Abstract
A practical and robust synthetic method to obtain the natural disaccharide sambubiose (2-O-β-D-xylopyranosyl-D-glucopyranose) is reported, exploring the key step in the synthesis, i.e., stereoselective O-glycosylation. Specifically, the best combinations of glycoside donors and acceptors were identified, stereospecific control of the reaction was achieved by screening several catalysts and protection/deprotection steps were evaluated and improved. The best result was obtained by coupling allyl 3,5,6-tri-O-benzyl-β-D-glucofuranoside with 2,3,4-tri-O-acetyl-D-xylopiranosyl-α-trichloro acetimidate in the presence of trimethylsilyl triflate as a catalyst giving the corresponding protected target compound as a correct single isomer. The latter was transformed accordingly into the desired final product by deprotection steps (deallylation, deacetylation, and debenzylation). Sambubiose was synthesized into a satisfactory and higher overall yield than previously reported and was also characterized.
1. Introduction
Chemopreventive phytochemicals (CPs) occur naturally in some plants and have been shown to inhibit all levels of carcinogenesis in both in vitro and in vivo models [1]. Among CPs, flavonoid 2-O-xylosylvitexin (Figure 1), which is constituted by carbohydrate (sambubiose, Figure 1) and aglycone (apigenin, Figure 1) components, was identified for the first time in Citrus sinensis leaves [2] and then in Beta vulgaris sub. cycla leaves and seeds [3] with the latter containing the most abundant and efficacious CPs.
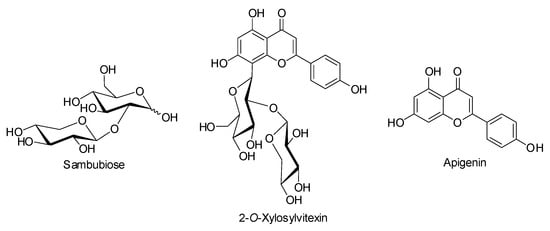
Figure 1.
Chemical structures of sambubiose, 2-O-xylosylvitexin, and apigenin.
2-O-xylosylvitexin shows strong antiproliferative activity against human colon cell cancer lines while also enhancing apoptosis, increasing cells in the G1 phase and reducing cells in the S phase as well as the proliferation of human fibroblasts [4].
Sambubiose, on the other hand, is formed by glucopyranose and xylopyranose units linked through a 1,2-trans bond in 2-position and was first isolated as a component of anthocyanins from elderberry (Sambucus nigra) [5], a plant with therapeutic properties [6,7]. In addition, sambubiose is also a constituent of the other major anthocyanins from black raspberries (Rubus occidentalis L.) [8], red currants, Davidson’s plums, and small red beans, and is linked to minor anthocyanins from açai and Vaccinium padifolium blueberries [9].
To date, to the best of our knowledge, only one synthesis of sambubiose has been reported [10]; however, the method followed by Erbing and Lindberg [10] (Scheme 1) and subsequently applied by Dick et al. [11] has several drawbacks and improvements could be made in the reagents and conditions.
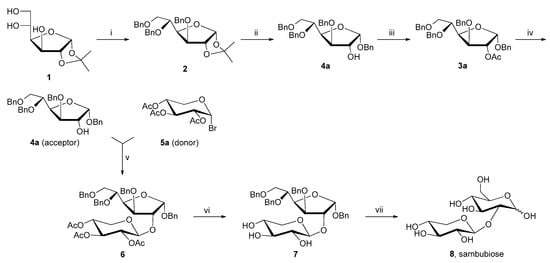
Scheme 1.
Reagents and conditions: (i) NaOH, BnCl, reflux, 7 h, 78%; (ii, iii) BnOH, HCl, reflux, 4 h then Ac2O, pyridine, rt, 24 h, 6%; (iv) MeONa, MeOH, rt, 12 h, 22%; (v, vi) Hg(CN)2/HgBr2, MeCN, rt, 4 h then MeONa, MeOH, rt, 12 h, 54%; (vii) H2, Pd/C 10%, EtOH, rt, 22 h, 43%.
Firstly, the purification of benzyl 3,5,6-tri-O-benzyl-α-D-glucofuranoside (4a) to eliminate high-boiling benzyl alcohol from the reaction crude mixture was extremely difficult both by distillation and/or by chromatography. Erbing and Lindberg [10], therefore, had to employ acetylation and deacetylation as additional synthetic steps via benzyl 2-O-acetyl-3,5,6-tri-O-benzyl-α-D-glucofuranoside (3a) to purify 4a (Scheme 1, “iii” and “iv” steps). Secondly, the use of the intrinsically unstable donor 2,3,4-tri-O-acetyl-α-D-xylopyranosyl bromide (5a), which must be freshly prepared and recrystallized because of its low stability, even when refrigerated, was also problematic. Moreover, Erbing and Lindberg [10] used a stoichiometric amount of mercuric cyanide as a glycosylation promoter. This reactant should not be used because of its high toxicity and dangerousness. Furthermore, Hg(CN)2 does not allow a stereospecific control of glycosylation: 6 [12] must be separated from its anomer since the output of the reaction is an α- and β-mixture. Finally, and importantly, the authors did not fully characterize the natural disaccharide sambubiose [10]. Hence, the aim of this investigation was to develop an improved efficient strategy to synthesize our target molecule overcoming the abovementioned drawbacks. The key synthetic step (i.e., the O-glycosylation between the appropriate protected glucose and activated/protected xylose) must be thoroughly explored. In particular, the leaving group of the donor should be screened, and the correct catalyst/promoter should be fine-tuned.
Herein, as a part of our ongoing investigations of disaccharide chemistry [13,14,15,16], we describe an efficient synthetic method that allowed us to obtain sambubiose in a satisfactory and higher overall yield than previously reported [10] as well as to achieve its full characterization. The proposed methodology ensured an effective stereospecific glycosylation control and exploited strategies of protection and deprotection of functional groups which also guide the regioselective reaction progress.
2. Results and Discussion
The study started with the optimization of the key step, consisting of the O-glycosylation reaction between the appropriately protected glucose (acceptor) and protected/activated xylose (donor) (Scheme 1, step “v”). In particular, our attention was first focused on the synthesis of several glucose acceptors and xylose donors to overcome the abovementioned problems [10]. With regard to the glucose acceptor, the synthesis of compounds 9–12 (Figure 2) was designed and explored aiming to avoid the acetylation and deacetylation steps necessary for the purification of 4a (Scheme 1).
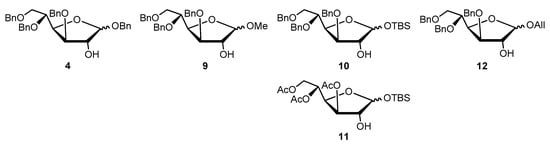
Figure 2.
Chemical structure of synthesized glycoside acceptors.
Methyl 3,5,6-tri-O-benzyl-D-glucofuranoside (9) is very easy to purify and relatively stable under basic or acid conditions. Compound 9 was synthesized starting from 3,5,6-tri-O-benzyl-1,2-O-isopropylidene-D-glucofuranoside (2, Scheme 2) (1% HCl, MeOH, 65 °C, 1.5 h, 91%; α/β 1:1.3), previously obtained by 1,2-O-isopropylidene-D-glucofuranoside (1, Scheme 2).
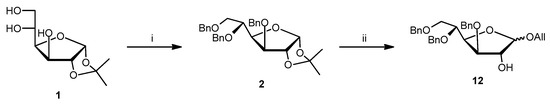
Scheme 2.
Reagents and conditions: (i) 80% NaH, BnBr, DMF, rt, 6 h, 70%; (ii) AllOH, TsOH, reflux, 3 h, 85%.
