
Assessment of ITS1, ITS2, 5′-ETS, and trnL-F DNA Barcodes for Metabarcoding of Poaceae Pollen

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. DNA Extraction
2.3. PCR and Primer Design
2.4. Library Preparation and Sequencing
2.5. Local Barcode Reference Database Construction
2.6. Data Analysis and Taxonomical Identification
3. Results
3.1. ETS Primers Design
3.2. Pollen DNA Extraction Optimization
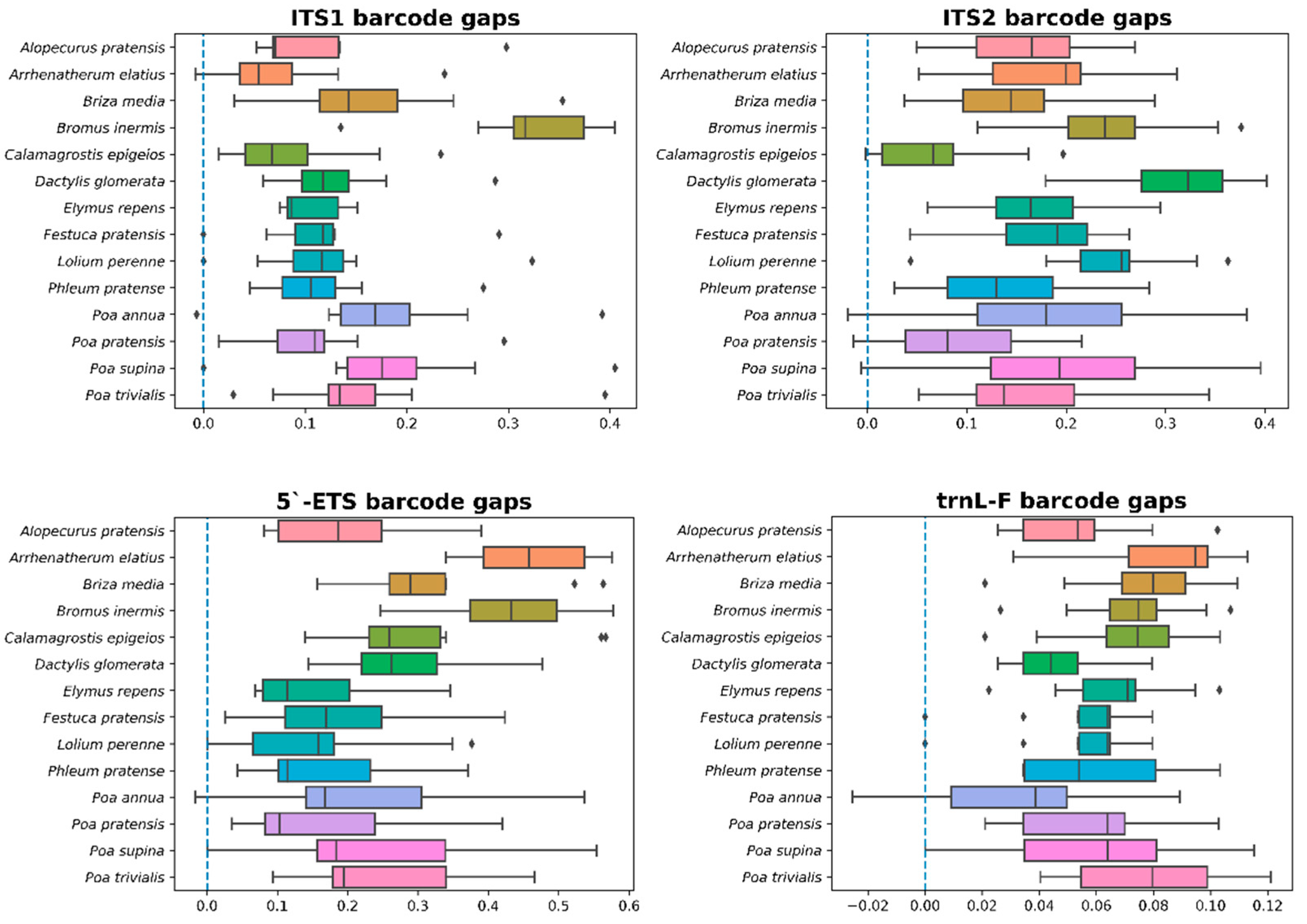
3.3. 5.’-ETS, ITS1, ITS2, and trnL-F Barcodes Comparison
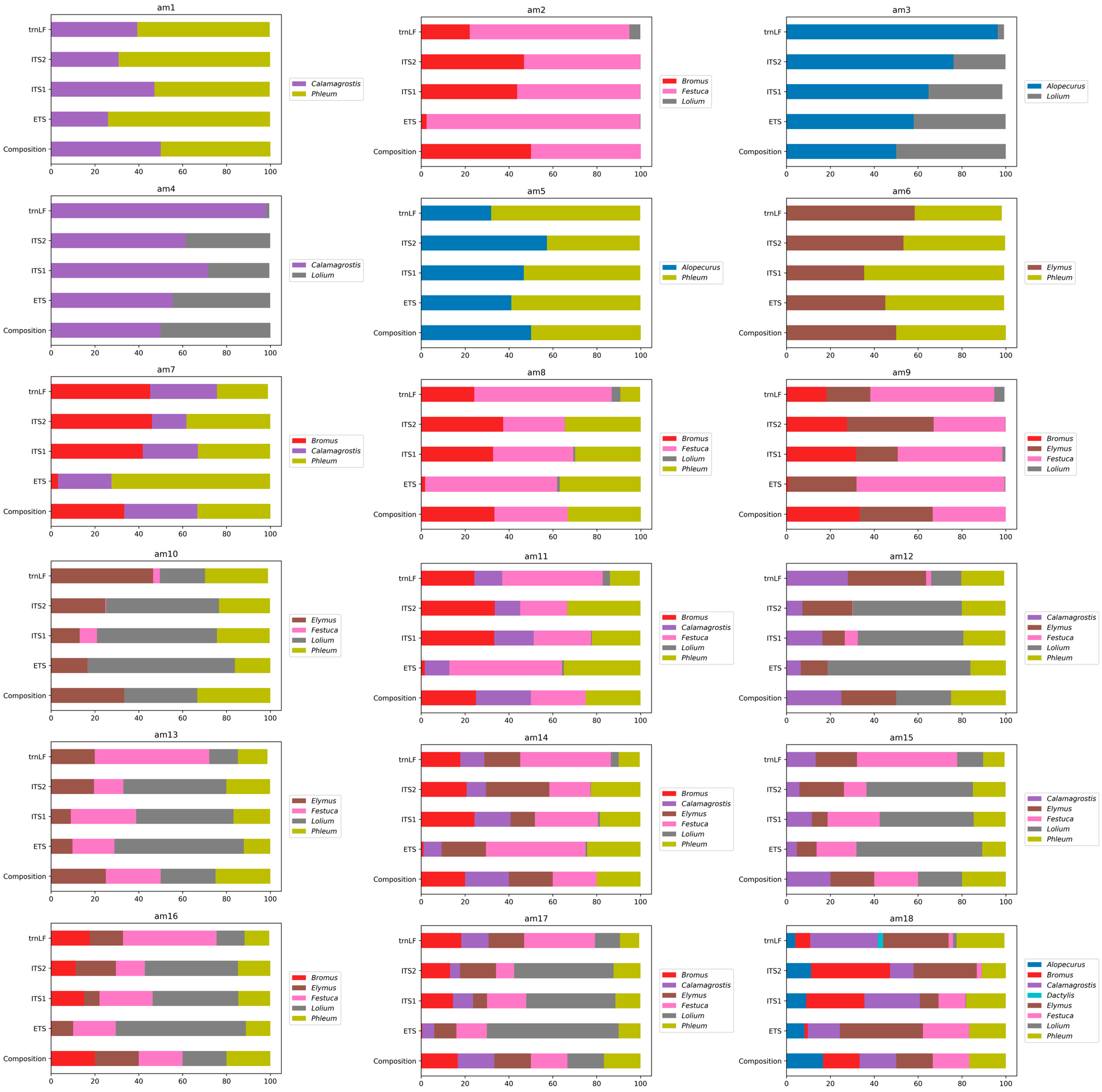
3.4. Metabarcoding Analysis of the Artificial Pollen Mixes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- García-Mozo, H. Poaceae Pollen as the Leading Aeroallergen Worldwide: A Review. Allergy 2017, 72, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- Damialis, A.; Traidl-Hoffmann, C.; Treudler, R. Climate Change and Pollen Allergies. In Biodiversity and Health in the Face of Climate Change; Marselle, M.R., Stadler, J., Korn, H., Irvine, K.N., Bonn, A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 47–66. ISBN 978-3-030-02318-8. [Google Scholar]
- D’Amato, G.; Cecchi, L.; Bonini, S.; Nunes, C.; Annesi-Maesano, I.; Behrendt, H.; Liccardi, G.; Popov, T.; Cauwenberge, P.V. Allergenic Pollen and Pollen Allergy in Europe. Allergy 2007, 62, 976–990. [Google Scholar] [CrossRef]
- Mayevsky, P.F. Flora of the Middle Zone of the European Part of Russia; KMK Scientific Press Ltd.: Moscow, Russia, 2014. [Google Scholar]
- Tree and Plant Allergy Info for Research—Allergen and Botanic Reference Library. Available online: http://www.pollenlibrary.com/ (accessed on 16 March 2020).
- Erdtman, G. Pollen Morphology and Plant Taxonomy: Angiosperms; E.J. Brill: Leiden, The Netherlands, 1986; ISBN 978-90-04-08122-2. [Google Scholar]
- Joly, C.; Barillé, L.; Barreau, M.; Mancheron, A.; Visset, L. Grain and Annulus Diameter as Criteria for Distinguishing Pollen Grains of Cereals from Wild Grasses. Rev. Palaeobot. Palynol. 2007, 146, 221–233. [Google Scholar] [CrossRef]
- Ünsal, S.G.; Çiftçi, Y.Ö.; Eken, B.U.; Velioğlu, E.; Di Marco, G.; Gismondi, A.; Canini, A. Intraspecific Discrimination Study of Wild Cherry Populations from North-Western Turkey by DNA Barcoding Approach. Tree Genet. Genomes 2019, 15, 16. [Google Scholar] [CrossRef]
