
Investigating the Diversity of Wolbachia across the Spiny Ants (Polyrhachis)


Abstract
1. Introduction
2. Materials and Methods
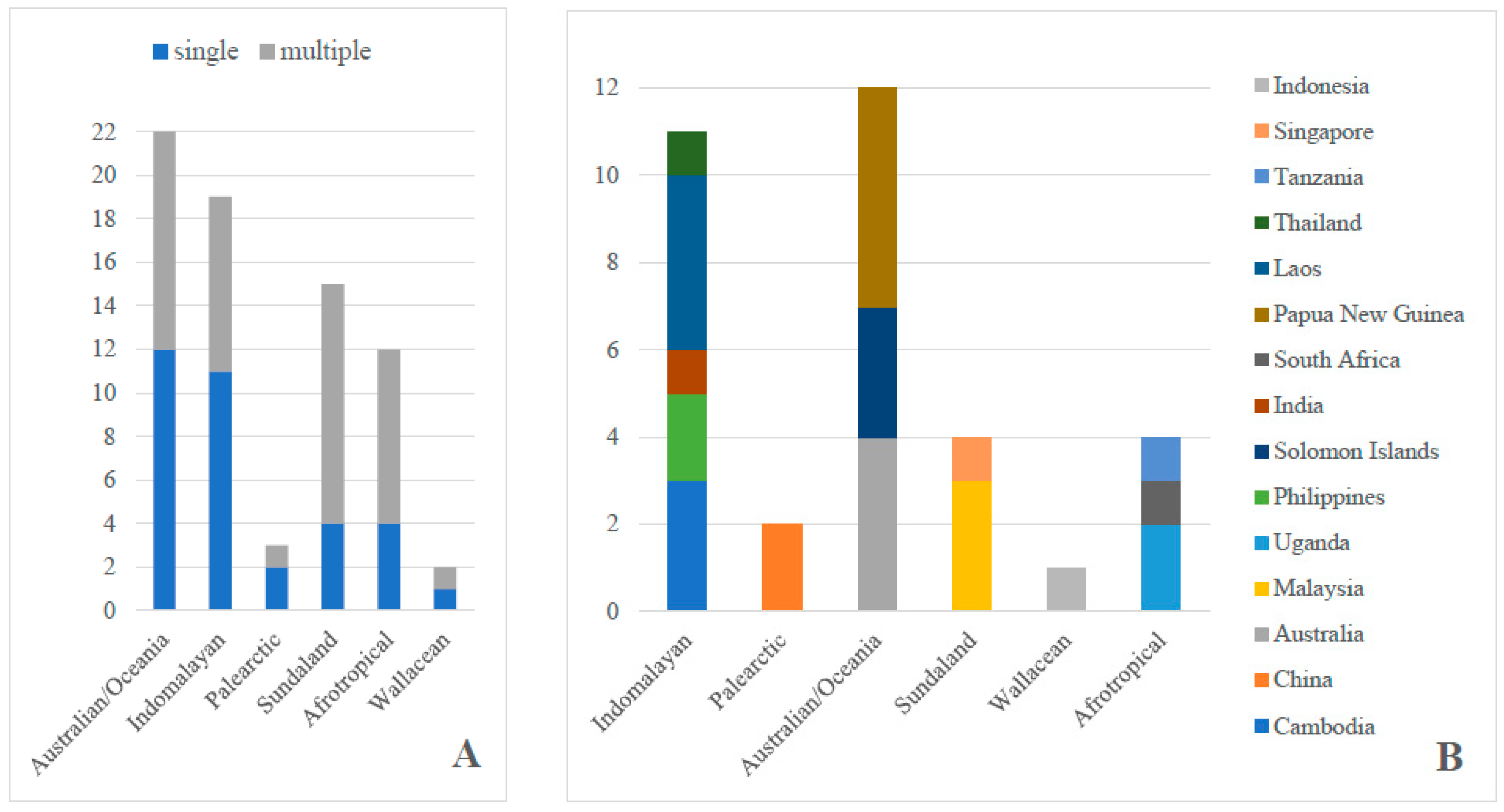
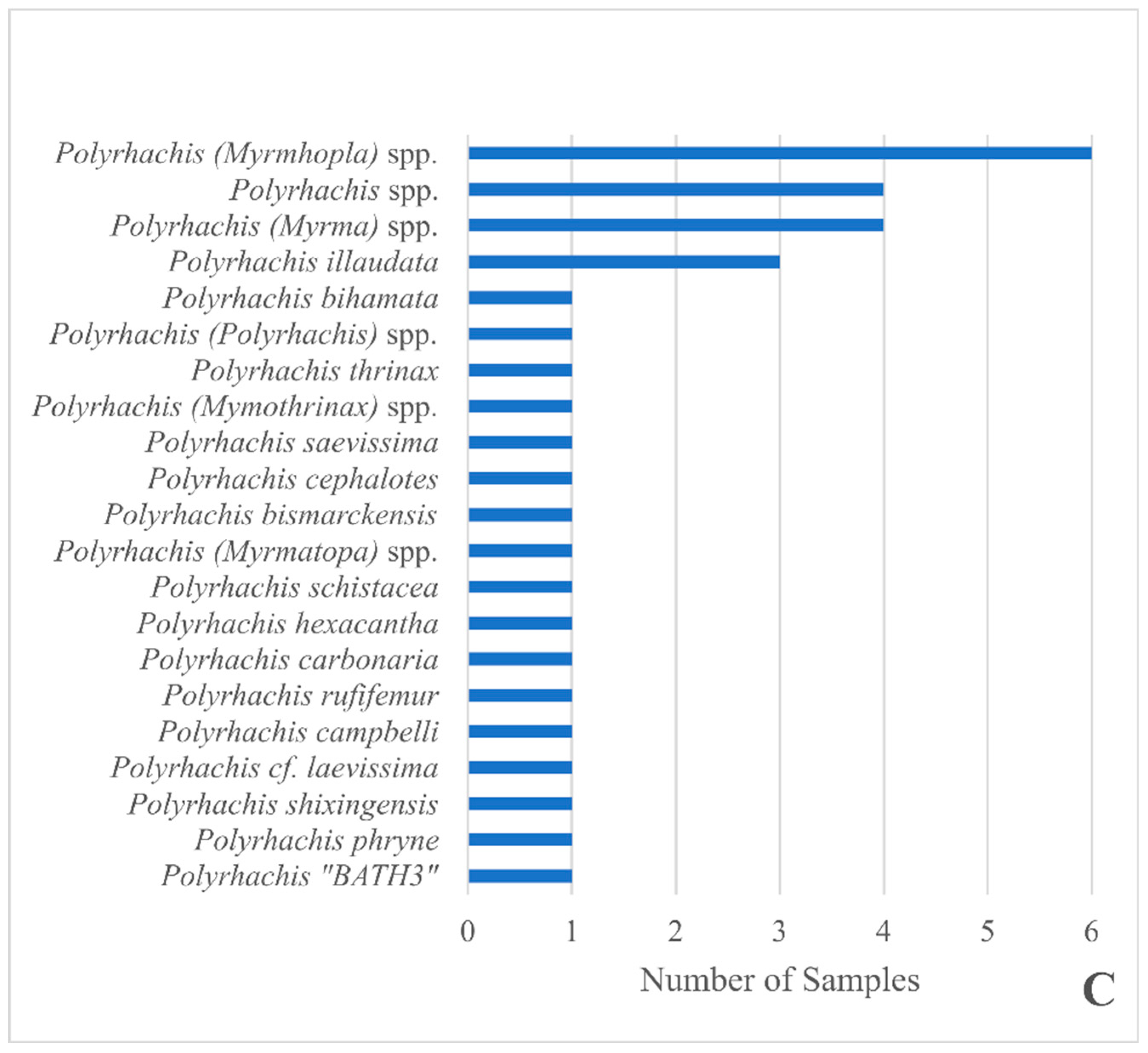
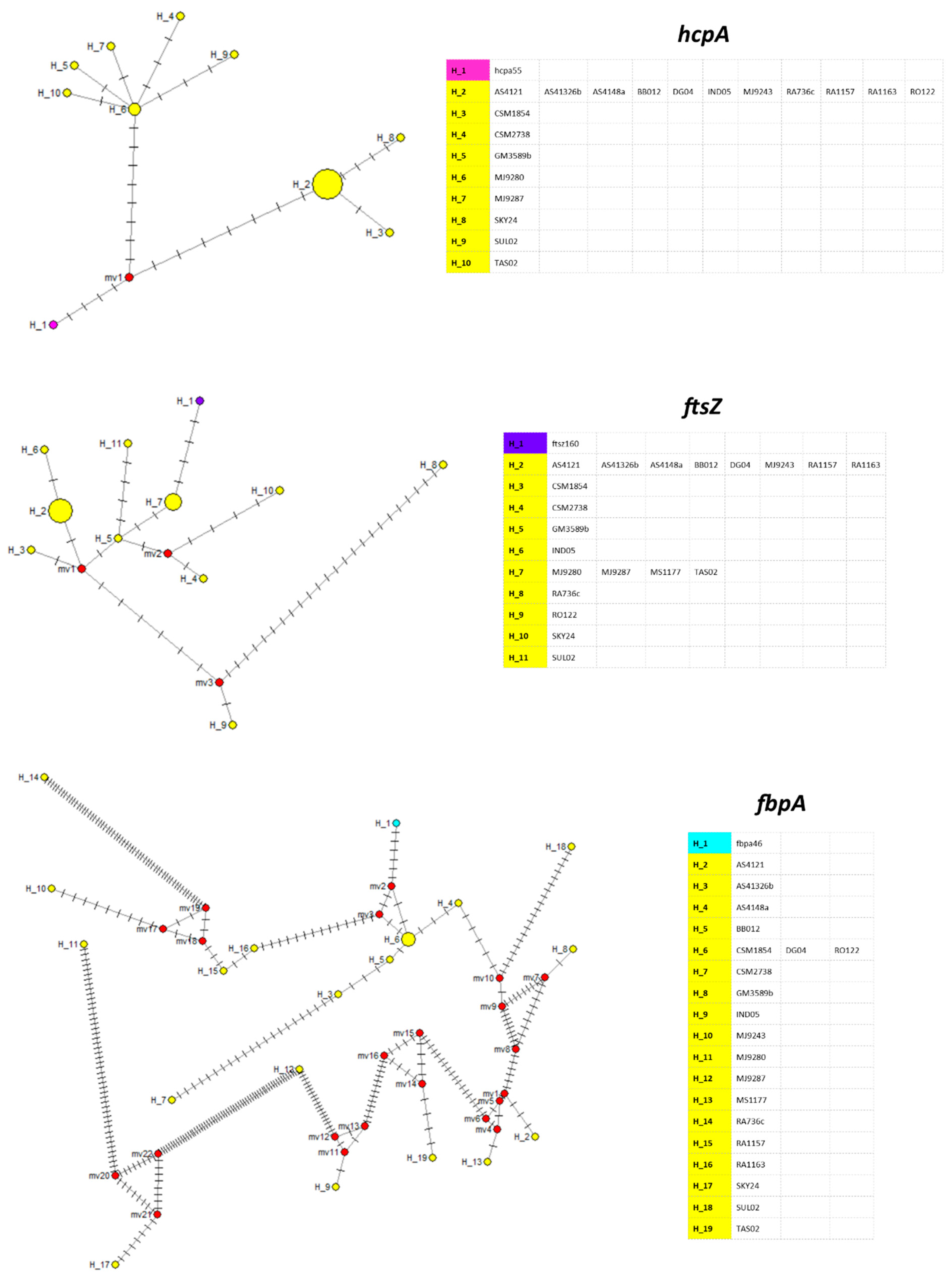
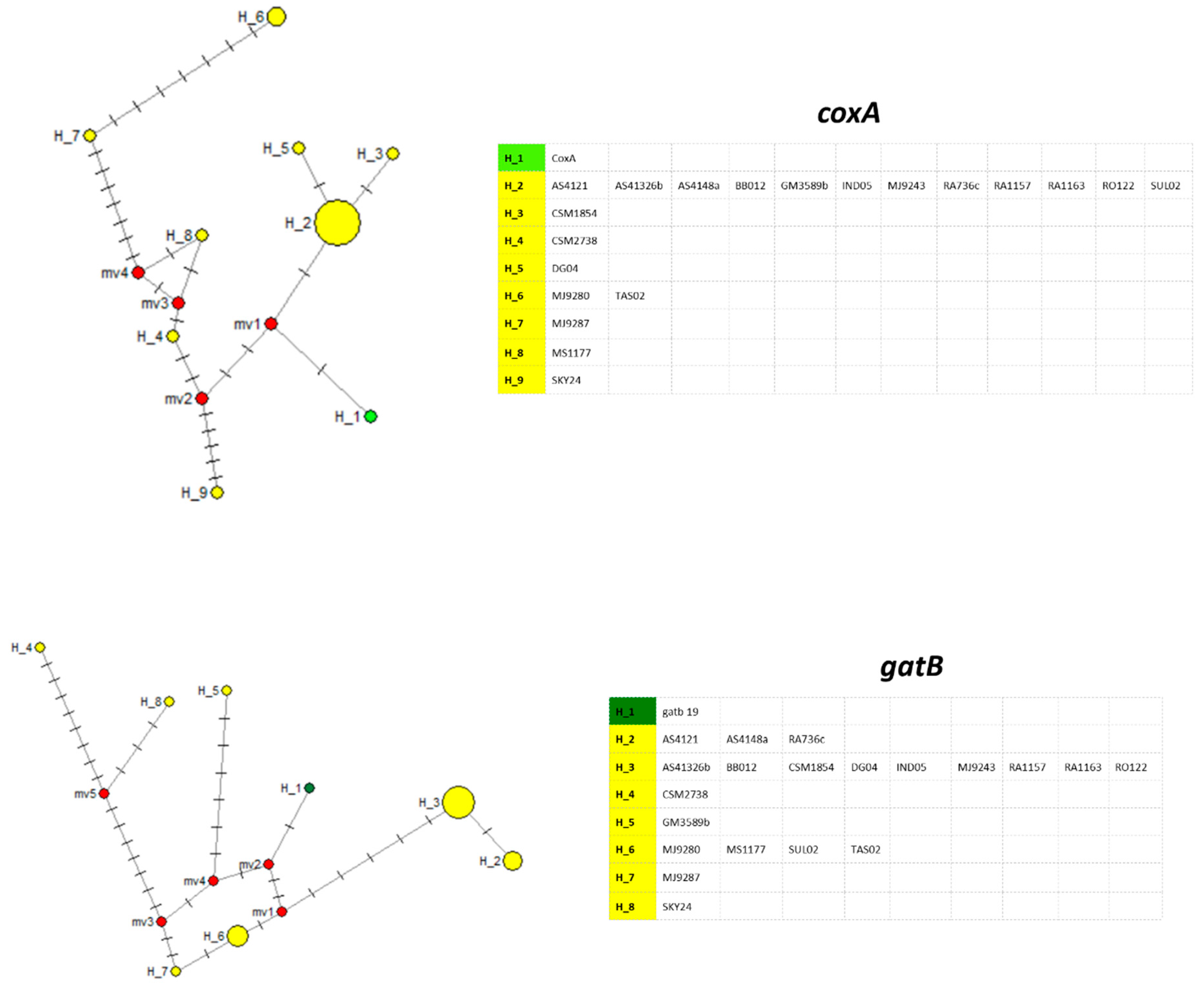
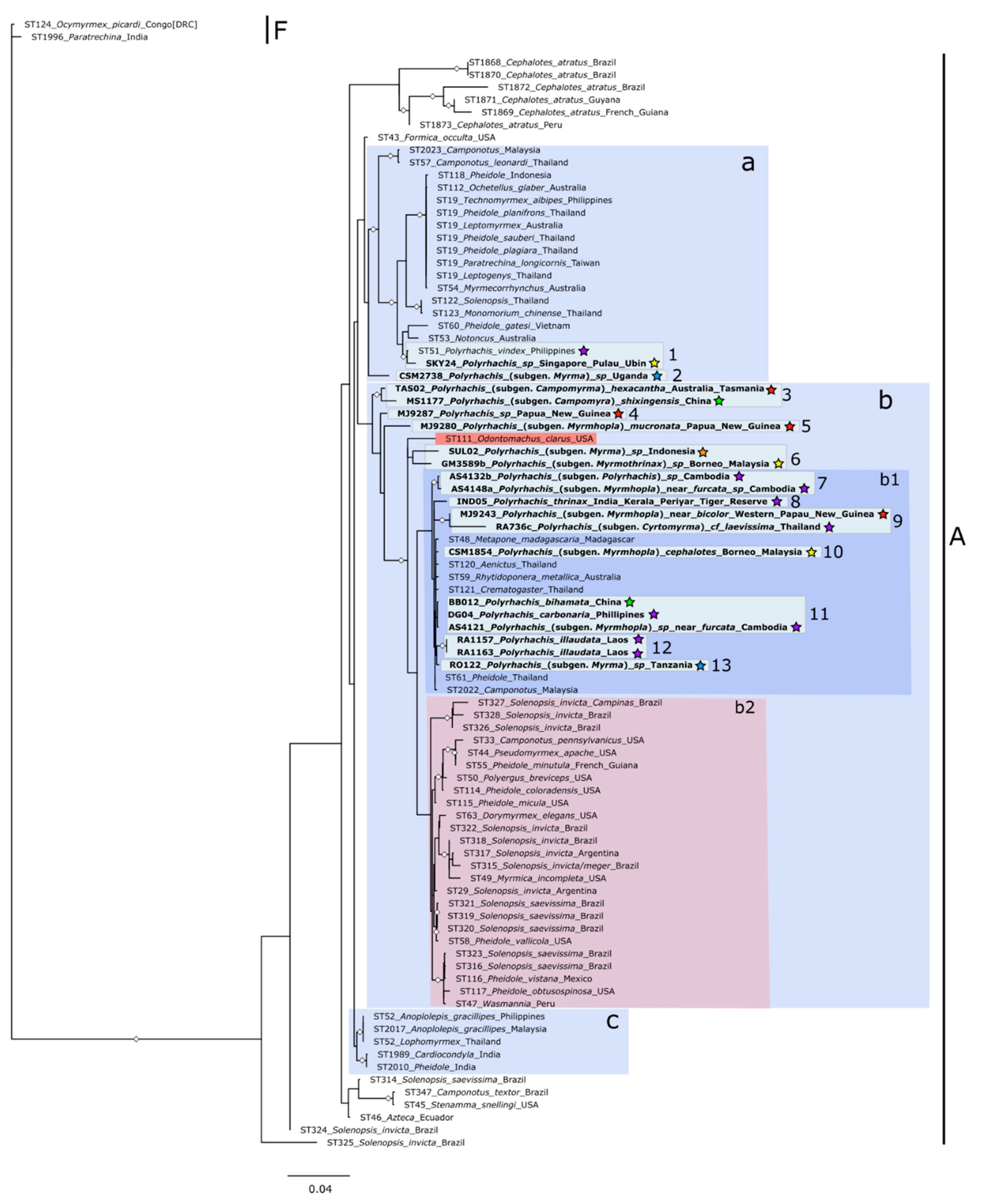
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moreau, C.S. Symbioses among ants and microbes. Curr. Opin. Insect Sci. 2020, 39, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sanders, J.G.; Łukasik, P.; D’Amelio, C.L.; Millar, J.S.; Vann, D.R.; Lan, Y.; Newton, J.A.; Schotanus, M.; Kronauer, D.J.; et al. Herbivorous turtle ants obtain essential nutrients from a conserved nitrogen-recycling gut microbiome. Nat. Commun. 2018, 9, 964. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.A. The ants (Hymenoptera: Formicidae) are unique and enigmatic hosts of prevalent Wolbachia (Alphaproteobacteria) symbionts. Myrmecol. News Myrmecol. News 2012, 16, 7–23. [Google Scholar]
- Werren, J.H.; Windsor, D.M. Wolbachia infection frequencies in insects: Evidence of a global equilibrium? Proc. R. Soc. B Biol. Sci. 2000, 267, 1277–1285. [Google Scholar] [CrossRef]
- Ramalho, M.O.; Vieira, A.S.; Pereira, M.C.; Moreau, C.S.; Bueno, O.C. Transovarian Transmission of Blochmannia and Wolbachia Endosymbionts in the Neotropical Weaver Ant Camponotus textor (Hymenoptera, Formicidae). Curr. Microbiol. 2018, 75, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Baldo, L.; Dunning Hotopp, J.C.; Jolley, K.A.; Bordenstein, S.R.; Biber, S.A.; Choudhury, R.R.; Hayashi, C.; Maiden, M.C.J.; Tettelin, H.; Werren, J.H. Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl. Environ. Microbiol. 2006, 72, 7098–7110. [Google Scholar] [CrossRef]
- Singh, R.; Linksvayer, T.A. Wolbachia-infected ant colonies have increased reproductive investment and an accelerated life cycle. J. Exp. Biol. 2020, 223, jeb220079. [Google Scholar] [CrossRef]
- Tseng, S.P.; Wetterer, J.K.; Suarez, A.V.; Lee, C.Y.; Yoshimura, T.; Shoemaker, D.W.; Yang, C.C.S. Genetic Diversity and Wolbachia Infection Patterns in a Globally Distributed Invasive Ant. Front. Genet. 2019, 10, 838. [Google Scholar] [CrossRef]
