
Complex Evolutionary History of the South American Fox Genus Lycalopex (Mammalia, Carnivora, Canidae) Inferred from Multiple Mitochondrial and Nuclear Markers

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Laboratory Procedures
2.2. Data Analyses
2.2.1. Mitochondrial Dataset
2.2.2. Nuclear Dataset
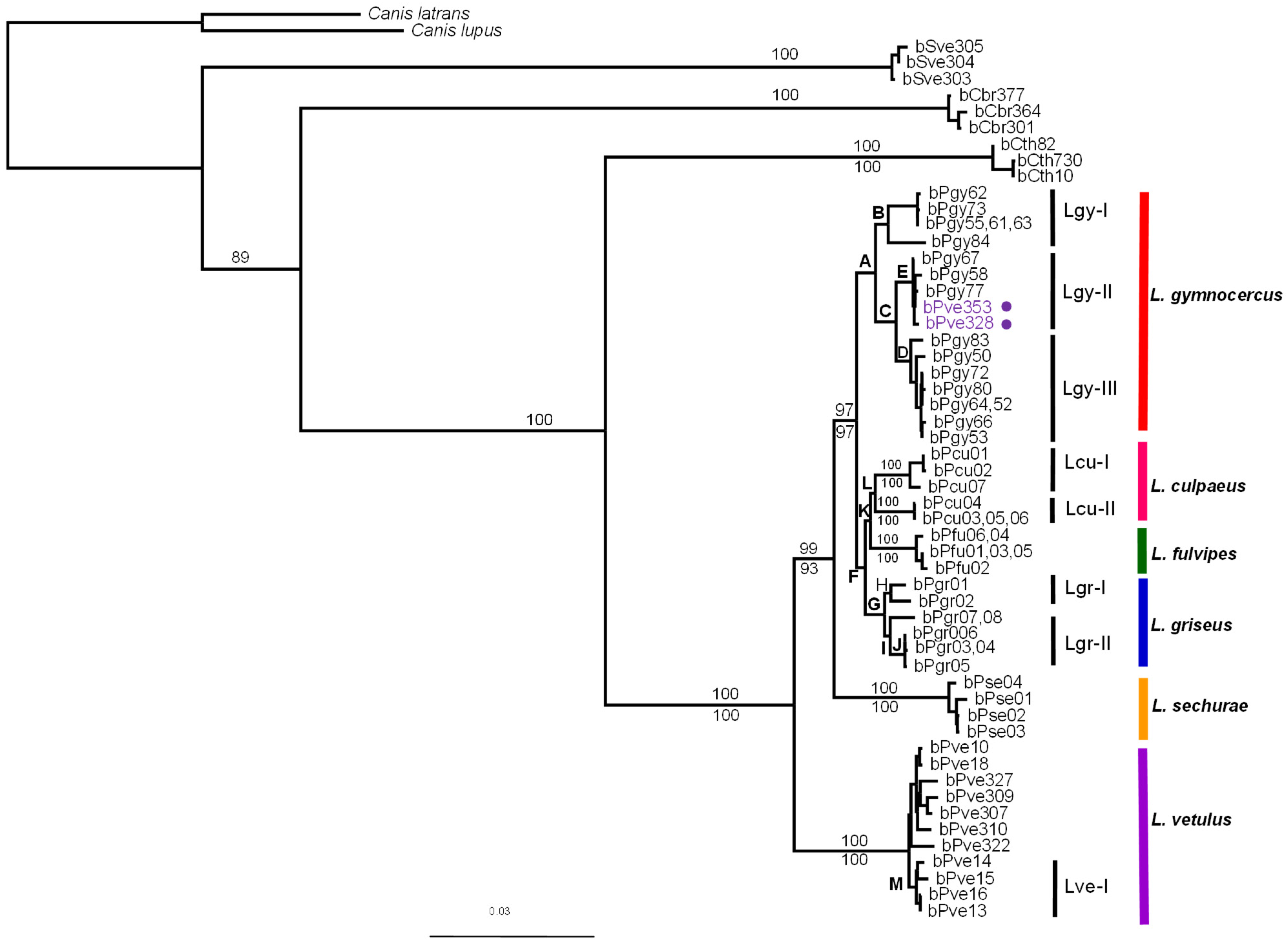
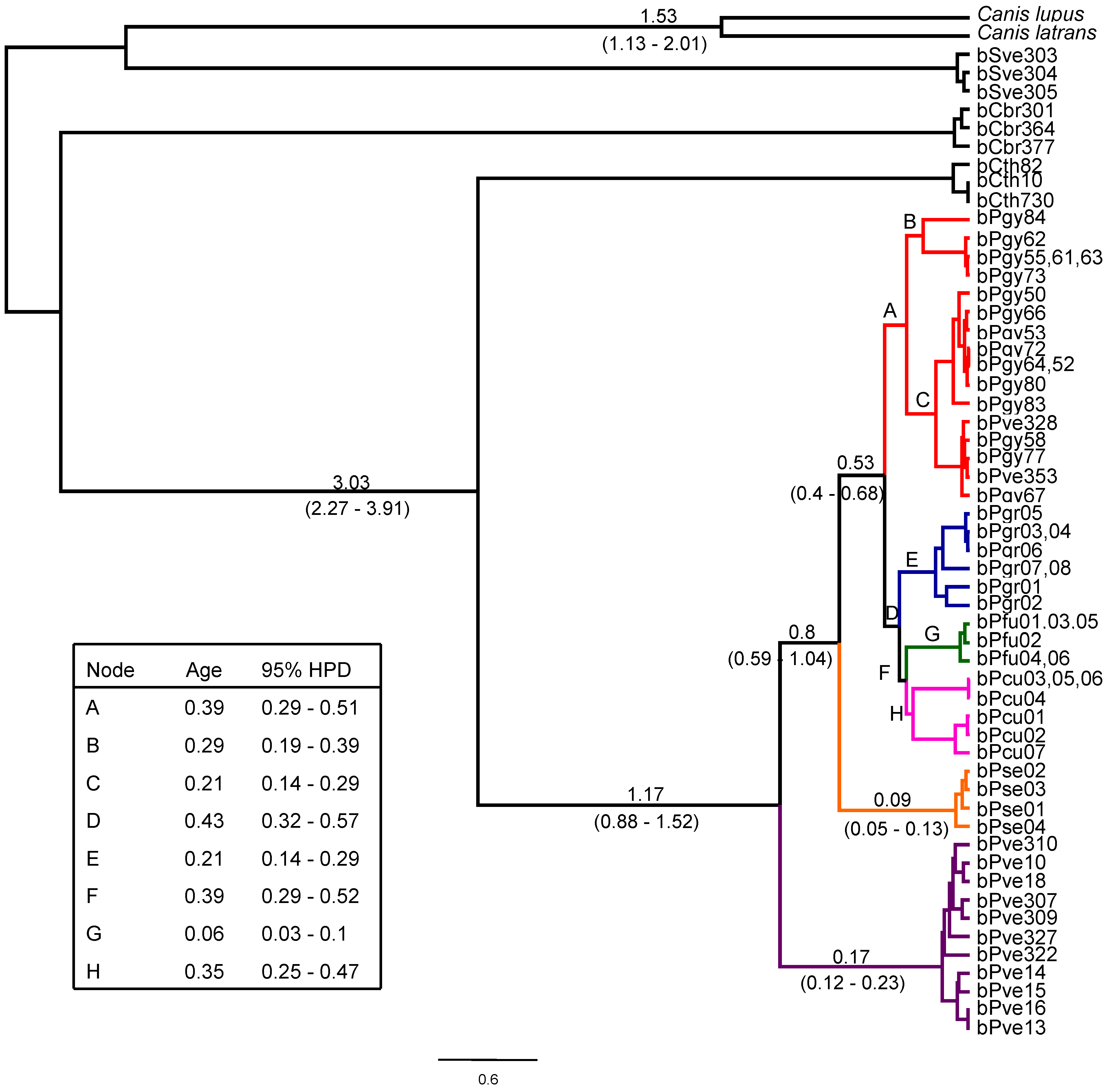
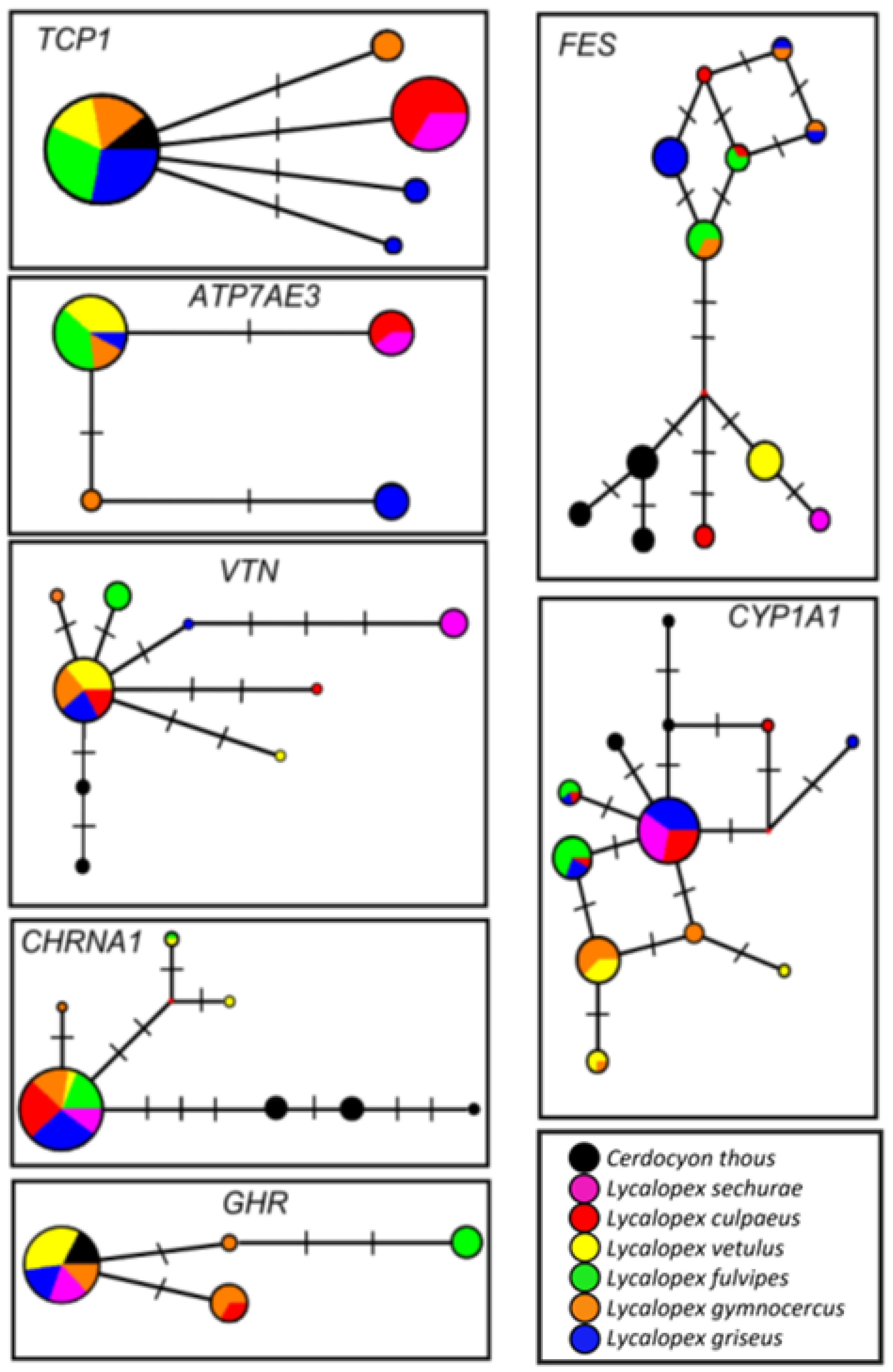
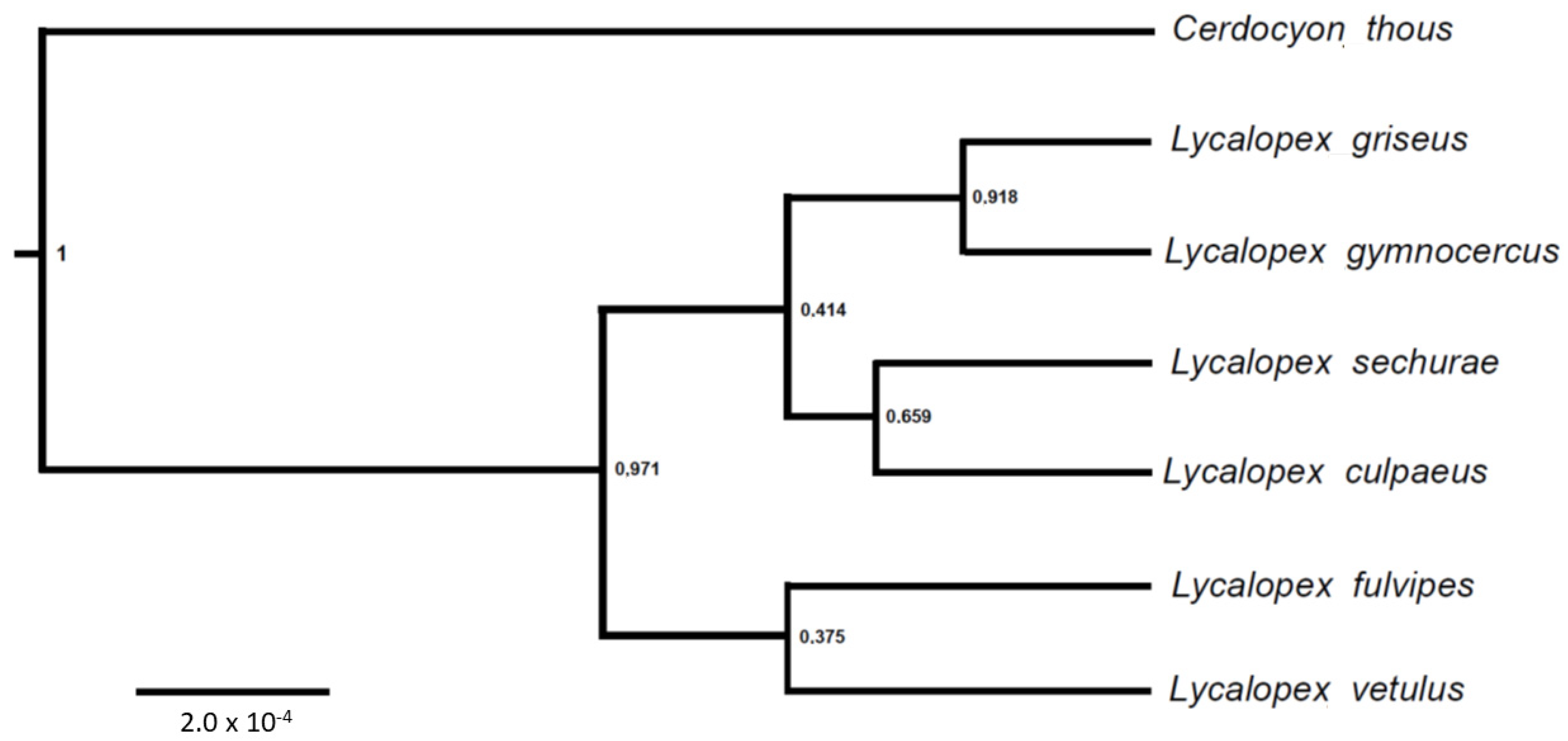
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemey, P.; Salemi, M.; Vandamme, A.M. (Eds.) The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Avise, J.C. Molecular Markers, Natural History and Evolution; Chapman & Hall: New York, NY, USA, 1994. [Google Scholar]
- Kapli, P.; Yang, Z.; Telford, M.J. Phylogenetic tree building in the genomic age. Nat. Rev. Genet. 2020, 21, 428–444. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific Phylogeography: The Mitochondrial DNA Bridge Between Population Genetics and Systematics. Annu. Rev. Ecol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Moritz, C. Defining ‘Evolutionarily Significant Units’ for conservation. Trends Ecol. Evol. 1994, 9, 373–375. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., III; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Johnson, W.E.; Eizirik, E.; Pecon-Slattery, J.; Murphy, W.J.; Antunes, A.; Teeling, E.; O’Brien, S.J. The late Miocene radiation of modern Felidae: A genetic assessment. Science 2006, 311, 73–77. [Google Scholar] [CrossRef]
