

Mitogenomic Phylogenetic Analyses Reveal New Insights into the Taxonomy and Evolution of Parnassiinae Swallowtail Butterflies (Lepidoptera: Papilionidae)

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, Identification, and DNA Extraction
2.2. Mitogenome Sequencing Assembly, Annotation, and Sequence Analysis
2.3. Phylogenetic Analyses
3. Results
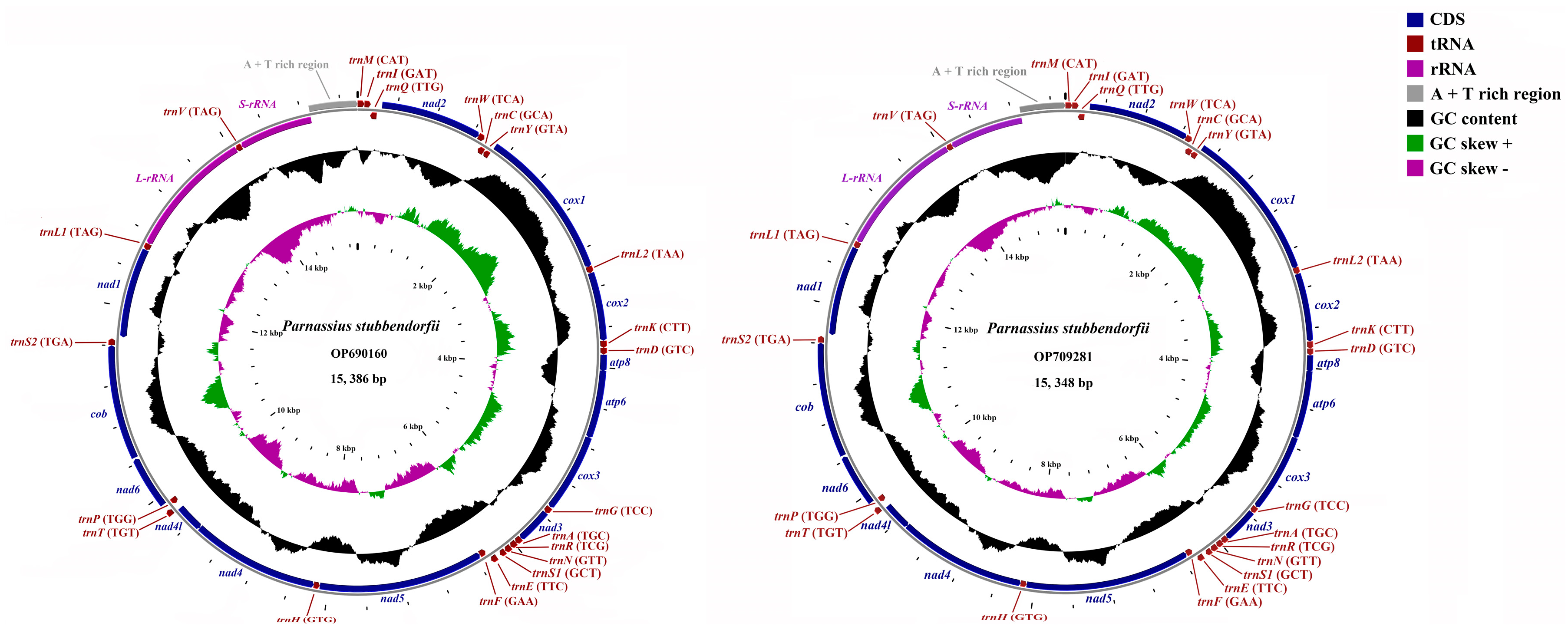
3.1. General Features of the Sequenced Mitogenomes
3.2. Protein-Coding Genes
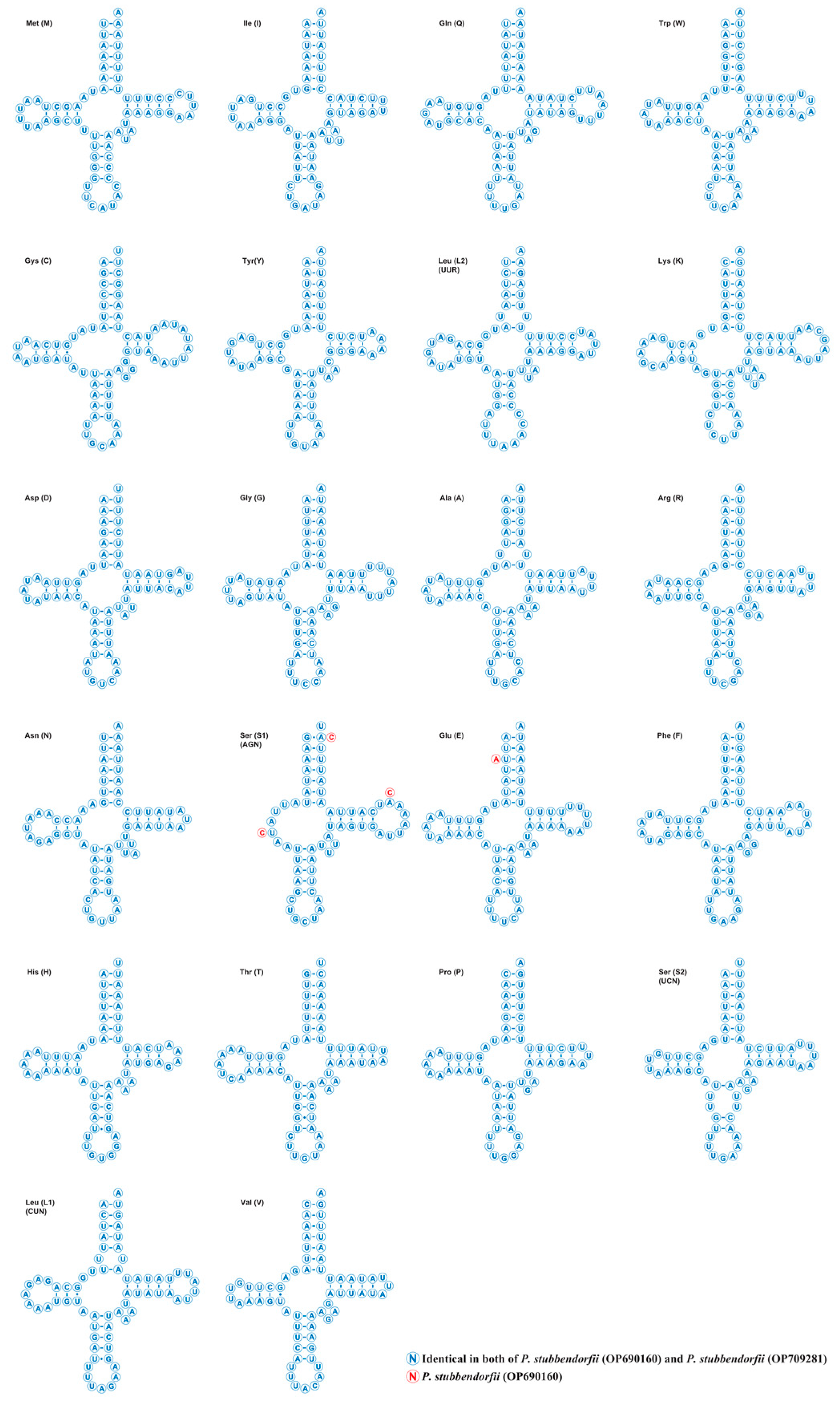
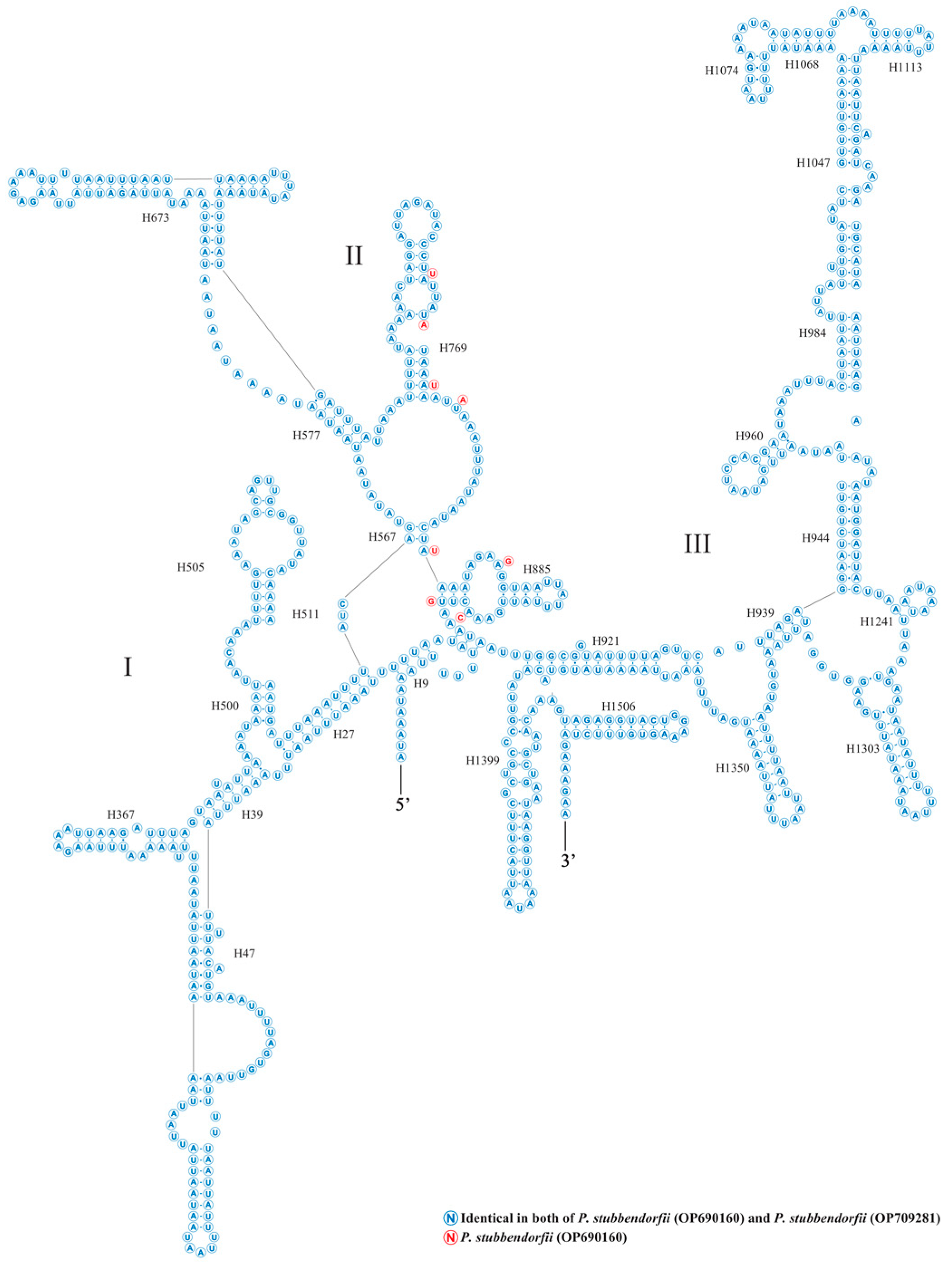
3.3. Transfer and Ribosomal RNA Genes
3.4. Gene Overlapping and Intergenic Regions
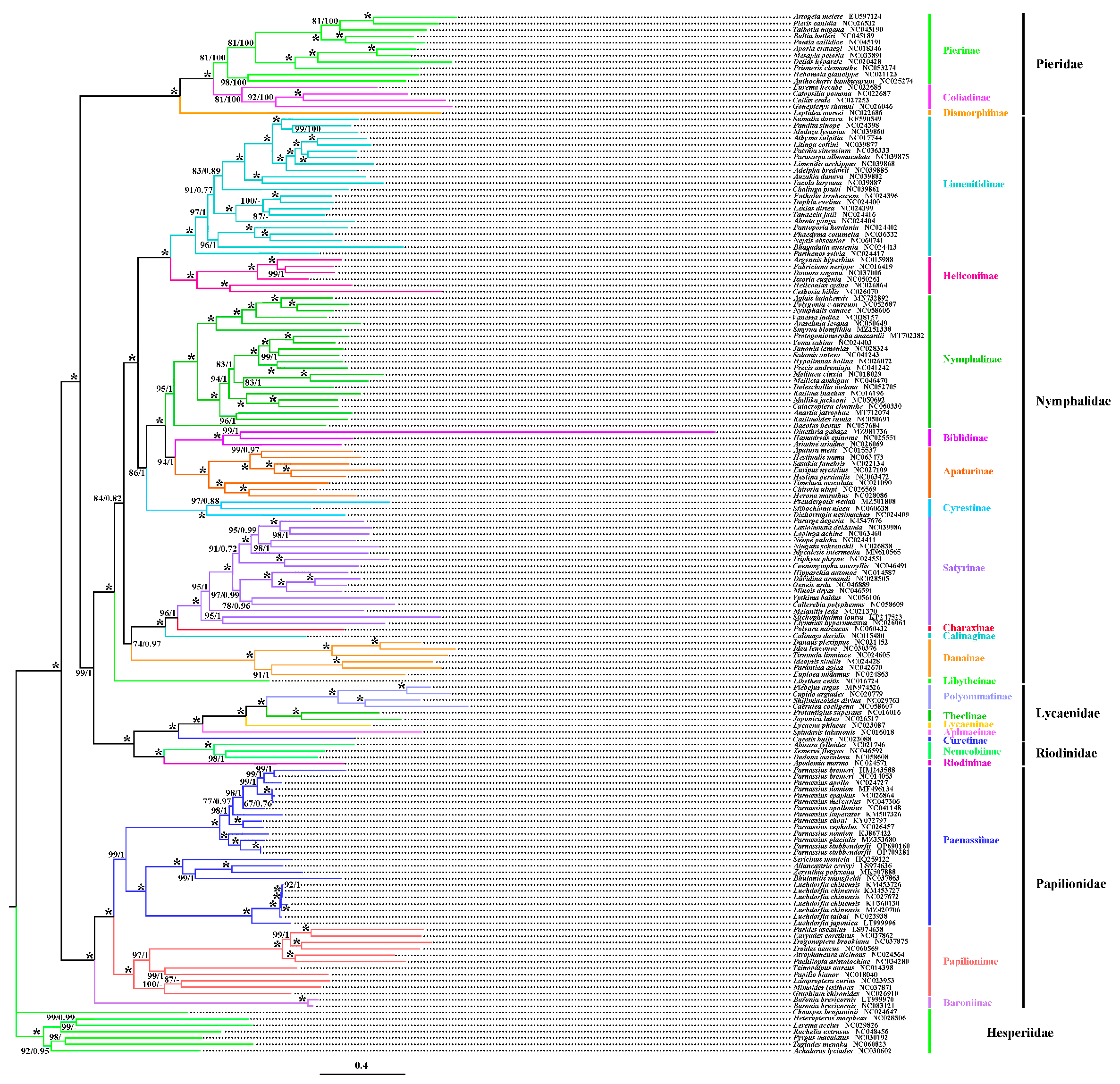
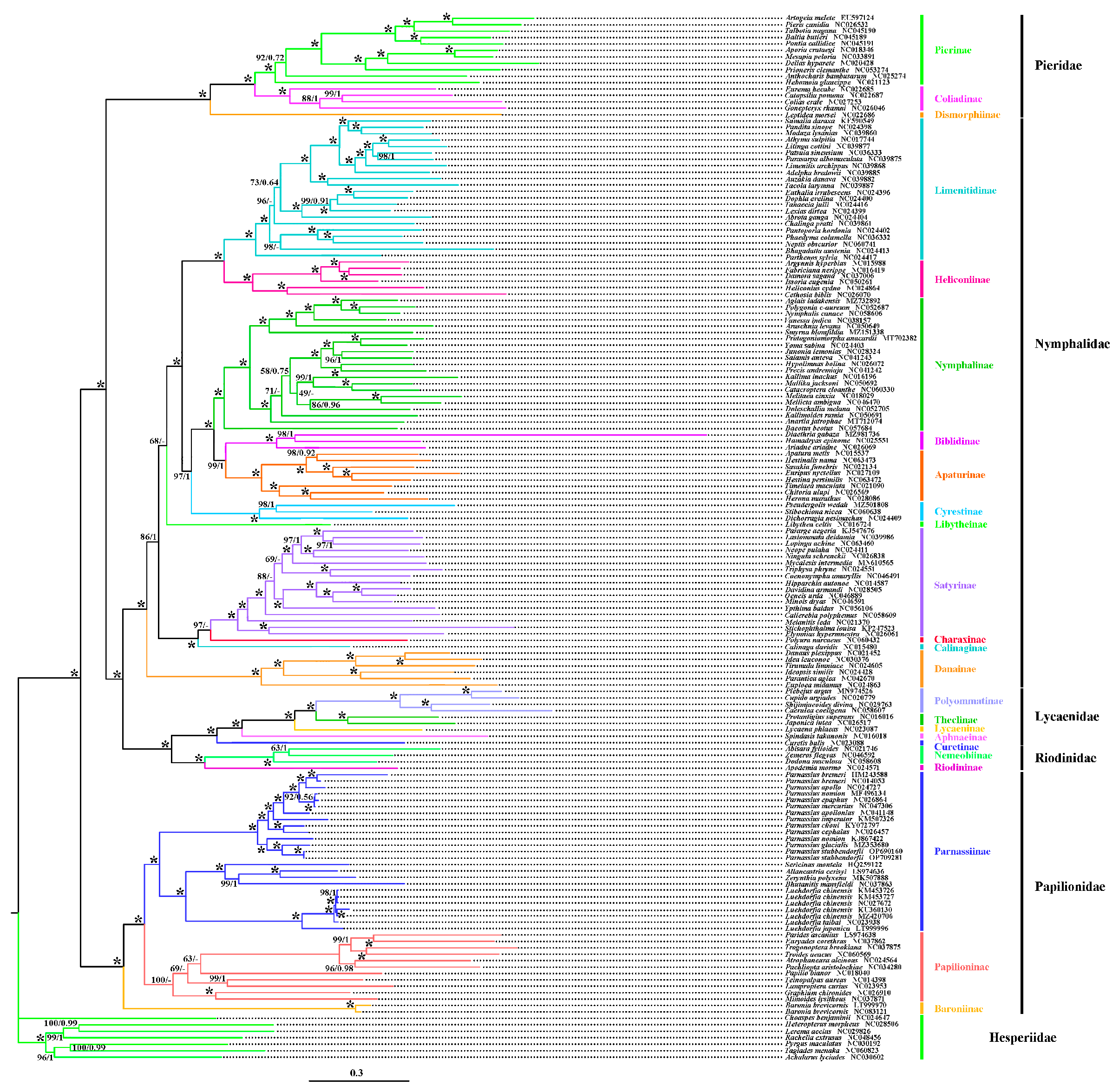
3.5. Phylogenetic Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Superfamily | Family | Subfamily | Species | GenBank Accession No. | Genome Size (bp) |
|---|---|---|---|---|---|
| Papilionoidea | Lycaenidae | Aphnaeinae | Spindasis takanonis | NC016018 | 15,349 |
| Curetinae | Curetis bulis | NC023088 | 15,162 | ||
| Lycaeninae | Lycaena phlaeas | NC023087 | 15,280 | ||
| Polyommatinae | Caerulea coeligena | NC058607 | 15,164 | ||
| Cupido argiades | NC020779 | 15,330 | |||
| Plebejus argus | MN974526 | 15,426 | |||
| Shijimiaeoides divina | NC029763 | 15,259 | |||
| Theclinae | Japonica lutea | NC026517 | 15,225 | ||
| Protantigius superans | NC016016 | 15,248 | |||
| Nymphalidae | Apaturinae | Apatura metis | NC015537 | 15,236 | |
| Chitoria ulupi | NC026569 | 15,279 | |||
| Euripus nyctelius | NC027109 | 15,417 | |||
| Herona marathus | NC028086 | 15,487 | |||
| Hestina persimilis | NC063472 | 15,252 | |||
| Hestinalis nama | NC063473 | 15,208 | |||
| Sasakia funebris | NC022134 | 15,233 | |||
| Timelaea maculata | NC021090 | 15,178 | |||
| Biblidinae | Ariadne ariadne | NC026069 | 15,179 | ||
| Diaethria gabaza | MZ981736 | 15,156 | |||
| Hamadryas epinome | NC025551 | 15,207 | |||
| Calinaginae | Calinaga davidis | NC015480 | 15,267 | ||
| Charaxinae | Polyura narcaeus | NC060432 | 15,319 | ||
| Cyrestinae | Dichorragia nesimachus | NC024409 | 15,355 | ||
| Pseudergolis wedah | MZ501808 | 15,079 | |||
| Stibochiona nicea | NC060638 | 15,298 | |||
| Danainae | Danaus plexippus | NC021452 | 15,314 | ||
| Euploea midamus | NC024863 | 15,187 | |||
| Idea leuconoe | NC030376 | 15,278 | |||
| Ideopsis similis | NC024428 | 15,200 | |||
| Parantica aglea | NC042670 | 15,219 | |||
| Tirumala limniace | NC024605 | 15,285 | |||
| Heliconiinae | Argynnis hyperbius | NC015988 | 15,156 | ||
| Cethosia biblis | NC026070 | 15,211 | |||
| Damora sagana | NC037006 | 15,151 | |||
| Fabriciana nerippe | NC016419 | 15,140 | |||
| Heliconius cydno | NC024864 | 15,367 | |||
| Issoria eugenia | NC050261 | 15,206 | |||
| Libytheinae | Libythea celtis | NC016724 | 15,164 | ||
| Limenitidinae | Abrota ganga | NC024404 | 15,356 | ||
| Adelpha bredowii | NC039885 | 15,187 | |||
| Athyma sulpitia | NC017744 | 15,268 | |||
| Auzakia danava | NC039882 | 15,367 | |||
| Bhagadatta austenia | NC024413 | 15,615 | |||
| Chalinga pratti | NC039861 | 15,290 | |||
| Dophla evelina | NC024400 | 15,320 | |||
| Euthalia irrubescens | NC024396 | 15,365 | |||
