


Pharmacokinetics and Anti-Tumor Efficacy of PEGylated Liposomes Co-Loaded with Cisplatin and Mifepristone

 and
and
Abstract
1. Introduction
2. Results
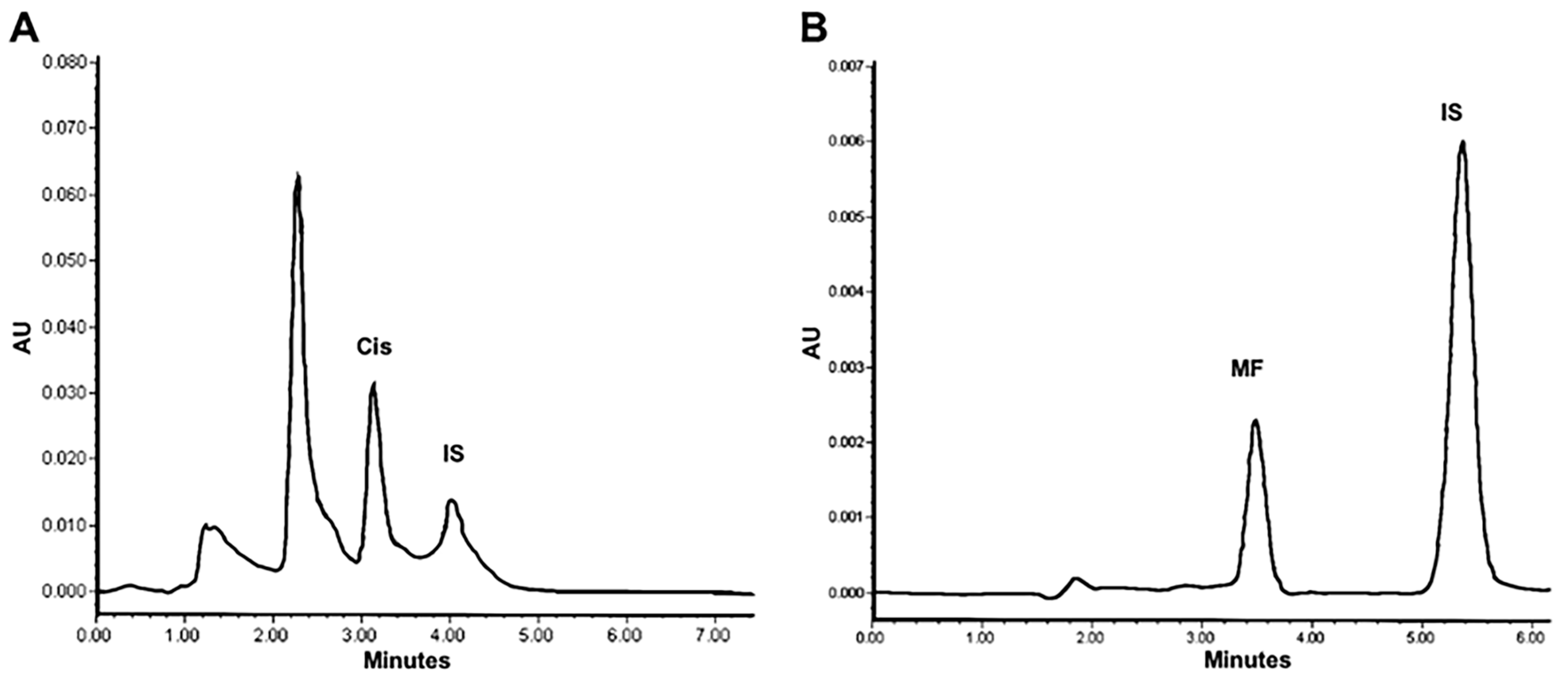
2.1. In Vitro Cisplatin and Mifepristone Release from L-Cis/MF
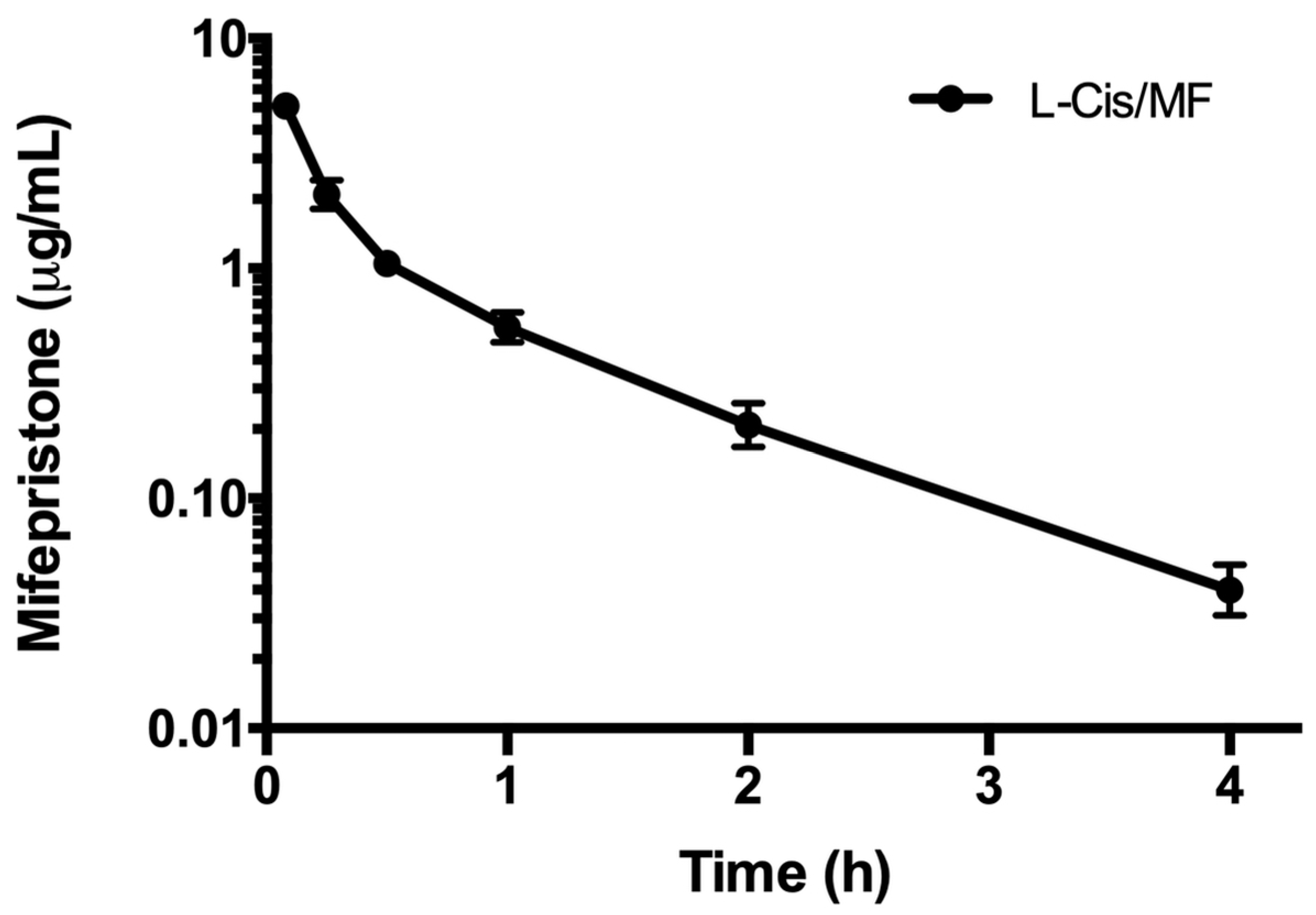
2.2. Pharmacokinetics of L-Cis/MF
2.3. Tumor Growth Inhibition and Systemic Toxicity
2.4. Quantitative Detection of VEGF in Tumors
3. Discussion
3.1. Strategies for Cancer Therapy with Cisplatin
3.2. Profile of the Release of Cisplatin and Mifepristone from L-Cis/MF
3.3. Pharmacokinetics of L-Cis/MF
3.4. The Therapeutic Efficacy of L-Cis/MF In Vivo
4. Materials and Methods
4.1. Drugs and Reagents
4.2. Animals
4.3. Preparation of Liposomes Co-Loaded with Cisplatin and Mifepristone
4.4. In Vitro Assay to Evaluate the Release of Cisplatin and Mifepristone from L-Cis/MF
4.5. Pharmacokinetics of L-Cis/MF
4.6. Tumor Xenografts and Systemic Toxicity
4.7. Quantitative Determination of VEGF in Tumors
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, A.; Kumar, S.; Tchounwou, P.B. Cisplatin-Based Chemotherapy of Human Cancers. J. Cancer Sci. Ther. 2019, 11, 97. [Google Scholar]
- Ranasinghe, R.; Mathai, M.L.; Zulli, A. Cisplatin for cancer therapy and overcoming chemoresistance. Heliyon 2022, 8, e10608. [Google Scholar] [CrossRef]
- Chi, R.A.; van der Watt, P.; Wei, W.; Birrer, M.J.; Leaner, V.D. Inhibition of Kpnbeta1 mediated nuclear import enhances cisplatin chemosensitivity in cervical cancer. BMC Cancer 2021, 21, 106. [Google Scholar] [CrossRef]
- Aktepe, O.H.; Sahin, T.K.; Guner, G.; Arik, Z.; Yalcin, S. Lycopene sensitizes the cervical cancer cells to cisplatin via targeting nuclear factor- kappa B (NF-kappaB) pathway. Turk. J. Med. Sci. 2021, 51, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Y.; Lan, X.; Yu, J.; Yang, C.; Sun, Z.; Kang, P.; Han, Y.; Yu, D. Halofuginone Sensitizes Lung Cancer Organoids to Cisplatin via Suppressing PI3K/AKT and MAPK Signaling Pathways. Front. Cell Dev. Biol. 2021, 9, 773048. [Google Scholar] [CrossRef]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 967. [Google Scholar] [CrossRef]
- Yardley, D.A. Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int. J. Breast Cancer 2013, 2013, 137414. [Google Scholar] [CrossRef]