Likewise, tert-butyldimethylsilyl 3,5,6-tri-O-benzyl-D-glucofuranoside (10, Figure 2) was synthesized from 2 via a vicinal diol intermediate. Unfortunately, 10 was obtained with a very low overall yield (1,4-dioxane, H2SO4, reflux, 3 h, 41%; TBSCl, imidazole, THF, 0 °C, 10 h, 10%; α/β 1:1). On the other hand, the attempt to change the “armed” protective groups with the “disarmed” tert-butyldimethylsilyl 3,5,6-tri-O-acetyl-D-glucofuranoside (11, Figure 2) also proved not to be effective in term of yields (Ac2O, Py, rt, 17 h, 85%; TFA, 50 °C, 3 h, 19%; TBSCl, imidazole, THF, 0 °C, 10 h; 10%). Both 10 and 11 were not assessed as acceptors in the glycosylation reaction. Finally, our attention was turned to the glycoside acceptor allyl 3,5,6-tri-O-benzyl-D-glucofuranoside (12, Figure 2) which was prepared in an acidic environment by treating 2 with allyl alcohol [17] giving an α/β 1:1.1 mixture (Scheme 2).
With regard to the donor glycoside, only compounds that are more stable than 5a (Scheme 1), such as fluoride [18] and trichloroacetimidate [19] derivatives, two of the most appealing and promising glycoside donors for O-glycosylation, were taken into account. Hence, 2,3,4-tri-O-acetyl-α-D-xylopyranosyl fluoride (13, Scheme 3) and 2,3,4-tri-O-acetyl-α-D-xylopyranosyl trichloroacetimidate (14a, Scheme 3) were synthesized.
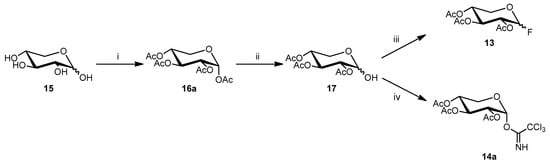
Scheme 3.
Reagents and conditions: (i) Ac2O, Py, 0 °C, 6 h, 75%; (ii) NH4OAc, DMF, rt, 22 h, 70%; (iii) DAST, CH2Cl2, −30 °C to rt, 1 h, 98%; (iv) CCl3CN, CH2Cl2, 0 °C, DBU, 1 h, 60%.
In the case of fluoride 13 [20], the desired hemiacetal intermediate 2,3,4-tri-O-acetyl-D-xylopyranoside (17, Scheme 3) was prepared starting from D-(+)-xylose (15) through an initial acetylation (Ac2O, Py, 0 °C, 6 h, 99%) followed by monohydrolysis of the corresponding D-xylopyranose 1,2,3,4-tetracetate (16a) [21] with ammonium acetate (DMF dry, rt, 22 h, 70%). Glycoside donor 13 was then obtained by fluorination of 17 with diethylaminosulfur trifluoride (DAST) (CH2Cl2, −30 °C to rt, 1 h, 98%) as a mixture of anomers (α/β 1:4), which could be easily isolated by a flash chromatography.
With regard to trichloroacetimidate 14a [22], this donor type showed some advantages such as a relatively high stability, easy purification, and, most importantly, a potential stereochemical control of the glycosylation reaction. In addition, 14a was synthesized by treating 17 with trichloroacetonitrile (CCl3CN) and 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) obtaining only α-anomer [23]. As previously reported, DBU promoted thermodynamic control of the reaction, which led to a more stable α-anomer 14a (Scheme 3). One-pot attempts to obtain 14a from 16a were also explored; however, such attempts proved not to be advantageous due to the fact of their low yields (data not shown).
Finally, the key synthetic step, consisting in the O-glycosylation reaction, was explored. The initial substitution of mercuric salts [10] with silver oxide or silver carbonate as an activator was ineffective any advantage because the yields in these conditions fell drastically (data not shown). The use of safer, greener catalysts such as boron trifluoride diethyl etherate (BF3 . OEt2) or trimethylsilyl trifluoromethanesulfonate (TMSOTf, particularly suitable for the glycoside donor activation bearing the trichloroacetoimidate group as the leaving group), warrant further investigation as alternatives to mercury compounds. Dichloromethane was used as a non-polar solvent; therefore, the stereoselection of glycosylation was led by the 2-O-acyl vicinal group on the donor (anchimeric assistance). All the prepared glycoside donors and acceptors were reacted in different conditions in the presence of powdered molecular sieves (MS) 4Å (Scheme 4) at −20 °C, and the main results are reported in Table 1.
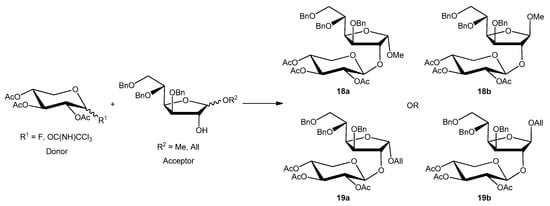
Scheme 4.
General scheme of O-glycosylation.

Table 1.
Optimization of the O-glycosylation reaction.
Firstly, the reactivity of fluoride glycoside 13b was explored with the methoxy derivative 9b or 9a as a glycoside acceptor, in the presence of BF3 . OEt2 at −20 °C to room temperature for 4 h. The reaction gave the corresponding β- or α-disaccharide in 80% or 50% yields, respectively, and a complete trans stereocontrol (entries 1 and 2).
A complete recovery of the acceptor (9b or 9a) was pointed out in both reactions. The lower yield for glycosylation obtained using the acetate 9a could be attributed to the steric hindrance of the methoxy group present in the same part of the plane of the sugar of the reactive 2-OH group. Similarly, the allyloxy acceptor 12b gave the corresponding coupling product in 30% yield, and its anomer 12a did not react at all and was completely recovered (entries 6 and 3, respectively). The absence of reactivity of 12a could be explained by the greater steric hindrance of its 1-allyloxy substituent compared to that of 1-methoxy present on 9a (entry 3 versus 2). On the other hand, the α-anomer fluoride donor 13a did not react with either highly reactive acceptor 9b or 12b in the same conditions (entries 4 and 5, respectively). The last donor to be investigated was trichloroacetimidate 14a, which was reacted with 9b (entry 7) in the presence of BF3 . OEt2, giving the corresponding β-linked disaccharide in 44% yield or with 12b (entry 8) using TMSOTf as a promoter and obtaining a better yield (60%), complete stereocontrol, and high reproducibility. Surprisingly, the use of the latter conditions (donor 14a and promoter TMSOTf) and very low reactive donor 12a allowed coupling with the formation of the corresponding disaccharide; however, with a lower yield (45%) and longer reaction time than anomer 12b (entry 9). Hence, with regard to O-glycosylation, the best overall result in terms of yield, stereocontrol, and reproducibility was obtained by reacting donor 14a and acceptor 12b promoted by TMSOTf.
We then focused on the deprotection steps of 3,5,6-tri-O-benzyl-2-O-(2,3,4-tri-O-acetyl-β-D-xylopiranosyl)-D-glucofuranoside 18a and 18b or 19a and 19b (Scheme 4) with the aim of maximizing the efficiency of the synthetic strategy to obtain sambubiose.
O-Methoxy group deprotection in the disaccharides 18a and 18b were first investigated using strong acids such as hydrochloride acid (HCl, rt or −15 °C, 10 h) or BF3 . OEt2 (0 °C, 10 h). Unfortunately, demethylation was not obtained in any of these cases, and degradation of the starting material or no reaction was observed. Also treating disaccharides with trityl tetrafluoroborate [(C6H5)3C.BF4] [24] (rt or −15 °C, 4–12 h), usually employed in the deprotection of benzyl and methyl ethers on anomeric carbons, was found to be detrimental (data not shown).
