133.1 (C-1′), 132.2 (C-8), 121.2 (C-7), 112.7 (C-6), 61.1 (4′-OCH3), 60.7 (4-OCH3), 56.5 (3′-,5′-OCH3), 56.4 (5-OCH3), 29.1 (C-3) ppm; HRMS (ESI+): m/z Anal. Calc. for C21H22O6 (M + H+) 371.14892, found 371.14859.
- (E)-2-(4-chlorobenzylidene)-4,5-dimethoxy-2,3-dihydro-1H-inden-1-one (7): Purified by crystallization from methanol. Yield: 65% as a white solid; 99.9% purity; mp 206- 209 °C (methanol); 1H NMR (CDCl3, 300.13 MHz) δ: 7.68 (d, J = 8.3 Hz, 1H, H-7), 7.61 (d, J = 8.6 Hz, 2H, H-2′, -6′), 7.56 (t, J = 2.2 Hz, 1H, H-1′′), 7.43 (d, J = 8.6 Hz, 2H, H-3′, -5′), 7.03 (d, J = 8.0 Hz, 1H, H-6), 3.98 (s, 3H, 5-OCH3), 3.97 (s, 3H, 4-OCH3), 3.96 (sl, 2H, H-3) ppm; 13C NMR (CDCl3, 75.47 MHz) δ: 192.8 (C=O), 157.9 (C-5), 145.3 (C-4), 142.5 (C-9), 135.8 (C-4′), 135.7 (C-2), 134.0 (C-1′), 132.1 (C-8), 132.0 (C-1′’), 131.9 (C-2′, -6′), 129.3 (C-3′,-5′), 121.4 (C-7), 112.7 (C-6), 61.5 (4-OCH3), 57.4 (5-OCH3), 28.6 (C-3) ppm; HRMS (ESI+): m/z Anal. Calc. for C18H16ClO3 (M + H+) 315.07825, found 315.07798.
- (E)-2-(3,4,5-trimethoxybenzylidene)-6,7-dimethoxy-3,4-dihydronaphthalen-1(2H)-one (10): Purified by crystallization from methanol. Yield: 35% as a light yellow solid; 99.9% purity; mp 145–147 °C (methanol); 1H NMR (CDCl3, 300.13 MHz) δ: 7.75 (sl, 1H, H-1′′), 7.62 (s, 1H, H-8), 6.68 (s, 1H, H-5), 6.66 (s, 2H, H-2′,-6′), 3.95 (s, 6H, 6-, 7-OCH3), 3.89 (s, 3H, 4′-OCH3), 3.88 (s, 6H, 3′-,5′-OCH3), 3.15 (td, J = 6.4; 1.8 Hz, 2H, H-3), 2.91 (t, J = 6.4 Hz, 2H, H-4) ppm; 13C NMR (CDCl3, 75.47 MHz) δ: 186.7 (C=O), 153.7 (C-7), 153.2 (C-3′,-5′), 148.4 (C-6), 138.6 (C-10), 138.2 (C-4′), 136.3 (C-1′′), 135.0 (C-2), 131.7 (C-1′), 126.7 (C-9), 110.0 (C-8), 109.7 (C-5), 107.3 (C-2′,-6′), 61.1 (4′-OCH3), 55.3 (6-, 7-OCH3), 55.2 (3′-,5′-OCH3), 28.7 (C-4), 27.7 (C-3) ppm. HRMS (ESI+): m/z Anal. Calc. for C22H25O6 (M + H+) 385.16156. found 385.16442.
- (Z)-3-(3,4,5-trimethoxybenzylidene)thiochroman-4-one (12): Purified by crystallization with methanol. Yield: 81% as light orange; 99.8% purity; mp 115–118 °C (methanol); 1H NMR (CDCl3, 300.13 MHz) δ: 8.60 (dt, J = 7.7; 1.4 Hz, 1H, H-8), 7.61–7.57 (m, 2H, H-6, -7). 7.56–7.52 (m, 1H, H-5), 7.42 (t, J = 1.2 Hz, 1H, H-1′′), 6.49 (s, 2H, H-2′, -6′), 3.94 (sl, 2H, H-3), 3.84 (s, 9H, 3′-,4′-,5′-OCH3) ppm; 13C NMR (CDCl3, 75.47 MHz) δ: 179.2 (C=O), 153.5 (C-3′,-5′), 137.4 (C-10), 136.7 (C-4′), 134.6 (C-2), 134.4 (C-1′′), 131.7 (C-9), 131.2 (C-6), 129.2 (C-8), 127.7 (C-5), 126.6 (C-7), 125.2 (C-1′), 106.6 (C-2′,-6′), 61.0 (4′-OCH3), 56.3 (3′-, 5′-OCH3), 38.1 (C-3) ppm. HRMS (ESI+): m/z Anal. Calc. for C19H19O4S (M + H+) 343.09986, found 343.09779.
- (E)-2-(4-chlorobenzylidene)-6,7-dimethoxy-3,4-dihydronaphthalen-1(2H)-one (14): Purified by crystallization from methanol. Yield: 73% as a light yellow solid; 99.8% purity; mp 124–125 °C (methanol); 1H NMR (CDCl3, 300.13 MHz) δ: 7.75 (t, J = 1.8 Hz, 1H, H-1′′), 7.62 (s, 1H, H-8), 7.40–7.33 (m, 4H, H-2′, H-3′,-5′,-6′), 6.68 (s, 1H, H-5), 3.95 (s, 6H, 6-, 7-OCH3), 3.08 (td, J = 6.5; 1.8 Hz, 2H, H-3), 2.89 (t, J = 6.4 Hz, 2H, H-4) ppm; 13C NMR (CDCl3, 75.47 MHz) δ: 186.8 (C=O), 153.8 (C-6), 148.4 (C-7), 138.3 (C-10), 136.1 (C-2), 134.7 (C-1′′), 134.6 (C-4′), 134.4 (C-1′), 131.2 (C-2′,-6′), 128.8 (C-3′,-5′), 126.6 (C-9), 110.9 (C-5), 109.7 (C-8), 58.1 (6-,7-OCH3), 28.3 (C-4), 27.2 (C-3) ppm. HRMS (ESI+): m/z Anal. Calc. for C19H17ClO3 (M + H+) 329.09390. found 329.09352.