| Lexias dirtea | NC024399 | 15,250 | |||
| Limenitis archippus | NC039868 | 15,220 | |||
| Litinga cottini | NC039877 | 15,205 | |||
| Moduza lysanias | NC039860 | 15,238 | |||
| Neptis obscurior | NC060741 | 15,172 | |||
| Pandita sinope | NC024398 | 15,257 | |||
| Pantoporia hordonia | NC024402 | 15,603 | |||
| Parasarpa albomaculata | NC039875 | 15,272 | |||
| Parthenos sylvia | NC024417 | 15,249 | |||
| Patsuia sinensium | NC036333 | 15,192 | |||
| Phaedyma columella | NC036332 | 15,197 | |||
| Sumalia daraxa | KF590549 | 15,173 | |||
| Tacola larymna | NC039887 | 15,503 | |||
| Tanaecia julii | NC024416 | 15,316 | |||
| Nymphalinae | Aglais ladakensis | MN732892 | 15,222 | ||
| Anartia jatrophae | MT712074 | 15,297 | |||
| Araschnia levana | NC050649 | 15,207 | |||
| Baeotus beotus | NC057684 | 15,131 | |||
| Catacroptera cloanthe | NC060330 | 15,204 | |||
| Doleschallia melana | NC052705 | 15,269 | |||
| Hypolimnas bolina | NC026072 | 15,260 | |||
| Junonia lemonias | NC028324 | 15,230 | |||
| Kallima inachus | NC016196 | 15,183 | |||
| Kallimoides rumia | NC050691 | 15,234 | |||
| Melitaea cinxia | NC018029 | 15,170 | |||
| Mallika jacksoni | NC050692 | 15,193 | |||
| Mellicta ambigua | NC046470 | 15,205 | |||
| Nymphalis canace | NC058606 | 15,216 | |||
| Polygonia c-aureum | NC052687 | 15,208 | |||
| Precis andremiaja | NC041242 | 15,239 | |||
| Protogoniomorpha anacardii | MT702382 | 15,220 | |||
| Salamis anteva | NC041243 | 15,201 | |||
| Smyrna blomfildia | MZ151338 | 15,149 | |||
| Vanessa indica | NC038157 | 15,191 | |||
| Yoma sabina | NC024403 | 15,330 | |||
| Satyrinae | Callerebia polyphemus | NC058609 | 15,156 | ||
| Coenonympha amaryllis | NC046491 | 15,125 | |||
| Davidina armandi | NC028505 | 15,214 | |||
| Elymnias hypermnestra | NC026061 | 15,167 | |||
| Hipparchia autonoe | NC014587 | 15,489 | |||
| Lasiommata deidamia | NC039986 | 15,244 | |||
| Lopinga achine | NC063460 | 15,284 | |||
| Melanitis leda | NC021370 | 15,122 | |||
| Minois dryas | NC046591 | 15,195 | |||
| Mycalesis intermedia | MN610565 | 15,386 | |||
| Neope pulaha | NC024411 | 15,209 | |||
| Ninguta schrenckii | NC026838 | 15,261 | |||
| Oeneis urda | NC046889 | 15,248 | |||
| Pararge aegeria | KJ547676 | 15,240 | |||
| Stichophthalma louisa | KP247523 | 15,721 | |||
| Triphysa phryne | NC024551 | 15,143 | |||
| Ypthima baldus | NC056106 | 15,304 | |||
| Baroniinae | Baronia brevicornis | LT999970 | 14,918 | ||
| Baronia brevicornis | NC083121 | 15,173 | |||
| Papilionidae | Papilioninae | Atrophaneura alcinous | NC024564 | 15,266 | |
| Euryades corethrus | NC037862 | 15,133 | |||
| Graphium chironides | NC026910 | 15,235 | |||
| Lamproptera curius | NC023953 | 15,277 | |||
| Mimoides lysithous | NC037871 | 15,038 | |||
| Pachliopta aristolochiae | NC034280 | 15,232 | |||
| Papilio bianor | NC018040 | 15,340 | |||
| Parides ascanius | LS974638 | 15,212 | |||
| Teinopalpus aureus | NC014398 | 15,242 | |||
| Troides aeacus | NC060569 | 15,224 | |||
| Trogonoptera brookiana | NC037875 | 15,005 | |||
| Parnassiinae | Allancastria cerisyi | LS974636 | 15,279 | ||
| Bhutanitis mansfieldi | NC037863 | 14,994 | |||
