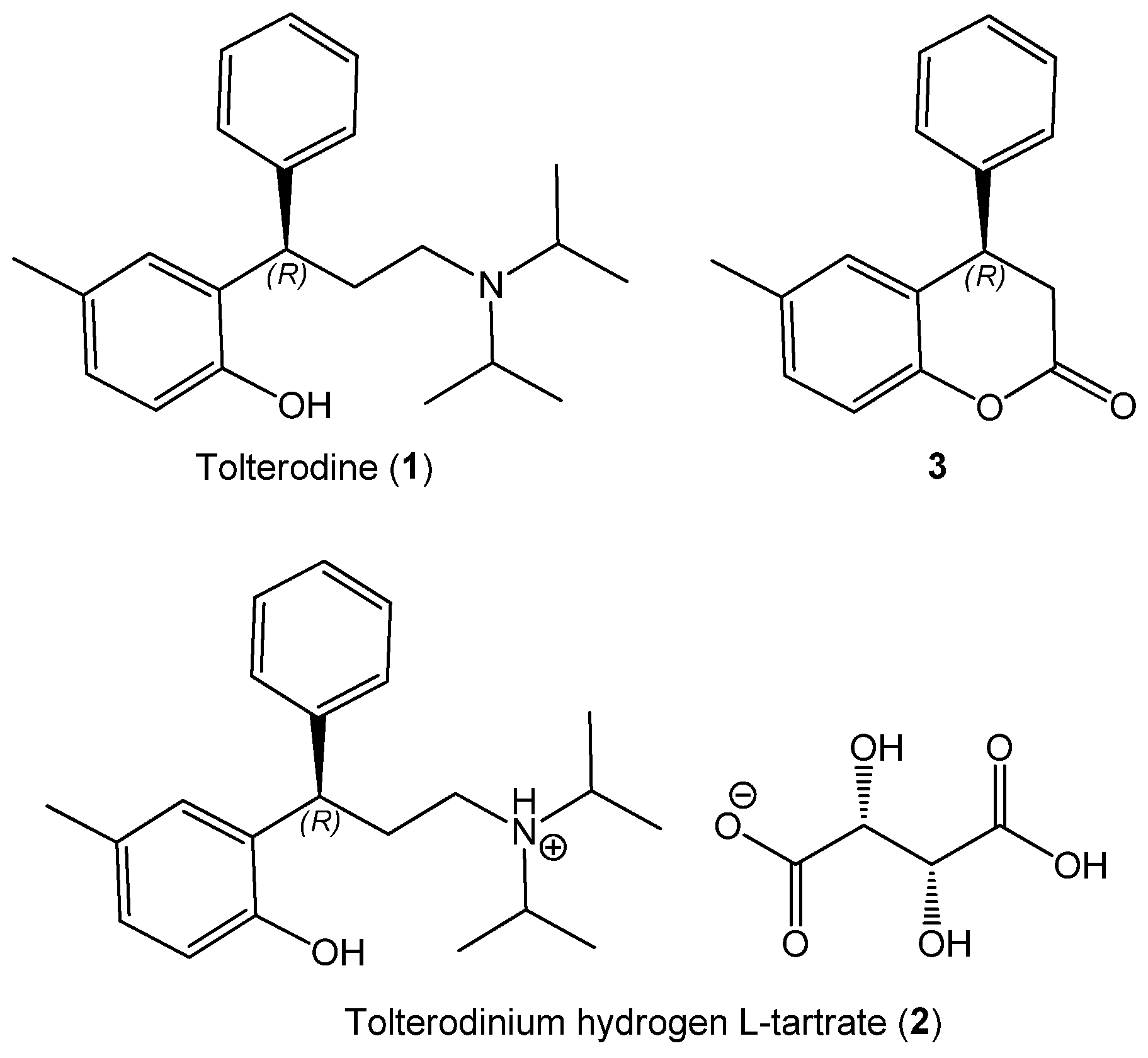
On the Absolute Stereochemistry of Tolterodine: A Circular Dichroism Study


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
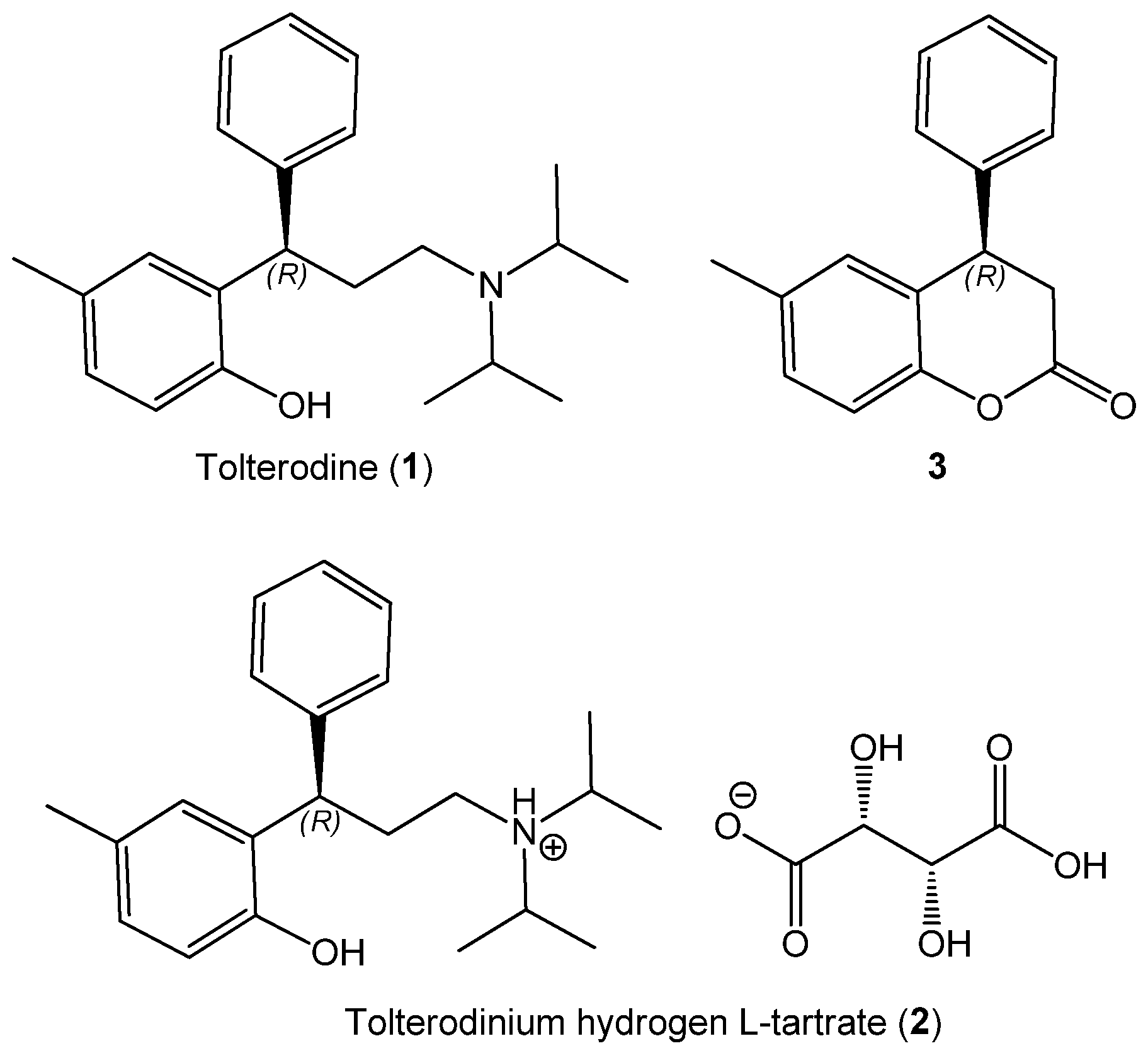
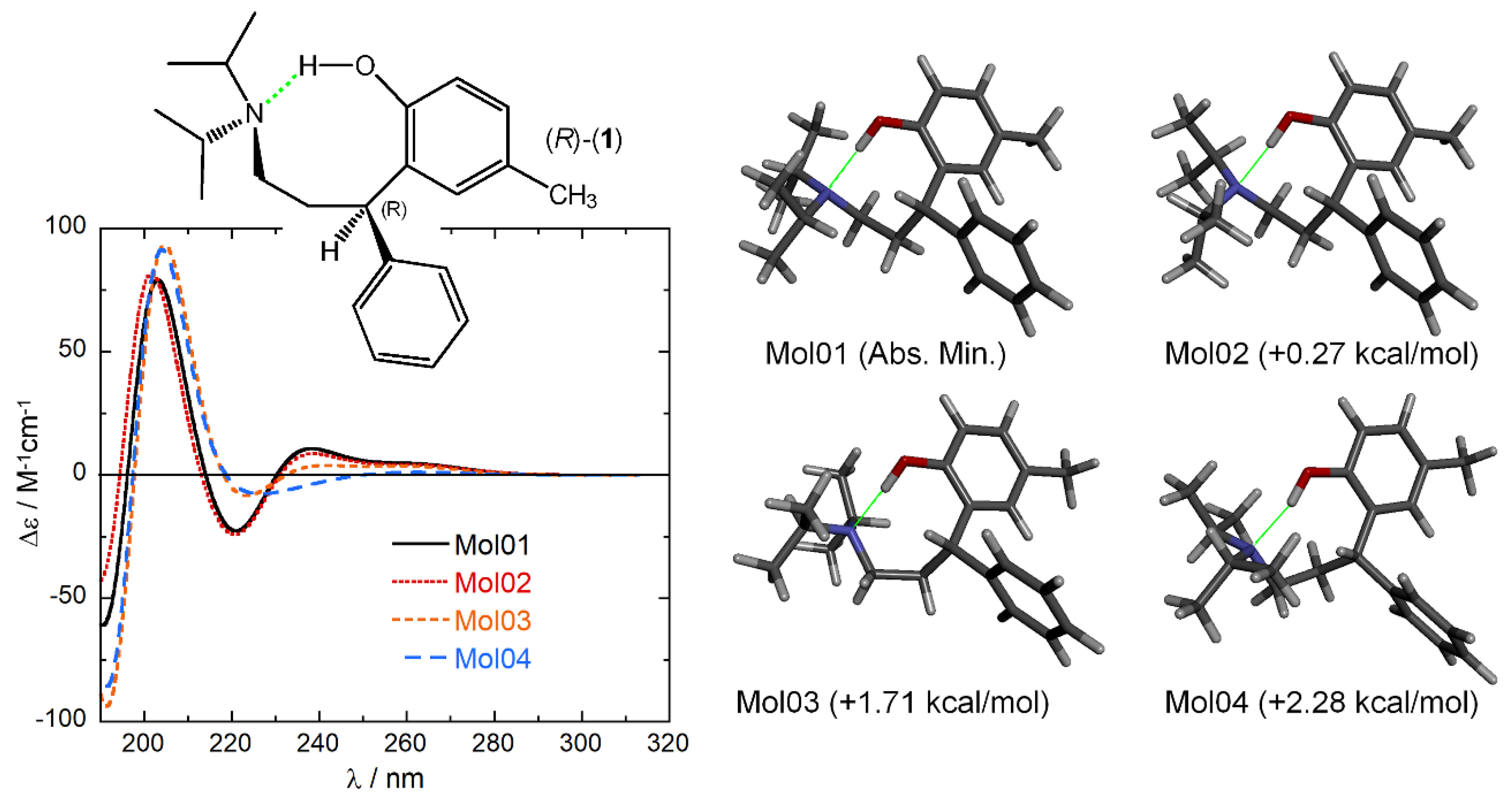
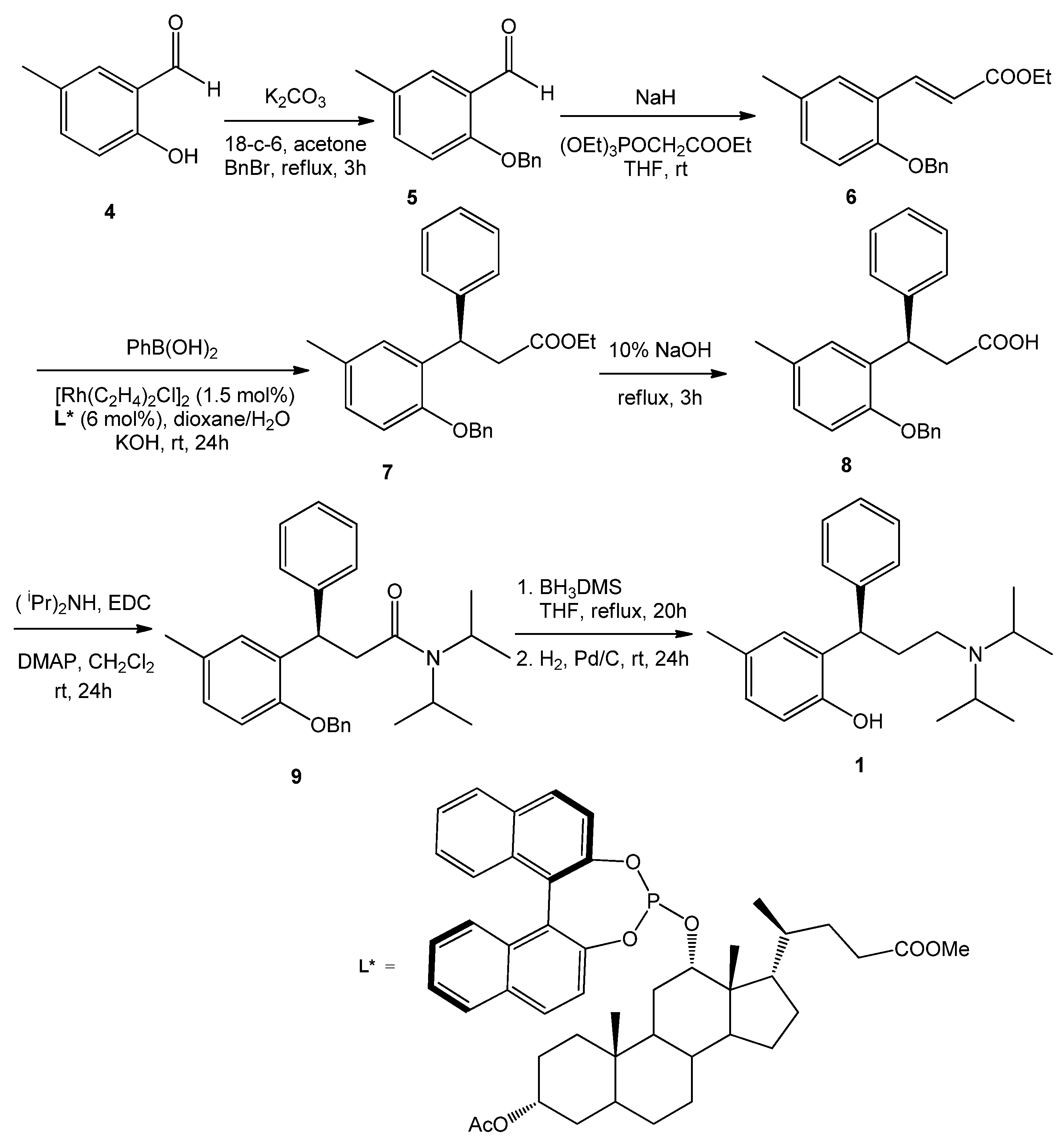
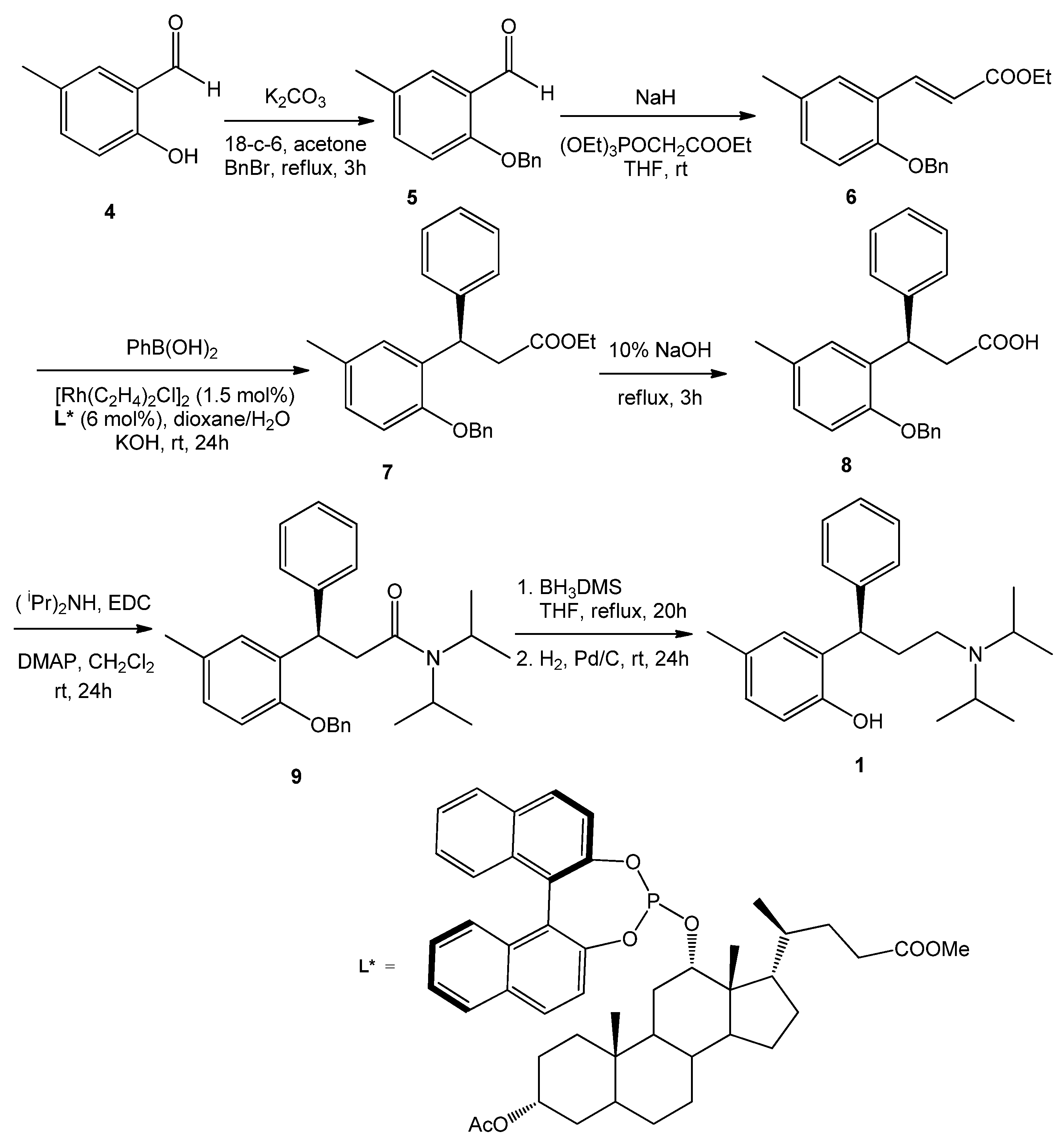