3.1.2. Synthesis of Compounds of Group A2
- Synthesis of 1-(2-hydroxy-4-methoxy-3-propylphenyl) ethan-1-one (25)
- Synthesis of (E)-3-(3,4,5-trimethoxy-chlorophenyl)-1-(2-hydroxy-4-methoxy-3-propylphenyl)prop-2-en-1-one (1)
- Synthesis of (E)-1-(4-methoxy-2-((3-methylbut-2-en-1-yl)oxy)-3-propylphenyl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (26)
- (E)-1-(4-methoxy-2-((3-methylbut-2-en-1-yl)oxy)-3-propylphenyl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (26): mp 100–103 °C (dichloromethane); 99.9% purity; 1H NMR (CDCl3, 300.13 MHz) δ: 7.64 (d, J = 15.7 Hz, 1H, H-β), 7.62 (d, J = 8.6 Hz, 1H, H-6), 7.50 (d, J = 15.7 Hz, 1H, H-α), 6.84 (s, 2H, H-2′,-6′), 6.73 (d, J = 8.7 Hz, 1H, H-5), 5.46–5.40 (m, 2H, H-2‴), 4.28 (d, J = 7.2 Hz, 2H, H-1‴), 3.90 (s, 6H, 3′-,5′-OCH3), 3.89 (s, 3H, 4′-OCH3), 3.88 (s, 3H, 4-OCH3), 2.66 (t, J = 7.8 Hz, 3H, H-2′’) 1.59–1.54 (m, 2H, H-1′’), 1.62 (s, 3H, H-4‴), 1.50 (s, 3H, H-5‴), 1.00 (t, J = 7.4 Hz, 3H, H-3′′) ppm; 13C NMR (CDCl3, 75.47 MHz) δ: 191.5 (C=O), 162.0 (C-4), 157.9 (C-2), 153.4 (C-3′,-5′), 142.8 (C-β), 140.1 (C-4′), 138.3 (C-4‴), 131.0 (C-1′), 129.8 (C-6), 126.7 (C-1), 126.1 (C-α), 125.4 (C-3), 120.1 (C-2‴), 106.4 (C-5), 105.6 (C-2′,-6′), 73.1 (C-1‴), 61.1 (4′-OCH3), 56.3 (3′-,5′-OCH3), 55.8 (4-OCH3), 26.1 (C-1′′), 25.8 (C-4‴), 23.0 (C-2′′), 18.0 (C-5‴), 14.7 (C-3′′) ppm; HRMS (ESI+): m/z Anal. Calc. for C27H34O6K (M + K+) 493.19870, found 493.19794.
3.1.3. General Procedure for the Synthesis of Compounds of Group A3 (17–24)
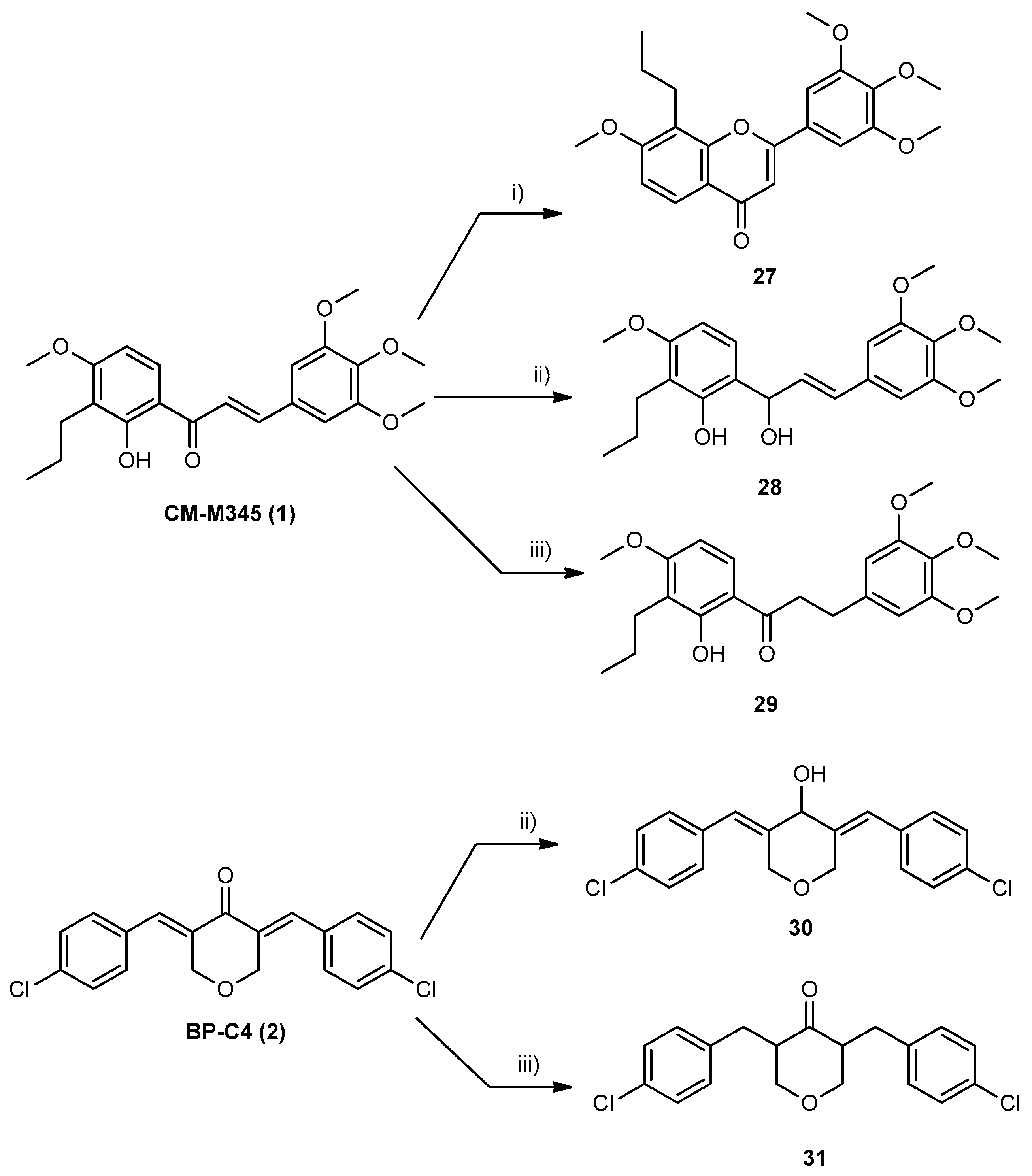
3.1.4. Synthesis of Compounds of Group B1
- Synthesis of 7-methoxy-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-4-one (27)
- 7-methoxy-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-4-one (27): mp 153–155 °C (dichloromethane); 99.8% purity; 1H NMR (CDCl3, 300.13 MHz) δ: 8.09 (d, J = 8.9 Hz, 1H, H-5), 7.16 (s, 2H, H-2′,-6′), 7.02 (d, J = 8.9 Hz, 1H, H-6), 6.72 (s, 1H, H-3), 3.96 (s, 3H, 4′-OCH3), 4.95 (s, 6H, 3′-5′-OCH3), 3.93 (s, 3H, 7-OCH3), 2.95 (t, J = 7.6 Hz, 2H, H-1′′), 1.75–1.67 (m, 2H, H-2′′), 1.03 (t, J = 7.4 Hz, 3H, H-3′′) ppm; 13C NMR (CDCl3, 75.47 MHz) δ: 178.7 (C=O), 162.7 (C-2), 161.6 (C-7), 155.3 (C-9), 153.7 (C-3′, -5′), 141.1 (C-4′), 127.5 (C-1′), 124.6 (C-5), 118.7 (C-8), 117.9 (C-10), 109.0 (C-6), 106.5 (C-3), 103.4 (C-2′, -6′), 61.2 (4′-OCH3), 56.3 (3′-,5′-OCH3), 56.2 (4-OCH3), 25.6 (C-1′′), 22.7 (C-2′′), 14.6 (C-3′′) ppm; HRMS (ESI+): m/z Anal. Calc. for C22H25O6 (M + H+) 385.16456, found 385.16371.