| Luehdorfia chinensis | MZ420706 | 15,443 | |||
| Luehdorfia chinensis | KU360130 | 15,550 | |||
| Luehdorfia chinensis | KM453726 | 15,580 | |||
| Luehdorfia chinensis | KM453727 | 15,580 | |||
| Luehdorfia chinensis | NC027672 | 16,028 | |||
| Luehdorfia japonica | LT999996 | 15,347 | |||
| Luehdorfia taibai | NC023938 | 15,553 | |||
| Parnassius apollo | NC024727 | 15,404 | |||
| Parnassius apollonius | NC041148 | 15,381 | |||
| Parnassius bremeri | NC014053 | 15,389 | |||
| Parnassius bremeri | HM243588 | 15,390 | |||
| Parnassius cephalus | NC026457 | 15,343 | |||
| Parnassius choui | KY072797 | 15,367 | |||
| Parnassius epaphus | NC026864 | 15,458 | |||
| Parnassius glacialis | MZ353680 | 15,353 | |||
| Parnassius imperator | KM507326 | 15,424 | |||
| Parnassius mercurius | NC047306 | 15,372 | |||
| Parnassius nomion | MF496134 | 15,362 | |||
| Parnassius nomion | KJ867422 | 14,557 | |||
| Parnassius stubbendorfii | OP690160 | 15,386 | |||
| Parnassius stubbendorfii | OP709281 | 15,348 | |||
| Sericinus montela | HQ259122 | 15,242 | |||
| Zerynthia polyxena | MK507888 | 15,092 | |||
| Pieridae | Coliadinae | Catopsilia pomona | NC022687 | 15,142 | |
| Colias erate | NC027253 | 15,184 | |||
| Eurema hecabe | NC022685 | 15,160 | |||
| Gonepteryx rhamni | NC026046 | 15,203 | |||
| Dismorphiinae | Leptidea morsei | NC022686 | 15,122 | ||
| Pierinae | Anthocharis bambusarum | NC025274 | 15,180 | ||
| Aporia crataegi | NC018346 | 15,140 | |||
| Artogeia melete | EU597124 | 15,140 | |||
| Baltia butleri | NC045189 | 15,124 | |||
| Delias hyparete | NC020428 | 15,186 | |||
| Hebomoia glaucippe | NC021123 | 15,701 | |||
| Mesapia peloria | NC033891 | 15,159 | |||
| Pieris canidia | NC026532 | 15,153 | |||
| Pontia callidice | NC045191 | 15,109 | |||
| Prioneris clemanthe | NC053274 | 15,131 | |||
| Talbotia nagana | NC045190 | 15,155 | |||
| Riodinidae | Nemeobiinae | Abisara fylloides | NC021746 | 15,301 | |
| Dodona maculosa | NC058608 | 15,486 | |||
| Zemeros flegyas | NC046592 | 15,219 | |||
| Riodininae | Apodemia mormo | NC024571 | 15,262 | ||
| Outgroup | |||||
| Hesperioidea | Hesperiidae | Coeliadinae | Choaspes benjaminii | NC024647 | 15,300 |
| Eudaminae | Achalarus lyciades | NC030602 | 15,612 | ||
| Hesperiinae | Lerema accius | NC029826 | 15,338 | ||
| Heteropterinae | Heteropterus morpheus | NC028506 | 15,769 | ||
| Pyrginae | Pyrgus maculatus | NC030192 | 15,346 | ||
| Tagiadinae | Tagiades menaka | NC060823 | 15,294 | ||
| Trapezitinae | Rachelia extrusus | NC048456 | 16,114 |
| Feature | Strand | Location | Start Codon | Stop Codon | Anticodon | Intergenic Nucleotides | |
|---|---|---|---|---|---|---|---|
| From | To | ||||||
| trnM | J | 1/1 | 68/68 | CAT/CAT | 0/0 | ||
| trnI | J | 69/69 | 132/132 | GAT/GAT | −3/−3 | ||
| trnQ | N | 130/130 | 198/198 | TTG/TTG | 47/47 | ||
| nad2 | J | 246/246 | 1259/1259 | ATC/ATC | TAA/TAA | −1/−1 | |
| trnW | J | 1259/1259 | 1324/1324 | TCA/TCA | −8/−8 | ||
| trnC | N | 1317/1317 | 1383/1383 | GCA/GCA | 3/3 | ||
| trnY | N | 1387/1387 | 1450/1450 | GTA/GTA | 3/3 | ||
| cox1 | J | 1454/1454 | 2984/2984 | CGA/CGA | T/T | 0/0 | |
| trnL2 | J | 2985/2985 | 3051/3051 | TAA/TAA | 0/0 | ||