- Eizirik, E.; Murphy, W.J.; Koepfli, K.P.; Johnson, W.E.; Dragoo, J.W.; Wayne, R.K.; O’Brien, S.J. Pattern and timing of diversification of the mammalian order Carnivora inferred from multiple nuclear gene sequences. Mol. Phylogenet. Evol. 2010, 56, 49–63. [Google Scholar] [CrossRef]
- Li, G.; Davis, B.W.; Eizirik, E.; Murphy, W.J. Phylogenomic evidence for ancient hybridization in the genomes of living cats (Felidae). Genome Res. 2016, 26, 1–11. [Google Scholar] [CrossRef]
- Perini, F.A.; Russo, C.A.M.; Schrago, C.G. The evolution of South American endemic canids: A history of rapid diversification and morphological parallelism. J. Evol. Biol. 2010, 23, 311–322. [Google Scholar] [CrossRef]
- Tchaicka, L.; de Freitas, T.R.O.; Bager, A.; Vidal, S.L.; Lucherini, M.; Iriarte, A.; Novaro, A.; Geffen, E.; Garcez, F.S.; Johnson, W.E.; et al. Molecular assessment of the phylogeny and biogeography of a recently diversified endemic group of South American canids (Mammalia: Carnivora: Canidae). Genet. Mol. Biol. 2016, 39, 442–451. [Google Scholar] [CrossRef]
- Wozencraft, W.C. Carnivora. In Mammal Species of the World: A Taxonomic and Geographic Reference, 3rd ed.; Wilson, D.E., Reeder, D.M., Eds.; Johns Hopkins University Press: Baltimore, MD, USA, 2005; pp. 532–628. [Google Scholar]
- Yahnke, C.J.; Johnson, W.E.; Geffen ESmith, D.; Hertel, F.; Roy, M.S.; Bonacic, C.F.; Fuller, T.K.; Valkenburgh, B.V.; Wayne, R.K. Darwin’s Fox: A Distinct Endangered Species in a Vanishing Habitat. Conserv. Biol. 1996, 10, 366–375. [Google Scholar] [CrossRef]