3.1.5. Synthesis of Compounds of Group B2
- Synthesis of (E)-6-(1-hydroxy-3-(3,4,5-trimethoxyphenyl)allyl)-3-methoxy-2-propylphenol (28) and 3,5-bis((E)-4-chlorobenzylidene)tetrahydro-2H-pyran-4-ol (30)
- 3,5-bis((E)-4-chlorobenzylidene)tetrahydro-2H-pyran-4-ol (30): Purified by TLC (SiO2, n-hexane: ethyl acetate, 7:3). Yield: 88% as a white solid; 98.9% purity; mp 219–222 °C (methanol); 1H NMR (CDCl3, 400.14 MHz) δ: 7.32 (d, J = 6.4 Hz, 4H, H-3′,-5′), 7.10 (d, J = 6.4 Hz, 4H, H-2′,-6′), 6.62 (sl, 2H, H-1′′), 4.91 (d, J = 4.0 Hz, 1-OH), 4.69 (dd, J = 10.8; 2.0 Hz, 2H, H-3ª,-5a), 4.51 (dd, J = 10.9; 2.0 Hz, 2H, H-3b,-5b) ppm; 13C NMR (CDCl3, 100.63 MHz) δ: 139.4 (C-2), 134.6 (C-1′), 133.3 (C-4′), 130.2 (C-3′,-5′), 128.8 (C-2′,-6′), 124.3 (C-1′’), 75.5 (C-1), 66.8 (C-3,-5) ppm. HRMS (ESI+): m/z Anal. Calc. for C19H16Cl2O2 (M + k+) 385.01598. found 385.01545.
3.1.6. Synthesis of Compounds of Group B3
- Synthesis of 1-(2-hydroxy-4-methoxy-3-propylphenyl)-3-(3,4,5-trimethoxyphenyl)propan-1-one (29) and 3,5-bis(4-chlorobenzyl)tetrahydro-4H-pyran-4-one (31)
- 1-(2-hydroxy-4-methoxy-3-propylphenyl)-3-(3,4,5-trimethoxyphenyl)propan-1-one (29): Purified by TLC (SiO2; dichloromethane: n-hexane, 8:2).Yield: 85% as a yellow gum; 99.8% purity; mp N.D. (gum); 1H NMR (CDCl3, 300.13 MHz) δ: 12.78 (s, 1H, 2-OH), 7.60 (d, J = 9.0 Hz, 1H, H-6), 6.45 (s, 2H, H-2′,-6′), 6.43 (d, J = 9.0 Hz, 1H, H-5), 3.87 (s, 3H, 4-OCH3), 3.84 (s, 6H, 3′-,5′-OCH3), 3.82 (s, 3H, 4′-OCH3), 3.24 (t, J = 7.5 Hz, 2H, H-1‴), 3.00 (t, J = 7.7 Hz, 2H, H-2‴), 2.63 (t, J = 7.6 Hz, 2H, H-1′′), 1.58–1.48 (m, 2H, H-2′’), 0.94 (t, J = 7.4 Hz, 3H, H-3′′) ppm; 13C NMR (CDCl3. 75.47 MHz) δ: 204.2 (C=O), 163.6 (C-4), 162.3 (C-2), 153.4 (C-3′, -5′), 137.0 (C-1′), 136.5 (C-4′), 129.4 (C-6),118.6 (C-3), 113.9 (C-1), 105.5 (C-2′, -6′), 102.1 (C-5), 61.0 (4′-OCH3), 56.2 (3′-,5′-OCH3), 55.8 (4-OCH3), 40.0 (C-1‴), 31.2 (C-2‴), 24.5 (C-1′′), 22.0 (C-2′′), 14.3 (C-3′′) ppm; HRMS (ESI+): m/z Anal. Calc. for C22H29O6 (M+H+) 389.19587, found 389.19580.
- 3,5-bis(4-chlorobenzyl)tetrahydro-4H-pyran-4-one (31): Purified by crystallization from methanol. Yield: 50% as a white solid; 98.2% purity; mp 145–146 °C (methanol); 1H NMR (CDCl3, 300.13 MHz) δ: 7.25 (dt, J = 8.8; 2.3 Hz, 4H, H-3′,-5′), 7.09 (dt, J = 8.8; 2.3 Hz, 4H, H-2′,-6′), 4.14 (qd, J = 5.4; 1.9 Hz, 2H, H-3a), 3.34 (t, J = 11.1 Hz, 2H, H-3b), 2.94–2.83 (m, 2H, H-2), 3.17 (q, J = 7.2 Hz, 2H, H-1′′a), 2.36 (q, J = 7.2 Hz, 2H, H-1′′b) ppm; 13C NMR (CDCl3. 75.47 MHz) δ: 208.0 (C=O), 137.7 (C-1′), 132.3 (C-4′), 130.3 (C-2′, -6′), 52.9 (C-2, -6), 128.7 (C-3′,-5′), 73.7 (C-3, -5), 30.5 (C-1′) ppm. HRMS (ESI+): m/z Anal. Calc. for C19H18Cl2O2 (M+k+) 387.03154. found 387.03260.
3.1.7. General Procedure for the Synthesis of Compounds of Group C1 (33–34)
- (E)-3-(4-fluorophenyl)-1-(2-hydroxy-4-methoxy-3-propylphenyl)prop-2-en-1-one (33): Purified by TLC (SiO2; n-hexane: ethyl acetate, 8:2). Yield: 12% as a yellow solid; 96.4% purity; mp 115–117 °C (ethyl acetate); H NMR (CDCl3, 300.13 MHz) δ: 13.33 (s, 1H, 2-OH), 7.84 (d, J = 15.4 Hz, 1H, H-β), 7.78 (d, J = 9.1 Hz, 1H, H-6), 7.67–7.62 (m, 2H, H-2′,-6′), 7.53 (d, J = 15.5 Hz, 1H, H-α), 7.16–7.08 (m, 2H, H-3′,-5′), 6.50 (d, J = 9.0 Hz, 1H, H-5), 3.90 (s, 3H, 4-OCH3), 2.66 (t, J = 7.7 Hz, 2H, H-1′’), 1.62–1.49 (m, 2H, H-2′’), 0.96 (t, J = 7.4 Hz, 3H, H-3′′) ppm; 13C NMR (CDCl3, 75.47 MHz) δ: 192.2 (C=O), 163.7 (d, J = 27.7 Hz, C-4′), 163.8 (C-4), 163.4 (C-2), 142.9 (C-β), 130.5 (d, J = 8.5 Hz, C-2′, -6′), 131.3 (d, J = 3.3 Hz, C-1′), 129.2 (C-6), 120.5 (C-α), 118.8 (C-3), 114.5 (C-1), 102.2 (C-5), 116.3 (d, J = 21.9 Hz, C-3′,-5′), 56.9 (4-OCH3), 24.6 (C-1′′), 22.0 (C-2′′), 14.4 (C-3′′) ppm; HRMS (ESI+): m/z Anal. Calc. for C19H19FO3 (M + H+) 315.13910, found 315.13906.