Hence, 18b was also first debenzylated by hydrogenolysis (10% Pd/C, EtOAc/EtOH 1:2, 32%) leading to the derivative β-methyl 2-O-(2,3,4-tri-O-acetyl-β-D-xylopyranosyl)-D-glucofuranoside (20b) which, in turn, was deacetylated (NaOMe, MeOH, 7 h, 70%) to give β-methyl 2-O-β-D-xylopyranosyl-D-glucofuranoside (21b). Attempts to obtain sambubiose by deprotection of 20b and 21b by cleaving the methoxy group also failed (data not shown).
In the case of allyloxy disaccharides 19a and 19b, sambubiose might theoretically be obtained by two different deprotection routes (compound 19b is reported in Scheme 5 as an example), both of which were explored. The first, on the left in Scheme 5, envisaged O-deacetylation, O-deallylation, and, finally, O-debenzylation. On the other hand, the second route called for O-deallylation before O-deacetylation. The initial deprotection route involved O-deacetylation of compounds 19a or 19b. The compounds were treated using a typical condition for the hydrolysis of the acetyl esters with sodium methoxide and gave the corresponding desired compounds α- and β-allyl 3,5,6-tri-O-benzyl-2-O-β-D-xylopyranosyl-D-glucofuranoside in good yield (22a, 70%; 22b, 90%). Subsequently, the deprotection of the allyl group was investigated with 22a and 22b. Several methods were explored, including palladium chloride [25] (MeCOONa, MeCOOH, PdCl2, H2O, rt, 12 h, 42%), Wilkinson’s catalyst [26] [(Ph3P)3RhCl, toluene:MeOH, rt to reflux, 20 h, 39%] or hydrogen activated iridium complex [27] {[Ir(COD)(PCH3Ph2)2]PF6, H2, TsOH, CH2Cl2/MeOH, 19%}, but only β-anomer 22b reacted, probably due to the steric hindrance of α-one. Therefore, 22b was the only possible anomer suitable for the synthesis of 23 (3,5,6-tri-O-benzyl-2-O-β-D-xylopyranosyl-α-D-glucofuranose and 3,5,6-tri-O-benzyl-2-O-β-D-xylopyranosyl-β-D-glucofuranose) which was obtained as an anomeric mixture.
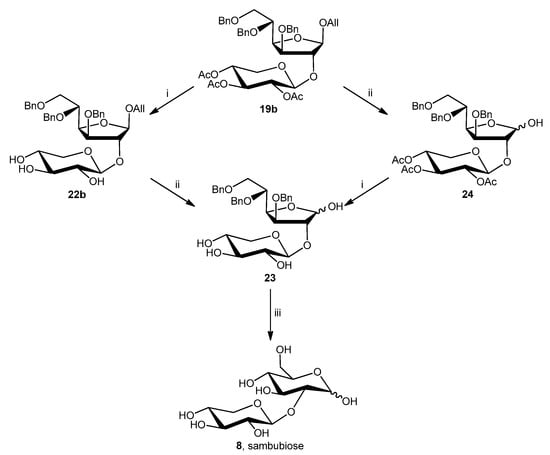
Scheme 5.
Reagents and conditions: (i) MeONa, MeOH, rt, 3 h, 90% (22b) or 38% (23); (ii) MeCOONa, MeCOOH, PdCl2, H2O, rt, 12 h, 42% (23) or 70% (24); (iii) H2, Pd/C 10%, EtOH, rt, 6 h, 51%.
On the other hand, using the second approach (on the right in Scheme 5), 19a and 19b were deallylated with palladium chloride or Wilkinson’s catalyst under the previously described conditions. Disaccharide 19b gave the corresponding product 3,5,6-tri-O-benzyl-2-O-(2,3,4-tri-O-acetyl-β-D-xylopiranosyl)-D-glucofuranose (24) in good yields with both methods. However, compound 19a was deprotected only by (Ph3P)3RhCl) in low yield (23%). Then, 24 was deacetylated using sodium methoxide and methanol to give 23. In conclusion, the deprotection scheme on the left used to obtain 23 was more effective in terms of overall yield than the one on the right (38% versus 27%). Finally, sambubiose was obtained by O-debenzylation of 23 which was treated with palladium under hydrogen atmosphere. During the last deprotection, the five-membered ring (glucofuranose) of compound 23 spontaneously transformed into a six-membered ring (glucopyranose).
The final four improved/developed steps are reported in Scheme 6.
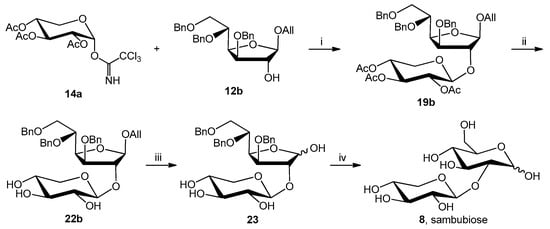
Scheme 6.
Reagents and conditions: (i) TMSOTf, CH2Cl2, –20 °C to rt, 3.5 h, 60%; (ii) MeONa, MeOH, rt, 3 h, 90%; (iii) MeCOONa, MeCOOH, PdCl2, H2O, rt, 12 h, 42%; (iv) H2, Pd/C 10%, EtOH, rt, 6 h, 51%.
Interestingly, sambubiose was obtained with a satisfactory overall yield (7%) starting from the acceptor precursor 1 compared to the 0.24% yield reported in the literature [10].
3. Materials and Methods
Chemicals, Materials, and Methods
All reagents were purchased from Sigma–Aldrich (St. Louis, MO, USA), Alfa Aesar (Haverhill, MA, USA), or TCI (Tokyo, Japan) in the highest quality commercially available. The solvents were RP grade. The melting points were determined using the Büchi (Flawil, Switzerland) B-540 apparatus. The purity of the products was evaluated unequivocally through MS, 1H NMR, 13C NMR, IR, and [α]20D. The MS (ESI) spectra were recorded with a Waters (Milford, MA, USA) Micromass ZQ spectrometer in a positive mode using a nebulizing nitrogen gas at 400 L/min and a temperature of 250 °C, cone flow 40 mL/min, capillary 3.5 K volts, and cone voltage 60 V; the revelation was performed either in ESI (+) and/or ESI (−), from 100 to 800 units of mass, spectrophotometrically measured using a diode array spectrophotometer. The 1H NMR and 13C NMR spectra were recorded on a Bruker (Billerica, MA, USA) AC 400 or 101 instrument and analyzed using the Top Spin 1.3 (2013) Bruker NMR software package. Chemical shifts were measured using the central peak of the solvent. Purification of the crude material was carried out by column chromatography on silica gel “flash” [230–400 mm, Merck (Darmstadt, Germany)]. The TLC analyses were performed on pre-coated aluminum oxide on aluminum sheets (60 F254, neutral; Merck). The IR spectra were obtained with a Thermo Fisher Scientific (Waltham, MA, USA) Nicolet Avatar 360 spectrophotometer. The optical rotations were measured on the Digital Perkin Elmer (Waltham, MA, USA) 241 polarimeter using a sodium lamp (589 nm) as a light source; concentrations were expressed in g/100 mL, and the length of the cell was 1 dm.