- Vilà, C.; Leonard, J.A.; Iriarte, A.; O’Brien, S.J.; Johnson, W.E.; Wayne, R.K. Detecting the vanishing populations of the highly endangered Darwin’s fox, Pseudalopex fulvipes. Anim. Conserv. 2004, 7, 147–153. [Google Scholar] [CrossRef]
- Eisenberg, J.F.; Redford, K.H. Mammals of the Neotropics. Ecuador, Peru, Bolivia, Brazil; University of Chicago Press: Chicago, IL, USA, 1999; Volume 3, pp. 13–18, 279–286, 339–348. [Google Scholar]
- MacDonald, D.W.; Zubiri, C.S. Biology and Conservation of Wild Canids; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Zunino, G.; Vaccaro, O.; Canevari, M.; Gardner, A. Taxonomy of the Genus Lycalopex (Carnivora, Canidae) in Argentina. Proc Biol. Soc. 1995, 108, 729–747. [Google Scholar]
- Prevosti, F.J.; Segura, V.; Cassini, G.; Martin, G.M. Revision of the systematic status of Patagonian and Pampean gray foxes (Canidae: Lycalopex griseus and L. gymnocercus) using 3D geometric morphometrics. Mastozool. Neotrop. 2013, 20, 289–300. [Google Scholar]
- Berta, A. Origin, diversification, and zoogeography of the South American Canidae. Fieldiana Zool. 1987, 39, 455–471. [Google Scholar]
- Wayne, R.K.; Geffen, E.; Girman, D.J.; Koepfli, K.P.; Lau, L.M.; Marshall, C.R. Molecular systematics of the Canidae. Syst. Biol. 1997, 46, 622–653. [Google Scholar] [CrossRef]
- Zrzavy, J.; Ricánková, V. Phylogeny of Recent Canidae (Mammalia, Carnivora): Relative reliability and utility of morphological and molecular datasets. Zool. Scr. 2004, 33, 311–333. [Google Scholar] [CrossRef]