| cox2 | J | 3052/3052 | 3733/3733 | ATG/ATG | T/T | 0/0 | |
| trnK | J | 3734/3734 | 3804/3804 | CTT/CTT | −1/−1 | ||
| trnD | J | 3804/3804 | 3870/3870 | GTC/GTC | 0/0 | ||
| atp8 | J | 3871/3871 | 4035/4035 | ATT/ATT | TAA/TAA | −7/−7 | |
| atp6 | J | 4029/4029 | 4706/4706 | ATG/ATG | TAA/TAA | −1/−1 | |
| cox3 | J | 4706/4706 | 5494/5494 | ATG/ATG | TAA/TAA | 3/3 | |
| trnG | J | 5498/5498 | 5564/5564 | TCC/TCC | 0/0 | ||
| nad3 | J | 5565/5565 | 5918/5918 | ATT/ATT | TAA/TAA | −1/−1 | |
| trnA | J | 5918/5918 | 5983/5983 | TGC/TGC | −1/−1 | ||
| trnR | J | 5983/5983 | 6048/6048 | TCG/TCG | 2/2 | ||
| trnN | J | 6051/6051 | 6116/6116 | GTT/GTT | 3/3 | ||
| trnS1 | J | 6120/6120 | 6180/6180 | GCT/GCT | 39/35 | ||
| trnE | J | 6220/6216 | 6285/6281 | TTC/TTC | −2/−2 | ||
| trnF | N | 6284/6280 | 6350/6346 | GAA/GAA | 1/1 | ||
| nad5 | N | 6352/6348 | 8085/8081 | ATT/ATT | TAA/TAA | 0/0 | |
| trnH | N | 8086/8082 | 8149/8145 | GTG/GTG | −1/−1 | ||
| nad4 | N | 8149/8145 | 9489/9485 | ATG/ATG | TAA/TAA | 0/0 | |
| nad4l | N | 9490/9486 | 9780/9776 | ATG/ATG | TAA/TAA | 2/2 | |
| trnT | J | 9783/9779 | 9848/9844 | TGT/TGT | 0/0 | ||
| trnP | N | 9849/9845 | 9913/9909 | TGG/TGG | 2/2 | ||
| nad6 | J | 9916/9912 | 10,446/10,442 | ATT/ATT | TAA/TAA | 13/13 | |
| cob | J | 10,460/10,456 | 11,611/11,604 | ATG/ATG | TAA/TAA | 1/1 | |
| trnS2 | J | 11,613/11,606 | 11,678/11,671 | TGA/TGA | 16/16 | ||
| nad1 | N | 11,695/11,688 | 12,633/12,626 | ATG/ATG | TAG/TAG | 1/1 | |
| trnL1 | N | 12,635/12,628 | 12,702/12,695 | TAG/TAG | 0/0 | ||
| rrnL | N | 12,703/12,696 | 14,056/14,049 | 0/0 | |||
| trnV | N | 14,057/14,050 | 14,120/14,113 | TAC/TAC | 0/0 | ||
| rrnS | N | 14,121/14,114 | 14,899/14,891 | 0/0 | |||
| A + T-rich region | 14,900/14,892 | 15,386/15,348 | |||||
| Size (bp) | A (%) | T (%) | C (%) | G (%) | AT Content (%) | AT-Skew | GC-Skew | |
|---|---|---|---|---|---|---|---|---|
| Genome | 15,377/ 15,348 | 40.42/ 40.64 | 40.85/ 40.92 | 11.19/ 11.02 | 7.53/ 7.42 | 81.27/ 81.56 | −0.00529/ −0.00342 | −0.19551/ −0.19523 |
| PCGs | 11,197/ 11,203 | 39.94/ 39.93 | 40.35/ 40.37 | 11.60/ 11.60 | 8.11/ 8.10 | 80.29/ 80.30 | −0.00511/ −0.00548 | −0.17707/ −0.17766 |
| tRNAs | 1455/ 1455 | 40.41/ 40.48 | 41.10/ 41.24 | 10.52/ 10.31 | 7.97/ 7.97 | 81.51/ 81.72 | −0.00847/ −0.00930 | −0.13791/ −0.12801 |
| rRNAs | 2133/ 2132 | 42.05/ 42.12 | 42.38/ 42.50 | 10.50/ 10.37 | 5.06/ 5.02 | 84.43/ 84.62 | −0.00391/ −0.00449 | −0.34961/ −0.34763 |
| A + T-rich region | 487/ 457 | 44.15/ 51.42 | 42.92/ 43.98 | 7.80/ 2.84 | 5.13/ 1.75 | 87.07/ 95.40 | 0.01413/ 0.07799 | −0.20650/ −0.23747 |
| Partitions | Models | Genes |
|---|---|---|
| P1 | GTR + I + G | c3p1; a6p1, cbp1 |
| P2 | GTR + I + G | c1p2, c2p2, a6p2 |
| P3 | TRN + G | a6p3, a8p3 |
| P4 | TIM + I + G | a8p1 |
| P5 | TIM + I + G | a8p2 |
| P6 | GTR + I + G | c2p1, c1p1 |
| P7 | GTR + G | c1p3 |
| P8 | GTR + I + G | c2p3, n6p3, c3p3, cbp3 |
| P9 | TVM + I + G | c3p2, cbp2 |
| P10 | TVM + I + G | n1p1, n5p1, n4p1, n4lp1 |
| P11 | GTR + I + G | n4lp2, n5p2, n4p2, n1p2 |
| P12 | TIM + I + G | n5pos3, n1p3, n4lp3 |
| P13 | GTR + I + G | n2p1 |
| P14 | TVM + I + G | n2p2, n6p2 |