2.1. Sample Preparation
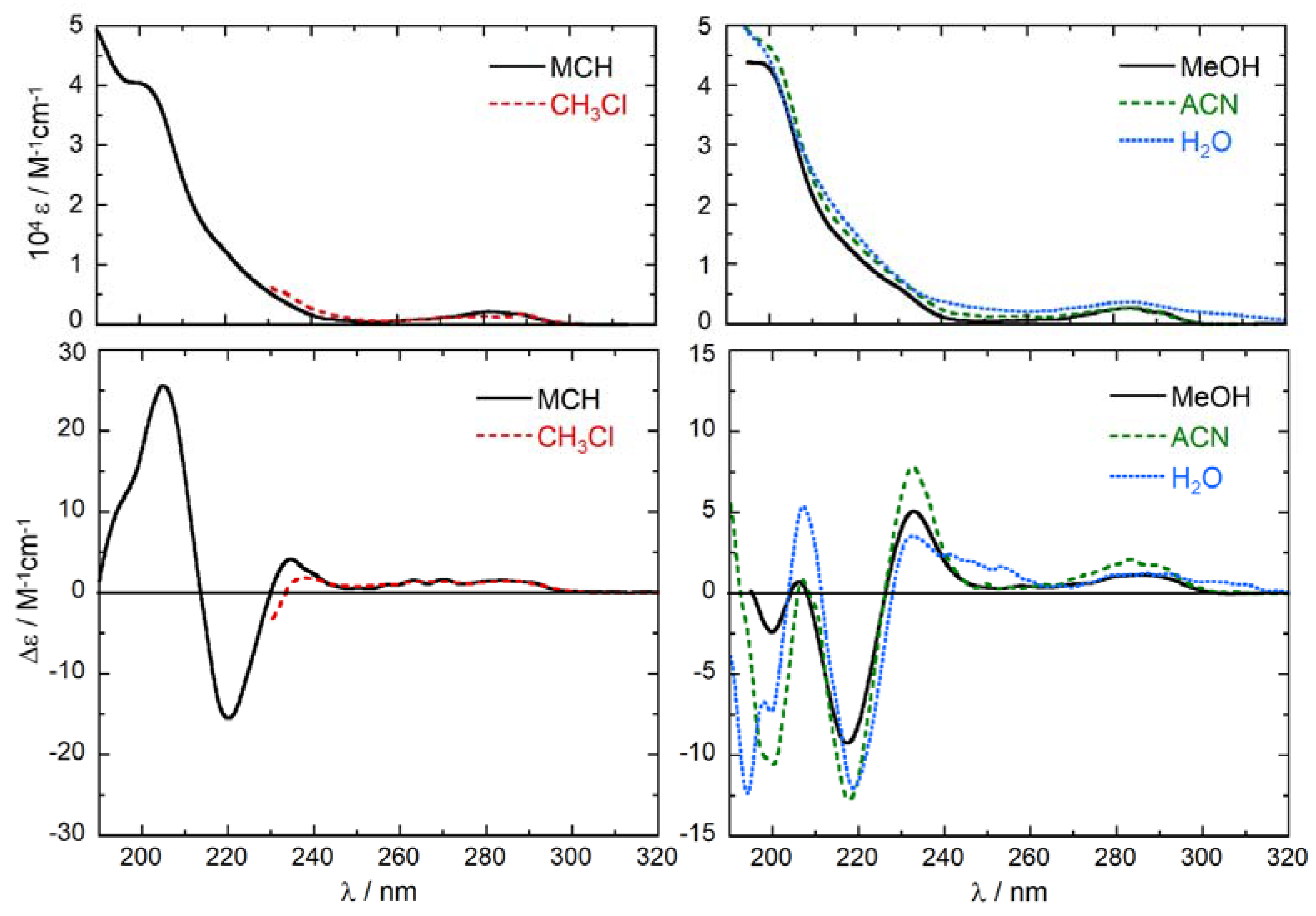
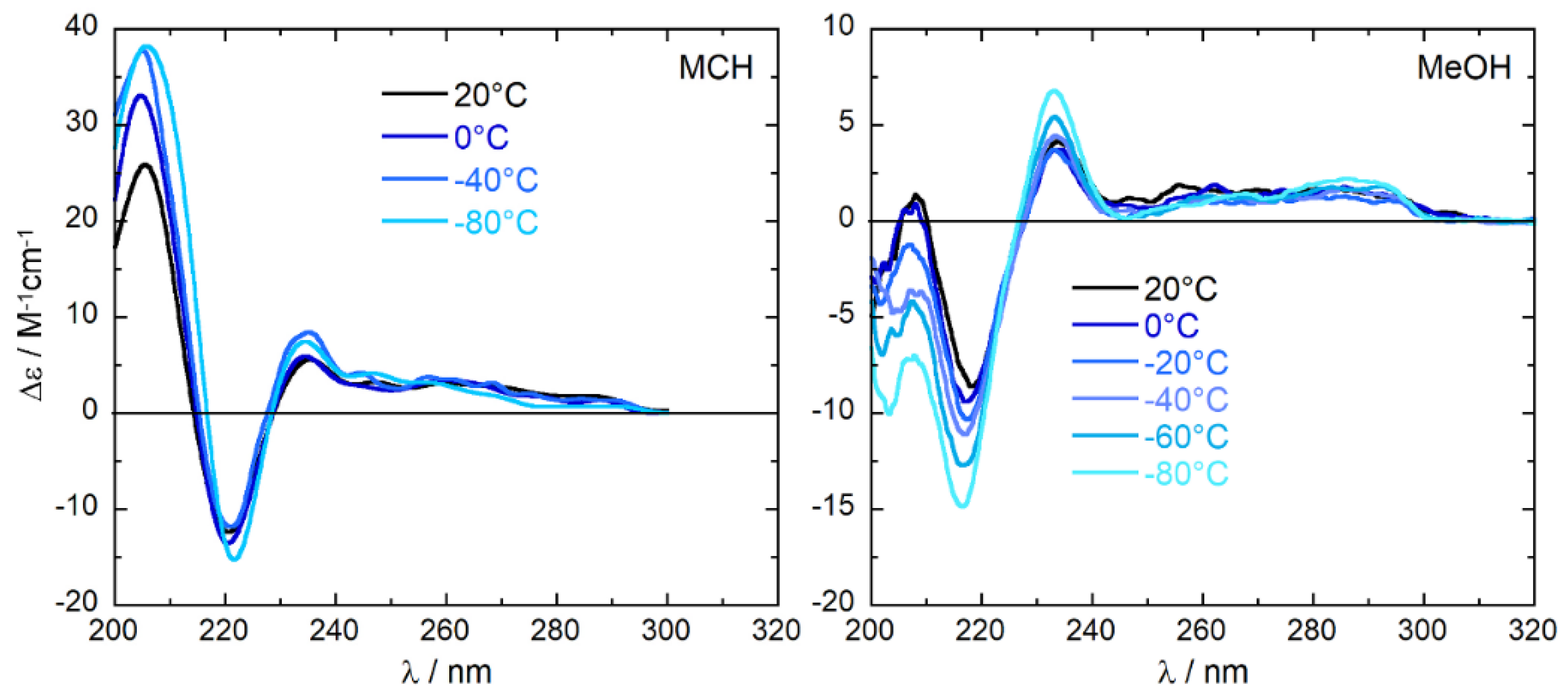
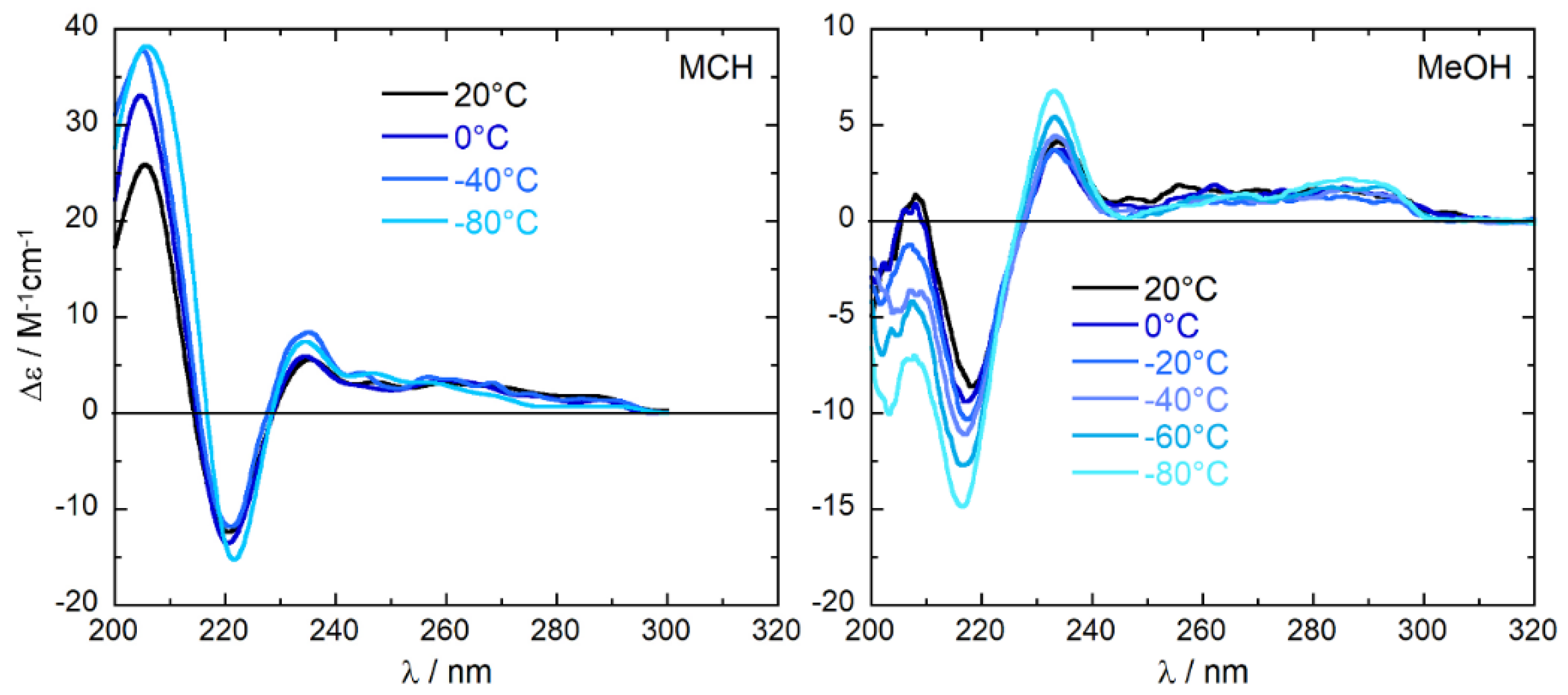
2.2. Experimental ECD Spectra
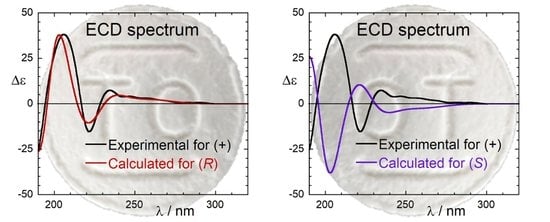
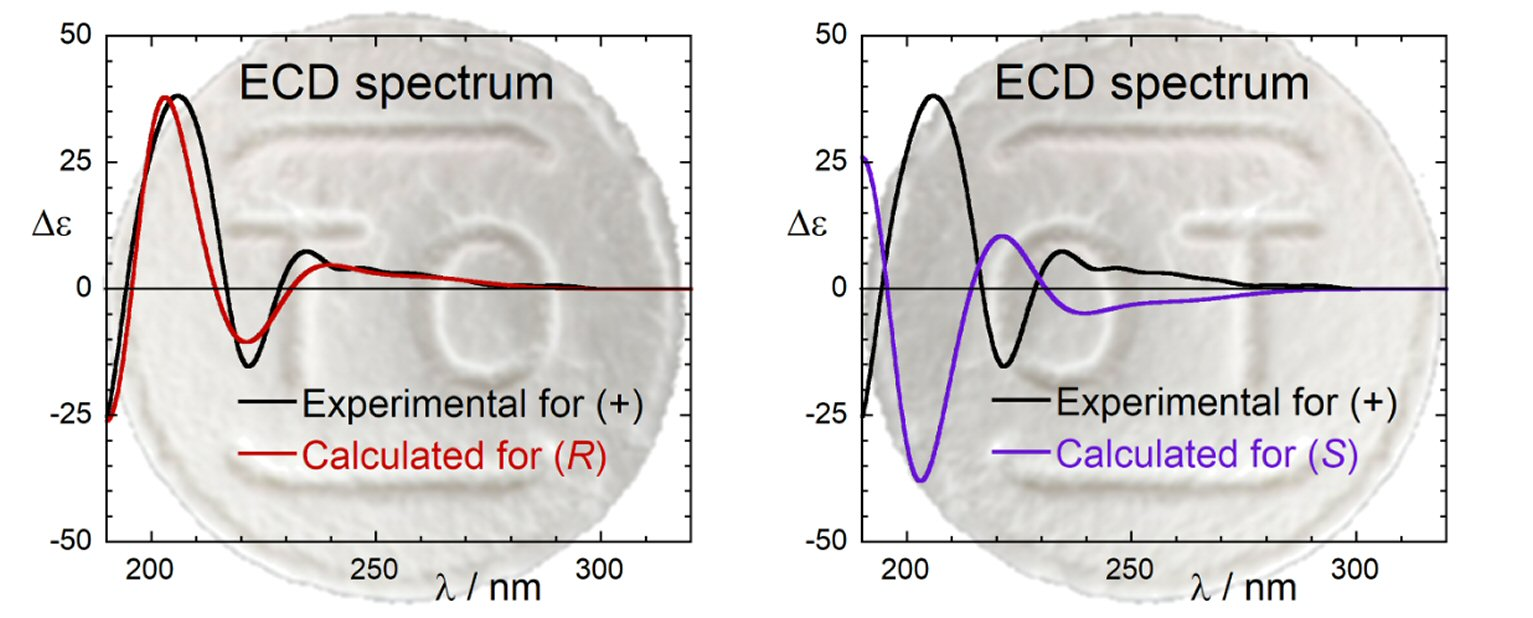
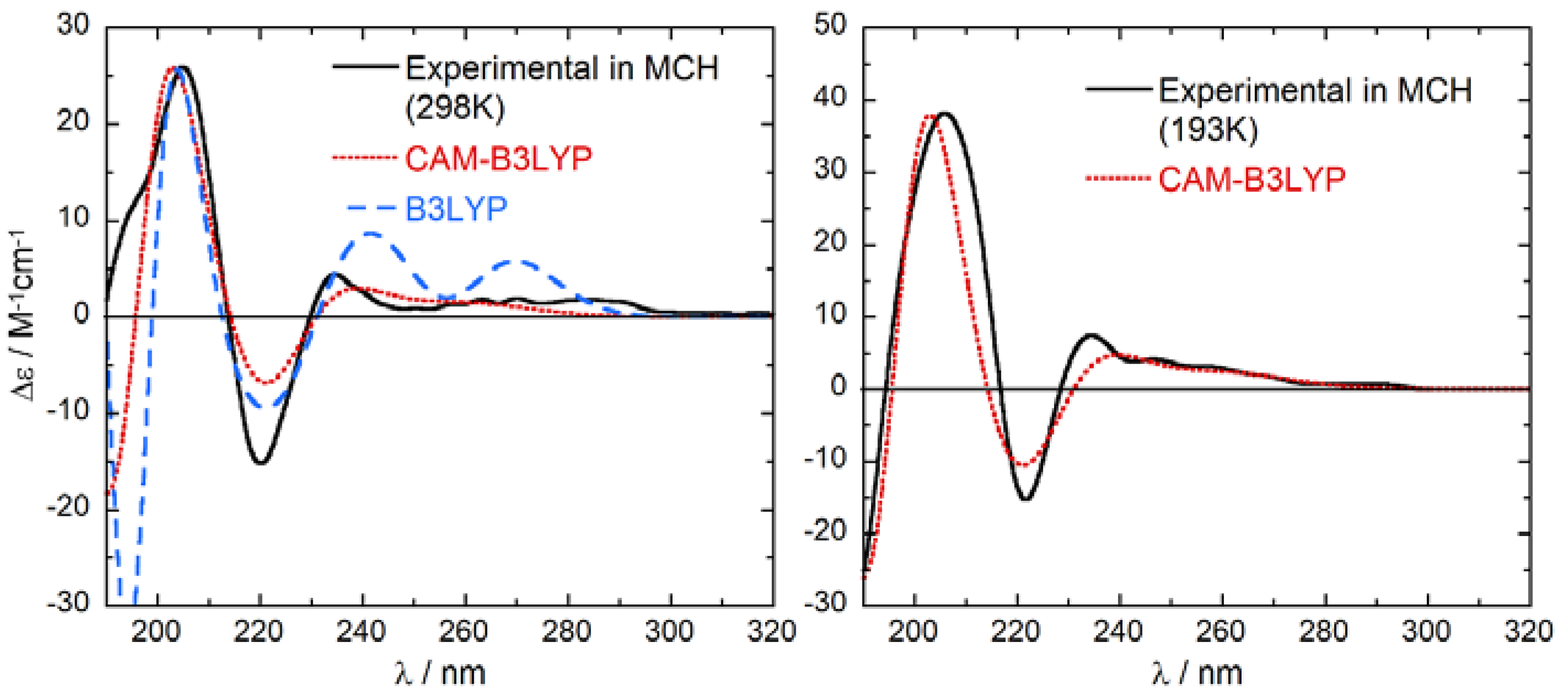