- Chemisquy, M.A.; Prevosti, F.J.; Martínez, P.; Raimondi, V.; Stom, J.E.C.; Acosta-Jamett, G.; Montoya-Burgos, J.I. How many species of grey foxes (Canidae, Carnivora) are there in southern South America? Mastozool. Neotrop. 2019, 26, 81–97. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Delisle, I.; Strobeck, C. Conserved Primers for Rapid Sequencing of the Complete Mitochondrial Genome from Carnivores, Applied to Three Species of Bears. Mol. Biol. Evol. 2002, 19, 357–361. [Google Scholar] [CrossRef]
- Irwin, D.M.; Kocher, T.D.; Wilson, A.C. Evolution of the cytochrome b gene of mammals. J. Mol. Evol. 1991, 32, 128–144. [Google Scholar] [CrossRef]
- Trigo, T.C.; Freitas, T.R.O.; Kunzler, G.; Cardoso, L.; Silva, J.C.R.; Johnson, W.E.; O’Brien, S.J.; Bonatto, S.L.; Eizirik, E. Interspecies hybridization among Neotropical cats of the genus Leopardus, and evidence for an introgressive hybrid zone between L. geoffroyi and L. tigrinus in southern Brazil. Mol. Ecol. 2008, 17, 4317–4333. [Google Scholar]
- Tchaicka, L.; Eizirik, E.; Oliveira, T.G.; Cândido, J.F., Jr.; Freitas, T.R.O. Phylogeography and population history of the crab-eating fox (Cerdocyon thous). Mol. Ecol. 2007, 16, 819–838. [Google Scholar] [CrossRef]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using phred II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Abajian, C.; Green, P. Consed: A Graphical Tool for Sequence Finishing. Genome Res. 1998, 8, 195–202. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 11, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4; Sinauer Associates: Suderland, MA, USA, 2000. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K. Modeltest: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef]
