3.1. Chlorine Sensor
Chlorine is a powerful oxidising agent that is used widely as a disinfectant in the treatment of industrial, recreational and potable drinking water. A variety of industrial processes are heavily dependant on the use of chlorine because of its potency as a sterilising agent for water, and it is essential that individual users and companies are able to measure chlorine concentrations to determine if adequate levels for disinfection are present. However, in addition to its benefits, the use of chlorine does incur some disadvantages and the presence of excessive concentrations of chlorine can in some cases be detrimental to human health and aquatic life. Concern over the environmental and health effects of chlorination have led to a raft of legislation relating to its determination (viz: European Economic Community Directive 80/778/EEC, 1998; US Environmental Protection Agency, 2000). It is clear that accurate, sensitive and simple procedures are required for the monitoring of chlorine by users and regulatory agents in order to assure compliance with regulations. The majority of chlorine testing within water is performed by colourimetric wet chemistry approaches and a wide range of commercial tests are available for different applications. These however suffer from a number of limitations including bleaching of the colour and a limited analytical range due to fluctuations in colour change. Colourimetric approaches also require skilled operators for use, the equipment is cumbersome and sampling/testing is time consuming [
34]. A technique which would permit easy, rapid, accurate and qualitative analysis of chlorine for both free and total determination over a wide analytical range would clearly be advantageous.
The microelectrode pore array could be utilised within an electrochemical sensor for chlorine. Chlorine in aqueous solution can exist as free chlorine (Cl2) or in a variety of combined forms such as hypochlorous acid (HOCl) or other forms upon reaction with organic materials. Ammonia will for example react with chlorine to give NH2Cl.
Experimental electrodes are based on commercial screen printed electrodes supplied by Microarray Ltd (Manchester) and contain two working electrode microarrays; one dedicated to the measurement of free chlorine and the other for measuring total chlorine. The electrochemical determination of chlorine is based on known titrimetric approaches. Chlorine (both free and combined) reacts with iodide ion to produce iodine which can then be titrated using sodium thiosulphate. Free chlorine can be directly detected electrochemically whereas combined chlorine will not directly reduce at an electrode surface and so acidified potassium iodide is added to the solution, preferentially oxidising both free chorine and combined chlorine according to
Equations 1a and
1b.
The iodine generated can be reduced at the working electrode to produce iodide, according to