- Segovia-Mendoza, M.; Jurado, R.; Mir, R.; Medina, L.A.; Prado-Garcia, H.; Garcia-Lopez, P. Antihormonal agents as a strategy to improve the effect of chemo-radiation in cervical cancer: In vitro and in vivo study. BMC Cancer 2015, 15, 21. [Google Scholar] [CrossRef]
- Jurado, R.; Lopez-Flores, A.; Alvarez, A.; Garcia-Lopez, P. Cisplatin cytotoxicity is increased by mifepristone in cervical carcinoma: An in vitro and in vivo study. Oncol. Rep. 2009, 22, 1237–1245. [Google Scholar] [CrossRef][Green Version]
- Zamboni, W.C.; Gervais, A.C.; Egorin, M.J.; Schellens, J.H.; Hamburger, D.R.; Delauter, B.J.; Grim, A.; Zuhowski, E.G.; Joseph, E.; Pluim, D.; et al. Inter- and intratumoral disposition of platinum in solid tumors after administration of cisplatin. Clin. Cancer Res. 2002, 8, 2992–2999. [Google Scholar]
- Ramon-Lopez, A.; Escudero-Ortiz, V.; Carbonell, V.; Perez-Ruixo, J.J.; Valenzuela, B. Population pharmacokinetics applied to optimising cisplatin doses in cancer patients. Farm. Hosp. 2012, 36, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, T.; Sibrian-Vazquez, M.; Strongin, R.M.; Steyger, P.S. Identification of cisplatin-binding proteins using agarose conjugates of platinum compounds. PLoS ONE 2013, 8, e66220. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Ornatsky, O.I.; Siddiqui, I.; Straus, R.; Baranov, V.I.; Hedley, D.W. Biodistribution of cisplatin revealed by imaging mass cytometry identifies extensive collagen binding in tumor and normal tissues. Sci. Rep. 2016, 6, 36641. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, N.N. Mifepristone: Bioavailability, pharmacokinetics and use-effectiveness. Eur. J. Obstet. Gynecol. Reprod. Biol. 2002, 101, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Ledezma-Gallegos, F.; Jurado, R.; Mir, R.; Medina, L.A.; Mondragon-Fuentes, L.; Garcia-Lopez, P. Liposomes Co-Encapsulating Cisplatin/Mifepristone Improve the Effect on Cervical Cancer: In Vitro and In Vivo Assessment. Pharmaceutics 2020, 12, 897. [Google Scholar] [CrossRef]
- Novais, M.V.M.; Gomes, E.R.; Miranda, M.C.; Silva, J.O.; Gomes, D.A.; Braga, F.C.; Pádua, R.M.; Oliveira, M.C. Liposomes co-encapsulating doxorubicin and glucoevatromonoside derivative induce synergic cytotoxic response against breast cancer cell lines. Biomed. Pharmacother. 2021, 136, 111123. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Kapperman, H.E.; Goyeneche, A.A.; Telleria, C.M. Mifepristone inhibits non-small cell lung carcinoma cellular escape from DNA damaging cisplatin. Cancer Cell Int. 2018, 18, 185. [Google Scholar] [CrossRef]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef]