- Zwickl, D.J. Genetic Algorithm Approaches for the Phylogenetic Analysis of Large Biological Sequence Datasets under the Maximum Likelihood Criterion. Ph.D. Thesis, The University of Texas at Austin, Austin, TX, USA, 2006. [Google Scholar]
- Goloboff, P.A.; Farris, J.S.; Nixon, K.C. TNT, a free program for phylogenetic analysis. Cladistics 2008, 24, 774–786. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Nylander, J.A.A. MrModeltest v2—Program Distributed by the Author; Evolutionary Biology Centre, Uppsala University: Uppsala, Sweden, 2004. [Google Scholar]
- McKenna, M.C.; Bell, S.K. Classification of Mammals above the Species Level; Columbia University Press: New York, NY, USA, 1997. [Google Scholar]
- Kurtén, B.; Anderson, E. Pleistocene Mammals of North America; Columbia University Press: New York, NY, USA, 1980. [Google Scholar]
- Stephens, M.; Smith, N.J.; Donnelly, P. A New Statistical Method for Haplotype Reconstruction from Population Data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Heled, J.; Drummond, A.J. Bayesian Inference of Species Trees from Multilocus Data. Mol. Biol. Evol. 2009, 27, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Prevosti, F. Phylogeny of the large extinct South American Canids (Mammalia, Carnivora, Canidae) using a “total evidence” approach. Cladistics 2010, 26, 456–481. [Google Scholar] [CrossRef]
- Langguth, A. Ecology and evolution in the South American canids. In The Wild Canids; Fox, M.W., Ed.; Litton Educational Publishing: New York, NY, USA, 1975; pp. 92–206. [Google Scholar]