Equation 2, thereby allowing electrochemical detection.
Provided sufficient iodine is made available initially, the current measured will be proportional to the concentration of total residual chlorine in the mixture. For this reason the microelectrode arrays were coated with a thin film of potassium iodide containing formulation using a BioDot AD 3200 dispensing platform, incorporating a BioJet Plus
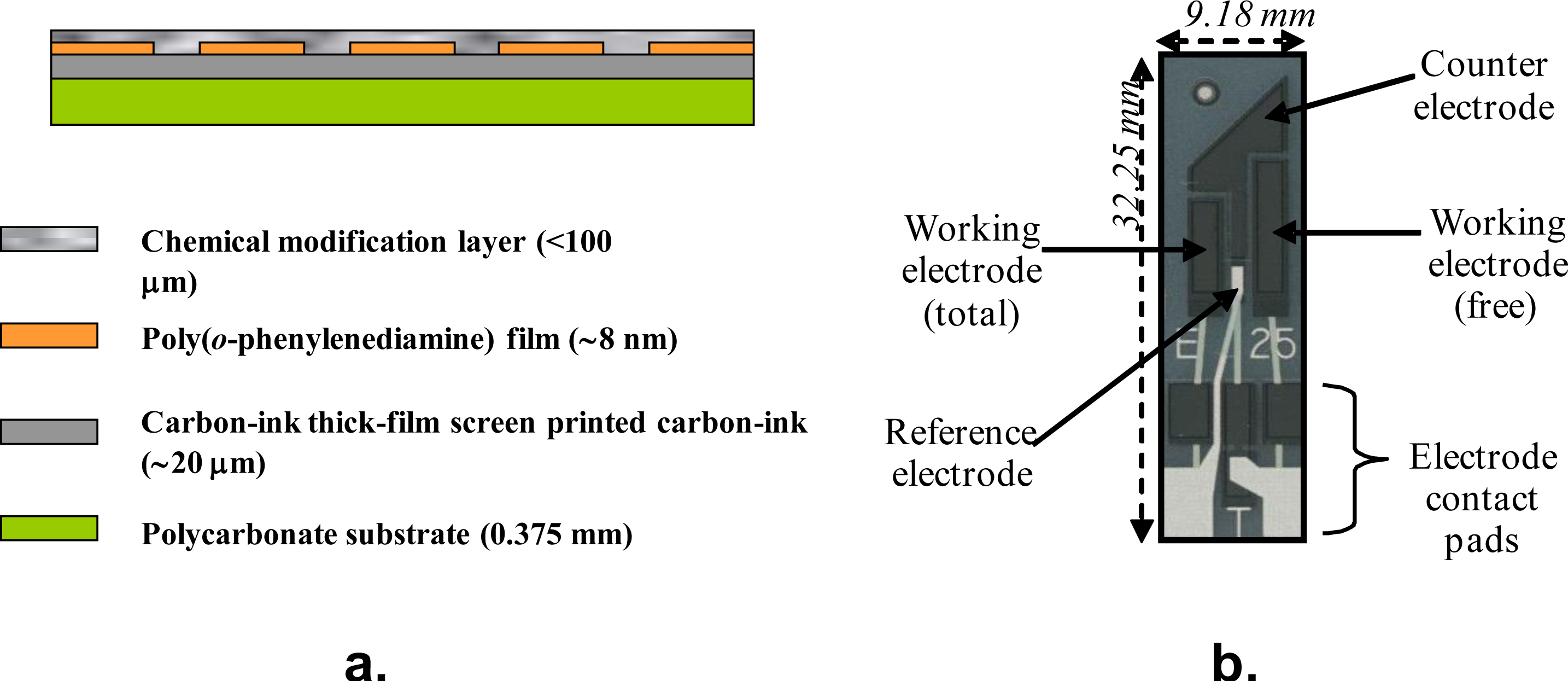
™ 3000 dispensing system. These electrodes were then used to measure free and total chlorine respectively. Subtraction of the free chlorine value from the total chlorine level gives the level of combined chlorine. A schematic of the final sensor construction is displayed in
Figure 8a, along with a photograph of a “base” electrode (
Figure 8b).
Although when placed in aqueous sample solutions, the iodide will dissolve, diffusion away from the electrode is relatively slow and a result for chlorine can be obtained with our system in 30 seconds, long before loss of iodide becomes a problem. Since these electrode assemblies have been developed for single-use only, it does not matter if the iodide is removed during the first measurement.
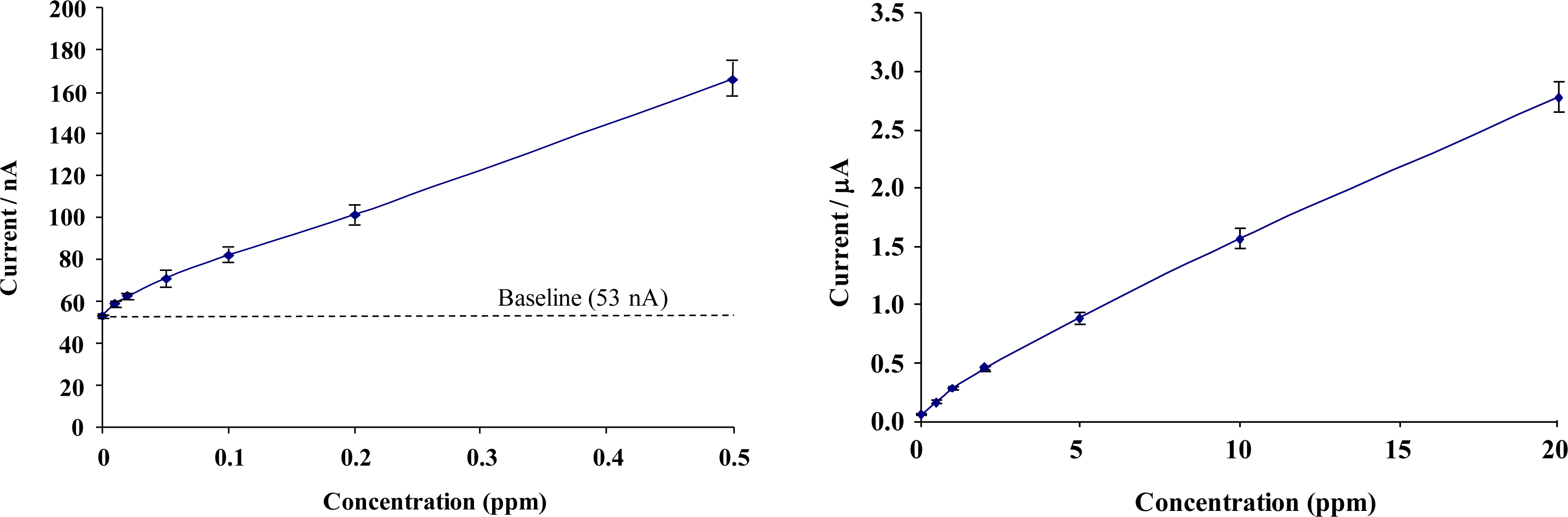
Figure 9 shows calibration profiles obtained for total chlorine solutions interrogated with chemically modified microelectrode array sensors (concentrations ranging from 0 to 20 ppm). Given the range of the current responses obtained, data is presented in low- and high-range calibration curves. Error bars represent the relative standard deviation (RSD) from the 5 sensors taken at each chlorine concentration. The full sensor calibration data exhibits a quasi-linear response to total chlorine solutions, resulting in diminishing escalation in response with increasing concentration.
A response of approximately 3 nA per 0.01 ppm for low range total chlorine and 1 nA per 0.01 ppm for high range concentrations was obtained – sufficiently large to permit the differentiation of 0.01 ppm total chlorine.
Figure 10 demonstrates similar calibration profiles for concentrations of 0–20 ppm free chlorine interrogated with modified screen printed carbon-ink based microelectrode array sensors. Once again, a quasi-linear calibration profile is obtained for the BioDot modified microelectrode arrays sensors with a response of approximately 2 nA per 0.02 ppm increment for low range free chlorine and 1 nA per 0.02 ppm for high range concentrations.
As expected, there is a considerably smaller amperometric response for free chlorine when compared to the total chlorine response. This process has proved suitable for scaling up for commercial production. Large sheets of screen-printed sensors containing hundreds of individual four electrode (two working, a counter and an Ag/AgCl reference) units can be easily constructed by industrial partners. Insulating layers can be simultaneously deposited onto all the working electrodes and the use of large sonic tanks allows the ablation of the electrode sheets. Thousands of chlorine sensors could be produced daily within our pilot scale facility.