- Fumoto, S.; Nishida, K. Co-delivery Systems of Multiple Drugs Using Nanotechnology for Future Cancer Therapy. Chem Pharm Bull 2020, 68, 603–612. [Google Scholar] [CrossRef]
- Al Bostami, R.D.; Abuwatfa, W.H.; Husseini, G.A. Recent Advances in Nanoparticle-Based Co-Delivery Systems for Cancer Therapy. Nanomaterials 2022, 12, 2672. [Google Scholar] [CrossRef]
- Yang, J.; Jin, R.M.; Wang, S.Y.; Xie, X.T.; Hu, W.; Tang, H.F.; Liu, B. Co-delivery of paclitaxel and doxorubicin using polypeptide-engineered nanogels for combination therapy of tumor. Nanotechnology 2022, 33. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.S.; Sun, J.H.; Yu, H.H.; Yu, S.Q. Co-delivery nanoparticles of anti-cancer drugs for improving chemotherapy efficacy. Drug Deliv. 2017, 24, 1909–1926. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, X.; Zheng, Z.; Chen, D.; Wang, X.; Shi, F.; Yu, D.; Wu, H. A single dose of dexamethasone encapsulated in polyethylene glycol-coated polylactic acid nanoparticles attenuates cisplatin-induced hearing loss following round window membrane administration. Int. J. Nanomed. 2015, 10, 3567–3579. [Google Scholar] [CrossRef]
- Mendes, L.P.; Gaeti, M.P.; de Avila, P.H.; de Sousa Vieira, M.; Dos Santos Rodrigues, B.; de Avila Marcelino, R.I.; Dos Santos, L.C.; Valadares, M.C.; Lima, E.M. Multicompartimental nanoparticles for co-encapsulation and multimodal drug delivery to tumor cells and neovasculature. Pharm. Res. 2014, 31, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Gu, F.X.; Langer, R.; Farokhzad, O.C.; Lippard, S.J. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc. Natl. Acad. Sci. USA 2008, 105, 17356–17361. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Jagan, L.; Isiah, R.; Rajesh, B.; Backianathan, S.; Subhashini, J. Nanotechnology in oncology: Characterization and in vitro release kinetics of cisplatin-loaded albumin nanoparticles: Implications in anticancer drug delivery. Indian. J. Pharmacol. 2011, 43, 409–413. [Google Scholar] [CrossRef]
- Yücel, Ç.; Değim, Z.; Yilmaz, Ş. Development of Cisplatin-loaded Liposome and Evaluation of Transport Prop-596 erties Through Caco-2 Cell Line. Turk. J. Pharm. Sci. 2016, 13, 95–108. [Google Scholar] [CrossRef]