| P15 | GTR + G | n2p3 |
| P16 | GTR + I + G | n3p1 |
| P17 | TVM + I + G | n3p2 |
| P18 | TRN + I + G | n3p3 |
| P19 | TIM + G | n4p3 |
| P20 | GTR + I + G | n6p1 |
| P21 | GTR + I + G | rrnS |
| P22 | GTR + I + G | rrnL |
| P23 | GTR + I + G | tRNAs |
| Partitions | Models | Genes |
|---|---|---|
| P1 | GTR + F + R6 | a6p1; c3p1; cbp1 |
| P2 | GTR + F + R4 | a6p2; c2p2; c3p2; cbp2 |
| P3 | TPM2 + F + R5 | a6p3 |
| P4 | TIM3 + F + I + G4 | a8p1 |
| P5 | TIM2 + F + I + G4 | a8p2; n3p1 |
| P6 | TPM2 + F + R5 | a8p3; n6p3 |
| P7 | GTR + F + R4 | c1p1; c2p1 |
| P8 | GTR + F + I + G4 | c1p2 |
| P9 | TVM + F + R7 | c1p3 |
| P10 | TIM + F + R7 | c2p3 |
| P11 | TIM2 + F + R7 | c3p3 |
| P12 | TN + F + R7 | cbp3; n3p3 |
| P13 | TVM + F + R6 | n1p1; n4p1; n4lp1; n5p1 |
| P14 | GTR + F + R5 | n1p2; n4p2; n4lp2; n5p2 |
| P15 | TIM + F + R8 | n1p3; n5p3 |
| P16 | GTR + F + R6 | n2p1; n6p1 |
| P17 | TVM + F + R4 | n2p2; n6p2 |
| P18 | GTR + F + R6 | n2p3 |
| P19 | GTR + F + I + G4 | n3p2 |
| P20 | TIM + F + R6 | n4p3; n4lp3; |
| P21 | GTR + F + R6 | rrnS |
| P22 | GTR + F + R6 | rrnL |
| P23 | GTR + F + R6 | tRNAs |
References
- Steffen, W.; Grinevald, J.; Crutzen, P.; McNeill, J. The Anthropocene: Conceptual and historical. Philos. Trans. R. Soc. A 2011, 369, 842–867. [Google Scholar] [CrossRef]
- Zalasiewicz, J.; Williams, M.; Haywood, A.; Ellis, M. The Anthropocene: A new epoch of geological time? Philos. Trans. R. Soc. A 2011, 369, 835–841. [Google Scholar] [CrossRef]
- Leichenko, R.; O’Brien, K. Teaching climate change in the Anthropocene: An integrative approach. Anthropocene 2020, 30, 100241. [Google Scholar] [CrossRef]
- Harvey, J.A.; Heinen, R.; Gols, R.; Thakur, M.P. Climate change-mediated temperature extremes and insects: From outbreaks to breakdowns. Glob. Chang. Biol. 2020, 26, 6685–6701. [Google Scholar] [CrossRef]
- Nordblad, J. On the difference between anthropocene and climate change temporalities. Crit. Inq. 2021, 47, 328–348. [Google Scholar] [CrossRef]
- Barua, M.; Gurdak, D.J.; Ahmed, R.A.; Tamuly, J. Selecting flagships for invertebrate conservation. Biodivers. Conserv. 2012, 21, 1457–1476. [Google Scholar] [CrossRef]
- Jepson, P.; Barua, M. A Theory of Flagship Species Action. Conserv. Soc. 2015, 13, 95–104. [Google Scholar] [CrossRef]
- Barbosa, W.F.; Smagghe, G.; Guedes, R.N.C. Pesticides and reduced-risk insecticides, native bees and pantropical stingless bees: Pitfalls and perspectives. Pest. Manag. Sci. 2015, 71, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, J.M.; Hogendoorn, K. How protection of honey bees can help and hinder bee conservation. Curr. Opin. Insect Sci. 2021, 46, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Botina, L.L.; Bernardes, R.C.; Barbosa, W.F.; Lima, M.A.P.; Martins, G.F. Toxicological assessments of agrochemical effects on stingless bees (Apidae, Meliponini). MethodsX 2020, 7, 100906. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, R.C.; Botina, L.L.; Araújo, R.D.S.; Guedes, R.N.C. Artificial Intelligence-Aided Meta-Analysis of Toxicological Assessment of Agrochemicals in Bees. Front. Ecol. Evol. 2022, 10, 845608. [Google Scholar] [CrossRef]