- Ramalho, M.O.; Moreau, C.S. The evolution and biogeography of wolbachia in ants (Hymenoptera: Formicidae). Diversity 2020, 12, 426. [Google Scholar] [CrossRef]
- Ramalho, M.D.O.; Kim, Z.; Wang, S.; Moreau, C.S. Wolbachia across Social Insects: Patterns and Implications. Ann. Entomol. Soc. Am. 2021, 114, 206–218. [Google Scholar] [CrossRef]
- Baldo, L.; Werren, J.H. Revisiting Wolbachia supergroup typing based on WSP: Spurious lineages and discordance with MLST. Curr. Microbiol. 2007, 55, 81–87. [Google Scholar] [CrossRef]
- Gerth, M. Classification of Wolbachia (Alphaproteobacteria, Rickettsiales): No evidence for a distinct supergroup in cave spiders. Infect. Genet. Evol. 2016, 43, 378–380. [Google Scholar] [CrossRef] [PubMed]
- Glowska, E.; Dragun-Damian, A.; Dabert, M.; Gerth, M. New Wolbachia supergroups detected in quill mites (Acari: Syringophilidae). Infect. Genet. Evol. 2015, 30, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.A.; Goldman-Huertas, B.; Moreau, C.S.; Baldo, L.; Stahlhut, J.K.; Werren, J.H.; Pierce, N.E. Specialization and geographic isolation among Wolbachia symbionts from ants and lycaenid butterflies. Evolution 2009, 63, 624–640. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Rousset, F.; O’Neill, S. Phylogeny and PCR–based classification of Wolbachia strains using wsp gene sequences. Proc. R. Soc. London B Biol. Sci. 1998, 265, 509–515. [Google Scholar] [CrossRef]
- Baldo, L.; Lo, N.; Werren, J.H. Mosaic nature of the wolbachia surface protein. J. Bacteriol. 2005, 187, 5406–5418. [Google Scholar] [CrossRef]
- Zhang, X.; Norris, D.E.; Rasgon, J.L. Distribution and molecular characterization ofWolbachia endosymbionts and larial nematodes inMaryland populations of the lone star tick (Amblyomma americanum). FEMS Microbiol. Ecol. 2011, 77, 50–56. [Google Scholar] [CrossRef]
- Salunke, B.K.; Salunkhe, R.C.; Dhotre, D.P.; Walujkar, S.A.; Khandagale, A.B.; Chaudhari, R.; Chandode, R.K.; Ghate, H.V.; Patole, M.S.; Werren, J.H.; et al. Determination of Wolbachia diversity in butterflies from Western Ghats, India, by a multigene approach. Appl. Environ. Microbiol. 2012, 78, 4458–4467. [Google Scholar] [CrossRef]