- Gismondi, A.; Di Marco, G.; Martini, F.; Sarti, L.; Crespan, M.; Martínez-Labarga, C.; Rickards, O.; Canini, A. Grapevine Carpological Remains Revealed the Existence of a Neolithic Domesticated Vitis Vinifera L. Specimen Containing Ancient DNA Partially Preserved in Modern Ecotypes. J. Archaeol. Sci. 2016, 69, 75–84. [Google Scholar] [CrossRef]
- Techen, N.; Parveen, I.; Pan, Z.; Khan, I.A. DNA Barcoding of Medicinal Plant Material for Identification. Curr. Opin. Biotechnol. 2014, 25, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Bruni, I.; De Mattia, F.; Galimberti, A.; Galasso, G.; Banfi, E.; Casiraghi, M.; Labra, M. Identification of Poisonous Plants by DNA Barcoding Approach. Int. J. Leg. Med. 2010, 124, 595–603. [Google Scholar] [CrossRef]
- Bruni, I.; Galimberti, A.; Caridi, L.; Scaccabarozzi, D.; De Mattia, F.; Casiraghi, M.; Labra, M. A DNA Barcoding Approach to Identify Plant Species in Multiflower Honey. Food Chem. 2015, 170, 308–315. [Google Scholar] [CrossRef]
- Prosser, S.W.J.; Hebert, P.D.N. Rapid Identification of the Botanical and Entomological Sources of Honey Using DNA Metabarcoding. Food Chem. 2017, 214, 183–191. [Google Scholar] [CrossRef]
- Taylor, H.R.; Harris, W.E. An Emergent Science on the Brink of Irrelevance: A Review of the Past 8 Years of DNA Barcoding. Mol. Ecol. Resour. 2012, 12, 377–388. [Google Scholar] [CrossRef]
- Coissac, E.; Riaz, T.; Puillandre, N. Bioinformatic Challenges for DNA Metabarcoding of Plants and Animals. Mol. Ecol. 2012, 21, 1834–1847. [Google Scholar] [CrossRef]
- CBOL Plant Working Group A DNA Barcode for Land Plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [CrossRef] [PubMed]
- Shneyer, V.S.; Rodionov, A.V. Plant DNA Barcodes. Biol. Bull Rev. 2019, 9, 295–300. [Google Scholar] [CrossRef]
- Beng, K.C.; Tomlinson, K.W.; Shen, X.H.; Surget-Groba, Y.; Hughes, A.C.; Corlett, R.T.; Slik, J.W.F. The Utility of DNA Metabarcoding for Studying the Response of Arthropod Diversity and Composition to Land-Use Change in the Tropics. Sci. Rep. 2016, 6, 24965. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.L.; de Vere, N.; Keller, A.; Richardson, R.T.; Gous, A.; Burgess, K.S.; Brosi, B.J. Pollen DNA Barcoding: Current Applications and Future Prospects. Genome 2016, 59, 629–640. [Google Scholar] [CrossRef]