- DiLeo, J.M.F.; Nair, A.; Kardos, M.; Saastamoinen, M. Demography and environment modulate the effects of genetic diversity on extinction risk in a butterfly metapopulation. Proc. Natl. Acad. Sci. USA 2024, 121, e2309455121. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, G.; Ekroos, J.; Persson, A.S.; Pettersson, L.B.; Ockinger, E. Intensive management reduces butterfly diversity over time in urban green spaces. Urban. Ecosyst. 2019, 22, 335–344. [Google Scholar] [CrossRef]
- Rija, A.A. Local habitat characteristics determine butterfly diversity and community structure in a threatened Kihansi gorge forest, Southern Udzungwa Mountains, Tanzania. Ecol. Process 2022, 11, 13. [Google Scholar] [CrossRef]
- Condamine, F.L.; Nabholz, B.; Clamens, A.L.; Dupuis, J.R.; Sperling, F.A.H. Mitochondrial phylogenomics, the origin of swallowtail butterflies, and the impact of the number of clocks in Bayesian molecular dating. Syst. Entomol. 2018, 43, 460–480. [Google Scholar] [CrossRef]
- Nieukerken, E.J.; Kaila, L.; Kitching, J.; Kristensen, N.P. Order Lepidoptera Linnaeus, 1758. In Animal Biodiversity: An Outline of Higher-Level Classification and Survey of Taxonomic Richness; Zhang, Z.-Q., Ed.; Zootaxa—Magnolia Press: Auckland, New Zealand, 2011; Volume 3148, pp. 212–221. [Google Scholar]
- Wang, Y.; Chen, Y.; Xia, C.; Xia, X.; Chen, X.; Hao, J. The complete mitochondrial genome of Parnassius imperator (Lepidoptera: Papilionidae: Parnassiinae). Mitochondrial Dna 2016, 27, 1900–1901. [Google Scholar] [CrossRef] [PubMed]
- Omoto, K.; Yonezawa, T.; Shinkawa, T. Molecular systematics and evolution of the recently discovered “Parnassian” butterfly (Parnassius davydovi Churkin, 2006) and its allied species (Lepidoptera, Papilionidae). Gene 2009, 441, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Todisco, V.; Gratton, P.; Zakharov, E.V.; Wheat, C.W.; Sbordoni, V.; Sperling, F.A.H. Mitochondrial phylogeography of the Holarctic Parnassius phoebus complex supports a recent refugial model for alpine butterflies. J. Biogeogr. 2012, 39, 1058–1072. [Google Scholar] [CrossRef]
- Todisco, V.; Gratton, P.; Cesaroni, D.; Sbordoni, V. Phylogeography of Parnassius apollo: Hints on taxonomy and conservation of a vulnerable glacial butterfly invader. Biol. J. Linn. Soc. 2010, 101, 169–183. [Google Scholar] [CrossRef]
- Fei, M.Z.; Mo, L.D. Competition index and application to conservation biology of Parnassius nomion. Acta Ecol. Sin. 2002, 10, 1695–1698. [Google Scholar]
- Minsu, K.; Junseok, L.; Cheolhak, K.; Seungsu, K.; Kyutek, P. Distributional data and ecological characteristics of Parnassius bremeri Bremer in Korea. Korean J. Appl. Entomol. 2004, 4, 7–14. [Google Scholar]
- Kim, Y.S. Illustrated Book of Korean Butterflies in Color; Kyo-Hak Pub. Co.: Seoul, Republioc of Korea, 2005. [Google Scholar]
- Heikkilä, M.; Kaila, L.; Mutanen, M.; Pena, C.; Wahlberg, N. Cretaceous origin and repeated tertiary diversification of the redefined butterflies. Proc. R. Soc. Lond. Biol. Sci. 2012, 279, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, A.Y.; Breinholt, J.W. Phylogenomics provides strong evidence for relationships of butterflies and moths. Proc. R. Soc. Lond. Biol. Sci. 2014, 281, 20140970. [Google Scholar]