Within this section, we have applied fabricated microelectrode arrays to the determination of chlorine, incorporating a method for localising the chemical reagents required for chlorine detection at the microelectrode array surface. Free, total (and thus combined) forms of chlorine were detected in solutions in the range of 0–20 ppm. Microelectrode array responses for both free and total chlorine analysis were found to show a measureable resolution of down to 0.02 ppm and 0.01 ppm respectively using chronoamperometric methods, well within the required limits defined under current legalisation. Particularly, the microelectrode arrays offer good signal differentiation at the low levels of chlorine concentration, for both forms, afforded by the dramatic increase of the signal/noise ratio that microelectrodes offer over conventional macroelectrodes.
3.3. Poly(siloxane) Coatings
One problem with sensor systems is a loss of signal linearity at high analyte concentrations (>10 mM). We describe here the modification of a sonochemically fabricated microelectrode glucose oxidase enzyme array via the deposition of a poly(siloxane) coating for the improvement of signal linearity.
The fabrication of microarrays with poly(aniline) protrusions were fabricated as described previously (section 2.1). Dimethyldichlorosilane, 0.5 mL, was placed in a watch glass at the bottom of a sealed reaction vessel kept at atmospheric humidity. A stage was designed to allow for the deposition of the poly(siloxane) directly at the working electrode surface (
Figure 16) thereby ensuring that the counter and reference electrodes, and all the electrode contacts were not exposed to the silane vapour. The electrode was then suspended 5 cm above the dichlorodimethylsilane liquid for pre-determined periods. The stage was then removed from the vessel and the electrode stored. Previous work within this group has been focused towards the development and exploitation of ultra-thin film composite membrane technology for the construction of microelectrodes. A technique for depositing poly(dimethylsiloxane) from dichlorodimethylsilane has been previously shown by us to give thin films which offer low diffusional resistance to glucose but which minimise surface biofouling [
47], thus offering protection against electrode passivation effects, and therefore allowing for the linearisation of sensor responses. Permittivity coefficients were measured for these films [
47] and showed a much higher permeability for oxygen than for glucose; this is important since oxygen is a co-factor in the glucose–glucose oxidase reaction and we do not wish it to become depleted at the electrode surface and cause the sensor to reach saturation.