- Nsairat, H.; Khater, D.; Sayed, U.; Odeh, F.; Al Bawab, A.; Alshaer, W. Liposomes: Structure, composition, types, and clinical applications. Heliyon 2022, 8, e09394. [Google Scholar] [CrossRef]
- Gupta, S.; De Mel, J.U.; Schneider, G.J. Dynamics of liposomes in the fluid phase. Curr. Opin. Colloid. Interface Sci. 2019, 42, 121–136. [Google Scholar] [CrossRef]
- Liu, P.; Chen, G.; Zhang, J. A Review of Liposomes as a Drug Delivery System: Current Status of Approved Products, Regulatory Environments, and Future Perspectives. Molecules 2022, 27, 1372. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, S.; Caliceti, P. Stealth properties to improve therapeutic efficacy of drug nanocarriers. J. Drug Deliv. 2013, 2013, 374252. [Google Scholar] [CrossRef] [PubMed]
- Papini, E.; Tavano, R.; Mancin, F. Opsonins and Dysopsonins of Nanoparticles: Facts, Concepts, and Methodological Guidelines. Front. Immunol. 2020, 11, 567365. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, J.; Wientjes, M.G.; Au, J.L. Delivery of nanomedicines to extracellular and intracellular compartments of a solid tumor. Adv. Drug Deliv. Rev. 2012, 64, 29–39. [Google Scholar] [CrossRef]
- Subhan, M.A.; Yalamarty, S.S.K.; Filipczak, N.; Parveen, F.; Torchilin, V.P. Recent Advances in Tumor Targeting via EPR Effect for Cancer Treatment. J. Pers. Med. 2021, 11, 571. [Google Scholar] [CrossRef]
- Dadpour, S.; Mehrabian, A.; Arabsalmani, M.; Mirhadi, E.; Askarizadeh, A.; Mashreghi, M.; Jaafari, M.R. The role of size in PEGylated liposomal doxorubicin biodistribution and anti-tumour activity. IET Nanobiotechnol. 2022, 16, 259–272. [Google Scholar] [CrossRef]
- Heikinheimo, O.; Pesonen, U.; Huupponen, R.; Koulu, M.; Lahteenmaki, P. Hepatic metabolism and distribution of mifepristone and its metabolites in rats. Hum. Reprod. 1994, 9 (Suppl. 1), 40–46. [Google Scholar] [CrossRef]
- Toro-Cordova, A.; Ledezma-Gallegos, F.; Mondragon-Fuentes, L.; Jurado, R.; Medina, L.A.; Perez-Rojas, J.M.; Garcia-Lopez, P. Determination of Liposomal Cisplatin by High-Performance Liquid Chromatography and Its Application in Pharmacokinetic Studies. J. Chromatogr. Sci. 2016, 54, 1016–1021. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, S.; Wei, D.; Zhai, J. Development of a high-performance liquid chromatographic method for the determination of mifepristone in human plasma using norethisterone as an internal standard: Application to pharmacokinetic study. Contraception 2007, 76, 228–232. [Google Scholar] [CrossRef]