- Medel, R.G.; Jiménez, J.E.; Jaksic, F.M.; Yáñez, J.L.; Armesto, J.J. Discovery of a continental population of the rare Darwin’s fox, Dusicyon fulvipes (Martin, 1837) in Chile. Biol. Cons. 1990, 51, 71–77. [Google Scholar] [CrossRef]
- Avise, J.C.; Walker, D.; Johns, G.C. Speciation durations and Pleistocene effects on vertebrate phylogeography. Proc. R. Soc. B 1998, 265, 1707–1712. [Google Scholar] [CrossRef]
- MacFadden, B.J. Extinct mammalian biodiversity of the ancient New World tropics. Trends Ecol. Evol. 2006, 21, 157–165. [Google Scholar] [CrossRef]
- Webb, S.D. A History of Savanna Vertebrates in The New World. Part I: South America and the Great Interchange. Ann. Rev. Ecol. Syst. 1977, 8, 355–380. [Google Scholar] [CrossRef]
- Cartelle, C. Pleistocene Mammals of the Cerrado and Caatinga of Brazil. Mammals of the Neotropics, the Central Neotropics: Volume 3, Ecuador, Peru, Bolivia, Brazil; Eisenberg, J.F., Redford, K.H., Eds.; The University of Chicago Press: Chicago, IL, USA, 1999; pp. 27–46. [Google Scholar]
- Webb, S.D. A History of Savanna Vertebrates in the New World. Part II: South America and the Great Interchange. Annu. Rev. Ecol. Syst. 1978, 9, 393–426. [Google Scholar] [CrossRef]
- Vivo, M.; Carmignotto, A.P. Holocene vegetation change and the mammal faunas of South America and Africa. J. Biogeogr. 2004, 31, 943–957. [Google Scholar] [CrossRef]
- Fariña, R.A.; Vizcaíno, S.F.; de Iuliis, G. Megafauna: Giant Beasts of Pleistocene South America; Indiana University Press: Bloomington, IN, USA, 2013. [Google Scholar]