- Kelly, M.; Price, S.L.; de Oliveira Ramalho, M.; Moreau, C.S. Diversity of Wolbachia Associated with the Giant Turtle Ant, Cephalotes atratus. Curr. Microbiol. 2019, 76, 1330–1337. [Google Scholar] [CrossRef]
- Ramalho, M.O.; Martins, C.; Silva, L.M.R.; Martins, V.G.; Bueno, O.C. Intracellular symbiotic bacteria of Camponotus textor, Forel (Hymenoptera, Formicidae). Curr. Microbiol. 2017, 74, 589–597. [Google Scholar] [CrossRef]
- Martins, C.; de Oliveira Ramalho, M.; Silva, L.M.R.; de Souza, R.F.; Bueno, O.C. New Strains of Wolbachia Unveiling the Complexity of This Symbiotic Interaction in Solenopsis (Hymenoptera: Formicidae). Microbiol. Res. 2021, 12, 40. [Google Scholar] [CrossRef]
- Guénard, B.; Weiser, M.D.; Gómez, K.; Narula, N.; Economo, E.P. The Global Ant Biodiversity Informatics (GABI) database: Synthesizing data on the geographic distribution of ant species (Hymenoptera: Formicidae). Myrmecol. News 2017, 24, 83–89. [Google Scholar]
- Blanchard, B.D.; Moreau, C.S. Defensive spines are associated with large geographic range but not diversification in spiny ants (Hymenoptera: Formicidae: Polyrhachis). Syst. Entomol. 2022, 1–13. [Google Scholar] [CrossRef]
- Mezger, D.; Moreau, C.S. Out of South-East Asia: Phylogeny and biogeography of the spiny ant genus Polyrhachis Smith (Hymenoptera: Formicidae). Syst. Entomol. 2016, 41, 369–378. [Google Scholar] [CrossRef]
- Blanchard, B.D.; Nakamura, A.; Cao, M.; Chen, S.T.; Moreau, C.S. Spine and dine: A key defensive trait promotes ecological success in spiny ants. Ecol. Evol. 2020, 10, 5852–5863. [Google Scholar] [CrossRef]
- Robson, S.K.A.; Kohout, R.J.; Beckenbach, A.T.; Moreau, C.S. Evolutionary transitions of complex labile traits: Silk weaving and arboreal nesting in Polyrhachis ants. Behav. Ecol. Sociobiol. 2015, 69, 449–458. [Google Scholar] [CrossRef]
- van Zweden, J.S.; Carew, M.E.; Henshaw, M.T.; Robson, S.K.A.; Crozier, R.H. Social and genetic structure of a supercolonial weaver ant, Polyrhachis robsoni, with dimorphic queens. Insectes Soc. 2007, 54, 34–41. [Google Scholar] [CrossRef]