An SEM of the working electrode (
Figure 17a) clearly shows one of the resulting polymer/enzyme protrusions. SEM images of the siloxane-coated enzyme microelectrodes are shown in
Figure 17b. A film can be seen to cover the entire electrode surface. In order to assess the performance of the ultra-thin film as a coating for the linearisation of sensor responses, the AC response to glucose of an enzyme microelectrode array/ultra-thin polymer film was compared to that of a similar electrode lacking an ultra-thin polymer film coating. AC impedimetric responses were determined over a range of frequencies from 0.1 Hz to 10 kHz. Again the responses were verified to be free of stirring (enforced convectional) effects. The Bode and Nyquist plots were almost identical to those published previously [
45] and therefore are not shown for brevity. Earlier work showed that the greatest variations in impedance occurred at 0.1 Hz [
45] and similar results were obtained for the siloxane-coated membranes. When the differences in impedance at frequency of 0.1 Hz are plotted for microelectrodes exposed to varying glucose concentrations in pH 7.4 buffer, a calibration plot is obtained (
Figure 18). The instrumentation required for commercial exploitation of biosensors may be greatly simplified if the sensor gives a linear response with respect to analyte concentration. The resulting calibration graph (
Figure 18) shows that the siloxane coating lowers the magnitude of sensor response compared to uncoated microelectrodes whilst also linearising the sensor responses between 0 and 60 mM glucose (r
2 = 0.671 (uncoated), r
2 = 0.995 (silane coated)). We have determined the reproducibility of sensor responses to offer <5% variability [
45].
We have combined two disparate biosensor technologies to form a novel enzyme-based electrochemical sensor. Initially we described the examination of a commercially available ceramic based disposable electrode as a suitable host for the fabrication of a microelectrode array. Microelectrode arrays were fabricated as described previously via sonochemical ablation; these arrays again displayed true stir-independent responses. Enzyme/polymer protrusions were then again electrochemically deposited [
45] at the host microelectrode templates so as to produce an enzyme (glucose oxidase) ultra-microelectrode array.
Enzyme microelectrode arrays were then coated with an ultra-thin film coating of poly(siloxane) (silicone) so as to provide a covering substrate diffusion limiting layer for the linearization of sensor responses. This coating has been shown to extend the useful analytical concentration range for the sensor via linearisation of its response.
3.4. Organophosphate Pesticide Detection Using Microarrays
Modern agricultural practices often involve the use of organophosphate pesticides to control insect infestation. However increasing concern is being shown towards their indiscriminate use due to the potential deleterious immediate and long term effects they may cause to the environment, livestock and human health [
48,
49]. Organophosphate pesticides (OPs) appear to have taken the place of many of the organochlorine pesticides previously used due to their lower persistence in the environment whilst still remaining effective. Unfortunately OPs are neurotoxins and therefore present a serious risk to human health. The possibility of OP contamination of our food and water supplies necessitates the development of analytical methods for the reliable detection of pesticides, to safeguard both food supplies and the environment. Possible contamination of water supplies is a hazard and legislation has now been passed (European Commission: EU Water Framework Directive 2000/60/EC, European Commission: Drinking Water Directive 98/83/EC, which recommends maximum levels within water supplies of 0.1 mg L
−1 for individual pesticides and 0.5 mg L
−1 for total pesticide) to restrict pesticides within water supplies. Due to concerns about these materials it appears probable that these permitted levels will decrease. More recently, along with concerns about accidental release there is also the possibility of deliberate use of these nerve agents, as happened for example, with the Sarin gas attack on the Tokyo underground in 1995.