Synthesis of allyl 3,5,6-tri-O-benzyl-α-D-glucofuranoside (12a) and allyl 3,5,6-tri-O-benzyl-β-D-glucofuranoside (12b). To a solution of 2 (1 g, 2.04 mmol) [obtained by treating 1 (2 g, 9.08 mmol) with 80% NaH (0.9 g, 30 mmol), BnBr (4.66 g, 27.25 mmol) in DMF (40 mL)] at room temperature for 6 h in allyl alcohol (14.6 mL) and under stirring, TsOH (0.062 g, 0.33 mmol) was added. After the reactants were refluxed for 3 h, the mixture was cooled, extracted with CH2Cl2, washed with NaHCO3 sat., dried over Na2SO4, filtered and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 9:1) gave 12a and 12b as light yellow oils, which were dried under N2 atmosphere because of their thermolability (Rf: 12a 0.4, 12b 0.3; cyclohexane/EtOAc 6:4). Yield: 12a 40% (0.4 g, 0.82 mmol); 12b 45% (0.45 g, 0.92 mmol). 12a: MS (ESI): 490 (M + H+). 1H NMR (CDCl3) δ: 3.61 (dd, 1H, J6b–5 = 5.8 Hz, J6b–6a = 10.6 Hz, H6b), 3.78 (dd, 1H, J6a–5 = 2.0 Hz, J6a–6b = 10.6 Hz, H6a), 3.95 (ddd, 1H, J5–6a = 2.0 Hz, J5–6b = 5.8 Hz, J5–4 = 8.4 Hz, H5), 4.00 (dd, 1H, J3–2 = 1.9 Hz, J3–4 = 4.4 Hz, H3), 4.01 (dddd, 1H, J7b–9a = J7b–9b = 1.5 Hz, J7b–8 = 6.2 Hz, J7b–7a = 12.8 Hz, H7b OCHHCHCH2), 4.22 (dddd, 1H, J7a–9a = J7a–9b = 1.5 Hz, J7a–8 = 5.2 Hz, J7a–7b = 12.8 Hz, H7a OCHHCHCH2), 4.26 (dd, 1H, J2–3 = 1.9 Hz, J2–1 = 4.5 Hz, H2), 4.27 (dd, 1H, J4–3 = 4.4 Hz, J4–5 = 8.4 Hz, H4), 4.45 (d, 1H, J = 11.5 Hz, OCH2C6H5), 4.47 (d, 1H, J = 11.5 Hz, OCH2C6H5), 4.49 (d, 1H, J = 12.2 Hz, OCH2C6H5), 4.53 (d, 1H, J = 12.2 Hz, OCH2C6H5), 4.63 (d, 1H, J = 11.5 Hz, OCH2C6H5), 4.72 (d, 1H, J = 11.5 Hz, OCH2C6H5), 5.11 (d, 1H, J1–2 = 4.5 Hz, H1), 5.12 (dddd, 1H, J9b–9a = J9b–7a = J9b–7b = 1.5 Hz, J9b–8 = 10.2 Hz, H9b OCH2CHCHH), 5.19 (dddd, 1H, J9a–9b = J9a–7a = J9a–7b = 1.5 Hz, J9a–8 = 17.2 Hz, H9a OCH2CHCHH), 5.83 (dddd, 1H, J8–7a = 5.2 Hz, J8–7b = 6.2 Hz, J8–b = 10.2 Hz, J8–9a = 17.2 Hz, H8 OCH2CHCH2), 7.24–7.37 (m, 15H, ArH) ppm. 13C NMR (CDCl3) δ: 69.2, 71.2, 71.7, 72.6, 73.4, 76.1, 77.9, 83.9, 100.4, 117.7, 127.5, 127.5, 127.6, 128.1, 128.2, 128.3, 133.7, 137.9, 138.6, 138.9 ppm. IR (Nujol) ν = 3550, 1454, 1372, 1206, 1057 cm−1. [α]20D = –56.8 (c 0.3, CHCl3). 12b: MS (ESI): 490 (M + H+). 1H NMR (CDCl3) δ: 3.63 (dd, 1H, J6b–5 = 5.3 Hz, J6b–6a = 10.7 Hz, H6b), 3.80 (dd, 1H, J6a–5 = 2.0 Hz, J6a–6b = 10.7 Hz, H6a), 3.90 (dddd, 1H, J7b–9a = J7b–9b = 1.5 Hz, J7b–8 = 6.0 Hz, J7b–7a = 13.0 Hz, H7b OCHHCHCH2), 4.00 (ddd, 1H, J5–6a = 2.0 Hz, J5–6b = 5.3 Hz, J5–4 = 8.8 Hz, H5), 4.00 (dd, 1H, J3–2 = 1.0 Hz, J3–4 = 5.0 Hz, H3), 4.14 (dddd, 1H, J7a–9a = J7a–9b = 1.5 Hz, J7a–8 = 5.0 Hz, J7a–7b = 13.0 Hz, H7a OCHHCHCH2), 4.21 (dd, 1H, J2–1 ≅ J2–3 = 1.0 Hz, H2), 4.33 (dd, 1H, J4–3 = 5.0 Hz, J4–5 = 8.8 Hz, H4), 4.43 (d, 1H, J = 11.5 Hz, OCH2C6H5), 4.47 (d, 1H, J = 11.5 Hz, OCH2C6H5), 4.52 (s, 2H, OCH2C6H5), 4.57 (d, 1H, J = 12.2 Hz, OCH2C6H5), 4.67 (d, 1H, J = 11.5 Hz, OCH2C6H5), 4.87 (d, 1H, J1–2 = 1.0 Hz, H1), 5.10 (dddd, 1H, J9b–9a = J9b–7a = J9b–7b = 1.5 Hz, J9b–8 = 10.5 Hz, H9b OCH2CHCHH), 5.22 (dddd, 1H, J9a–9b = J9a–7a = J9a–7b = 1.5 Hz, J9a–8 = 17.0 Hz, H9a OCH2CHCHH), 5.83 (dddd, 1H, J8–7a = 5.0 Hz, J8–7b = 6.0 Hz, J8–9b = 10.5 Hz, J8–9a = 17.0 Hz, H8 OCH2CHCH2), 7.24–7.37 (m, 15H, ArH) ppm. 13C NMR (CDCl3) δ: 68.9, 70.8, 72.0, 72.5, 73.4, 76.7, 77.0, 77.3, 77.3, 78.6, 79.9, 83.0, 107.8, 117.0, 127.6, 127.7, 128.2, 128.3, 128.3, 134.2, 134.3, 138.0, 138.6, 138.9 ppm. IR (Nujol) ν = 3423, 1462, 1398, 1061 cm−1. [α]20D = +19.5 (c 0.2, CHCl3). The physico-chemical data are in agreement with those reported in the literature [28].
Synthesis of 1,2,3,4-tetra-O-acetyl-α-D-xylopyranose (16a). To a solution of 15 (1 g, 6.66 mmol) in pyridine (5 g, 4.5 mL, 62 mmol), Ac2O was added (5.5 g, 4.5 mL, 52.74 mmol). The mixture was stirred at 0 °C for 6 h, extracted with Et2O and washed with H2O and CuSO4 saturated solution. The combined organic layers were dried with Na2SO4, filtered and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 9:1) gave 16a as a yellow oil which was dried under N2 atmosphere because of its thermolability. Yield: 75% (1.59 g, 5 mmol). MS (ESI): 336 (M + NH4+). 1H NMR (CDCl3) δ: 2.04–2.19 (m, 12H, CH3CO), 3.72 (dd, 1H, J5b–4 = J5b–5a = 11.0 Hz, H5b), 3.95 (dd, 1H, J5a–4 = 6.0 Hz, J5a–5b = 11.0 Hz, H5a), 5.04 (dd, 1H, J2–1 = 3.5 Hz, J2–3 = 10.0 Hz, H2), 5.05 (ddd, 1H, J4–5a = 6.0 Hz, J4–3 = 10.0 Hz, J4–5b = 11.0 Hz, H4), 5.48 (dd, 1H, J3–2 = J3–4 = 10.0 Hz, H3), 6.27 (d, 1H, J1–2 = 3.5 Hz, H1) ppm. 13C NMR (CDCl3) δ: 20.4, 20.6, 20.7, 20.8, 60.6, 68.7, 69.3, 89.2, 168.9, 169.6, 169.7, 170.1 ppm. IR (film) ν = 1766, 1232, 1128 cm−1. [α]20D = +88.0 (c 0.3, CHCl3). 1H NMR, 13C NMR, and IR data are in agreement with those reported in the literature [21].