- Bell, K.L.; Fowler, J.; Burgess, K.S.; Dobbs, E.K.; Gruenewald, D.; Lawley, B.; Morozumi, C.; Brosi, B.J. Applying Pollen DNA Metabarcoding to the Study of Plant–Pollinator Interactions. Appl. Plant Sci. 2017, 5, 1600124. [Google Scholar] [CrossRef]
- Bell, K.L.; Burgess, K.S.; Botsch, J.C.; Dobbs, E.K.; Read, T.D.; Brosi, B.J. Quantitative and Qualitative Assessment of Pollen DNA Metabarcoding Using Constructed Species Mixtures. Mol. Ecol. 2019, 28, 431–455. [Google Scholar] [CrossRef]
- Baksay, S.; Pornon, A.; Burrus, M.; Mariette, J.; Andalo, C.; Escaravage, N. Experimental Quantification of Pollen with DNA Metabarcoding Using ITS1 and TrnL. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Peterson, P.M.; Romaschenko, K.; Soreng, R.J. A Laboratory Guide for Generating DNA Barcodes in Grasses: A Case Study of Leptochloa s.l. (Poaceae: Chloridoideae). Webbia 2014, 69, 1–12. [Google Scholar] [CrossRef]
- Kraaijeveld, K.; de Weger, L.A.; García, M.V.; Buermans, H.; Frank, J.; Hiemstra, P.S.; Dunnen, J.T. den Efficient and Sensitive Identification and Quantification of Airborne Pollen Using Next-Generation DNA Sequencing. Mol. Ecol. Resour. 2015, 15, 8–16. [Google Scholar] [CrossRef]
- Naciri, Y.; Caetano, S.; Salamin, N. Plant DNA Barcodes and the Influence of Gene Flow. Mol. Ecol. Resour. 2012, 12, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Columbus, J.; Cerros-Tlatilpa, R.; Kinney, M.; Siqueiros-Delgado, M.E.; Bell, H.; Griffith, M.; Refulio-Rodriguez, N. Phylogenetics of Chloridoideae (Gramineae): A Preliminary Study Based on Nuclear Ribosomal Internal Transcribed Spacer and Chloroplast TrnL–F Sequences. Aliso J. Syst. Evol. Bot. 2007, 23, 565–579. [Google Scholar] [CrossRef][Green Version]
- Lloyd, K.; Hunter, A.; Orlovich, D.; Draffin, S.; Stewart, A.; Lee, W. Phylogeny and Biogeography of Endemic Festuca (Poaceae) from New Zealand Based on Nuclear (ITS) and Chloroplast (TrnL–TrnF) Nucleotide Sequences. Aliso J. Syst. Evol. Bot. 2007, 23, 406–419. [Google Scholar] [CrossRef][Green Version]
- Da Silva, L.N.; Essi, L.; Iganci, J.R.V.; Souza-Chies, T.T.D. Advances in the Phylogeny of the South American Cool-Season Grass Genus Chascolytrum (Poaceae, Pooideae): A New Infrageneric Classification. Bot. J. Linn. Soc. 2019, 192, 97–120. [Google Scholar] [CrossRef]