Current methods for determination and/or the monitoring of pesticides include gas and liquid chromatography and various spectroscopic techniques [
50]. Many of these methods are time consuming, not sufficiently sensitive and/or require complex sample preparation and are not suitable for continuous monitoring. The development of a simplified analytical approach would prove highly beneficial.
Biosensor technology has been utilised as a potential solution to this problem, especially electrochemical biosensing techniques since they are amongst the easiest and most inexpensive methods for detection of binding events. Many groups have previously demonstrated the fabrication of biosensors for the detection of pesticides [
51]. Usually these do not directly detect the pesticides itself. Instead it is known that pesticides, even at very low concentrations, can inhibit enzyme reactions, usually by “poisoning” of the enzyme. It is possible to first construct an enzyme-electrode, measure its response when exposed to a suitable concentration of its substrate and then expose it to a dilute pesticide solution. Further exposure to the initial substrate concentration and comparison with the response prior to pesticide exposure allows quantification of the inhibition which can then be correlated with pesticide concentration.
Cholinesterases, such as acetyl cholinesterase (AChE) catalyse the hydrolysis of choline esters to the corresponding carboxylic acid and choline,
Equation 6.
The inhibition of this reaction has been used for the detection of organophosphate and other pesticides due to its high specificity and sensitivity [
51]. Usually the simple amperometric detection of the product of the enzyme catalysed ester hydrolysis reaction is utilised [
52]. Screen-printed carbon electrodes, often containing electrochemical catalysts, are the favoured substrate for these sensors due to their inexpensiveness and ease of mass-production. Enzymes can be immobilised onto these substrates using a wide variety of methods. A discussion of the field of pesticide detection using biosensors is outside the scope of this paper but is reviewed here [
51].
3.4.1. Application of Microelectrodes
Microelectrode arrays have also been used for the detection of organophosphates [
51,
53,
54]. Acetylcholine esterase hydrolyses acetylthiocholine chloride to give thiocholine which can be easily detected using screen-printed carbon electrodes doped with cobalt phthalocyanine (CoPC) [
53,
54]. The use of the CoPC catalyst reduces the working potential to approximately +100 mV (
versus Ag/AgCl), thereby minimising interference from other electroactive compounds. We therefore utilised our sonochemical fabrication technique to form micropore arrays from which microarrays of poly(aniline) containing entrapped acetylcholine esterases were grown [
55]. A genetically modified AChE, designed to maximise sensitivity was incorporated, as was an I
125 labelled AChE to quantify the amount of enzyme deposited, which in this instance corresponded to 0.15 units activity [
53]. Measuring the amperometric response of the electrode in 2 mmol L
−1 acetylthiocholine before and following exposure to paraoxon solutions of various concentrations allowed calibration profiles of the inhibition of enzyme activity against pesticide concentration to be constructed (
Figure 19a). A typical current transient response for an AChE-modified electrode to 2 mmol L
−1 acetylthiocholine chloride is shown in
Figure 19b. Levels as low as 10
−17 mol L
−1 paraoxon could be reproducibly detected and a current transient response after exposure to this ultra-low concentration is shown (
Figure 19c). Other workers have also reported pesticide detection at comparable levels using acetylcholine esterase immobilised on microporous conductive carbon [
56].
These AChE containing micro-electrode arrays could also be utilised within a flow injection system [
52]. A sample was separated and flushed simultaneously through eight cells, each containing a screen-printed electrode and fitted with a separate bespoke mini-potentiostat (
Figure 20). This offers the possibility of allowing multiple measurements to be made on a single water sample. Each electrode could be designed to be specific for a different pesticide due to the availability of different AChE mutants. Alternatively native enzymes could also be installed as controls to show the presence of other interfering pollutants such as heavy metals. These arrays again proved successful with a high sensitivity for pesticide detection, limits of detection for dichlorvos as low as 1 × 10
−17 mol L
−1 and parathion and azinphos both at concentrations as low as 1 × 10
−16 mol L
−1 [
54].
We have developed sensitive and selective enzyme electrodes based on poly(aniline) microarrays capable of measuring concentrations of several pesticides down to levels hitherto undetectable (1 × 10−17 mol L−1). There is the potential for the use of multiple electrodes, pattern recognition software and flow injection techniques to enable determination of different pesticides from mixtures as well as subtraction of matrix effects such as heavy metals from the system. The automation within this system allows use of the instrumentation by unskilled personal, thereby removing the sensing platform from specialized laboratories and making it directly available to the end-users. The relative low cost of electrochemical technology and mass produced screen printed electrodes (which can be utilised as single shot sensors) makes it an attractive, inexpensive alternative to other technologies. This low cost, coupled to equipment portability, means it could conceivably be utilised in the field as well as under laboratory conditions.