- Wernegreen, J.J.; Kauppinen, S.N.; Brady, S.G.; Ward, P.S. One nutritional symbiosis begat another: Phylogenetic evidence that the ant tribe Camponotini acquired Blochmannia by tending sap-feeding insects. BMC Evol. Biol. 2009, 9, 292. [Google Scholar] [CrossRef]
- Ramalho, M.O.; Bueno, O.C.; Moreau, C.S. Microbial composition of spiny ants (Hymenoptera: Formicidae: Polyrhachis) across their geographic range. BMC Evol. Biol. 2017, 17, 96. [Google Scholar] [CrossRef]
- Ramalho, M.O.; Bueno, O.C.; Moreau, C.S. Species-specific signatures of the microbiome from Camponotus and Colobopsis ants across developmental stages. PLoS ONE 2017, 12, e0187461. [Google Scholar] [CrossRef]
- Braig, H.R.; Zhou, W.; Dobson, S.L.; O’Neill, S.L. Cloning and characterization of a gene encoding the major surface protein of the bacterial endosymbiont Wolbachia pipientis. J. Bacteriol. 1998, 180, 2373–2378. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.S. A practical guide to DNA extraction, PCR, and gene-based DNA sequencing in insects. Halteres 2014, 5, 32–42. [Google Scholar]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications [version 1; referees: 2 approved]. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Chernomor, O.; Von Haeseler, A.; Minh, B.Q. Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B. The vegan package. Community Ecol. 2007, 10, 631–637. [Google Scholar]
- Salunke, B.K.; Salunkhe, R.C.; Dhotre, D.P.; Khandagale, A.B.; Walujkar, S.A.; Kirwale, G.S.; Ghate, H.V.; Patole, M.S.; Shouche, Y.S. Diversity of Wolbachia in Odontotermes spp. (Termitidae) and Coptotermes heimi (Rhinotermitidae) using the multigene approach. FEMS Microbiol. Lett. 2010, 307, 55–64. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | MLST Allele Number | ST | Country | Polyrhachis | ||||
|---|---|---|---|---|---|---|---|---|
| coxA | fbpA | ftsZ | gatB | hcpA | Host Species | |||
| AS4121 | 2 | 51 | 45 | 20 | 47 | 61 * | Cambodia | P. (Myrmhopla) sp. |
| AS4132b | 2 | 51 | 45 | 20 | 47 | 61 * | Cambodia | P. (Polyrhachis) sp. |
| AS4148a | 2 | 51 | 45 | 20 | 47 | 61 * | Cambodia | P. (Myrmhopla) sp. |