Synthesis of 2,3,4-tri-O-acetyl-D-xylopyranose (17). To a solution of 16a (1 g, 3.14 mmol) in dry DMF (9 mL), AcONH4 (0.485 g, 6.29 mmol) was added and the mixture was stirred at room temperature for 22 h, extracted with EtOAc, and washed with H2O and saturated aq. NH4Cl solution. The combined organic layers were dried with Na2SO4 and concentrated. Purification of the oily residue by column chromatography (cyclohexane/EtOAc 7:3) gave 17 as a white solid. Yield 70% (0.607 g, 2.2 mmol). Mp: 142–144 °C (EtOAc/petroleum ether). MS (ESI): 299 (M + Na+). [α]20D = +69.0 (c 0.2, CHCl3). 1H NMR, 13C NMR, and IR data are in agreement with those reported in the literature [29].
Synthesis of 2,3,4-tri-O-acetyl-D-xylopyranosyl-α-trichloroacetimidate (14a). To a solution of 17 (0.407 g, 1.48 mmol) and CCl3CN (1.25 g, 0.86 mL, 8.83 mmol) in dry CH2Cl2 (14 mL) at 0 °C and under N2 atmosphere, DBU was added (0.09 g, 0.88 mL, 0.59 mmol) and the mixture was stirred at 0 °C for 1 h and then concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 9:1) gave 14a as a yellow oil (Rf: 0.5, cyclohexane/EtOAc 7:3). Yield: 60% (0.37 g, 0.86 mmol). MS (ESI): 437 (M + NH4+). 1H NMR (CDCl3) δ: 2.03 (s, 3H, CH3CO), 2.06 (s, 3H, CH3CO), 2.07 (s, 3H, CH3CO), 3.82 (dd, 1H, J5a–4 = J5a–5b = 11.0 Hz, H5a), 4.00 (dd, 1H, J5b–4 = 5.9 Hz, J5b–5a = 11.0 Hz, H5b), 5.08 (dd, 1H, J2–1 = 3.6 Hz, J2–3 = 10.0 Hz, H2), 5.11 (ddd, 1H, J4–5b = 5.9 Hz, J4–5a = 7.5 Hz, J4–3 = 11.0 Hz, H4), 5.58 (dd, 1H, J3–2 = J3–4 = 10.0 Hz, H3), 6.50 (d, 1H, J1–2 = 3.6 Hz, H1), 8.67 [s, 1H, C(NH)CCl3] ppm. 13C NMR (CDCl3) δ: 20.5, 20.6, 20.7, 60.8, 68.6, 69.3, 69.9, 76.7, 77.0, 77.3, 93.1, 160.9, 169.8 ppm. IR (film): ν = 3481, 2500, 1757, 1487, 1218, 1046 cm−1. [α]20D = +45.2 (c 0.2, CHCl3). The physico-chemical data are in agreement with those reported in the literature [22].
Synthesis of α-allyl 3,5,6-tri-O-benzyl-(2,3,4-tri-O-acetyl-2-O-β-D-xylopyranosyl)-D-glucofuranoside (19a). A mixture of 12a (0.35 g, 0.71 mmol) and 14a (0.358 g, 0.86 mmol) in dry CH2Cl2 (8.75 mL) under N2 atmosphere and in the presence of activated 4Å molecular sieves (0.8 g) was stirred for 30 min at room temperature, cooled at −20 °C and TMSOTf (6 mg, 5 μL, 0.03 mmol) was then added. The mixture was stirred again at room temperature for 6 h, quenched with Et3N, filtered on Celite®, and concentrated. Purification of the residue by column chromatography (petroleum ether/Et2O 6:4) gave 19a as a colorless oil (Rf 0.4, petroleum ether/Et2O 1:1). Yield: 45% (0.24 g, 0.32 mmol). MS (ESI): 766 (M + NH4+). 1H NMR (CDCl3) δ: 1.99 (s, 3H, CH3CO), 2.03 (s, 3H, CH3CO), 2.08 (s, 3H, CH3CO), 3.37 (dd, 1H, J5’b–4’ = 7.0 Hz, J5’b–5’a = 12.2 Hz, H5’b), 3.71 (dd, 1H, J6b–5 = 6.1 Hz, J6b–6a = 10.5 Hz, H6b) 3.84 (dd, 1H, J6a–5 = 2.4 Hz, J6a–6b = 10.5 Hz, H6a), 3.97–4.05 (m, 3H, H2, H5, H7b), 4.18–4.23 (m, 1H, H7a), 4.22 (dd, 1H, J5’a–4’ = 4.5 Hz, J5’a–5’b = 12.2 Hz, H5’a), 4.28 (dd, 1H, J3–2 = 5.0 Hz, J3–4 = 6.5 Hz, H3), 4.33 (dd, 1H, J4–3 ≅ J4–5 = 6.5 Hz, H4), 4.51–4.58 (m, 5H, OCH2C6H5), 4.63 (d, 1H, J1’–2’ = 5.9 Hz, H1’) 4.80 (d, 1H, J = 11.6 Hz, OCH2C6H5), 4.93 (ddd, 1H, J4’–5’a = 4.5 Hz, J4’–3’ ≅ J4’–5’b = 7.0 Hz, H4’), 4.99 (dd, 1H, J2’–1’ = 5.9 Hz, J2’–3’ = 8.0 Hz, H2’), 5.02 (d, 1H, J1–2 = 4.3 Hz, H1), 5.12 (dd, 1H, J3’–4’ = 7.0 Hz, J3’–2’ = 8.0 Hz, H3’), 5.16 (dddd, 1H, J9b–9a = J9b–7a = J9b–7b = 1.6 Hz, J9b–8 = 10.5 Hz, H9b OCH2CHCHH), 5.30 (dddd, 1H, J9a–9b = J9a–7a = J9a–7b = 1.6 Hz, J9a–8 = 17.2 Hz, H9a OCH2CHCHH), 5.89 (dddd, 1H, J8–7a = 5.1 Hz, J8–7b = 6.1 Hz, J8–9b = 10.5 Hz, J8–9a = 17.2 Hz, H8 OCH2CHCH2), 7.26–7.33 (m, 15 H, ArH) ppm. 13C NMR (CDCl3) δ: 20.6, 20.7, 20.8, 61.7, 68.6, 68.8, 70.1, 70.6, 72.5, 72.6, 73.3, 76.0, 76.7, 76.8, 81.0, 84.9, 99.5, 100.7, 117.0, 122.4, 127.5, 127.6, 128.2, 128.4, 134.2, 137.8, 138.6, 138.8, 169.1, 169.9, 170.0 ppm. IR (CHCl3): ν = 3064, 1756, 1454, 1372, 1221, 1057 cm−1. [α]20D = +17.6 (c = 0.2, MeOH).