3.5. Immunosensors Based on Poly(aniline) Microarrays
The interaction of antibodies and antigens leads to formation of tightly bound complexes with a very high degree of both sensitivity and selectivity. This selectivity has led to the development of such widely used assays such as the Enzyme Linked Immunosensor Assay (ELISA). There has been a wide interest in exploiting the selectivity of antibody-antigen interaction within electrochemical based immunosensors. The development of immunosensors which give electrochemical responses is a widely studied topic as detailed in several recent reviews [
57–
59].
Our group has also pioneered the development of sonochemically fabricated microarrays of conductive polymers for use in enzyme based sensors and also the studied the development of immunosensors which utilise either polymer entrapment or an affinity based protocol. Our first venture into this field involved the development of immunosensors for bovine serum albumin (BSA) and digoxin. Poly(pyrrole) was laid down on a carbon electrode by simple electrochemical polymerisation in solutions that contained antibodies to either BSA or digoxin. Studies using a radioactive labelled antibody showed that up to 2–3 μg of antibody may be successfully entrapped into the conducting poly(pyrrole) films [
60].
Unfortunately the interaction between antibody and antigen does not release any electrochemically active species as observed for the enzyme reactions, so simple voltammetry will not measure a response upon binding. Instead we utilised an AC impedance protocol [
61] and showed that the binding of antigen gave measurable changes in the impedance, allowing the development of immunosensors for digoxin and BSA. However problems exist with this technique. The first complication is with the deposition of a conductive polymer film which may require conditions such as a low pH under which the biological species of interest may suffer some loss of activity or even be completely denatured. Also entrapped antibodies may experience steric effects from the matrix or be wrongly orientated to bind the antigen, especially in the case of bulky antigens such as proteins. Therefore in later work we utilised a method in which poly(aniline) coated screen-printed carbon electrodes were used as substrates for antibody immobilisation via avidin-biotin interactions.
High quality poly(aniline) films could be deposited at low pH and then biotinylated using commercial biotinylation reactions. The interaction between biotin units and avidin proteins is well known as a method of selective immobilisation of various species. Therefore treating the biotinylated poly(aniline) with a solution of neutravidin allowed the deposition of a monolayer of the protein onto the conductive polymer. Meanwhile in a separate procedure, the same biotinylation reagent is used to treat antibodies to generate antibodies with a number, usually between two and four, of biotin units per antibody on average. Exposure of the avidin coated poly(aniline) electrode to these moieties caused deposition of orientated antibodies on the electrode surface as shown schematically in
Figure 21.
We successfully utilised this approach to construct immunosensors for the antibiotic ciprofloxacin which could detect levels of 1 ng mL
−1 of the antibiotic, not only in PBS [
62] but also in milk [
63]. We have also developed electrochemical immunosensors for myelin basic protein—a marker for stroke and multiple sclerosis [
64] and Internalin B, a surface protein marker for Listeria [
65].
Different methods of immobilisation were also investigated, a programme of work which is described in more detail elsewhere [
66]. Initial studies had utilised antibodies immobilised in planar polymer films but our work on the affinity modified planar electrodes gave higher sensitivities than noted for these films. It was therefore decided to directly compare entrapped and affinity immobilised immunosensors and also to compare planar electrodes with microelectrodes. Immunosensors containing anti-digoxin were fabricated by utilising the neutravidin-biotin procedure and shown to have limits of detection approximately 1,000 times less [
66] than electrodes where the antibody was simply entrapped within a polymer film. Similarly entrapment of BSA in microelectrode arrays led to almost a 10,000 fold lowering in detection limit compared to entrapment within planar films.