2.3. ECD Calculations and AC Assignment
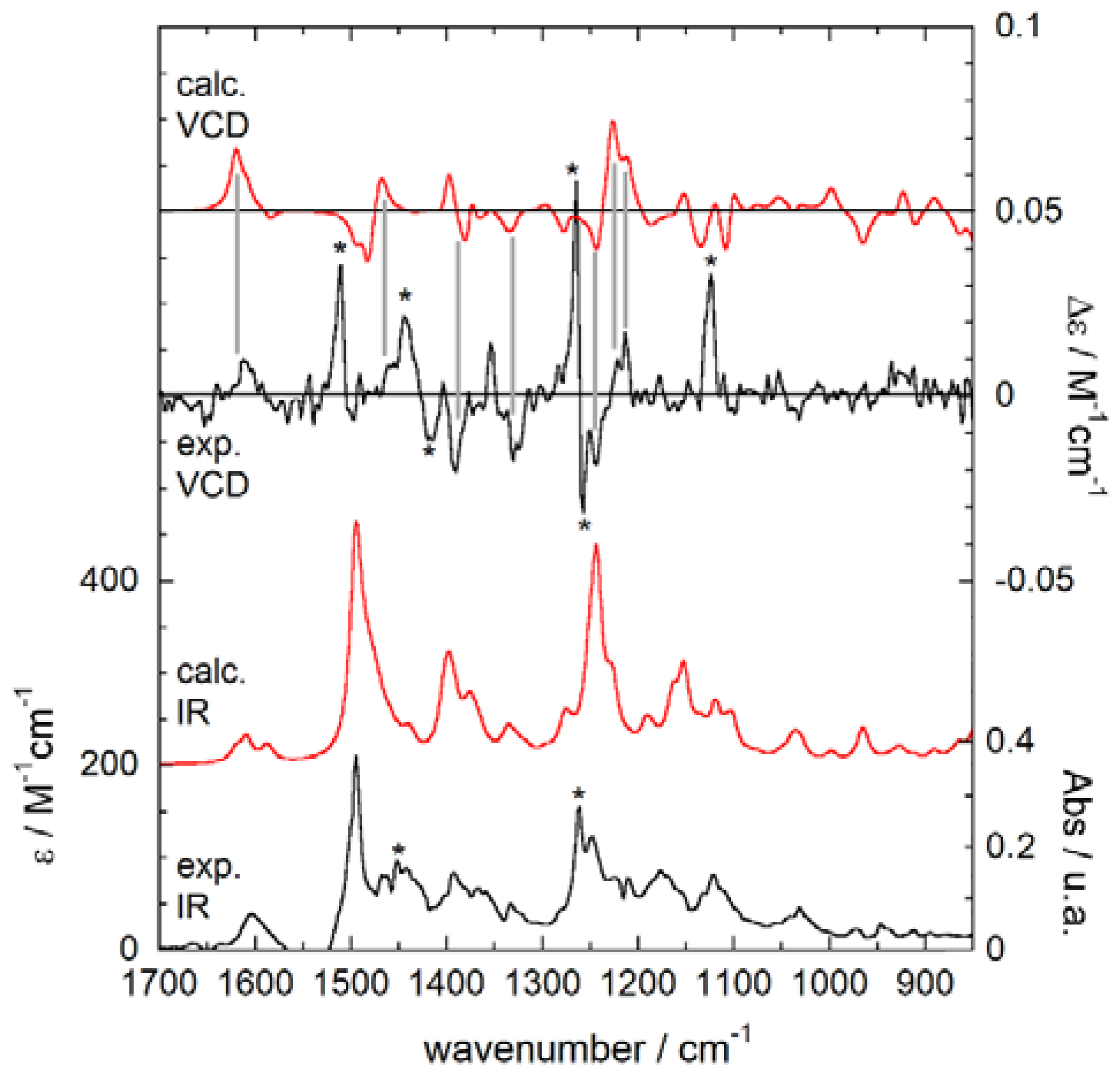
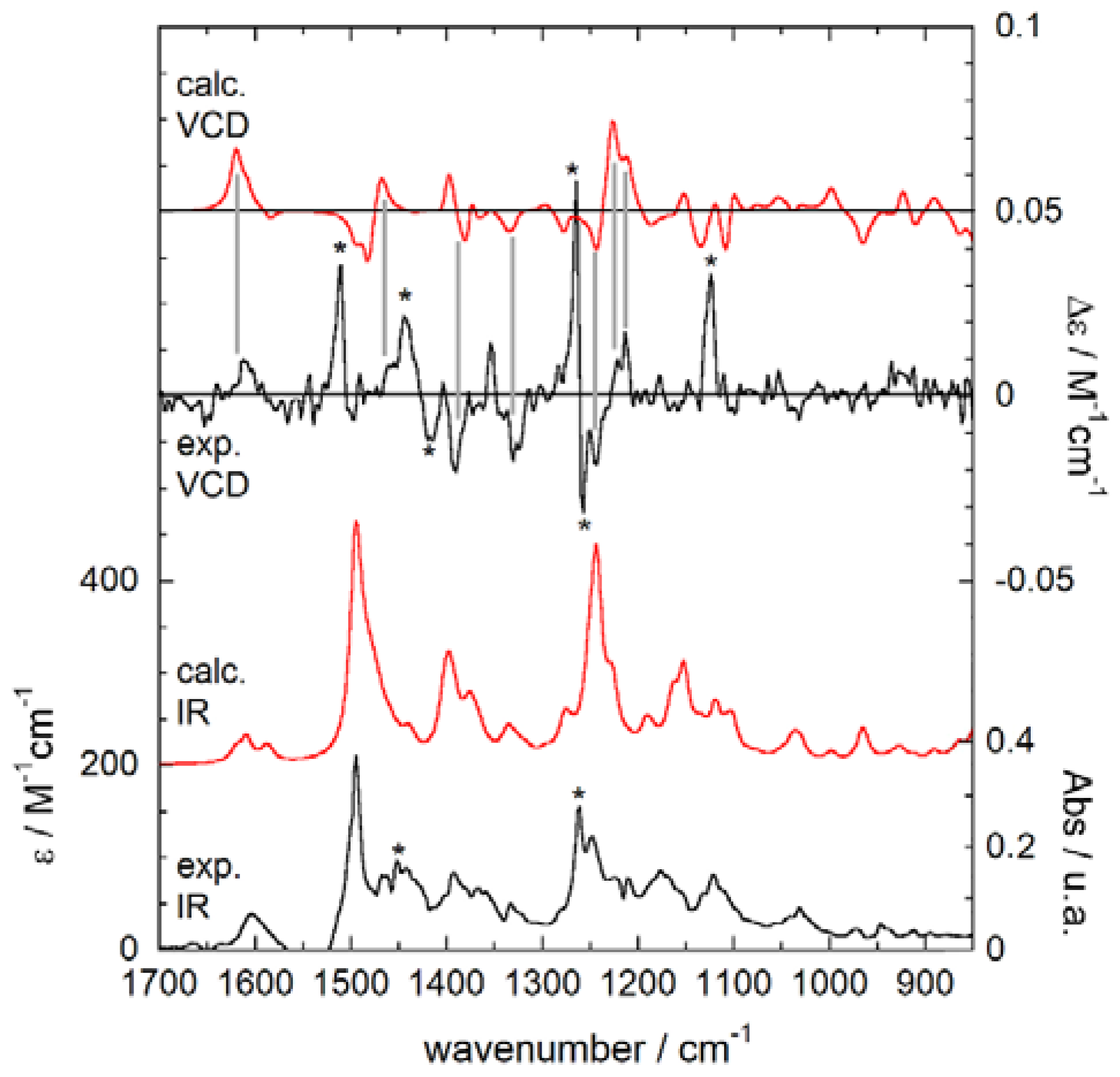
2.4. Experimental and Calculated IR/VCD Spectra
3. Materials and Methods
3.1. General Information
3.2. Computational Section
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Ananchenko, G.; Novakovic, J. Tolterodine Tartrate. In Profiles of Drug Substances, Excipients and Related Methodology; Brittain, H.G., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 42, pp. 339–403. [Google Scholar]
- Hills, C.J.; Winter, S.A.; Balfour, J.A. Tolterodine. Drugs 1998, 55, 813–820. [Google Scholar] [CrossRef]
- Appell, R.A. Clinical efficacy and safety of tolterodine in the treatment of overactive bladder: A pooled analysis. Urology 1997, 50, 90–96. [Google Scholar] [CrossRef]
- Chiral Drugs: Chemistry and Biological Action; Lin, G.-Q.; You, Q.-D.; Cheng, J.-F. (Eds.) Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Jönsson, N.A.; Sparf, B.A.; Mikiver, L.; Moses, P.; Nilvebrant, L.; Glas, G. New Amines, Their Use and Preparation. EP0325571B1, 22 January 1988. [Google Scholar]
- Detrol (tolterodine l-tartrate) Tablets. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/98/20771_Detrol.cfm (accessed on 24 January 2019).