- Wang, A.; Gopurenko, D.; Wu, H.; Lepschi, B. Evaluation of Six Candidate DNA Barcode Loci for Identification of Five Important Invasive Grasses in Eastern Australia. PLoS ONE 2017, 12, e0175338. [Google Scholar] [CrossRef]
- Alonso, A.; Bull, R.D.; Acedo, C.; Gillespie, L.J. Design of Plant-Specific PCR Primers for the ETS Region with Enhanced Specificity for Tribe Bromeae and Their Application to Other Grasses (Poaceae). Botany 2014, 92, 693–699. [Google Scholar] [CrossRef]
- Logacheva, M.D.; Valiejo-Roman, C.M.; Degtjareva, G.V.; Stratton, J.M.; Downie, S.R.; Samigullin, T.H.; Pimenov, M.G. A Comparison of NrDNA ITS and ETS Loci for Phylogenetic Inference in the Umbelliferae: An Example from Tribe Tordylieae. Mol. Phylogenetics Evol. 2010, 57, 471–476. [Google Scholar] [CrossRef]
- Cai, Z.-M.; Zhang, Y.-X.; Zhang, L.-N.; Gao, L.-M.; Li, D.-Z. Testing Four Candidate Barcoding Markers in Temperate Woody Bamboos (Poaceae: Bambusoideae). J. Syst. Evol. 2012, 50, 527–539. [Google Scholar] [CrossRef]
- Su, X.; Liu, Y.P.; Chen, Z.; Chen, K.L. Evaluation of Candidate Barcoding Markers in Orinus (Poaceae). Genet. Mol. Res. GMR 2016, 15. [Google Scholar] [CrossRef]
- Krinitsina, A.A.; Sizova, T.V.; Zaika, M.A.; Speranskaya, A.S.; Sukhorukov, A.P. A Rapid and Cost-Effective Method for DNA Extraction from Archival Herbarium Specimens. Biochemistry 2015, 80, 1478–1484. [Google Scholar] [CrossRef]
- Omelchenko, D.O.; Speranskaya, A.S.; Ayginin, A.A.; Khafizov, K.; Krinitsina, A.A.; Fedotova, A.V.; Pozdyshev, D.V.; Shtratnikova, V.Y.; Kupriyanova, E.V.; Shipulin, G.A.; et al. Improved Protocols of ITS1-Based Metabarcoding and Their Application in the Analysis of Plant-Containing Products. Genes 2019, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Gielly, L.; Pautou, G.; Bouvet, J. Universal Primers for Amplification of Three Non-Coding Regions of Chloroplast DNA. Plant Mol. Biol. 1991, 17, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Speranskaya, A.S.; Khafizov, K.; Ayginin, A.A.; Krinitsina, A.A.; Omelchenko, D.O.; Nilova, M.V.; Severova, E.E.; Samokhina, E.N.; Shipulin, G.A.; Logacheva, M.D. Comparative Analysis of Illumina and Ion Torrent High-Throughput Sequencing Platforms for Identification of Plant Components in Herbal Teas. Food Control 2018, 93, 315–324. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K. Estimation of the Number of Nucleotide Substitutions When There Are Strong Transition-Transversion and G+C-Content Biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A Simple Method for Estimating Evolutionary Rates of Base Substitutions through Comparative Studies of Nucleotide Sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; Volume 445, pp. 56–61. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.L. Seaborn: Statistical Data Visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Gillespie, L.J.; Soreng, R.J.; Paradis, M.; Bull, R.D. Phylogeny and Reticulation in Subtribe Poinae and Related Subtribes (Poaceae) Based on NrITS, ETS, and TrnTLF Data. Diversity, Phylogeny, and Evolution in the Monocotyledons; Aarhus University Press: Aarhus, Denmark, 2010; p. 29. [Google Scholar]
- Consaul, L.L.; Gillespie, L.J.; Waterway, M.J. Evolution and Polyploid Origins in North American Arctic Puccinellia (Poaceae) Based on Nuclear Ribosomal Spacer and Chloroplast DNA Sequences. Am. J. Bot. 2010, 97, 324–336. [Google Scholar] [CrossRef]
- Leontidou, K.; Vernesi, C.; De Groeve, J.; Cristofolini, F.; Vokou, D.; Cristofori, A. DNA Metabarcoding of Airborne Pollen: New Protocols for Improved Taxonomic Identification of Environmental Samples. Aerobiologia 2018, 34, 63–74. [Google Scholar] [CrossRef]
- Ghitarrini, S.; Pierboni, E.; Rondini, C.; Tedeschini, E.; Tovo, G.R.; Frenguelli, G.; Albertini, E. New Biomolecular Tools for Aerobiological Monitoring: Identification of Major Allergenic Poaceae Species through Fast Real-Time PCR. Ecol. Evol. 2018, 8, 3996–4010. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Charmet, G.; Ravel, C.; Balfourier, F. Phylogenetic Analysis in the Festuca-Lolium Complex Using Molecular Markers and ITS RDNA. Theor. Appl. Genet. 1997, 94, 1038–1046. [Google Scholar] [CrossRef]
- Gaut, B.S.; Tredway, L.P.; Kubik, C.; Gaut, R.L.; Meyer, W. Phylogenetic Relationships and Genetic Diversity among Members of TheFestuca-Lolium Complex (Poaceae) Based on ITS Sequence Data. Pl. Syst. Evol. 2000, 224, 33–53. [Google Scholar] [CrossRef]
- Ghesquière, M.; Humphreys, M.W.; Zwierzykowski, Z. Festulolium. In Fodder Crops and Amenity Grasses; Boller, B., Posselt, U.K., Veronesi, F., Eds.; Handbook of Plant Breeding; Springer: New York, NY, USA, 2010; pp. 288–311. ISBN 978-1-4419-0760-8. [Google Scholar]