| BB012 | 2 | 51 | 45 | 20 | 47 | 61 * | China | P. bihamata |
| CSM1854 | 2 | 51 | 45 | 20 | 47 | 61 * | Malaysia | P. cephalotes |
| CSM2738 | 33 | 61 | 47 | 34 | 195 | ** | Uganda | P. (Myrma) sp. |
| DG04 | 2 | 51 | 45 | 20 | 47 | 61 * | Philippines | P. carbonaria |
| GM3589b | 2 | 356 | 258 | 22 | 343 | 52 * | Malaysia | P. (Mymothrinax) sp. |
| IND05 | 2 | 52 | 45 | 20 | 47 | 61 | India | P. thrinax |
| MJ9243 | 2 | 51 | 45 | 20 | 47 | 61 * | Papua New Guinea | P. (Myrmhopla) sp. |
| MJ9280 | 33 | 465 | 17 | 3 | 343 | ** | Papua New Guinea | P. (Myrmhopla) sp. |
| MJ9287 | 33 | 463 | 17 | 130 | 343 | ** | Papua New Guinea | Polyrhachis sp. |
| MS1177 | 33 | 463 | 17 | 3 | 343 | ** | China | P. shixingensis |
| RA1157 | 2 | 51 | 45 | 20 | 47 | 61 * | Laos | P. illaudata |
| RA1163 | 2 | 51 | 45 | 20 | 47 | 61 * | Laos | P. illaudata |
| RA736c | 2 | 51 | 45 | 20 | 47 | 61 * | Thailand | P. cf. laevissima |
| RO122 | 2 | 51 | 45 | 20 | 47 | 61 * | Tanzania | P. (Myrma) sp. |
| SKY24 | 32 | 48 | 6 | 57 | 50 | 51 * | Singapore | Polyrhachis sp. |
| SUL02 | 296 | 97 | 258 | 3 | 343 | ** | Indonesia | P. (Myrma) sp. |
| TAS02 | 33 | 277 | 17 | 3 | 343 | 481 * | Australia | P. hexacantha |
| CSM0655 | 32 | - | 6 | 57 | 50 | - | Australia | P. rufifemur |
| DG11 | 2 | - | 258 | 22 | 343 | - | Philippines | P. saevissima |
| EMS2584 | 218 | 6 | - | 158 | 141 | - | Solomon Islands | P. campbelli |
| EMS2617 | 33 | - | 17 | 3 | 343 | - | Solomon Islands | P. bismarckensis |
| FH1101 | 2 | - | 261 | 20 | 47 | - | Uganda | P. (Myrma) sp. |
| GM894 | 32 | - | 6 | 57 | 50 | - | Malaysia | P. (Myrmhopla) sp. |
| KATE02 | - | - | - | - | - | - | South Africa | P. schistacea |
| MJ8291 | - | - | - | - | - | - | Papua New Guinea | Polyrhachis sp. |
| MJ9286 | 109 | - | 261 | 191 | 83 | - | Papua New Guinea | Polyrhachis sp. |
| RA0755 | 32 | - | - | 57 | 50 | - | Australia | Polyrhachis “BATH3” |
| RA0784 | 33 | - | 17 | 3 | 343 | - | Solomon Islands | P. (Myrmatopa) sp. |
| RA1158 | 2 | - | 258 | 182 | 343 | - | Laos | P. (Myrmhopla) sp. |
| RA1160 | 2 | - | 45 | 20 | 47 | - | Laos | P. illaudata |
| TAS03 | 33 | - | 17 | 3 | 343 | - | Australia | P. phryne |