One shortcoming of immunosensors is that they effectively measure deposition of material on the electrode surface. However many materials, either the antigen itself or interfering species found especially in complex media like blood can undergo non-specific interactions with surfaces. This can interfere with immunosensor performance, leading to erroneously elevated results. In an attempt to correct for non specific interactions, identical sets of immunosensors were therefore fabricated containing a non-specific IgG antibody (AIgG) in place of the specific antibodies. Results for these electrodes were obtained in exactly the same manner as for the specific electrodes. Therefore within this work we will only detail results which have been corrected. To obtain a “corrected” calibration profile, for an APSA immunosensor, non-specific responses were subtracted from the specific responses over the entire analytical concentration range.
Since both the affinity protocol and the use of microelectrodes led to increases in sensitivity, these two techniques were combined. This study was performed using antibodies for prostate specific antigen (PSA). Prostate cancer is a disease most frequently encountered in men over fifty and second only to lung cancer for the number of male deaths in the US, 30,350 deaths in 2005 [
67] and the UK, 10,000 deaths in 2005 [
68]. Levels of a 34 kD glycoprotein PSA, which is manufactured almost exclusively by the prostate gland are often (but not always) elevated in the presence of prostate cancer and other prostate disorders [
69]. We will only give an overview of the construction and utilisation of microarray based biosensors for PSA but a much more detailed description has been published [
70].
As before, poly(aniline) microarrays were fabricated using the methods described. In some cases, antibodies to PSA (APSA) were entrapped by growing the microelectrodes from solutions containing APSA. Alternatively microarrays of poly(aniline) were treated with the biotinylating agent, followed by avidin and finally biotinylated APSA to give microarrays with immobilised antibodies. Impedance analyses were performed from 1 Hz to 10,000 Hz (±5 mV amplitude perturbation) offset to the formal potential of the redox probe (+0.12 V versus Ag/AgCl). A redox mediator mixture, 5 mmol L−1 each of [Fe(CN)6]3−/4−, was utilised to enable the performance of faradaic impedance spectroscopy. Baseline traces were obtained for the APSA doped sensors in phosphate buffer containing the mediator but no antigen—these were subsequently exposed to a range of concentrations from 1 to 300 ng or pg mL−1 PSA in pH 7.4 phosphate buffer containing [Fe(CN)6] 3−/4−.
A series of typical Nyquist plots (
Figure 22) are shown for the sensors containing (a) affinity immobilised APSA and (b) entrapped APSA. The immobilised sensors display large changes in the Nyquist plots at much lower concentrations of antigen than occurred for the entrapped microelectrode arrays. It appears that the effects of affinity binding and the use of microelectrodes are complimentary and combining them leads to further enhancements in sensitivity and lowering of detection limits.
The semicircular shape of the Nyquist plots is characteristic of a surface modified electrode system where the electron transfer is slow and the impedance is controlled by the interfacial electron transfer [
71]. From these curves it is possible to extract the surface electron transfer resistance. Relative electron transfer resistance changes from the baseline response can be plotted against the antigen concentration to give calibration profiles. Binding of antigen appears to hinder the access of the redox probe to the surface, thereby insulating it and increasing the electron transfer resistance. Entrapped APSA immunosensors gave linear responses to the analyte from 1 ng mL
−1 to 200 ng mL
−1 PSA and then reached a plateau. Affinity based immunosensors gave a linear response from 1 pg mL
−1 to 100 pg mL
−1 PSA.
Measurements of the AIgG electrodes also gave changes in Nyquist curves and calibration plots could also be drawn, indicating that about 20% of the increase in electron transfer resistance was in fact due to non-specific effects [
70]. Therefore these results were subtracted from the specific binding results to give the corrected results (average of 10 specific and 10 non-specific electrodes) as shown in
Figure 23. As can be seen the immobilised immunosensors display much higher sensitivities and lower detection limits, with a linear range from 1 to 100 pg mL
−1 PSA and the entrapped immunosensors from 1–100 ng mL
−1 PSA. Insets in the figures show expanded plots for the lower concentrations.
