Abstract
Pravastatin is a popular statin agent with applications in the treatment of cardiovascular diseases for patients with an abnormal lipid panel. The starting form of pravastatin is amorphous and following recrystallization, it becomes a crystalline solid with tert-butylamine salt molecules embedded within the lattice. Its molecular and crystal structure were elucidated based on single-crystal X-ray diffraction and characterized by ss-NMR.
1. Introduction
Statins represent a family of agents with applications in the treatment of cardiovascular diseases, which work via the cholesterol-lowering pathway [1,2]. Pravastatin [(3R,5R)-7-[(1S,2S,6S,8S,8aR)-6-hydroxy-2-methyl-8-[(2S)-2-methylbutanoyl]oxy-1,2,6,7,8,8a-hexahydronaphthalen-1-yl]-3,5-dihydroxyheptanoic acid] (Figure 1a) is a medication included in this class that is medically used in the treatment of dyslipidemia [3,4].
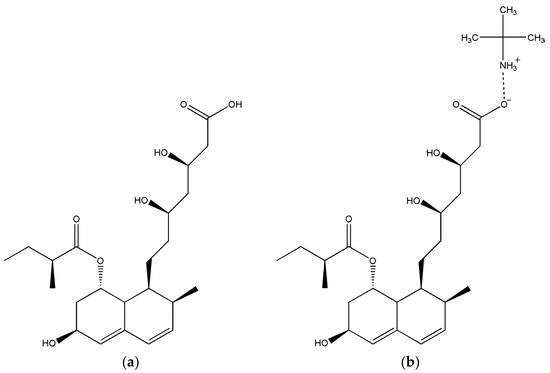
Figure 1.
Molecular structure of pravastatin (a); pravastatin tert-butylamine salt (b).
In the investigation of active pharmaceutical ingredients from a pharmaceutical point of view, certain parameters such as bioavailability, pharmacokinetics, dissolution rate and solubility are frequently studied [5]. Improving one of these aspects can be achieved using various methods including the formation of salts [6], preparation of nano emulsions [7], complexation with cyclodextrins [8,9,10,11], lipid formulations [12] and preparation of cocrystals and polymorphs [13]. Solid forms of pharmaceutical compounds are found in crystalline and amorphous forms and crystalline forms are thermodynamically more stable than amorphous forms. In general, it has been observed that the amorphous forms of pharmaceutics exhibit higher solubility in comparison with crystalline solids [14].
The aim of this study was to achieve the preparation of pravastatin in a crystalline form, which was successfully prepared as a tert-butylamine salt (denoted Prav-tbua, Figure 1b), the elucidation of its crystal structure via X-ray crystallography and its characterization from a structural point of view.
2. Results
2.1. X-ray Structure Analysis of Prav-tbua
The analysis of Prav-tbua by single-crystal X-ray diffraction revealed that the structure belongs to the orthorhombic P212121 space group (Table 1). The asymmetric unit is depicted in Figure 2a and the compound crystallizes in a stoichiometric ratio of 1:1 pravastatin to solvent molecules. It should be noted that the carboxyl group of pravastatin is deprotonated and the amino group of the solvent molecule gains the hydrogen. The crystal packing and formation of supramolecular self-arrangements are driven by trifurcated N-H···O hydrogen bonds (1.864 Å, 1.874 Å and 1.941 Å) between the protonated amino of solvent molecules with the deprotonated carboxyl group, which are arranged in layers (Figure 2b) and adopt a R34 (10) graph set motif. O5-H5···O4 interactions (2.068 Å) also take place between hydroxyl groups that connect the neighboring layers. The overall packing perspective along the a-axis is illustrated in Figure 2c.

Table 1.
Crystallographic details related to the investigated compound.
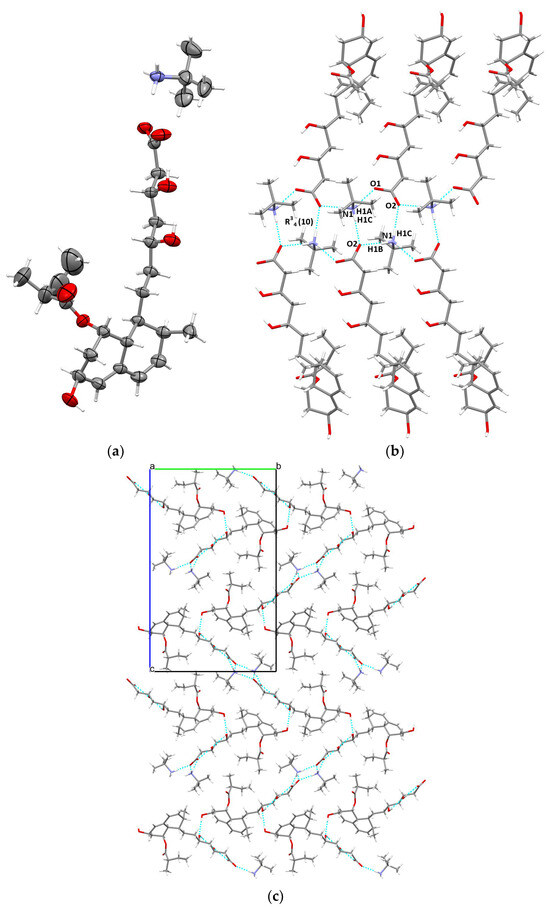
Figure 2.