- Stahl, M.M.S.; Ekström, B.; Sparf, B.; Mattiasson, A.; Andersson, K.-E. Urodynamic and other effects of tolterodine: A novel antimuscarinic drug for the treatment of detrusor overactivity. Neurourol. Urodyn. 1995, 14, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Nilvebrant, L.; Hallén, B.; Larsson, G. Tolterodine-a new bladder selective muscarinic receptor antagonist: Preclinical pharmacological and clinical data. Life Sci. 1997, 60, 1129–1136. [Google Scholar] [CrossRef]
- Alberg, G. S(−)-tolterodine in the treatment of Urinary and Gastrointestinal Disorders. U.S. Patent 6310103B1, 19 July 1996. [Google Scholar]
- Novotna, A.; Kamenickova, A.; Pecova, M.; Korhonova, M.; Bartonkova, I.; Dvorak, Z. Profiling of enantiopure drugs towards aryl hydrocarbon (AhR), glucocorticoid (GR) and pregnane X (PXR) receptors in human reporter cell lines. Chem. Biol. Interact. 2014, 208, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Doricakova, A.; Theile, D.; Weiss, J.; Vrzal, R. Differential effects of the enantiomers of tamsulosin and tolterodine on P-glycoprotein and cytochrome P450 3A4. Naunyn Schmiedebergs Arch. Pharmacol. 2017, 390, 49–59. [Google Scholar] [CrossRef]
- Krasulova, K.; Siller, M.; Holas, O.; Dvorak, Z.; Anzenbacher, P. Enantiospecific effects of chiral drugs on cytochrome P450 inhibition in vitro. Xenobiotica 2016, 46, 315–324. [Google Scholar] [CrossRef]
- Dakarapu, V.V.; Allaka, T.R.; Uppalla, L.K.; Jha, A. Design, Synthesis, and Molecular Modeling of Asymmetric Tolterodine Derivatives as Anticancer Agents. J. Heterocycl. Chem. 2018, 55, 2157–2167. [Google Scholar] [CrossRef]
- Gupta, S.; Sathyan, G.; Mori, T. New perspectives on the overactive bladder: Pharmokinetics and bioavailability11Suneel Gupta, Gayatri Sathyan, and Timothy Mori hold stock in Johnson & Johnson and the ALZA Corporation. Urology 2002, 60, 78–80. [Google Scholar] [CrossRef]
- Gage, J.R.; Cabaj, J.E. Process to Prepare Tolterodine. U.S. Patent 5,922,914A, 31 December 1996. [Google Scholar]
- Košutić-Hulita, N.; Žegarac, M. Tolterodinium (+)-(2R,3R)-hydrogen tartrate. Acta Crystallogr. Sect. C 2005, 61, o171–o173. [Google Scholar] [CrossRef] [PubMed]
- Cianci, M.; Helliwell, J.R.; Helliwell, M.; Kaucic, V.; Logar, N.Z.; Mali, G.; Tusar, N.N. Anomalous scattering in structural chemistry and biology. Crystallogr. Rev. 2005, 11, 245–335. [Google Scholar] [CrossRef]
- Smith, G.; Wermuth, U.D.; White, J.M. Proton-transfer and non-transfer in compounds of quinoline and quinaldic acid with l-tartaric acid. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2006, 62, o694–o698. [Google Scholar] [CrossRef] [PubMed]
- Ünal, A.; Şentürk, S.; Şenyel, M. Vibrational spectroscopic and thermal studies of some 3-phenylpropylamine complexes. Vib. Spectrosc 2009, 51, 299–307. [Google Scholar] [CrossRef]
- Osborne, R.; Clarke, N.; Glossop, P.; Kenyon, A.; Liu, H.; Patel, S.; Summerhill, S.; Jones, L.H. Efficient conversion of a nonselective norepinephrin reuptake inhibitor into a dual muscarinic antagonist-β2-agonist for the treatment of chronic obstructive pulmonary disease. J. Med. Chem. 2011, 54, 6998–7002. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, O.; Ulgheri, F.; Marchetti, M. Enantioslective Synthesis of Enantiomerically Enriched Compounds. WO2005005356A2, 2 July 2003. [Google Scholar]
- Ulgheri, F.; Marchetti, M.; Piccolo, O. Enantioselective Synthesis of (S)- and (R)-Tolterodine by Asymmetric Hydrogenation of a Coumarin Derivative Obtained by a Heck Reaction. J. Org. Chem. 2007, 72, 6056–6059. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Tokunaga, N.; Hayashi, T. Rhodium-Catalyzed Asymmetric 1,4-Addition of Arylboronic Acids to Coumarins: Asymmetric Synthesis of (R)-Tolterodine. Org. Lett. 2005, 7, 2285–2288. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, C.; Andersson, P.G. Catalytic Asymmetric Total Synthesis of the Muscarinic Receptor Antagonist (R)-Tolterodine. Adv. Synth. Catal. 2005, 347, 662–666. [Google Scholar] [CrossRef]
- Kobayashi, K.; Nishikata, T.; Yamamoto, Y.; Miyaura, N. Stepwise Palladium-Catalyzed 1,4-Addition of Arylboronic Acids to Enones and Regioselective Baeyer–Villiger Oxidation for Enantioselective Synthesis of β-Diaryl Esters and (+)-(R)-Tolterodine. Bull. Chem. Soc. Jpn. 2008, 81, 1019–1025. [Google Scholar] [CrossRef]
- Gallagher, B.D.; Taft, B.R.; Lipshutz, B.H. Asymmetric Conjugate Reductions of Coumarins. A New Route to Tolterodine and Related Coumarin Derivatives. Org. Lett. 2009, 11, 5374–5377. [Google Scholar] [CrossRef]
- Korenaga, T.; Maenishi, R.; Osaki, K.; Sakai, T. Highly active rhodium catalyst with electron-poor diphosphine enables efficient synthesis of chiral 4-aryl-δ-lactones. Heterocycles 2010, 80, 157–162. [Google Scholar] [CrossRef]
- Kim, H.; Yun, J. Copper-Catalyzed Asymmetric 1,4-Hydroboration of Coumarins with Pinacolborane: Asymmetric Synthesis of Dihydrocoumarins. Adv. Synth. Catal. 2010, 352, 1881–1885. [Google Scholar] [CrossRef]
- Luo, Y.; Carnell, A.J. Chemoenzymatic Synthesis and Application of Bicyclo[2.2.2]octadiene Ligands: Increased Efficiency in Rhodium-Catalyzed Asymmetric Conjugate Additions by Electronic Tuning. Angew. Chem. Int. Ed. 2010, 49, 2750–2754. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, D.; Fain, M.; Yang, J.; Trehy, M. Enantiomeric impurity analysis using circular dichroism spectroscopy with United States Pharmacopeia liquid chromatographic methods. J. Pharmacol. Biomed. Anal. 2018, 156, 366–371. [Google Scholar] [CrossRef] [PubMed]
- The original paper only contains a single CD value at 231 nm. The full ECD spectrum was kindly made available to us by the authors.