It appears that the best sensitivities can be obtained by combining poly(aniline) microarrays with affinity based attachment protocols. The detection limit for PSA (three times the standard deviation of the baseline value) was 1 pg mL
−1. It was therefore decided to study a range of other antigens. Two which were of interest were Neuron Specific Enolase (NSE), a 78 KDa protein and S100[β], a 21 KDa protein. Both of these proteins, when found in blood, are markers for transient ischemic events such as strokes. Stroke is the third leading cause of death in the USA and the UK after heart disease and all cancers [
72,
73]. Delays of above three hours after stroke onset in medical intervention may contribute to patient deterioration and disability. To help identify those individuals at risk, there is a need for a rapid, sensitive and specific diagnostic assay for stroke events. Both NSE and S100[β] concentrations in blood are elevated after stroke or other cerebral injury. We therefore instigated a study into the detection of stroke marker proteins. Again detailed descriptions of this work have been published [
74,
75] so an overview will be given.
Antibodies to NSE and S100[β] were biotinylated and immobilised at the surface of poly(aniline) microarrays as described for PSA. The analysis was carried out as before and strong specific responses noted for both proteins. Non-specific binding was also observed for both proteins, of a similar level to that noted for PSA and was therefore subtracted. Corrected calibration plots for both proteins are shown in
Figure 24. Both sensors were capable of detecting low levels of the requisite antigen. For NSE a linear relationship was observed between concentrations of 1–50 pg mL
−1 (
Figure 24a) and the response tends towards a plateau as the sensor approaches saturation. A limit of detection for the immunosensor (three times the standard deviation of the baseline value) of 0.5 pg mL
−1 is obtained. The sensitivity of the S100[β] is somewhat lower than the other two immunosensors as shown in
Figure 24b, however it still gives good response at extremely low levels of antigen with the limit of detection of the AS100 immunosensor (three times the standard deviation of the baseline value) being 1 pg mL
−1 S-100[β].
Attempts were made to measure the effect on other proteins in the samples and whether they would interfere. Affinity bound immunosensors for all three antigens were fabricated and the testing protocol repeated except that in each case the solution, besides containing the antigen also contained a cocktail of 10 ng mL−1 each of several interfering proteins (in the case of PSA these were BSA, HSA, CA125 protein (tumour marker), S100[β] and NSE) along with the antigen. With the other two antigens, PSA 10 ng mL−1 replaced the antigen in the cocktail. For all three antigens the mean “corrected” electron transfer changes showed a variation of less than 5% from the unadulterated samples. This indicates either minimal adsorption of potential interfering proteins or if it does occur it does not significantly affect the biorecognition events (stable immunocomplex formation) and its effect on impedance.
The regeneration and stability of these affinity bound complexes were also tested. Samples of all three immunosensors were tested over the range of concentrations shown as before, given three 1 minute washes in 0.1 mol L
−1 HCl and then retested.
Figure 25a shows the overall results for the response at 100 pg mL
−1 antigen for the immunosensors. As can be seen the PSA sensors could be rinsed and reused once with no loss of response, a second wash lowered it to 85% and further washes caused further loss of performance. Similar behaviour was observed for NSE and S100[β] in that the first regeneration did not affect them but after a second washing the performance had dropped to 60% and 15% of the original respectively. Washing immunosensors containing embedded APSA with 0.1 mol L
−1 HCl completely destroyed their activity.
Samples of all three immunosensors were stored dry at 4 °C for up to twelve weeks and two of each tested at two week intervals. As
Figure 25b shows, the performance did not deteriorate with storage. Samples containing embedded PSA were also tested, and were found to be stable for up to six weeks but after 12 weeks their performance had deteriorated to about a third of “fresh” sensors [
66].
This section provides an overview towards the development of a viable approach towards the labeless sensing of markers for cancer and stroke, thus demonstrating the generic nature of the technology. The combination of microarrays and affinity binding protocols have given sensors with good linear ranges and limits of detection as low as 1 pg mL−1 for PSA, NSE and S-100[β].
The immunosensors were reversible for one regeneration attempt although after this further acid treatment reduces the performance, probably due to either loss of or denaturation of the immobilised antibody. Negligible loss of sensitivity after storage for twelve weeks at 4 °C in the dry state was observed. Minimal interference was observed upon exposure to relatively high levels of other proteins, encouraging for possible future applications.
These initial studies show great promise and it should be noted the limits of detection of the affinity based sensors are far below the levels found in clinical samples. This high sensitivity means that clinical samples can be highly diluted in PBS before measurement, thereby greatly reducing the amount of initial sample required, simplifying sample handling and minimising the effects of potential interferents.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



