| Sample ID | Species | Country | Sample ID | Species | Country |
|---|---|---|---|---|---|
| DG06 | (Polyrhachis (Myrmatopa) sp. | Phillipines | RA0766 | Polyrhachis flavibasis | Australia |
| ISR_06 | Polyrhachis (Myrma) sp. | Thailand | SUL02 | Polyrhachis (Myrma) sp. 1 | Indonesia |
| GM 894 | Polyrhachis (Myrmhopla) sp. 2 | Malaysia | SKY20 | Polyrhachis sp. | Singapore |
| GM3990 | Polyrhachis (Myrmhopla) sp. 4 | Malaysia | SL_28_2 | Polyrhachis illaudata | Malaysia |
| GM3589b | Polyrhachis (Myrmothrinax) sp. | Malaysia | SKY24 | Polyrhachis sp. | Singapore |
| AS4132a | Polyrhachis (Polyrhachis) sp. | Cambodia | LEA04 | Polyrachis schistaceae | Mozambique |
| CSM0776 | Polyrhachis abbreviata | Australia | MS1177 | Polyrhachis shixingensis | China |
| DG10 | Polyrhachis armata | Philippines | RA0784 | Polyrhachis sp. | Solomon Islands |
| DG14 | Polyrhachis armata | Phillipines | RA1157 | Polyrhachis illaudata | Laos |
| CSM0761 | Polyrhachis australis | Australia | MJ9286 | Polyrhachis sp. | Papua New Guinea |
| DG26 | Polyrhachis bicolor | Philippines | RA1163 | Polyrhachis illaudata | Laos |
| BB012 | Polyrhachis bihamata | China | MJ 8277 | Polyrhachis sp. | Papua New Guinea |
| CSM1806a | Polyrhachis bihamata | Malaysia | PH09 | Polyrhachis afrc_cd03 | Democratic Republic of the Congo |
| CSM1806b | Polyrhachis bihamata | Malaysia | PH11 | Polyrhachis laboriosa | Democratic Republic of the Congo |
| DG08 | Polyrhachis bihamata | Phillipines | RA0769 | Polyrhachis “chario5” | Australia |
| CSM1846 | Polyrhachis boltoni | Malaysia | PH14 | Polyrhachis gagates | South Africa |
| EMS2584 | Polyrhachis campbelli | Solomon Islands | RA736a | Polyrhachis dives-group sp. | Thailand |
| DG04 | Polyrhachis carbonaria | Phillipines | RA0765 | Polyrhachis ammon | Australia |
| CSM1854 | Polyrhachis cephalotes | Malaysia | PH15 | Polyrhachis afr_cd01 | Democratic Republic of the Congo |
| EMS2617 | Polyrhachis cf. bismarckensis | Solomon Islands | RA736c | Polyrhachis cf. laevissima | Thailand |
| CSM1841 | Polyrhachis danum | Malaysia | PH12 | Polyrhachis revoili | Democratic Republic of the Congo |