Synthesis of β-allyl 3,5,6-tri-O-benzyl-(2,3,4-tri-O-acetyl-2-O-β-D-xylopyranosyl)-D-glucofuranoside (19b). A mixture of 12b (0.35 g, 0.71 mmol) and 14a (0.358 g, 0.86 mmol) in dry CH2Cl2 (8.75 mL) under N2 atmosphere and in the presence of activated 4Å molecular sieves (0.8 g) was stirred for 30 min at room temperature, cooled at −20 °C and TMSOTf (6 mg, 5 μL, 0.03 mmol) was then added. The mixture was stirred again at room temperature for 3.5 h, quenched with Et3N, filtered on Celite®, and concentrated. Purification of the residue by column chromatography (petroleum ether/Et2O 6:4) gave 19b as a colorless oil (Rf 0.4, petroleum ether/Et2O 1:1). Yield: 60% (0.321 g, 0.43 mmol). MS (ESI): 766 (M + NH4+). 1H NMR (CDCl3) δ: 1.97 (s, 3H, CH3CO), 2.02 (s, 3H, CH3CO), 2.06 (s, 3H, CH3CO), 3.23 (dd, 1H, J5’b–4’ = 9.2 Hz, J5’b–5’a = 12.0 Hz, H5’b), 3.68 (dd, 1H, J6b–5 = 5.2 Hz, J6b–6a = 10.5 Hz, H6b), 3.88 (dd, 1H, J6a–5 = 2.0 Hz, J6a–6b = 10.5 Hz, H6a), 3.94 (dd, 1H, J3–2 = 1.2 Hz, J3–4 = 5.0 Hz, H3), 3.97 (dddd, 1H, J7b–9a = J7b–9b = 1.5 Hz, J7b–8 = 6.0 Hz, J7b–7a = 13.0 Hz, H7b, OCHHCHCH2), 4.06 (dd, 1H, J5’a–4’ = 5.2 Hz, J5’a–5’b = 12.0 Hz, H5’a), 4.06 (ddd, 1H, J5–6a = 2.0 Hz, J5–6b = 5.2, J5–4 = 9.5 Hz, H5), 4.14 (dd, 1H, J2–1 = Hz, J2–3 = 1.2 Hz, H2), 4.19 (dd, 1H, J4–3 = 5.0 Hz, J4–5 = 9.5 Hz, H4), 4.19 (dddd, 1H, J7a–9a = J7a–9b = 1.5 Hz, J7a–8 = 5.0, J7a–7b = 13.0 Hz, H7a OCHHCHCH2), 4.24 (d, 1H, J1’–2’ = 7.2 Hz, H1’), 4.52 (d, 1H, J = 11.2 Hz, OCH2C6H5), 4.53 (d, 1H, J = 12.0 Hz, OCH2C6H5), 4.59 (s, 2H, OCH2C6H5), 4.60 (d, 1H, J = 12.0 Hz, OCH2C6H5), 4.79 (d, 1H, J = 11.2 Hz, OCH2C6H5), 4.83 (dd, 1H, J2’–1’ = 7.2 Hz, J2’–3’ = 8.5 Hz, H2’), 4.91 (ddd, 1H, J4’–5’a = 5.2 Hz, J4’–3’ = 8.5 Hz, J4’–5’b = 9.2 Hz, H4’), 5.01 (d, 1H, J1–2 = 1.2 Hz, H1), 5.08 (dd, 1H, J3’–2’ = J3’–4’ = 8.5 Hz, H3’), 5.18 (dddd, 1H, J9b–9a = J9b–9a = J9b–7b = 1.5 Hz, J9b–8 = 10.5 Hz, H9b OCH2CHCHH), 5.28 (dddd, 1H, J9a–9b = J9a–7a = J9a–7b = 1.5 Hz, J9a–8 = 17.0 Hz, H9a OCH2CHCHH), 5.89 (dddd, 1H, J8–7a = 5.0 Hz, J8–7b = 6.0 Hz, J8–9b = 10.5 Hz, J8–9a = 17.0 Hz, H8 OCH2CHCH2), 7.25–7.35 (m, 15 H, ArH) ppm. 13C NMR (MeOD) δ: 19.1, 19.1, 19.3, 61.9, 68.5, 68.8, 69.8, 70.9, 71.8, 71.8, 71.8, 73.0, 76.4, 79.9, 80.4, 84.6, 100.1, 106.6, 115.7, 117.1, 127.2, 127.4, 127.4, 127.5, 127.8, 127.9, 127.9, 128.1, 134.2, 137.9, 138.4, 138.7, 169.6, 170.0, 170.2 ppm. IR (CHCl3): ν = 3064, 1756, 1454, 1372, 1221, 1057 cm−1. [α]20D = −12.8 (c 0.2, MeOH).
Synthesis of β-allyl 3,5,6-tri-O-benzyl-2-O-β-D-xylopyranosyl-D-glucofuranoside (22b). To a solution of 19b (0.21 g, 0.29 mmol) in dry MeOH (62 mL) MeONa was added (0.038 g, 0.70 mmol) and the mixture was stirred at room temperature for 3 h, then concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 1:9) gave 22b as a pale-yellow oil (Rf: 0.3, EtOAc). Yield: 90% (0.161 g, 0.26 mmol). MS (ESI): 605 (M + H+). 1H NMR (CDCl3) δ: 2.51 (br s, 1H, OH), 2.77 (br s, 1H, OH), 2.94 (br s, 1H, OH), 3.19 (dd, 1H, J5’b-4’ = 9.2, J5’b-5’a = 12.0 Hz, H5’b), 3.29 (dd, 1H, J2’-1’ = 6.5 Hz, J2’-3’ = 8.0 Hz, H2’), 3.43 (dd, 1H, J3’-2’ = J3’-4’ = 8.0 Hz, H3’), 3.67 (ddd, J4’-5’a = 4.8 Hz, J4’-3’ = 8.0 Hz, J4’-5’b = 9.2 Hz, H4’), 3.70 (dd, 1H, J6b-5 = 4.5 Hz, J6b–6a = 10.8 Hz, H6b), 3.88 (dd, 1H, J6a–5 = 2.0 Hz, J6a–6b = 10.8 Hz, H6a), 3.95 (dd, 1H, J5’a–4’ = 4.8 Hz, J5’a–5’b = 12.0 Hz, H5’a), 3.98 (dddd, 1H, J7b–9a = J7b–9b = 1.5 Hz, J7b–8 = 6.0 Hz, J7b–7a = 13.0 Hz, H7b OCHHCHCH2), 4.04 (dd, 1H, J3–2 = 1.0 Hz, J3–4 = 4.8 Hz, H3), 4.06 (ddd, 1H, J5–6a = 2.0 Hz, J5–6b = 4.5 Hz, J5–4 = 9.2 Hz, H5), 4.08 (d, 1H, J1’–2’ = 6.5 Hz, H1’), 4.19 (dddd, 1H, J7a–9a = J7a–9b = 1.5 Hz, J7a–8 = 5.0 Hz, J7a–7b = 13.0 Hz, H7a OCHHCHCH2), 4.23 (dd, 1H, J2–1 = J2–3 = 1.0 Hz, H2), 4.35 (dd, 1H, J4–3 = 4.8 Hz, J4–5 = 9.2 Hz, H4), 4.51 (d, 1H, J = 11.2 Hz, OCH2C6H5), 4.56 (d, 1H, J = 12.5 Hz, OCH2C6H5), 4.58 (d, 1H, J = 12.5 Hz, OCH2C6H5), 4.61 (d, 1H, J = 12.5 Hz, OCH2C6H5), 4.62 (d, 1H, J = 12.5 Hz, OCH2C6H5), 4.75 (d, 1H, J = 11.2 Hz, OCH2C6H5), 5.06 (d, 1H, J1-2 = 1.0 Hz, H1), 5.17 (dddd, 1H, J9b–9a = J9b–7a = J9b–7b = 1.6 Hz, J9b–8 = 10.5 Hz, H9b OCH2CHCHH), 5.28 (dddd, 1H, J9a–9b = J9a–7a = J9a–7b = 1.6 Hz, J9a–8 = 17.0 Hz, H9a OCH2CHCHH), 5.90 (dddd, 1H, J8–7a = 5.0 Hz, J8–7b = 6.0 Hz, J8–9b = 10.5 Hz, J8–9a = 17.0 Hz, H8 OCH2CHCH2), 7.28–7.37 (m, 15H, ArH) ppm. 13C NMR (CDCl3) δ: 64.7, 68.9, 69.4, 70.3, 72.4, 72.4, 72.4, 72.6, 73.4, 75.1, 79.9, 80.5, 84.6, 101.9, 106.6, 117.0, 127.5, 127.6, 127.7, 127.7, 127.9, 128.0, 128.3, 128.3, 128.4, 134.1, 137.7, 138.7, 138.7 ppm. IR (CHCl3): ν = 3415, 3019, 1785, 1450, 1368, 1045 cm−1. [α]20D = –19.6 (c = 0.2, MeOH).