- (E)-3-(4-bromophenyl)-1-(2-hydroxy-4-methoxy-3-propylphenyl)prop-2-en-1-one (34): Purified by TLC (SiO2; dichloromethane: n-hexane, 5:5). Yield: 10% as an orange solid; 96.2% purity; mp 115–116 °C (dichloromethane); 1H NMR (CDCl3, 300.13 MHz) δ: 13.29 (s, 1H, 2-OH), 7.80 (d, J = 15.5 Hz, 1H, H-β), 7.77 (d, J = 9.0 Hz, 1H, H-6), 7.59 (d, J = 15.5 Hz, 1H, H-α), 7.57–7.54 (m, 2H, H-2, -6), 7.52–7.49 (m, 2H, H-3, -5), 6.50 (d, J = 9.0 Hz, 1H, H-5), 3.90 (s, 3H, 4-OCH3), 2.66 (t, J = 7.7 Hz, 2H, H-1″), 1.59–1.49 (m, 2H, H-2″), 0.96 (t, J = 7.4 Hz, 3H, H-3″) ppm; 13C NMR (CDCl3, 75.47 MHz) δ: 192.1 (C=O), 163.9 (C-4), 163.5 (C-2), 142.7 (C-β), 134.0 (C-1′), 132.4 (C-3′,-5′), 130.0 (C-2′,-6′), 129.2 (C-6), 125.0 (C-4′), 121.4 (C-α), 118.8 (C-3), 114.5 (C-1), 102.2 (C-5), 56.4 (4-OCH3), 24.6 (C-1″), 22.0 (C-2″), 14.4 (C-3″) ppm; HRMS (ESI+): m/z Anal. Calc. for C19H20BrO3 (M + H+) 375.05903, found 375.05889.
3.1.8. General Procedure for the Synthesis of Compounds of Group C2 (35 and 36)
- (E)-3-(4-(dimethylamino)phenyl)-1-(2-hydroxy-4-methoxy-3-propylphenyl)prop-2-en-1-one (35): Purified by flash column chromatography (SiO2; n-hexane: ethyl acetate, 9:1). Yield: 15% as an orange solid; 98.7% purity; mp 126–128 °C (ethyl acetate); 1H NMR (CDCl3, 300.13 MHz) δ: 13.71 (s, 1H, 2-OH), 7.87 (d, J = 15.2 Hz, 1H, H-β), 7.80 (d, J = 9.0 Hz, 1H, H-6), 7.56 (dt, J = 9.3; 2.2 Hz, 2H, H-2′,-6′), 7.41 (d, J = 15.2 Hz, 1H, H-α), 6.70 (dt, J = 9.4; 2.2 Hz, 2H, H-3′, -5′), 6.48 (d, J = 9.1 Hz, 1H, H-5), 3.89 (s, 3H, 4-OCH3), 3.05 (s, 6H, 4′-NCH3), 2.66 (t, J = 7.7 Hz, 2H, H-1″), 1.62–1.50 (m, 2H, H-2″), 0.97 (t, J = 7.3 Hz, 3H, H-3″) ppm; 13C NMR (CDCl3, 75.47 MHz) δ: 192.5 (C=O), 163.3 (C-2), 163.2 (C-4), 152.2 (C-4′), 145.2 (C-β), 130.7 (C-2′, -6′), 128.8 (C-6), 122.8 (C-1′), 118.5 (C-3), 115.1 (C-α), 114.8 (C-1), 111.9 (C-3′,-5′), 101.8 (C-5), 55.8 (4-OCH3), 40.3 (4′-NCH3), 24.6 (C-1″), 22.1 (C-2″), 14.4 (C-3″) ppm; HRMS (ESI+): m/z Anal. Calc. for C21H26NO3 (M+H+) 340.19072, found 340.19093.
- (E)-1-(2-hydroxy-4-methoxy-3-propylphenyl)-3-(4-morpholinophenyl)prop-2-en-1-one (36): Purified by flash column chromatography (SiO2; n-hexane: ethyl acetate, 9:1). Yield: 15% as an orange solid; 99.1% purity; mp 129–131 °C (ethyl acetate); 1H NMR (CDCl3, 300.13 MHz) δ: 13.56 (s, 1H, 2-OH), 7.85 (d, J = 15.3 Hz, 1H, H-β), 7.80 (d, J = 9.1 Hz, 1H, H-6), 7.58 (d, J = 8.8 Hz, 2H, H-2′,-6′), 7.47 (d, J = 15.3 Hz, 1H, H-α), 6.90 (d, J=8.9 Hz, 2H, H-3′, -5′), 6.49 (d, J = 9.0 Hz, 1H, H-5), 3.90 (s, 3H, 4-OCH3), 3.87 (t, J = 4.8 Hz, 2H, H-8′), 3.28 (t, J = 4.9 Hz, 2H, H-7′), 2.66 (t, J = 7.7 Hz, 2H, H-1″), 1.59–1.52 (m, 2H, H-2″), 0.96 (t, J = 7.4 Hz, 3H, H-3″) ppm; 13C NMR (CDCl3. 75.47 MHz) δ: 192.5 (C=O), 163.5 (C-4), 163.4 (C-2), 152.9 (C-4′), 144.4 (C-β), 130.3 (C-2′,-6′), 129.0 (C-6), 126.0 (C-1′), 118.6 (C-3), 117.1 (C-α), 114.8 (C-1, -3′, -5′), 102.0 (C-5), 66.8 (C-8′), 55.8 (4-OCH3), 48.1 (C-7′), 24.6 (C-1″), 22.1 (C-2″), 14.4 (C-3″) ppm. HRMS (ESI+): m/z Anal. Calc. for C23H28NO4 (M + H+) 382.20128, found 382.20104.
3.1.9. General Procedure for the Synthesis of Compounds of Group C3
- 3,5-bis((E)-2-((3-methylbut-2-en-1-yl)oxy)benzylidene)tetrahydro-4H-pyran-4-one (38): Purified by TLC (SiO2, n-hexane: ethyl acetate, 8:2). Yield: 50% as an orange gum; 99.9% purity; mp N.D. (gum); 1H NMR (CDCl3, 400.14 MHz) δ: 8.10 (sl, 2H, H-1′′), 7.32 (td, J = 7.9; 1.8 Hz, 2H, H-4′), 7.06 (dd, J = 7.5; 1.8 Hz, 2H, H-6′), 6.97–6.91 (m, 4H, H-3′, -5′), 5.51–5.47 (m, 2H, H-2‴), 4.80 (d, J = 2.0 Hz, 4H, H-3, -5), 4.57 (d, J = 6.8 Hz, 4H, H-1′’’), 1.79 (d, J = 1.3 Hz, 6H, H-4‴), 1.74 (d, J = 1.3 Hz, 6H, H-5‴) ppm; 13C NMR (CDCl3, 100.13 MHz) δ: 185.9 (C=O), 157.9 (C-2′), 137.8 (C-3‴), 133.2 (C-2, -6), 132.7 (C-1′’), 130.9 (C-4′), 130.8 (C-6′), 124.5 (C-1′), 119.8 (C-2′’’), 120.1 (C-5′), 112.4 (C-3′), 69.1 (C-3, -5), 65.6 (C-1‴), 25.9 (C-4‴), 18.4 (C-5‴) ppm; HRMS (ESI+): m/z Anal. Calc. for C29H31O4 (M-H+) 443.22278, found 443.22146.
- 3,5-bis((E)-4-((3-methylbut-2-en-1-yl)oxy)benzylidene)tetrahydro-4H-pyran-4-one (40): Purified by crystallization from methanol. Yield: 25% as a yellow solid; 99.9% purity; mp 145–147 °C (methanol); 1H NMR (CDCl3, 300.13 MHz) δ: 7.79 (sl, 2H, H-1′′), 7.28 (d, J = 8.7 Hz, 4H, H-2′,-6′), 6.95 (d, J = 8.7 Hz, 4H, H-3′,-5′), 5.52–5.47 (m, 2H, H2‴), 4.93 (d, J = 1.9 Hz, 4H, H-3,-5), 4.55 (d, J = 6.8 Hz, 4H, H-1‴), 1.89 (sl, 6H, H-4‴), 1.76 (sl, 6H, H-5‴) ppm; 13C NMR (CDCl3, 75.47 MHz) δ: 185.6 (C=O), 160.0 (C-4′), 138.9 (C-3‴), 136.1 (C-1′′), 132.6 (C-2′,-6′), 131.3 (C-2, -6), 127.6 (C-1′), 119.3 (C-2‴), 115.0 (C-3′,-5′), 68.8 (C-3,-5), 65.5 (C-1‴), 26.0 (C-4‴), 18.4 (C-5‴) ppm; HRMS (ESI+): m/z Anal. Calc. for C29H33O4 (M + H+) 445.23734, found 445.23760.