- Jiang, Z.; Priat, C.; Galibert, F. Traced orthologous amplified sequence tags (TOASTs) and mammalian comparative maps. Mamm. Genome 1988, 9, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Lyons, L.A.; Laughlin, T.F.; Copeland, N.G.; Jenkins, N.A.; Womack, J.E.; O’Brien, S.J. Comparative anchor tagged sequences (CATS) for Integrative mapping of mammalian genomes. Nat. Genet. 1997, 15, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Venta, P.J.; Brouillette, J.A.; Yuzbasiyan-Gurkan, V.; Brewer, G.J. Gene-specific universal mammalian sequence-tagged sites: Application to the canine genome. Biochem. Genet. 1996, 34, 321–341. [Google Scholar] [CrossRef] [PubMed]
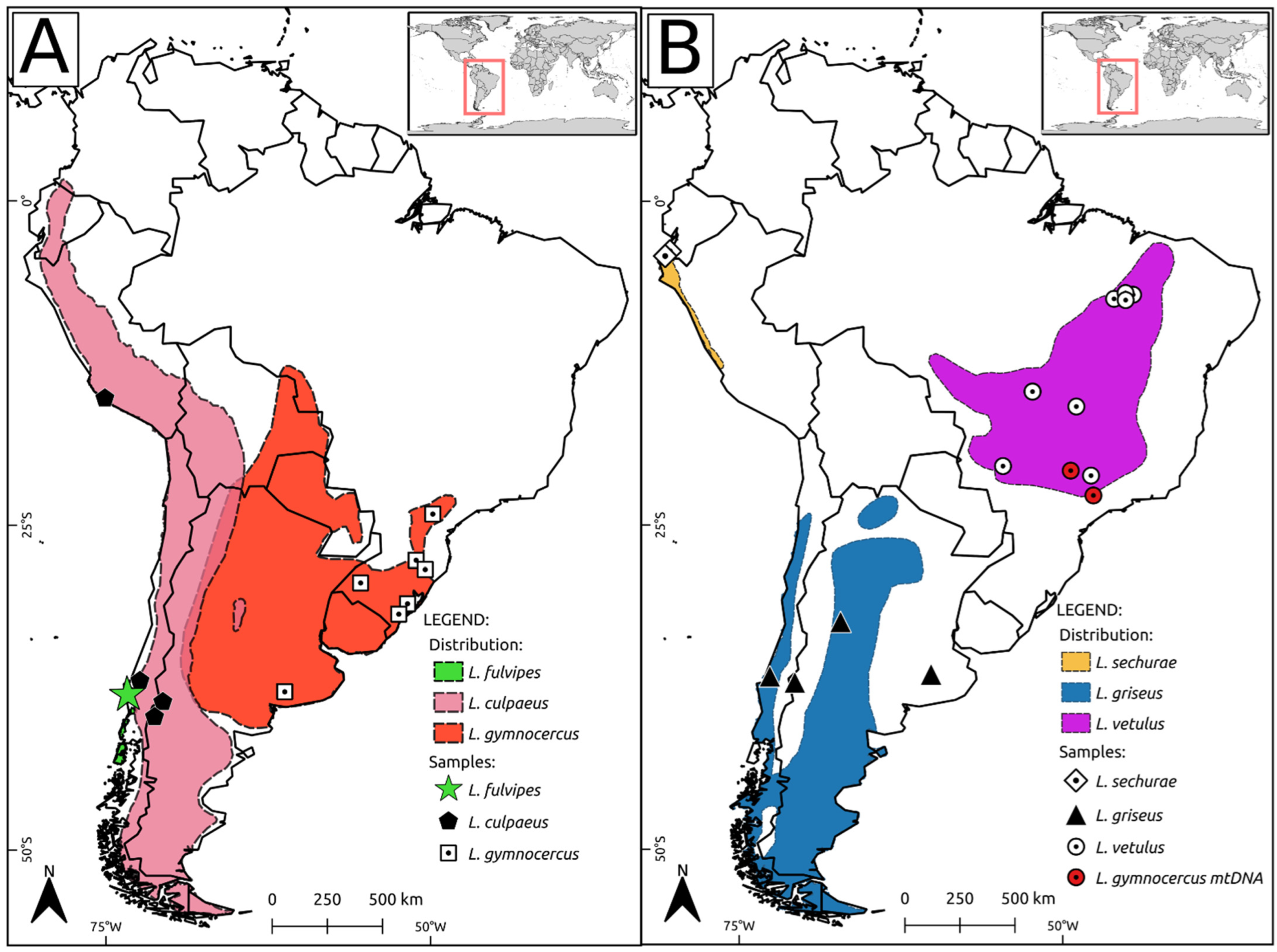
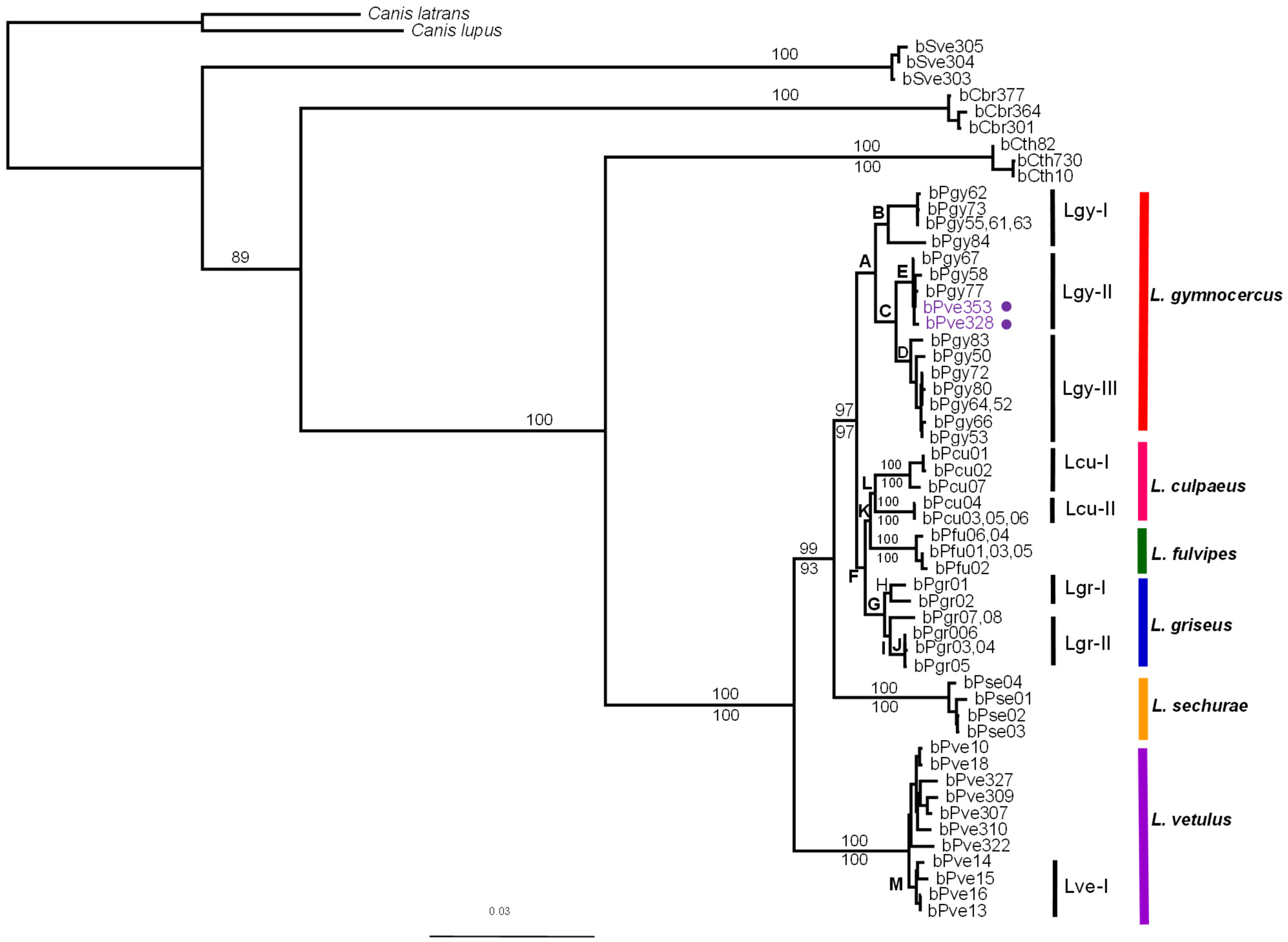
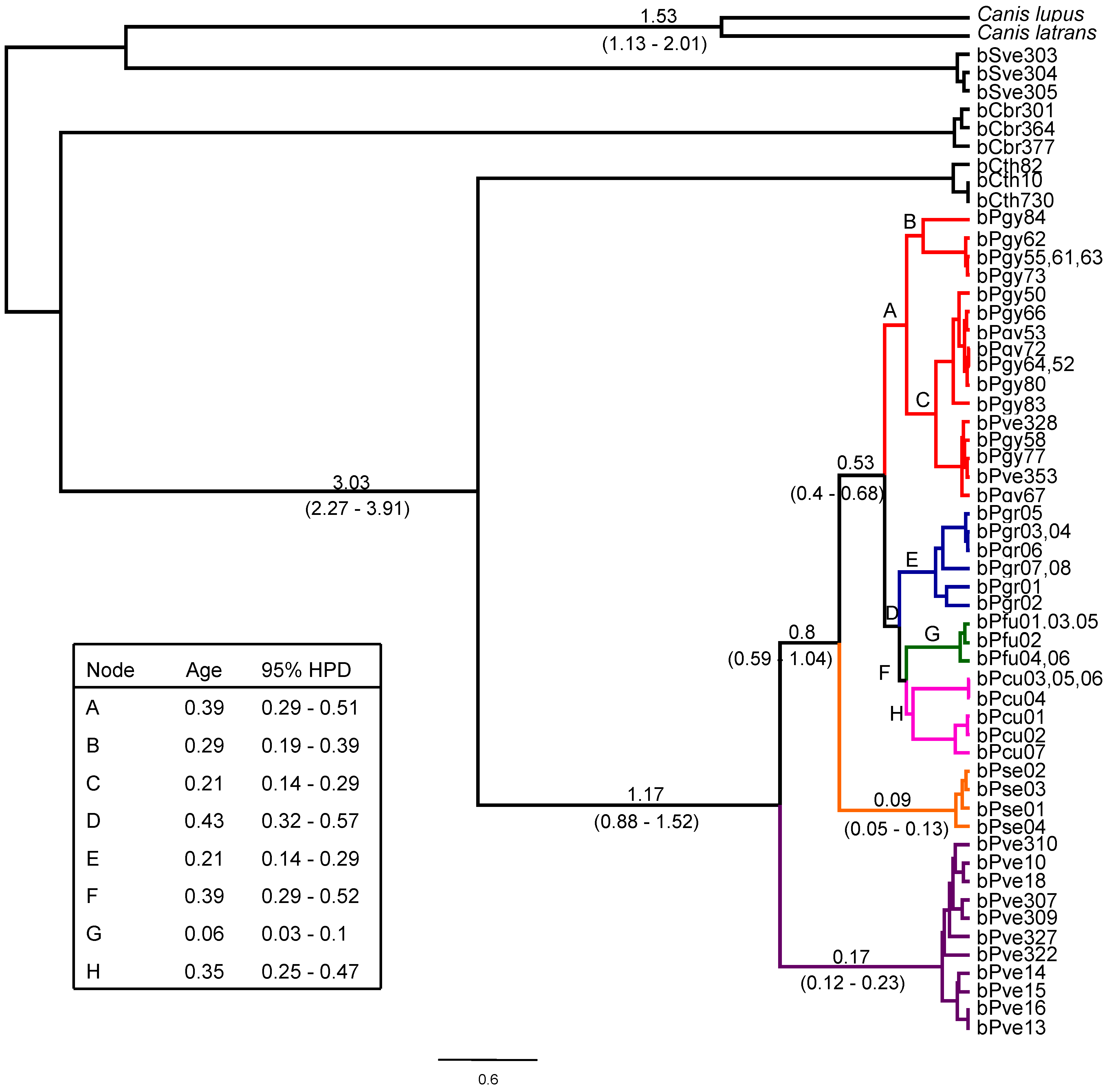



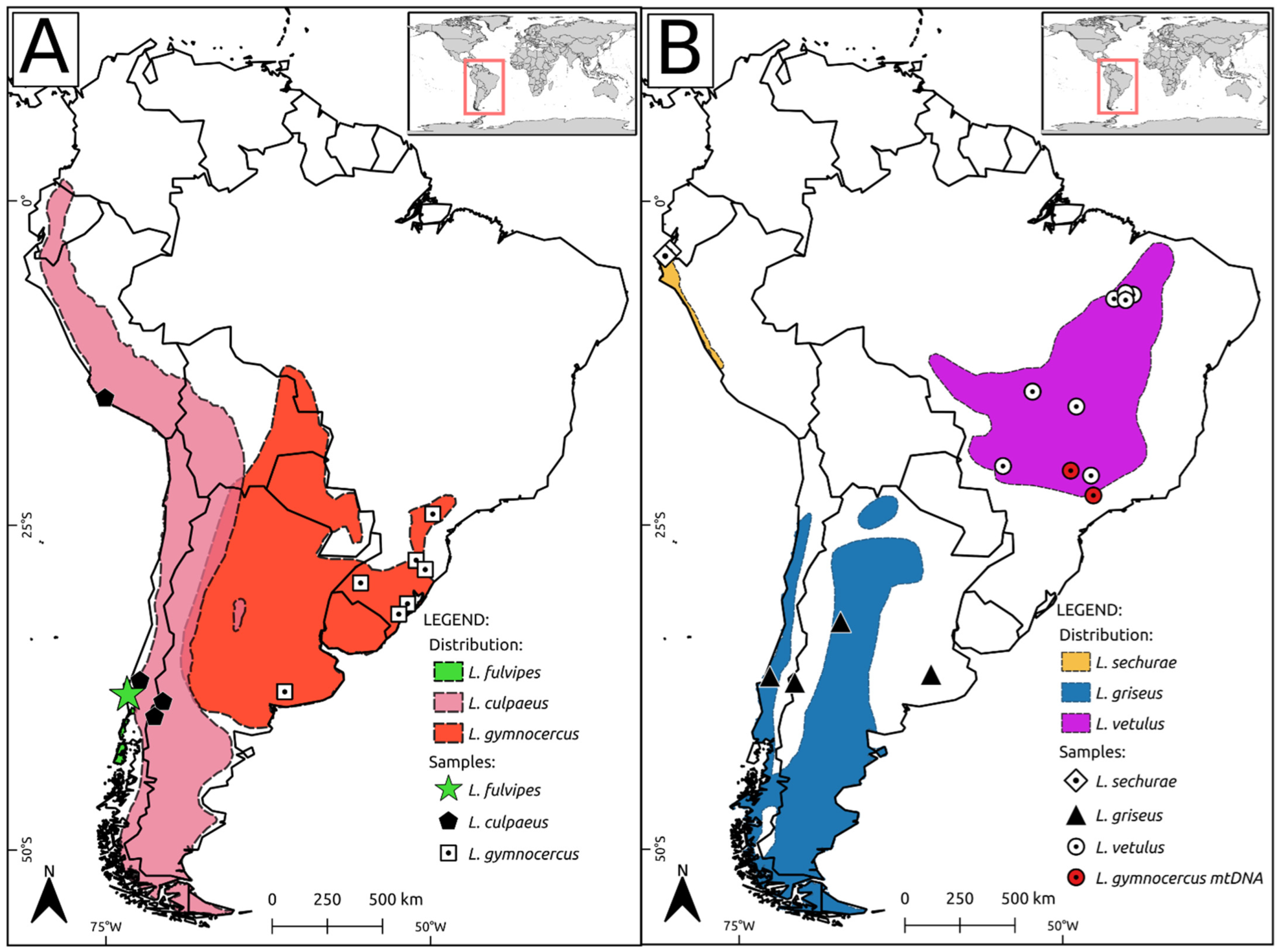{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Node | Phylogeny | ||||
|---|---|---|---|---|---|
| MP | ML | ||||
| PAUP | TNT | PAUP | Garli | Beast | |
| A | 100 | 99 | 99 | 100 | 100 |
| B | 91 | 83 | 96 | 97 | 100 |
| C | 100 | 99 | 99 | 100 | 100 |
| D | 100 | 99 | 99 | 99 | 100 |
| E | 100 | 100 | 100 | 100 | 100 |
| F | 70 | 69 | 87 | 83 | 100 |
| G | 100 | 99 | 100 | 100 | 100 |
| H | 94 | 87 | 93 | 97 | 99 |
| I | 96 | 90 | 90 | 94 | 100 |
| J | 100 | 99 | 100 | 100 | 99 |
| K | 55 | 55 | 76 | 86 | 100 |
| L | 81 | 81 | 75 | 92 | 98 |