- Liu, N.Y.; Wu, Y.H.; Yang, X.J.; Wu, J.; Zheng, S.Z.; Fang, J. Complete Mitochondrial Genome of Papilio protenor (Lepidoptera, Papilionidae) and Implications for Papilionidae Taxonomy. J. Insect Sci. 2017, 6, 144–145. [Google Scholar] [CrossRef]
- Häuser, C.L. Critical comments on the phylogenetic relationships within the family Papilionidae (Lepidoptera). Nota Lepid. 1993, 16, 34–43. [Google Scholar]
- Yagi, T.; Sasaki, G.; Takebe, H. Phylogeny of Japanese papilionid butterflies inferred from nucleotide sequences of the mitochondrial ND5 gene. J. Mol. Evol. 1999, 48, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Caterino, M.S.; Reed, R.D.; Kuo, M.M.; Sperling, A.F. A partitioned likelihood analysis of swallowtail butterfly phylogeny (Lepidoptera:Papilionidae). Syst. Biol. 2001, 1, 106–127. [Google Scholar] [CrossRef]
- Wang, C.S.; Sinica, F. Lepidoptera; Papilionidae; Papilioninae, Zerynthiinae, Parnassiinae. In Fauna Sinica: Insecta; Science Press: Beijing, China, 2001; Volume 25. [Google Scholar]
- Condamine, F.L.; Sperling, F.; Kergoat, G.J. Global biogeographical pattern of swallowtail diversification demonstrates alternative colonization routes in the Northern and Southern hemispheres. J. Biogeogr. 2013, 40, 9–23. [Google Scholar] [CrossRef]
- Kim, M.I.; Baek, J.Y.; Kim, M.J.; Jeong, H.C.; Kim, K.G.; Bae, C.H.; Han, Y.S.; Jin, B.R.; Kim, I. Complete nucleotide sequence and organization of the mitogenome of the red-spotted apollo butterfly, Parnassius bremeri (Lepidoptera: Papilionidae) and comparison with other lepidopteran insects. Mol. Cells 2009, 28, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.J.; Zhang, X.; Duan, K. Complete mitochondrial genomes of two insular races of Pazala swordtails from Taiwan, China (Lepidoptera: Papilionidae: Graphium). Mitochondrial DNA Part B 2021, 6, 1557–1559. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Zhu, X.-Q.; Barker, S.C.; Herd, K. Evolution of extensively fragmented mitochondrial genomes in the lic of humans. Genome Biol. Evol. 2012, 4, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Jeng, M.L.; Chen, M.Y.; Wu, L.W. Two complete mitochondrial genomes of papilio butterflies obtained from historical specimens (Lepidoptera: Papilionidae). Mitochondrial DNA Part B 2021, 6, 1341–1343. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Cong, Q.; Grishin, N.V. The complete mitochondrial genome of Papilio glaucus and its phylogenetic implications. Meta Gene 2015, 5, 68–83. [Google Scholar] [CrossRef]
- Breinholt, J.W.; Earl, C.; Lemmon, A.R.; Lemmon, E.M.; Xiao, L.; Kawahara, A.Y. Resolving relationships among the megadiverse butterflies and moths with a novel pipeline for anchored phylogenomics. Syst. Biol. 2018, 67, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, r12. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Stadler, P. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Huang, D.Y.; Wang, Y.L.; Zhu, C.D.; Hao, J.S. The complete mitochondrial genome of the endangered Apollo butterfly, Parnassius apollo (Lepidoptera: Papilionidae) and its comparison to other Papilionidae species. J. Asia-Pac. Entomol. 2014, 17, 663–671. [Google Scholar] [CrossRef]