3.2. Biological Activity
3.2.1. Human Cell Lines and Growth Conditions
3.2.2. Cell Proliferation and Viability Assays
3.2.3. Apoptosis Analysis
3.2.4. Western Blot Analysis
3.2.5. Statistical Analysis
3.3. In Silico Studies
3.3.1. Predicting Druglikeness
3.3.2. ADMET Properties Prediction
3.3.3. Docking Studies
3.3.4. Molecular Dynamics Simulation (MD)
3.3.5. Quantitative Structure–Activity Relationship (QSAR)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liebl, M.C.; Hofmann, T.G. The role of p53 signaling in colorectal cancer. Cancers 2021, 13, 2125. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Toshiyuki, M.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Hong, B.; van den Heuvel, P.J.; Prabhu, V.V.; Zhang, S.; El-Deiry, W.S. Targeting tumor suppressor p53 for cancer therapy: Strategies, challenges and opportunities. Curr. Drug Targets 2014, 15, 80–89. [Google Scholar] [CrossRef]
- Waning, D.L.; Lehman, J.A.; Batuello, C.N.; Mayo, L.D. Controlling the Mdm2-Mdmx-p53 Circuit. Pharmaceuticals 2010, 3, 1576–1593. [Google Scholar] [CrossRef]
- Huang, L.; Yan, Z.; Liao, X.; Li, Y.; Yang, J.; Wang, Z.-G.; Zuo, Y.; Kawai, H.; Shadfan, M.; Ganapathy, S. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12001–12006. [Google Scholar] [CrossRef]
- Lu, Y.; Wu, M.; Xu, Y.; Yu, L. The Development of p53-Targeted Therapies for Human Cancers. Cancers 2023, 15, 3560. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Bharti, S.K. Therapeutic potential of chalcones as cardiovascular agents. Life Sci. 2016, 148, 154–172. [Google Scholar] [CrossRef]
- Kontogiorgis, C.; Mantzanidou, M.; Hadjipavlou-Litina, D. Chalcones and their potential role in inflammation. Mini Rev. Med. Chem. 2008, 8, 1224–1242. [Google Scholar] [CrossRef]
- Boumendjel, A.; Ronot, X.; Boutonnat, J. Chalcones derivatives acting as cell cycle blockers: Potential anti cancer drugs? Curr. Drug Targets 2009, 10, 363–371. [Google Scholar] [PubMed]
- Go, M.; Wu, X.; Liu, X. Chalcones: An update on cytotoxic and chemoprotective properties. Curr. Med. Chem. 2005, 12, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. Chalcones and their therapeutic targets for the management of diabetes: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2015, 92, 839–865. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Chalcone scaffolds as anti-infective agents: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 101, 496–524. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef]
- Orlikova, B.; Tasdemir, D.; Golais, F.; Dicato, M.; Diederich, M. Dietary chalcones with chemopreventive and chemotherapeutic potential. Genes Nutr. 2011, 6, 125. [Google Scholar] [CrossRef]
- Zhou, B.; Xing, C. Diverse molecular targets for chalcones with varied bioactivities. Med. Chem. 2015, 5, 388. [Google Scholar] [CrossRef]
- Moreira, J.; Almeida, J.; Saraiva, L.; Cidade, H.; Pinto, M. Chalcones as Promising Antitumor Agents by Targeting the p53 Pathway: An Overview and New Insights in Drug-Likeness. Molecules 2021, 26, 3737. [Google Scholar] [CrossRef]
- Moreira, J.; Saraiva, L.; Pinto, M.M.; Cidade, H. Bioactive Diarylpentanoids: Insights into the Biological Effects beyond Antitumor Activity and Structure–Activity Relationships. Molecules 2022, 27, 6340. [Google Scholar] [CrossRef]
- Moreira, J.; Saraiva, L.; Pinto, M.M.; Cidade, H. Diarylpentanoids with antitumor activity: A critical review of structure–activity relationship studies. Eur. J. Med. Chem. 2020, 192, 112177. [Google Scholar] [CrossRef]
- Moreira, J.; Almeida, J.; Loureiro, J.B.; Ramos, H.; Palmeira, A.; Pinto, M.M.; Saraiva, L.; Cidade, H. A Diarylpentanoid with Potential Activation of the p53 Pathway: Combination of in silico Screening Studies, Synthesis, and Biological Activity Evaluation. ChemMedChem 2021, 16, 2969–2981. [Google Scholar] [CrossRef] [PubMed]
- Novais, P.; Silva, P.; Moreira, J.; Palmeira, A.; Amorim, I.; Pinto, M.; Cidade, H.; Bousbaa, H. BP-M345, a New Diarylpentanoid with Promising Antimitotic Activity. Molecules 2021, 26, 7139. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.; Lima, R.T.; Palmeira, A.; Seca, H.; Soares, J.; Gomes, S.; Raimundo, L.; Maciel, C.; Pinto, M.; Sousa, E. Design and synthesis of new inhibitors of p53–MDM2 interaction with a chalcone scaffold. Arab. J. Chem. 2019, 12, 4150–4161. [Google Scholar] [CrossRef]
- Aramini, A.; Brinchi, L.; Germani, R.; Savelli, G. Reductions of α, β-Unsaturated Ketones by NaBH4 or NaBH4 + CoCl2: Selectivity Control by Water or by Aqueous Micellar Solutions. Eur. J. Org. Chem. 2000, 2000, 1793–1797. [Google Scholar] [CrossRef]
- Jesus, A.; Durães, F.; Szemerédi, N.; Freitas-Silva, J.; da Costa, P.M.; Pinto, E.; Pinto, M.; Spengler, G.; Sousa, E.; Cidade, H. BDDE-Inspired Chalcone Derivatives to Fight Bacterial and Fungal Infections. Mar. Drugs. 2022, 20, 315. [Google Scholar] [CrossRef] [PubMed]
- Narang, G.; Jindal, D.P.; Jit, B.; Bansal, R.; Potter, B.S.; Palmer, R.A. Formation of Dimers of Some 2-Substituted Indan-1-one Derivatives during Base-Mediated Cross-Aldol Condensation. Helv. Chim. Acta 2006, 89, 258–264. [Google Scholar] [CrossRef]
- Hu, J.; Yan, J.; Chen, J.; Pang, Y.; Huang, L.; Li, X. Synthesis, biological evaluation and mechanism study of a class of benzylideneindanone derivatives as novel anticancer agents. MedChemComm 2015, 6, 1318–1327. [Google Scholar] [CrossRef]