| BB28 | Polyrhachis hippomanes | China | MJ 9243 | Polyrhachis sp. near bicolor | Papua New Guinea |
| JRNG01 | Polyrhachis hookeri | Australia | TAS 02 | Polyrhachis hexacantha | Australia |
| DG03 | Polyrhachis illaudata | Phillipines | RA0755 | Polyrhachis “BATH3” | Australia |
| GM3551 | Polyrhachis illaudata | Malaysia | SKY21 | Polyrhachis nigropilosa | Singapore |
| DG25 | Polyrhachis inermis | Philippines | RA1162 | Polyrhachis illaudata | Laos |
| EMS2637 | Polyrhachis kaipi | Solomon Islands | RO 122 | Polyrhachis sp. | Tanzania |
| ISR_03 | Polyrhachis lacteipennis | Israel | SOH 02 | Polyrhachis beccari | Singapore |
| CSM1868 | Polyrhachis lepida | Malaysia | PH21 | Polyrhachis schistacea | Mozambique |
| DG16 | Polyrhachis near lilianae | Philippines | RA1158 | Polyrhachis mucronata-group sp. | Laos |
| BB48 | Polyrhachis proxima | China | MJ 9280 | Polyrhachis mucronata-group sp. | |
| CSM0655 | Polyrhachis rufifemur | Australia | MJ8280 | Polyrhachis sp. | Papua New Guinea |
| CSM0740 | Polyrhachis rufifemur | Australia | MJ9242 | Polyrhachis sexspi-sa group | Papua New Guinea |
| DG11 | Polyrhachis saevissima | Phillipines | RA1154 | Polyrhachis mucronata-group sp. | Laos |
| DG17 | Polyrhachis saevissima | Phillipines | RA1160 | Polyrhachis illaudata? | Laos |
| KATE02 | Polyrhachis schistacea | South Africa | TAS04 | Polyrhachis semipolita | Australia |
| AS4132b | Polyrhachis sp. | Cambodia | LEA05 | Polyrachis schistaceae | Mozambique |
| BB026 | Polyrhachis sp. | China | MJ 8282 | Polyrhachis sexspi-sa group | Papua New Guinea |
| CSM1860 | Polyrhachis sp. | Malaysia | PH16 | Polyrhachis latharis | Democratic Republic of the Congo |
| CSM2632 | Polyrhachis sp. | Uganda | PH22 | Polyrhachis schistacea | Tanzania |
| CSM2738 | Polyrhachis sp. | Uganda | PSW5403 | Polyrhachis andromache | Australia |
| CSM2745 | Polyrhachis sp. | Uganda | PSW6454 | Polyrhachis obesior | Malaysia |
| CSM2831 | Polyrhachis sp. | Australia | RA0735 | Polyrhachis abdominalis | Singapore |
| FH1085 | Polyrhachis sp. | Uganda | RO538 | Polyrhachis sp. | Tanzania |
| FH1101 | Polyrhachis sp. | Uganda | SKY05 | Polyrhachis frustorferi | Indonesia |