- Wang, Y.L.; Chen, Y.H.; Xia, C.C.; Xia, X.Q.; Tao, R.S.; Hao, J.S. The complete mitochondrial genome of the Common Red Apollo, Parnassius epaphus (Lepidoptera: Papilionidae: Parnassiinae). J. Asia-Pac. Entomol. 2015, 18, 239–248. [Google Scholar] [CrossRef]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 2000. [Google Scholar]
- Wright, F. The effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Federico, A.; Rafael, Z.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, 7–13. [Google Scholar]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Mihn, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Haeseler, A.V.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution formolecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 2008, 408, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.F.; Su, T.J.; Luo, A.R.; Zhu, C.D.; Wu, C.S. Characterization of the complete mitochondrion genome of diurnal moth Amata emma (Butler) (Lepidoptera: Erebidae) and its phylogenetic implications. PLoS ONE 2013, 8, e72410. [Google Scholar] [CrossRef] [PubMed]
- Fenn, J.D.; Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome sequence of the Mormon cricket (Anabrus simplex: Tettigoniidae: Orthoptera) and an analysis of control region variability. Insect Mol. Biol. 2007, 16, 239–252. [Google Scholar] [CrossRef]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Zhu, B.Y. Atlas of Chinese Butterflies; Shanghai Far East Publisher: Shanghai, China, 1992. [Google Scholar]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Miller, J.S. Phylogenetic studies in the Papilioninae (Lepidoptera, Papilionidae). Bull. Am. Mus. Nat. Hist. 1987, 186, 365–512. [Google Scholar]
- Caterino, M.S.; Sperling, F. Papilio phylogeny based on mitochondrial cytochrome oxidase I and II genes. Mol. Phylogenetics Evol. 1999, 11, 122–137. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, L.; Chen, X.; Li, X.; Guedes, R.N.C.; Dewer, Y.; Shang, S.; Zhou, J. Mitogenomic Phylogenetic Analyses Reveal New Insights into the Taxonomy and Evolution of Parnassiinae Swallowtail Butterflies (Lepidoptera: Papilionidae). Diversity 2025, 17, 19. https://doi.org/10.3390/d17010019
Song L, Chen X, Li X, Guedes RNC, Dewer Y, Shang S, Zhou J. Mitogenomic Phylogenetic Analyses Reveal New Insights into the Taxonomy and Evolution of Parnassiinae Swallowtail Butterflies (Lepidoptera: Papilionidae). Diversity. 2025; 17(1):19. https://doi.org/10.3390/d17010019
Chicago/Turabian StyleSong, Lu, Xiaoxiao Chen, Xiushan Li, Raul Narciso C. Guedes, Youssef Dewer, Suqin Shang, and Jingjiang Zhou. 2025. "Mitogenomic Phylogenetic Analyses Reveal New Insights into the Taxonomy and Evolution of Parnassiinae Swallowtail Butterflies (Lepidoptera: Papilionidae)" Diversity 17, no. 1: 19. https://doi.org/10.3390/d17010019
APA StyleSong, L., Chen, X., Li, X., Guedes, R. N. C., Dewer, Y., Shang, S., & Zhou, J. (2025). Mitogenomic Phylogenetic Analyses Reveal New Insights into the Taxonomy and Evolution of Parnassiinae Swallowtail Butterflies (Lepidoptera: Papilionidae). Diversity, 17(1), 19. https://doi.org/10.3390/d17010019