- Karanjit, S.; Tamura, A.; Kashihara, M.; Ushiyama, K.; Shrestha, L.K.; Ariga, K.; Nakayama, A.; Namba, K. Hydrotalcite-Supported Ag/Pd Bimetallic Nanoclusters Catalyzed Oxidation and One-Pot Aldol Reaction in Water. Catalysts 2020, 10, 1120. [Google Scholar] [CrossRef]
- Lokhande, P.D.; Hasanzadeh, K.; Khaledi, H.; Mohd Ali, H. Aromatization and Halogenation of 3,3a,4,5-Tetrahydro-3-aryl-2-phenyl-2H-benzo [g] indazole Using I2/DMSO, CuCl2/DMSO, and N-Bromosuccinimide. J. Heterocycl. Chem. 2012, 49, 1398–1406. [Google Scholar] [CrossRef]
- Gaspar, A.; Silva, T.; Yánez, M.; Vina, D.; Orallo, F.; Ortuso, F.; Uriarte, E.; Alcaro, S.; Borges, F. Chromone, a privileged scaffold for the development of monoamine oxidase inhibitors. J. Med. Chem. 2011, 54, 5165–5173. [Google Scholar] [CrossRef]
- Sk, M.; Kumar, A.; Das, J.; Banerjee, D. A Simple Iron-Catalyst for Alkenylation of Ketones Using Primary Alcohols. Molecules 2020, 25, 1590. [Google Scholar] [CrossRef] [PubMed]
- Magar, T.B.T.; Seo, S.H.; Kadayat, T.M.; Jo, H.; Shrestha, A.; Bist, G.; Katila, P.; Kwon, Y.; Lee, E.-S. Synthesis and SAR study of new hydroxy and chloro-substituted 2, 4-diphenyl 5H-chromeno [4, 3-b] pyridines as selective topoisomerase IIα-targeting anticancer agents. Bioorg. Med. Chem. 2018, 26, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.M.; Nogueira, C.E.; Almeida-Neto, F.W.Q.; Santos, H.S.; Teixeira, A.M.; de Lima-Neto, P.; Marinho, E.S.; de Moraes, M.O.; Pessoa, C.; Barros-Nepomuceno, F.W.A. Full Spectroscopic Characterization and Cytotoxicity Activity of Synthetic Dibenzalacetone Derivatives. J. Mol. Struct. 2021, 1231, 129670. [Google Scholar] [CrossRef]
- Abaee, M.; Mojtahedi, M.; Sharifi, R.; Zahedi, M.; Abbasi, H.; Tabar-Heidar, K. Facile synthesis of bis (arylmethylidene) cycloalkanones mediated by lithium perchlorate under solvent-free conditions. J. Iran. Chem. Soc. 2006, 3, 293–296. [Google Scholar] [CrossRef]
- Kar, S.; Ramamoorthy, G.; Sinha, S.; Ramanan, M.; Pola, J.K.; Golakoti, N.R.; Nanubolu, J.B.; Sahoo, S.K.; Dandamudi, R.B.; Doble, M. Synthesis of diarylidenecyclohexanone derivatives as potential anti-inflammatory leads against COX-2/mPGES1 and 5-LOX. New J. Chem. 2019, 43, 9012–9020. [Google Scholar] [CrossRef]
- Abaee, M.S.; Mojtahedi, M.M.; Zahedi, M.M.; Sharifi, R. A highly efficient method for solvent-free synthesis of bisarylmethylidenes of pyranones and thiopyranones. Heteroat. Chem. 2007, 18, 44–49. [Google Scholar] [CrossRef]
- da Silva, A.A.; da Silva Maia, P.I.; Lopes, C.D.; de Albuquerque, S.; Valle, M.S. Synthesis, characterization and antichagasic evaluation of thiosemicarbazones prepared from chalcones and dibenzalacetones. J. Mol. Struct. 2021, 1232, 130014. [Google Scholar] [CrossRef]
- Wei, X.; Du, Z.-Y.; Zheng, X.; Cui, X.-X.; Conney, A.H.; Zhang, K. Synthesis and evaluation of curcumin-related compounds for anticancer activity. Eur. J. Med. Chem. 2012, 53, 235–245. [Google Scholar] [CrossRef]
- Almeida, J.; Moreira, J.; Pereira, D.; Pereira, S.; Antunes, J.; Palmeira, A.; Vasconcelos, V.; Pinto, M.; Correia-da-Silva, M.; Cidade, H. Potential of synthetic chalcone derivatives to prevent marine biofouling. Sci. Total Environ. 2018, 643, 98–106. [Google Scholar] [CrossRef]
- Rahimpour, K.; Nikbakht, R.; Aghaiepour, A.; Teimuri-Mofrad, R. Synthesis of 2-(4-amino substituted benzylidene) indanone analogues from aromatic nucleophilic substitution (SNAr) reaction. Synth. Commun. 2018, 48, 2253–2259. [Google Scholar] [CrossRef]
- Fonseca, J.; Marques, S.; Silva, P.; Brandão, P.; Cidade, H.; Pinto, M.M.; Bousbaa, H. Prenylated chalcone 2 acts as an antimitotic agent and enhances the chemosensitivity of tumor cells to paclitaxel. Molecules 2016, 21, 982. [Google Scholar] [CrossRef] [PubMed]
- Leão, M.; Soares, J.; Gomes, S.; Raimundo, L.; Ramos, H.; Bessa, C.; Queiroz, G.; Domingos, S.; Pinto, M.; Inga, A. Enhanced cytotoxicity of prenylated chalcone against tumour cells via disruption of the p53–MDM2 interaction. Life Sci. 2015, 142, 60–65. [Google Scholar] [CrossRef]
- Raimundo, L.; Espadinha, M.; Soares, J.; Loureiro, J.B.; Alves, M.G.; Santos, M.M.; Saraiva, L. Improving anticancer activity towards colon cancer cells with a new p53-activating agent. Br. J. Pharmacol. 2018, 175, 3947–3962. [Google Scholar] [CrossRef] [PubMed]
- Bade, R.; Chan, H.-F.; Reynisson, J. Characteristics of known drug space. Natural products, their derivatives and synthetic drugs. Eur. J. Med. Chem. 2010, 45, 5646–5652. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef]
- Anwar-Mohamed, A.; El-Kadi, A. P-glycoprotein effects on drugs pharmacokinetics and drug-drug-interactions and their clinical implications. Libyan J. Pharm. Clin. Pharmacol. 2012, 1, 48154. [Google Scholar]
- Ma, B.; Pan, Y.; Gunasekaran, K.; Keskin, O.; Venkataraghavan, R.B.; Levine, A.J.; Nussinov, R. The contribution of the Trp/Met/Phe residues to physical interactions of p53 with cellular proteins. Phys. Bio. 2005, 2, S56. [Google Scholar] [CrossRef]
- Fu, T.; Min, H.; Xu, Y.; Chen, J.; Li, G. Molecular dynamic simulation insights into the normal state and restoration of p53 function. Int. J. Mol. Sci. 2012, 13, 9709–9740. [Google Scholar] [CrossRef]