| M | 86 | 81 | 83 | 83 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Favarini, M.O.; Simão, T.L.L.; Macedo, G.S.; Garcez, F.S.; Oliveira, L.R.; Cárdenas-Alayza, S.; Cardeña Mormontoy, M.; Angulo, F.; Kasper, C.B.; Johnson, W.E.; et al. Complex Evolutionary History of the South American Fox Genus Lycalopex (Mammalia, Carnivora, Canidae) Inferred from Multiple Mitochondrial and Nuclear Markers. Diversity 2022, 14, 642. https://doi.org/10.3390/d14080642
Favarini MO, Simão TLL, Macedo GS, Garcez FS, Oliveira LR, Cárdenas-Alayza S, Cardeña Mormontoy M, Angulo F, Kasper CB, Johnson WE, et al. Complex Evolutionary History of the South American Fox Genus Lycalopex (Mammalia, Carnivora, Canidae) Inferred from Multiple Mitochondrial and Nuclear Markers. Diversity. 2022; 14(8):642. https://doi.org/10.3390/d14080642
Chicago/Turabian StyleFavarini, Marina O., Taiz L. L. Simão, Gabriel S. Macedo, Fabrício S. Garcez, Larissa R. Oliveira, Susana Cárdenas-Alayza, Marco Cardeña Mormontoy, Fernando Angulo, Carlos Benhur Kasper, Warren E. Johnson, and et al. 2022. "Complex Evolutionary History of the South American Fox Genus Lycalopex (Mammalia, Carnivora, Canidae) Inferred from Multiple Mitochondrial and Nuclear Markers" Diversity 14, no. 8: 642. https://doi.org/10.3390/d14080642
APA StyleFavarini, M. O., Simão, T. L. L., Macedo, G. S., Garcez, F. S., Oliveira, L. R., Cárdenas-Alayza, S., Cardeña Mormontoy, M., Angulo, F., Kasper, C. B., Johnson, W. E., & Eizirik, E. (2022). Complex Evolutionary History of the South American Fox Genus Lycalopex (Mammalia, Carnivora, Canidae) Inferred from Multiple Mitochondrial and Nuclear Markers. Diversity, 14(8), 642. https://doi.org/10.3390/d14080642