Synthesis of 3,5,6-tri-O-benzyl-2-O-β-D-xylopyranosyl-α-D-glucofuranoside and 3,5,6-tri-O-benzyl-2-O-β-D-xylopyranosyl-β-D-glucofuranoside (23). To a solution of 22b (0.07 g, 0.12 mmol) in MeCOONa (0.034 g, 0.42 mmol) and MeCOOH (0.22 mL), PdCl2 (0.04 g, 0.22 mmol), and H2O (11 μL) were added. The mixture was stirred at room temperature for 12 h, filtered on Celite®, and washed with EtOAc. The filtrate was washed with a saturated NaHCO3 solution and extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. Purification of the residue by column chromatography (EtOAc/cyclohexane 8:2) gave 23 (α/β 2:3) as a colorless oil (Rf: 23a 0.2, 23b 0.3; EtOAc/MeOH 99:1). Yield: 42% (0.027 g, 0.05 mmol). MS (ESI): 600 (M + NH4+). 1H NMR (MeOD) δ: 3.11–3.21 (m, 3H), 3.23–3.30 (m, 3H), 3.44–3.54 (m, 3H), 3.66–3.73 (m, 2H), 3.82–3.91 (m, 3H), 3.93–3.98 (m, 2H), 4.08–4.11 (m, 1H), 4.11–4.14 (4H), 4.19 (dd, 1H, J1 = 2.6 Hz, J2 = 4.0 Hz), 4.25 (dd, 1H, J1 = 2.5 Hz, J2 = 4.6 Hz), 4.28 (d, 1H, J = 4.0 Hz), 4.30 (dd, 1H, J1 = J2 = 2.1 Hz), 4.38 (dd, 1H, J1 = 4.6 Hz, J2 = 7.9 Hz), 4.50–4.77 (m, 10H, OCH2C6H5), 5.25 (d, 1H, J = 1.0 Hz, H1 β-anomer), 5.39 (d, 1H, J = 4.0 Hz, H1 α-anomer), 7.26–7.38 (m, 30H, ArH) ppm. 13C NMR (MeOD) δ: 65.6, 65.6, 69.7, 69.7, 70.2, 70.2, 71.8, 71.9, 71.9, 71.9, 72.9, 73.0, 73.0, 73.3, 75.9, 76.4, 76.5, 76.9, 79.8, 80.7, 80.8, 81.6, 85.0, 85.0, 96.5, 102.3, 102.8, 103.0, 127.1, 127.1, 127.2, 127.3, 127.3, 127.4, 127.4, 127.4, 127.5, 127.7, 127.8, 127.8, 127.9, 127.9, 128.0, 128.0, 128.1, 128.1, 137.9, 138.0, 138.4, 138.4, 138.8, 138.8 ppm. IR (Nujol): ν = 3399, 1458, 1376, 1041 cm−1. [α]20D = −32.6 (c = 0.3, MeOH).
Synthesis of 2-O-β-D-xylopyranosyl-D-α-glucopyranose and 2-O-β-D-xylopyranosyl-D-β-glucopyranose (8, sambubiose). To a solution of 23 (0.09 g, 0.15 mmol) in EtOH (9 mL), Pd/C 10% (0.018 g) was added and the mixture was hydrogenated under stirring at room temperature at 1 atm for 6 h, then filtered on Celite®, washed with EtOH, and kept overnight. The crystals obtained were then filtered and triturated to obtain 8 (sambubiose) as a white solid (Rf: 0.2, CH3CN/H2O 9:1). Yield 51% (0.024 g, 0.08 mmol). Mp: 132–134 °C (petroleum ether). MS (ESI): 313 (M + 1), 311 (M-1). 1H NMR (DMSO-d6) δ: 3.02–3.05 (m, 2H, H5, H2’), 3.08–3.17 (m, 3H, H2, H3’, H5’a), 3.23–3.28 (m, 1H, H4’), 3.41–3.47 (m, 1H, H6a), 3.54–3.68 (m, 4H, H3, H4, H6b, H5’b), 4.25 (d, 1H, J1’–2’ = 7.2 Hz, H1’), 4.39 (dd, 1H, JOH6–6a ≅ JOH6–6b = 5.5 Hz, OH6), 4.77 (d, 1H, J = 3.2 Hz, OH), 4.93 (d, 1H, J = 5.6 Hz, OH), 4.98 (d, 1H, J = 5.2 Hz, OH4’), 5.00 (d, 1 H, J = 4.8 Hz, OH), 5.02–5.05 (m, 2H, H1, OH) 6.34 (d, 1H, J = 4.5 Hz, OH1) ppm. 13C NMR (DMSO-d6) δ: 61.5 (C6), 66.1 (C5’), 69.9 (C4’), 70.5 (C5), 72.1 (C3), 72.4 (C4), 74.2 (C2’), 76.7 (C3’), 82.7 (C2), 92.0 (C1), 106.4 (C1’). α-anomer 13C NMR (D2O) δ: 60.4 (C6), 65.0 (C5’), 69.2 (C4’), 69.4 (C5), 71.0 (C3), 71.7 (C4), 73.1 (C2’), 75.5 (C3’), 80.7 (C2), 91.7 (C1), 104.7 (C1’). β-anomer 13C NMR (D2O) δ: 60.6 (C6), 65.1 (C5’), 69.1 (C4’), 69.4 (C5), 73.4 (C2’), 75.5, 75.69, 75.64, 81.8 (C2), 94.6 (C1), 103.6 (C1’). IR (Nujol): ν = 3400, 2840, 1070 cm−1. [α]20D = −5.0 (c = 0.03, MeOH). [10]
4. Conclusions
An efficient method for the total synthesis of natural disaccharide sambubiose was developed. Our investigation enabled us to identify the best combination of donor and acceptor glycosides as well as a catalyst that allowed a complete stereocontrol of the key O-glycosylation step. The protocol described herein was carried out using stable, safe, non-toxic agents and a step-by-step economical synthetic route. At the same time, a satisfactory overall yield and the characterization of sambubiose were achieved, thus overcoming the drawbacks associated with the current method reported in the literature [10].
Author Contributions
Conceptualization, S.L. and A.D.; methodology, S.L. and A.D.; validation, S.L. and A.D.; formal analysis, S.L. and A.D.; investigation, M.G.C. and P.M.; resources, S.L. and A.D.; data curation, S.L. and A.D.; writing—original draft preparation, S.L. and A.D.; writing—review and editing, M.G.C. and P.M.; visualization, S.L. and A.D.; supervision, S.L. and A.D.; project administration, S.L. and A.D.; funding acquisition, S.L. and A.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the University of Urbino Carlo Bo.