- Espadinha, M.; Barcherini, V.; Lopes, E.A.; Santos, M.M. An update on MDMX and dual MDM2/X inhibitors. Curr. Top. Med. Chem. 2018, 18, 647–660. [Google Scholar] [CrossRef]
- Bharatham, N.; Bharatham, K.; Shelat, A.A.; Bashford, D. Ligand binding mode prediction by docking: mdm2/mdmx inhibitors as a case study. J. Chem. Inf. Model. 2014, 54, 648–659. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2–p53 protein–protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment: Miniperspective. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.; Mohammad, T.; Hasan, G.M.; Hassan, M.I. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr. Top. Med. Chem. 2018, 18, 1755–1768. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.H.; Ferreira, R.S.; Caffarena, E.R. Integrating molecular docking and molecular dynamics simulations. In Docking Screens for Drug Discovery; Humana Press: Totowa, NJ, USA, 2019; pp. 13–34. [Google Scholar]
- Das, P.; Mattaparthi, V.S.K. Computational Investigation on the p53–MDM2 Interaction Using the Potential of Mean Force Study. ACS Omega 2020, 5, 8449–8462. [Google Scholar] [CrossRef] [PubMed]
- Riedinger, C.; McDonnell, J.M. Inhibitors of MDM2 and MDMX: A structural perspective. Future Med. Chem. 2009, 1, 1075–1094. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef]
- Dudek, A.Z.; Arodz, T.; Gálvez, J. Computational methods in developing quantitative structure-activity relationships (QSAR): A review. Comb. Chem. High Throughput Screen. 2006, 9, 213–228. [Google Scholar] [CrossRef]
- Liu, P.; Long, W. Current mathematical methods used in QSAR/QSPR studies. Int. J. Mol. Sci. 2009, 10, 1978–1998. [Google Scholar] [CrossRef]
- Kubinyi, H. QSAR: Hansch Analysis and Related Approaches; VcH Weinheim: Weenheim, Germany, 1993; Volume 1. [Google Scholar]
- Alexander, D.L.; Tropsha, A.; Winkler, D.A. Beware of R 2: Simple, unambiguous assessment of the prediction accuracy of QSAR and QSPR models. J. Chem. Inf. Model. 2015, 55, 1316–1322. [Google Scholar] [CrossRef]
- Gramatica, P. On the development and validation of QSAR models. In Computational Toxicology; Humana Press: Totowa, NJ, USA, 2013; Volume II, pp. 499–526. [Google Scholar]
- Veerasamy, R.; Rajak, H.; Jain, A.; Sivadasan, S.; Varghese, C.P.; Agrawal, R.K. Validation of QSAR models-strategies and importance. Int. J. Drug Des. Discov 2011, 3, 511–519. [Google Scholar]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.-D.; Lee, K.-H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Todeschini, R.; Consonni, V. Molecular descriptors for chemoinformatics. In Methods and Principles in Medicinal Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Hoboken, NJ, USA, 2009; Volume 41. [Google Scholar]
- Brandão, P.; Loureiro, J.B.; Carvalho, S.; Hamadou, M.H.; Cravo, S.; Moreira, J.; Pereira, D.; Palmeira, A.; Pinto, M.; Saraiva, L.; et al. Targeting the MDM2-p53 protein-protein interaction with prenylchalcones: Synthesis of a small library and evaluation of potential antitumor activity. Eur. J. Med. Chem. 2018, 156, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.P.; Cidade, H.; Pinto, M.; Silva, A.M.; Gales, L.; Damas, A.M.; Lima, R.T.; Vasconcelos, M.H.; Nascimento, M.d.S.J. Prenylated derivatives of baicalein and 3, 7-dihydroxyflavone: Synthesis and study of their effects on tumor cell lines growth, cell cycle and apoptosis. Eur. J. Med. Chem. 2011, 46, 2562–2574. [Google Scholar] [CrossRef]
- Leao, M.; Pereira, C.; Bisio, A.; Ciribilli, Y.; Paiva, A.M.; Machado, N.; Palmeira, A.; Fernandes, M.X.; Sousa, E.; Pinto, M. Discovery of a new small-molecule inhibitor of p53–MDM2 interaction using a yeast-based approach. Biochem. Pharmacol. 2013, 85, 1234–1245. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Le, J.; Abraham, M.H.; Hersey, A.; Eddershaw, P.J.; Luscombe, C.N.; Boutina, D.; Beck, G.; Sherborne, B.; Cooper, I. Evaluation of human intestinal absorption data and subsequent derivation of a quantitative structure–activity relationship (QSAR) with the Abraham descriptors. J. Pharm. Sci. 2001, 90, 749–784. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Furubayashi, T.; Kataoka, M.; Sakane, T.; Sezaki, H.; Tokuda, H. Optimized conditions for prediction of intestinal drug permeability using Caco-2 cells. Eur. J. Pharm. Sci. 2000, 10, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37 (Suppl. S2), W623–W633. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Anil, B.; Riedinger, C.; Endicott, J.A.; Noble, M.E. The structure of an MDM2–Nutlin-3a complex solved by the use of a validated MDM2 surface-entropy reduction mutant. Acta Crystallogr. Sect. D. Biol. Crystallogr. 2013, 69, 1358–1366. [Google Scholar] [CrossRef]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using PyMOL. J. Comput. Aided Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | GI50 | SI1 | SI2 | |||
|---|---|---|---|---|---|---|
| HCT116 p53+/+ | HCT116 p53−/− | HFF-1 | ||||
| CM-M345 (1) | 2.6 ± 0.2 * | 3.4 ± 0.1 * | 7.7 ± 0.8 | 1.31 | 2.96 | |
| Group A | 3 | 4.6 ± 0.30 | 3.2 ± 0.47 | 8.1 ± 0.1 | 0.70 | 1.76 |
| 4 | 3.8 ± 0.32 | 3.1 ± 0.22 | 5.1 ± 0.9 | 0.82 | 1.34 | |
| 5 | 6.1 ± 0.12 | 4.1 ± 0.36 | 3.2 ± 0.07 | 0.67 | 0.52 | |
| 9 | 5.9 ±1.28 | 7.8 ± 0.9 | 5.5 ± 0.03 | 1.32 | 0.93 | |
| 10 | 4.0 ± 0.23 | 2.9 ± 0.11 | 5.4 ± 0.1 | 0.73 | 1.35 | |