- Czaban, A.; Sharma, S.; Byrne, S.L.; Spannagl, M.; Mayer, K.F.; Asp, T. Comparative Transcriptome Analysis within the Lolium/Festuca Species Complex Reveals High Sequence Conservation. BMC Genom. 2015, 16, 249. [Google Scholar] [CrossRef]
- Loera-Sánchez, M.; Studer, B.; Kölliker, R. DNA Barcode TrnH-PsbA Is a Promising Candidate for Efficient Identification of Forage Legumes and Grasses. BMC Res. Notes 2020, 13, 35. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhou, K.; Humphreys, M.W.; Harper, J.A.; Ma, X.; Zhang, X.; Yan, H.; Huang, L. Phylogenetic Relationships in the Festuca-Lolium Complex (Loliinae; Poaceae): New Insights from Chloroplast Sequences. Front. Ecol. Evol. 2016, 4, 89. [Google Scholar] [CrossRef]
- Wu, S.; Yin, L.; Deng, Z.; Chen, Q.; Fu, Y.; Xue, H. Using DNA Barcoding to Identify the Genus Lolium. Not. Bot. Horti Agrobot. Cluj-Napoca 2015, 43, 536–541. [Google Scholar] [CrossRef]
- Sodmergen; Suzuki, T.; Kawano, S.; Nakamura, S.; Tano, S.; Kuroiwa, T. Behavior of Organelle Nuclei (Nucleoids) in Generative and Vegetative Cells during Maturation of Pollen InLilium Longiflorum AndPelargonium Zonale. Protoplasma 1992, 168, 73–81. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Artificial Pollen Mix Name | Species |
|---|---|
| Pollen mixes with two species, 50% pollen of each type | |
| am1 | Calamagrostis epigeios, Phleum pretense |
| am2 | Bromus inermis, Festuca pratensis |
| am3 | Alopecurus pratensis, Lolium perenne |
| am4 | Calamagrostis epigeios, Lolium perenne |
| am5 | Phleum pratense, Alopecurus pratensis |
| am6 | Phleum pratense, Elymus repens |
| Pollen mixes with three species, 33.3% pollen of each type | |
| am7 | Calamagrostis epigeios, Phleum pratense, Bromus inermis |
| am8 | Phleum pratense, Bromus inermis, Festuca pratensis |
| am9 | Bromus inermis, Festuca pratensis, Elymus repens |
| am10 | Phleum pratense, Lolium perenne, Elymus repens |
| Pollen mixes with four species, 25% pollen of each type | |
| am11 | Calamagrostis epigeios, Phleum pratense, Bromus inermis, Festuca pratensis |
| am12 | Phleum pratense, Calamagrostis epigeios, Elymus repens, Lolium perenne |
| am13 | Phleum pratense, Festuca pratensis, Lolium perenne, Elymus repens |
| Pollen mixes with five species, 20% pollen of each type | |