- Zullo, V.; Iuliano, A. Rh-catalyzed enantioselective conjugate addition of arylboronic acids to 3-arylpropenoates: Enantioselective synthesis of (R)-Tolterodine. Eur. J. Org. Chem. 2018, in press. [Google Scholar] [CrossRef]
- Jaffé, H.H.; Orchin, M. Theory and Applications of Ultraviolet Spectroscopy; Wiley: New York, NY, USA, 1962. [Google Scholar]
- Herzberg, G. Molecular Spectra and Molecular Structure. III. Electronic Spectra and Electronic Structure of Polyatomic Molecules; D. Van Nostrand Co.: Princeton, NJ, USA, 1966. [Google Scholar]
- Doub, L.; Vandenbelt, J.M. The Ultraviolet Absorption Spectra of Simple Unsaturated Compounds. I. Mono- and p-Disubstituted Benzene Derivatives. J. Am. Chem. Soc. 1947, 69, 2714–2723. [Google Scholar] [CrossRef]
- Dearden, J.C.; Forbes, W.F. Light Absoprtion Studies: Part XIV. The Ultraviolet Absoprtion Spectra of Phenols. Can. J. Chem. 1959, 37, 1294–1304. [Google Scholar] [CrossRef]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Pescitelli, G.; Di Bari, L.; Berova, N. Conformational Aspects in the Studies of Organic Compounds by Electronic Circular Dichroism. Chem. Soc. Rev. 2011, 40, 4603–4625. [Google Scholar] [CrossRef]
- Pescitelli, G.; Bruhn, T. Good Computational Practice in the Assignment of Absolute Configurations by TDDFT Calculations of ECD Spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef]
- Superchi, S.; Scafato, P.; Górecki, M.; Pescitelli, G. Absolute Configuration Determination by Quantum Mechanical Calculation of Chiroptical Spectra: Basics and Applications to Fungal Metabolites. Curr. Med. Chem. 2018, 25, 287–320. [Google Scholar] [CrossRef]
- Matsumoto, K.; Miki, K.; Inagaki, T.; Nehira, T.; Pescitelli, G.; Hirao, Y.; Kurata, H.; Kawase, T.; Kubo, T. “Marking” the nitrogen atoms of phenyl-(2-pyridyl)-(3-pyridyl)-(4-pyridyl)-methane. Synthesis and absolute configuration of the corresponding tris(pyridine N-oxide). Chirality 2011, 23, 543–548. [Google Scholar] [CrossRef]
- Matsumoto, K.; Miki, K.; Kurata, H.; Rikitake, N.; Nehira, T.; Inagaki, T.; Pescitelli, G.; Hirao, Y.; Kawase, T.; Oda, M.; et al. Chiral Tetrakis(2-thienyl)methane Derivative: A Possible Precursor for Cryptochiral Tetraalkylmethanes. Chem. Lett. 2008, 37, 1236–1237. [Google Scholar] [CrossRef]
- Matsumoto, K.; Inagaki, T.; Nehira, T.; Kannami, M.; Inokuchi, D.; Kurata, H.; Kawase, T.; Pescitelli, G.; Oda, M. Phenyl-(2-pyridyl)-(3-pyridyl)-(4-pyridyl)methane: Synthesis, chiroptical properties, and theoretical calculation of its absolute configuration. Chem. As. J. 2007, 2, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Padula, D.; Pescitelli, G. How and how much molecular conformation affects electronic circular dichroism: The case of 1,1-diarylcarbinols. Molecules 2018, 23, 128. [Google Scholar] [CrossRef] [PubMed]
- Mennucci, B.; Cappelli, C.; Cammi, R.; Tomasi, J. Modeling solvent effects on chiroptical properties. Chirality 2011, 23, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Štěpánek, P.; Bouř, P. Multi-scale modeling of electronic spectra of three aromatic amino acids: Importance of conformational averaging and explicit solute–solvent interactions. Phys. Chem. Chem. Phys. 2014, 16, 20639–20649. [Google Scholar] [CrossRef]
- Pikulska, A.; Hopmann, K.H.; Bloino, J.; Pecul, M. Circular Dichroism and Optical Rotation of Lactamide and 2-Aminopropanol in Aqueous Solution. J. Phys. Chem. B 2013, 117, 5136–5147. [Google Scholar] [CrossRef] [PubMed]
- Mazzeo, G.; Santoro, E.; Andolfi, A.; Cimmino, A.; Troselj, P.; Petrovic, A.G.; Superchi, S.; Evidente, A.; Berova, N. Absolute Configurations of Fungal and Plant Metabolites by Chiroptical Methods. ORD, ECD, and VCD Studies on Phyllostin, Scytolide, and Oxysporone. J. Nat. Prod. 2013, 76, 588–599. [Google Scholar] [CrossRef]
- Cappelli, C.; Bronco, S.; Monti, S. Computational study of conformational and chiroptical properties of (2R,3S,4R)-(+)-3,3′,4,4′,7-flavanpentol. Chirality 2005, 17, 577–589. [Google Scholar] [CrossRef]
- Parac, M.; Grimme, S. A TDDFT study of the lowest excitation energies of polycyclic aromatic hydrocarbons. Chem. Phys. 2003, 292, 11–21. [Google Scholar] [CrossRef]
- Pescitelli, G.; Barone, V.; Di Bari, L.; Rizzo, A.; Santoro, F. Vibronic Coupling Dominates the Electronic Circular Dichroism of the Benzene Chromophore L-1(b) band. J. Org. Chem. 2013, 78, 7398–7405. [Google Scholar] [CrossRef]
- Pescitelli, G.; Di Bari, L.; Caporusso, A.M.; Salvadori, P. The prediction of the circular dichroism of the benzene chromophore: TDDFT calculations and sector rules. Chirality 2008, 20, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Rode, J.E.; Górecki, M.; Witkowski, S.; Frelek, J. Solvation of 2-(hydroxymethyl)-2,5,7,8-tetramethyl-chroman-6-ol revealed by circular dichroism: A case of chromane helicity rule breaking. Phys. Chem. Chem. Phys. 2018, 20, 22525–22536. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Merten, C. Vibrational optical activity as probe for intermolecular interactions. Phys. Chem. Chem. Phys. 2017, 19, 18803–18812. [Google Scholar] [CrossRef] [PubMed]
- Merten, C.; Amkreutz, M.; Hartwig, A. Determining the structure of α-phenylethyl isocyanide in chloroform by VCD spectroscopy and DFT calculations—simple case or challenge? Phys. Chem. Chem. Phys. 2010, 12, 11635–11641. [Google Scholar] [CrossRef] [PubMed]
- Bünnemann, K.; Merten, C. Solvation of a chiral carboxylic acid: Effects of hydrogen bonding on the IR and VCD spectra of α-methoxyphenylacetic acid. Phys. Chem. Chem. Phys. 2017, 19, 18948–18956. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Pescitelli, G. SpecDis Version 1.71; Berlin, Germany, 2017. Available online: https://specdis-software.jimdo.com/ (accessed on 24 January 2019).



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Górecki, M.; Zullo, V.; Iuliano, A.; Pescitelli, G. On the Absolute Stereochemistry of Tolterodine: A Circular Dichroism Study. Pharmaceuticals 2019, 12, 21. https://doi.org/10.3390/ph12010021
Górecki M, Zullo V, Iuliano A, Pescitelli G. On the Absolute Stereochemistry of Tolterodine: A Circular Dichroism Study. Pharmaceuticals. 2019; 12(1):21. https://doi.org/10.3390/ph12010021
Chicago/Turabian StyleGórecki, Marcin, Valerio Zullo, Anna Iuliano, and Gennaro Pescitelli. 2019. "On the Absolute Stereochemistry of Tolterodine: A Circular Dichroism Study" Pharmaceuticals 12, no. 1: 21. https://doi.org/10.3390/ph12010021
APA StyleGórecki, M., Zullo, V., Iuliano, A., & Pescitelli, G. (2019). On the Absolute Stereochemistry of Tolterodine: A Circular Dichroism Study. Pharmaceuticals, 12(1), 21. https://doi.org/10.3390/ph12010021