- Medina, L.A.; Herrera-Penilla, B.I.; Castro-Morales, M.A.; Garcia-Lopez, P.; Jurado, R.; Perez-Cardenas, E.; Chanona-Vilchis, J.; Brandan, M.E. Use of an orthovoltage X-ray treatment unit as a radiation research system in a small-animal cancer model. J. Exp. Clin. Cancer Res. 2008, 27, 57. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Conventional Cisplatin | Liposomal Cisplatin |
|---|---|---|
| AUC0→t (μg·h/mL·kg) | 5.27 ± 0.66 | 801.76 ± 77.88 * |
| t1⁄2 (h) | 5.73 ± 2.25 | 17.26 ± 3.73 * |
| Cmax (μg/mL) | 9.62 ± 0.66 | 61.17 ± 3.07 * |
| Vd (mL/kg) | 636.73 ± 39.64 | 169.11 ± 16.35 * |
| Cl (mL/h) | 1286.53 ± 248.37 | 7.95 ± 0.76 * |
| Parameter | Conventional Mifepristone | Liposomal Mifepristone |
|---|---|---|
| AUC0→t (μg·h/mL·kg) | Not detectable | 2.03 ± 0.26 |
| t1⁄2 (h) | Not detectable | 0.4 ± 0.16 |
| Cmax (μg/mL) | Not detectable | 7.4 ± 1.01 |
| Vd (mL/kg) | Not detectable | 939.82 ± 105.40 |
| Cl (mL/h) | Not detectable | 3290.54 ± 351.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ocaña-Arakachi, K.; Martínez-Herculano, J.; Jurado, R.; Llaguno-Munive, M.; Garcia-Lopez, P. Pharmacokinetics and Anti-Tumor Efficacy of PEGylated Liposomes Co-Loaded with Cisplatin and Mifepristone. Pharmaceuticals 2023, 16, 1337. https://doi.org/10.3390/ph16101337
Ocaña-Arakachi K, Martínez-Herculano J, Jurado R, Llaguno-Munive M, Garcia-Lopez P. Pharmacokinetics and Anti-Tumor Efficacy of PEGylated Liposomes Co-Loaded with Cisplatin and Mifepristone. Pharmaceuticals. 2023; 16(10):1337. https://doi.org/10.3390/ph16101337
Chicago/Turabian StyleOcaña-Arakachi, Karen, Julio Martínez-Herculano, Rafael Jurado, Monserrat Llaguno-Munive, and Patricia Garcia-Lopez. 2023. "Pharmacokinetics and Anti-Tumor Efficacy of PEGylated Liposomes Co-Loaded with Cisplatin and Mifepristone" Pharmaceuticals 16, no. 10: 1337. https://doi.org/10.3390/ph16101337
APA StyleOcaña-Arakachi, K., Martínez-Herculano, J., Jurado, R., Llaguno-Munive, M., & Garcia-Lopez, P. (2023). Pharmacokinetics and Anti-Tumor Efficacy of PEGylated Liposomes Co-Loaded with Cisplatin and Mifepristone. Pharmaceuticals, 16(10), 1337. https://doi.org/10.3390/ph16101337