| FH205 | Polyrhachis sp. | Kenya | SKY11 | Polyrhachis lamellidens | Japan |
| FH987 | Polyrhachis sp. | Uganda | SKY17 | Polyrhachis hector | Indonesia |
| JCM120P | Polyrhachis sp. | Palau | TAS 01 | Polyrhachis hexacantha | Australia |
| JRNG02 | Polyrhachis sp. | Australia | TAS03 | Polyrhachis phryne | Australia |
| LD01 | Polyrhachis sp. | Ghana | LEA03 | Polyrachis schistaceae | Mozambique |
| AS4121 | Polyrhachis sp. near furcata | Cambodia | MJ 8263 | Polyrhachis sp. | Papua New Guinea |
| AS4148a | Polyrhachis sp. near furcata | Cambodia | MJ 8291 | Polyrhachis sp. | Papua New Guinea |
| BB_075 | Polyrhachis sp. near sixspi-sa | China | MJ9275 | Polyrhachis sp. | Papua New Guinea |
| CSM0746 | Polyrhachis thais | Australia | SL32 | Polyrhachis furcata | Malaysia |
| IND05 | Polyrhachis thrinax | India | MJ 9287 | Polyrhachis sp. | Papua New Guinea |
| CAB01 | Polyrhachis ypsilon | Malaysia |
| Partition | Gene(s) | Position in Concatenation (bp) | Length of Gene(s) in Partition (bp) | Model |
|---|---|---|---|---|
| 1 | coxA | 1–403 | 403 | HKY + F+G4 |
| 2 | fbpA | 404–840 | 437 | HKY + F+G4 |
| 3 | hcpA, ftsZ | 1651–2098, 841–1277 | 448, 437 | TIM3 + F+I+G4 |
| 4 | gatB | 1278–1650 | 373 | TIM + F+I+G4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webb, J.L.; Graber, L.C.; Ramalho, M.O.; Moreau, C.S. Investigating the Diversity of Wolbachia across the Spiny Ants (Polyrhachis). Diversity 2023, 15, 348. https://doi.org/10.3390/d15030348
Webb JL, Graber LC, Ramalho MO, Moreau CS. Investigating the Diversity of Wolbachia across the Spiny Ants (Polyrhachis). Diversity. 2023; 15(3):348. https://doi.org/10.3390/d15030348
Chicago/Turabian StyleWebb, Jenna L., Leland C. Graber, Manuela O. Ramalho, and Corrie S. Moreau. 2023. "Investigating the Diversity of Wolbachia across the Spiny Ants (Polyrhachis)" Diversity 15, no. 3: 348. https://doi.org/10.3390/d15030348
APA StyleWebb, J. L., Graber, L. C., Ramalho, M. O., & Moreau, C. S. (2023). Investigating the Diversity of Wolbachia across the Spiny Ants (Polyrhachis). Diversity, 15(3), 348. https://doi.org/10.3390/d15030348