| am14 | Calamagrostis epigeios, Phleum pratense, Bromus inermis, Festuca pratensis, Elymus repens |
| am15 | Phleum pratense, Calamagrostis epigeios, Festuca pratensis, Lolium perenne, Elymus repens |
| am16 | Phleum pratense, Festuca pratensis, Bromus inermis, Lolium perenne, Elymus repens |
| Pollen mixes with six species, 16.7% pollen of each type | |
| am17 | Phleum pratense, Calamagrostis epigeios, Festuca pratensis, Bromus inermis, Lolium perenne, Elymus repens |
| am18 | Calamagrostis epigeios, Phleum pratense, Bromus inermis, Festuca pratensis, Elymus repens, Alopecurus pratensis |
| Name | 5′-3′ | Tm (Q5), °C | Binomial Species Name |
|---|---|---|---|
| ETS-allF | GCYDTTGGTYYHGGATG | 53–70 | |
| ETS-1F | GCTATTGGTCTCGGATG | 59 | Poa palustris |
| ETS-2F | GCTGTTGGTCTCGGATG | 63 | Poa trivialis, Poa pratensis, Alopecurus pratensis, Lolium perenne, Festuca pratensis, Festuca arundinacea, Poa annua, Poa supina, Elymus repens |
| ETS-3F | GCCGTTGGTCTCGGATG | 66 | Phleum pratense |
| ETS-4F | GCTTTTGGTCTAGGATG | 56 | Bromus inermis |
| ETS-5F | GCTGTTGGTTTCGGATG | 61 | Briza media |
| ETS-6F | GCTGTTGGTTTTGGATG | 58 | Calamagrostis epigeios, Arrhenatherum elatius |
| ETS-7F | GCCGTTGGTCCTGGATG | 66 | Dactylis glomerata |
| Lysis Buffer | Proteinase K mg per Sample | 1 H Lysis | 2 H Lysis | |||||
|---|---|---|---|---|---|---|---|---|
| Yield, ng ∗ µL−1 | 260/280 | 260/230 | Yield, ng ∗ µL−1 | 260/280 | 260/230 | |||
| P. pratense DNA | ||||||||
| CTAB | 0.2 | 12.50 ± 0.66 | 2.06 ± 0.01 | 1.99 ± 0.05 | 13.79 ± 0.49 | 2.03 ± 0.03 | 2.00 ± 0.01 | |
| 0.4 | 7.34 ± 0.22 | 1.99 ± 0.11 | 1.99 ± 0.06 | 6.29 ± 0.29 | 2.01 ± 0.05 | 1.96 ± 0.06 | ||
| CTAB + 0.04% SDS | 0.2 | 16.57 ± 0.61 | 2.03 ± 0.004 | 1.95 ± 0.09 | 16.88 ± 0.19 | 2.05 ± 0.05 | 1.99 ± 0.09 | |
| 0.4 | 9.51 ± 0.88 | 2.07 ± 0.01 | 1.99 ± 0.01 | 9.08 ± 0.96 | 2.04 ± 0.10 | 1.98 ± 0.08 | ||
| CTAB + 0.4% SDS | 0.2 | 4.41 ± 1.04 | 2.01 ± 0.03 | 1.92 ± 0.09 | 4.43 ± 0.93 | 1.97 ± 0.06 | 1.99 ± 0.03 | |
| 0.4 | 2.91 ± 0.11 | 2.01 ± 0.01 | 1.89 ± 0.05 | 2.78 ± 0.6 | 1.99 ± 0.03 | 2.00 ± 0.03 | ||
| B. inermis DNA | ||||||||
| CTAB | 0.2 | 9.84 ± 0.34 | 2.01 ± 0.03 | 1.89 ± 0.09 | 11.41 ± 0.42 | 2.05 ± 0.05 | 2.00 ± 0.01 | |
| 0.4 | 7.72 ± 0.44 | 2.01 ± 0.18 | 1.99 ± 0.003 | 8.31 ± 0.31 | 2.06 ± 0.09 | 1.89 ± 0.09 | ||