Acknowledgments
The authors wish to thank Giovanni Piersanti for his helpful suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Landis-Piwowar, K.R.; Iyer, N.R. Cancer chemoprevention: Current state of the art. Cancer Growth Metastasis 2014, 7, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Gentili, B.; Horowitz, R.M. Flavonoids of citrus. IX. Some new C-glycosylflavones and nuclear magnetic resonance method for differentiating 6- and 8-C-glycosyl isomers. J. Org. Chem. 1968, 33, 1571–1577. [Google Scholar] [CrossRef]
- Gil, M.; Ferreres, F.; Tomás-Barberán, F.A. Effect of modified atmosphere packaging on the flavonoids and vitamin C content of minimally processed Swiss chard (Beta vulgaris subspecies cycla). J. Agric. Food Chem. 1998, 46, 2007–2012. [Google Scholar] [CrossRef]
- Sato, F.; Matsukawa, Y.; Matsumoto, K.; Nishino, H.; Sakai, T. Apigenin induces morphological differentiation and G2-M arrest in rat neuronal cells. Biochem. Biophys. Res. Commun. 1994, 204, 578–584. [Google Scholar] [CrossRef]
- Reichel, L.; Reichwald, W. Ueber die farbstoffe der schawarzen holunderbeere. Naturwissenschaften 1960, 47, 40–41. [Google Scholar] [CrossRef]
- Olejnik, A.; Kowalska, K.; Olkowicz, M.; Rychlik, J.; Juzwa, W.; Myszka, K.; Dembczynski, R.; Bialas, W. Anti-inflammatory effects of gastrointestinal digested Sambucus nigra L. fruit extract analysed in co-cultured intestinal epithelial cells and lipopolysaccharide-stimulated macrophages. J. Funct. Foods 2015, 19, 649–660. [Google Scholar] [CrossRef]
- Loizzo, M.R.; Pugliese, A.; Bonesi, M.; Tenuta, M.C.; Menichini, F.; Xiao, J.; Tundis, R. Edible flowers: A rich source of phytochemicals with antioxidant and hypoglycemic properties. J. Agric. Food Chem. 2016, 64, 2467–2474. [Google Scholar] [CrossRef]
- Wu, X.; Pittman, H.E., III; Prior, R.L. Fate of anthocyanins and antioxidant capacity in contents of the gastrointestinal tract of weanling pigs following black raspberry consumption. J. Agric. Food Chem. 2006, 54, 583–589. [Google Scholar] [CrossRef]
- Socaciu, C. Food Colorants. Chemical and Functional Properties, 1st ed.; CRC Press: Boca Raton, FL, USA, 2007; p. 648. [Google Scholar]
- Erbing, B.; Lindberg, B. Synthesis of sambubiose. Acta Chem. Scand. 1969, 23, 2213–2215. [Google Scholar] [CrossRef][Green Version]
- Dick, W.R., Jr.; Hodge, J.E.; Inglett, G.E. Structure-taste relationships in (1→2)-linked disaccharides. Carbohydr. Res. 1974, 36, 319–329. [Google Scholar] [CrossRef]
- Helferich, B.; Ost, W. Synthese einiger β-d-xylopyranoside. Chem. Ber. 1962, 95, 2612–2615. [Google Scholar] [CrossRef]
- Lucarini, S.; Fagioli, L.; Campana, R.; Cole, H.; Duranti, A.; Baffone, W.; Vllasaliu, D.; Casettari, L. Unsaturated fatty acids lactose esters: Cytotoxicity, permeability enhancement and antimicrobial activity. Eur. J. Pharm. Biopharm. 2016, 107, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Perinelli, D.R.; Lucarini, S.; Fagioli, L.; Campana, R.; Vllasaliu, D.; Duranti, A.; Casettari, L. Lactose oleate as new biocompatible surfactant for pharmaceutical applications. Eur. J. Pharm. Biopharm. 2018, 124, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lucarini, S.; Fagioli, L.; Cavanagh, R.; Liang, W.; Perinelli, D.; Campana, M.; Stolnik, S.; Lam, J.; Casettari, L.; Duranti, A. Synthesis, structure–activity relationships and in vitro toxicity profile of lactose-based fatty acid monoesters as possible drug permeability enhancers. Pharmaceutics 2018, 10, 81. [Google Scholar] [CrossRef]
- Campana, R.; Merli, A.; Verboni, M.; Biondo, F.; Favi, G.; Duranti, A.; Lucarini, S. Synthesis and evaluation of saccharide-based aliphatic and aromatic esters as antimicrobial and antibiofilm agents. Pharmaceuticals 2019, 12, 186. [Google Scholar] [CrossRef]
- Das, S.N.; Chowdhury, A.; Tripathi, N.; Jana, P.K.; Mandal, S.B. Exploitation of in situ generated sugar-based olefin keto-nitrones: Synthesis of carbocycles, heterocycles, and nucleoside derivatives. J. Org. Chem. 2015, 80, 1136–1148. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Murai, Y.; Shoda, S. An efficient method for glucosylation of hydroxy compounds using glucopyranosyl fluoride. Chem. Lett. 1981, 10, 431–432. [Google Scholar] [CrossRef]
- Schmidt, R.R.; Kinzy, W. Anomeric-oxygen activation for glycoside synthesis: The trichloroacetimidate method. Adv. Carbohydr. Chem. Biochem. 1994, 50, 21. [Google Scholar]
- Hayashi, M.; Hashimoto, S.-I.; Noyori, R. Simple synthesis of glycosyl fluorides. Chem. Lett. 1984, 13, 1747–1750. [Google Scholar] [CrossRef]
- Kam, B.L.; Barascut, J.-L.; Imbach, J.-L. A general method of synthesis and isolation, and an NMR-spectroscopic study, of tetra-O-acetyl-d-aldopentofuranoses. Carbohydr. Res. 1979, 69, 135–142. [Google Scholar] [CrossRef]
- Mori, M.; Ito, Y.; Ogawa, T. Total synthesis of the mollu-series glycosyl ceramides α-d-Manp-(1→3)-β-d-Manp-(1→4)-β-d-Glcp-(1→1)-Cer and α-d-Manp-(1→3)-[β-d-Xylp-(1→2)]-β-d-Manp-(1→4)-β-d-Glcp-(1→1)-Cer. Carbohydr. Res. 1990, 195, 199–224. [Google Scholar] [CrossRef]
- Chang, C.-W.T.; Hui, Y.; Elchert, B.; Wang, J.; Li, J.; Ra, R. Pyranmycins, a novel class of aminoglycosides with improved acid stability: The SAR of d-pyranoses on ring III of pyranmycin. Org. Lett. 2002, 26, 4603–4606. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Doddi, V.R.; Vankar, Y.D. Mild and efficient chemoselective deprotection of anomeric O-methyl glycosides with trityl tetrafluoroborate. J. Org. Chem. 2008, 73, 5993–5995. [Google Scholar] [CrossRef] [PubMed]
- Hohgardt, H.; Dietrich, W.; Kühne, H.; Müller, D.; Grzelak, D.; Welzel, P. Synthesis of two structural analogues of the smallest antibiotically active degradation product of moenomycin a. Tetrahedron 2008, 44, 5771–5790. [Google Scholar] [CrossRef]
- Maranduba, A.; Veyrieres, A. Glycosylation of lactose. Synthesis of methyl O-(2-acetamido-2-deoxy-β-d-glucopyranosyl)-(1→3)-O-β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside and methyl O-β-d-galactopyranosyl-(1→4)-O-(2-acetamido-2-deoxy-β-d-glucopyranosyl)-(1→3)-O-β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside. Carbohydr. Res. 1985, 135, 330–336. [Google Scholar]
- Boonyarattanakalin, S.; Michieletti, M.; Lepenies, B.; Seeberger, P.H. Chemical synthesis of all phosphatidylinositol mannoside (PIM) glycans from mycobacterium tuberculosis. J. Am. Chem. Soc. 2008, 130, 16791–16799. [Google Scholar] [CrossRef]
- Listkowski, A.; Ing, P.; Cheaib, R.; Chambert, S.; Doutheau, A.; Queneau, Y. Carboxymethylglycoside lactones (CMGLs): Structural variations on the carbohydrate moiety. Tetrahedron: Asymmetry 2007, 18, 2201–2210. [Google Scholar] [CrossRef]
- Yan, Y.-L.; Guo, J.-R.; Liang, C.-F. Sequential Dy(OTf)3-catalyzed solvent-free per-O-acetylation and regioselective anomeric de-O-acetylation of carbohydrates. Chem. Asian J. 2017, 12, 2471–2479. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).