| 11 | 6.3 ±2.18 | 6.1 ± 1.0 | 8.3 ± 1.4 | 0.97 | 1.32 | |
| 12 | 5.7 ± 0.51 | 3.9 ± 0.26 | 6.7 ± 0.4 | 0.68 | 1.18 | |
| 26 | 6.0 ± 0.93 | 4.6 ± 1.7 | 7.3 ± 2.0 | 0.77 | 1.22 | |
| 21 | >30 | >30 | >30 | ----- | ----- | |
| 22 | 0.09 ± 0.03 | 0.07 ± 0.01 | 0.135 ± 0.004 | 0.78 | 1.50 | |
| 23 | 2.3 ± 0.03 | 0.56 ± 0.07 | 1.65 ± 0.04 | 0.24 | 0.72 | |
| 24 | 0.31 ± 0.03 | 0.57 ± 0.009 | 0.23 ± 0.02 | 1.84 | 0.74 | |
| Group B | 27 | >30 | >30 | >30 | ---- | ---- |
| 28 | 15 ± 1.73 | 12 ± 5.0 | 17 ± 4.4 | 0.80 | 1.13 | |
| 29 | 18.7 ± 2.19 | 11.18 ± 5.3 | 22.1 ± 7.2 | 0.60 | 1.18 | |
| Group C | 32 | 9.5 ± 0.5 | 9.5 ± 0.1 | 11.12 ± 0.12 | 1 | 1.17 |
| 33 | 9.25 ± 0.75 | 7.2 ± 0.32 | 8.23 ± 0.08 | 0.77 | 0.89 | |
| 34 | 18.51 ± 0.35 | 11.45 ± 0.145 | 14.89 ± 0.232 | 0.62 | 0.80 | |
| 35 | 10.7 ± 2.86 | 16.3 ± 2.6 | 14 ± 3.6 | 1.52 | 1.31 | |
| 36 | 5.1 ± 0.32 | 14.3 ± 1.1 | 15.7 ± 2.8 | 2.65 | 3.08 | |
| BP-C4 (2) | 6.25 ± 1.18 ** | 10.13 ± 0.47 ** | 36.2 ± 5.54 ** | 1.62 | 5.79 | |
| Group A | 6 | 21.50 ± 1.19 | >30 | >30 | ----- | ----- |
| 7 | 7.4 ± 0.56 | 4.5 ± 0.13 | 4.7 ± 0.5 | 0.61 | 0.64 | |
| 8 | 7.5 ± 0.45 | 4.5 ± 0.28 | 5.1 ± 0.9 | 0.60 | 0.68 | |
| 13 | >30 | >30 | >30 | ----- | ----- | |
| 14 | 6.7 ± 0.21 | 4.3 ± 0.15 | 7.7 ± 0.2 | 0.64 | 1.15 | |
| 15 | 8.60 ± 0.50 | 5.29 ± 0.19 | 4.25 ± 0.32 | 0.62 | 0.49 | |
| 16 | 0.69 ± 0.07 | 7.95 ± 0.85 | 3.62 ± 0.99 | 11.5 | 5.25 | |
| 17 | 2.18 ± 0.59 | 3.55 ± 0.05 | 27.10 ± 1.25 | 1.63 | 12.43 | |
| 18 | >30 | >30 | >30 | ----- | ----- | |
| 19 | 3.10 ± 0.10 | 2.95 ± 0.15 | 14.10 ± 0.12 | 0.95 | 4.55 | |
| 20 | 1.13 ± 0.17 | 1.05 ± 0.05 | 2.27 ± 0.09 | 0.93 | 2.01 | |
| Group B | 30 | >30 | >30 | >30 | ----- | ----- |
| 31 | >30 | >30 | >30 | ----- | ----- | |
| Group C | 38 | >30 | >30 | >30 | ----- | ----- |
| 40 | 9.53 ± 0.25 | 5.8 ± 0.21 | 14.1 ± 0.08 | 0.61 | 1.48 | |
| Molecular Descriptor | 16 | 17 | Guidelines |
|---|---|---|---|
| Formula | C16H11ClOS | C17H12Cl2O | - |
| MW | 286.78 | 303.18 | 150 < MW < 500 * x̄ = 325.2 g mol−1 ** x̄ = 313 g mol−1 *** |
| Nº HA | 19 | 20 | - |
| Nº AHA | 12 | 12 | - |
| Far | 0.63 | 0.60 | - |
| Fsp3 | 0.06 | 0.00 | 0.25 < Fps3 < 1 ** |
| RB | 1 | 4 | 0 < RB < 9 ** |
| Nº HBA | 1 | 1 | HBA ≤ 10 * x̄ of HBA = 4.8 ** |
| Nº HBD | 0 | 0 | HBD ≤ 5 * x̄ of HBD = 1.6 ** |
| TPSA (a) | 42.37 | 17.07 | 20 < PSA < 125 Å2 ** |
| Log P (iLOGP) (a) | 3.02 | 3.39 | Log P < 5 * 0 < Log P < 5 *** |
| Log P (XLOGP3) (a) | 4.50 | 5.51 | |
| Log P (WLOGP) (a) | 4.60 | 5.07 | |
| Log P (MLOGP) (a) | 4.08 | 4.86 | |
| Log P (SILICOS-IT) (a) | 5.19 | 5.78 | |
| Log P (Consensus) (a) | 4.28 | 4.92 | |
| Mean of Log P values | 4.28 | 4.92 | - |
| Log S (ESOL) (a) | −4.85 | −5.37 | Log S > −4 *** |
| Log S (Ali) (a) | −5.11 | −5.63 | |
| Log S (SILICOS-IT) (a) | −6.38 | −6.26 | |
| Mean of Log S values | −5.45 | −5.75 | - |
| 16 | 17 | |
|---|---|---|
| Medicinal chemistry rules | ||
| Lipinski (a) | 0 | 1 |
| Ghose (a) | 0 | 0 |
| Veber (a) | 0 | 0 |
| Egan (a) | 0 | 0 |
| Muegge (a) | 0 | 2 |
| CMC-like (b) | 0 | 0 |
| Lead-like (b) | 1 | 1 |
| MDDR-like (b) | 1 | 2 |
| WDI-like (b) | 0 | 2 |
| Bioavailability score (a) | 0.55 | 0.55 |
| Synthetic accessibility (a) | 3.17 | 2.48 |
| 16 | 17 | |
|---|---|---|
| Pharmacokinetic properties | ||
| GI absorption (a) | High | High |
| HIA (%) (b) | 97.88 | 100 |
| Caco-2 cell (nm·s−1) (b) | 57.50 | 56.90 |
| BBB permeant (a) | Yes | Yes |
| Pgp substrate (a) | No | No |
| PPB (%) (b) | 100 | 100 |
| CYP1A2 Inhibitor (a) | Yes | Yes |
| CYP2C19 Inhibitor (a) | Yes | Yes |
| CYP2C9 Inhibitor (a) | Yes | Yes |
| CYP2D6 Inhibitor (a) | No | No |
| CYP3A4 Inhibitor (a) | No | No |
| CYP2C19 Inhibitor (b) | Yes | Yes |
| CYP2C9 Inhibitor (b) | Yes | Yes |
| CYP2D6 Inhibitor (b) | No | No |
| CYP3A4 Inhibitor (b) | No | No |
| Carcinogenic activity mouse (b) | Negative | Positive |
| Carcinogenic activity rat (b) | Negative | Negative |
| hERG-inhibitor (b) | Medium risk | Medium risk |
| Log Kp (a) | −4.85 cm/s | −4.24 cm/s |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, J.; Loureiro, J.B.; Correia, D.; Palmeira, A.; Pinto, M.M.; Saraiva, L.; Cidade, H. Structure–Activity Relationship Studies of Chalcones and Diarylpentanoids with Antitumor Activity: Potency and Selectivity Optimization. Pharmaceuticals 2023, 16, 1354. https://doi.org/10.3390/ph16101354
Moreira J, Loureiro JB, Correia D, Palmeira A, Pinto MM, Saraiva L, Cidade H. Structure–Activity Relationship Studies of Chalcones and Diarylpentanoids with Antitumor Activity: Potency and Selectivity Optimization. Pharmaceuticals. 2023; 16(10):1354. https://doi.org/10.3390/ph16101354
Chicago/Turabian StyleMoreira, Joana, Joana B. Loureiro, Danilo Correia, Andreia Palmeira, Madalena M. Pinto, Lucília Saraiva, and Honorina Cidade. 2023. "Structure–Activity Relationship Studies of Chalcones and Diarylpentanoids with Antitumor Activity: Potency and Selectivity Optimization" Pharmaceuticals 16, no. 10: 1354. https://doi.org/10.3390/ph16101354
APA StyleMoreira, J., Loureiro, J. B., Correia, D., Palmeira, A., Pinto, M. M., Saraiva, L., & Cidade, H. (2023). Structure–Activity Relationship Studies of Chalcones and Diarylpentanoids with Antitumor Activity: Potency and Selectivity Optimization. Pharmaceuticals, 16(10), 1354. https://doi.org/10.3390/ph16101354