| CTAB + 0.04% SDS | 0.2 | 13.62 ± 0.64 | 2.04 ± 0.02 | 1.95 ± 0.08 | 15.55 ± 0.59 | 2.09 ± 0.10 | 1.99 ± 0.02 | |
| 0.4 | 6.46 ± 0.53 | 1.99 ± 0.01 | 1.97 ± 0.09 | 9.69 ± 0.80 | 2.00 ± 0.02 | 1.99 ± 0.05 | ||
| CTAB + 0.4% SDS | 0.2 | 4.15 ± 0.53 | 2.00 ± 0.05 | 1.93 ± 0.08 | 5.36 ± 0.19 | 2.00 ± 0.05 | 1.99 ± 0.09 | |
| 0.4 | 3.34 ± 0.91 | 2.01 ± 0.04 | 1.99 ± 0.02 | 3.54 ± 0.49 | 1.95 ± 0.05 | 1.98 ± 0.04 | ||
| Pollen Amount, Dilution Factor | Approximate Pollen Grain Count | Concentration, ng ∗ µL−1 |
|---|---|---|
| 10 mg | 150,000 | 14.7 ± 0.7 |
| 1:4 | 37,500 | 3.88 ± 0.28 |
| 1:16 | 10,000 | 1.07 ± 0.12 |
| 1:64 | 2350 | too low |
| 1:256 | 600 | too low |
| 1:1024 | 150 | too low |
| Barcode | Interspecific | Intraspecific | ||||
|---|---|---|---|---|---|---|
| Minimum | Maximum | Median | Minimum | Maximum | Median | |
| ITS1 | 0.0000 | 0.4112 | 0.1514 | 0.0000 | 0.0756 | 0.0000 |
| ITS2 | 0.0000 | 0.4549 | 0.2345 | 0.0000 | 0.0726 | 0.0000 |
| ETS | 0.0000 | 0.6142 | 0.2897 | 0.0000 | 0.0431 | 0.0000 |
| trnL-F | 0.0000 | 0.1278 | 0.0698 | 0.0000 | 0.0254 | 0.0000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omelchenko, D.O.; Krinitsina, A.A.; Kasianov, A.S.; Speranskaya, A.S.; Chesnokova, O.V.; Polevova, S.V.; Severova, E.E. Assessment of ITS1, ITS2, 5′-ETS, and trnL-F DNA Barcodes for Metabarcoding of Poaceae Pollen. Diversity 2022, 14, 191. https://doi.org/10.3390/d14030191
Omelchenko DO, Krinitsina AA, Kasianov AS, Speranskaya AS, Chesnokova OV, Polevova SV, Severova EE. Assessment of ITS1, ITS2, 5′-ETS, and trnL-F DNA Barcodes for Metabarcoding of Poaceae Pollen. Diversity. 2022; 14(3):191. https://doi.org/10.3390/d14030191
Chicago/Turabian StyleOmelchenko, Denis O., Anastasia A. Krinitsina, Artem S. Kasianov, Anna S. Speranskaya, Olga V. Chesnokova, Svetlana V. Polevova, and Elena E. Severova. 2022. "Assessment of ITS1, ITS2, 5′-ETS, and trnL-F DNA Barcodes for Metabarcoding of Poaceae Pollen" Diversity 14, no. 3: 191. https://doi.org/10.3390/d14030191
APA StyleOmelchenko, D. O., Krinitsina, A. A., Kasianov, A. S., Speranskaya, A. S., Chesnokova, O. V., Polevova, S. V., & Severova, E. E. (2022). Assessment of ITS1, ITS2, 5′-ETS, and trnL-F DNA Barcodes for Metabarcoding of Poaceae Pollen. Diversity, 14(3), 191. https://doi.org/10.3390/d14030191