Asymmetric unit illustrating the atoms as thermal ellipsoids at a 50% probability level (a); intermolecular interactions in the crystal (b); unit cell packing along the a-axis (c).
Recent studies have focused on the crystal structure of tert-octylamine salt of pravastatin, which monoclinically crystallizes in the P21 space group where the migration of protons from carboxylic acid is similar to the migration in the title crystal [15].
2.2. Solid-State NMR Analysis
The 1H MAS NMR spectrum (Figure 3) showed strongly overlapped lines in the downfield region (0–9 ppm) and one deshielded signal for the protons involved in strong hydrogen bonds. This hydrogen is gained by the amino group of the tert-butylamine molecule as indicated by the single-crystal X-ray structural model and confirmed by the fact that C1–O1 and C1–O2 bond lengths have similar values, i.e., 1.246 and 1.260 Å. Geometrical features of neutral carboxylic groups (C–O bonds are longer than C=O) are different from those of a carboxylate anion, where depending on the extent of delocalization of negative charge between the oxygen atoms, there can be practically no difference between the C–O and C=O distances [16].
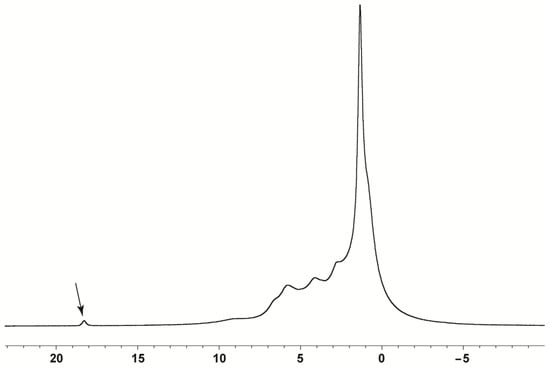
Figure 3.
1H MAS NMR spectrum of pravastatin tert-butylamine salt.
The 13C spectrum shown in Figure 4 presents very narrow spectral lines specific to a highly crystalline solid.
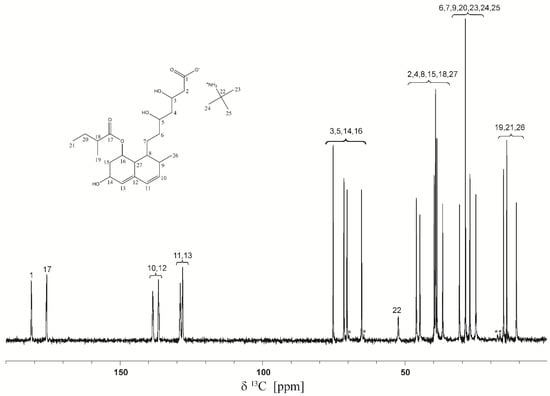
Figure 4.
13C CP-MAS NMR spectrum of pravastatin tert-butylamine salt. The spinning side-bands are shown by asterisks. The 22–25 labels correspond to tert-butyl-amine molecules.
3. Materials and Methods
3.1. General and Sample Preparations
Pravastatin found as amorphous white powder was purchased from AHH Chemical Co., Ltd., Changzhou, China. The solvents (tert-butylamine, ethanol and hexane) were received from Sigma Aldrich and were used without further purification.
Suitable single crystals of Prav-tbua for X-ray analysis were successfully grown as transparent crystals using vapor diffusion of solids. First, 50 mg (0.12 mmol) of pravastatin was weighed in a 4 mL vial, which was placed in a 20 mL beaker containing 5 mL of ethanol and tert-butylamine mixture in a 1:1 ratio. The beaker was covered with parafilm and held at room temperature for 7 days. The crystals obtained were filtrated and washed with 2 × 1 mL hexane.
3.2. X-ray Diffraction and Structure Refinement
The diffraction data were collected at room temperature using a SuperNova diffractometer equipped with dual X-ray sources (Mo and Cu) (Rigaku, Tokyo, Japan), an Eos CCD detector and an operating tube at 50 kV and 0.08 mA. The crystallographic data were collected and corrected for Lorentz, adsorption and polarization effects using the CrysAlis PRO program [17]. Using Olex2 software, version 1.2.10 [18], the crystal structure was solved with the Superflip solution program [19] using Charge Flipping and refined with ShelXL [20] via least squares minimization.
Crystal Data: Empirical formula: C27H47NO7 (M = 497.65 g/mol): orthorhombic, space group P212121 (no. 19), a = 5.98809(13) Å, b = 17.4955(3) Å, c = 28.0358(6) Å, V = 2937.17(10) Å3, Z = 4, T = 293(2) K, μ(CuKα) = 0.648 mm−1, Dcalc = 1.125 g/cm3, 12,408 reflections measured (6.306° ≤ 2Θ ≤ 143.05°); 5198 unique (Rint = 0.0350, Rsigma = 0.0643), which were used in all calculations. The final R1 was 0.0646 (I > 2σ(I)) and wR2 was 0.1890 (all data). The data are available as Supplementary Materials (CIF file).
3.3. 13C Solid-State NMR
The 1H and 13C solid-state NMR spectra were acquired with a Bruker Avance III 500 wide-bore spectrometer (Bruker Biospin, Ettlingen, Germany) operating at 499.98 MHz Larmor frequency for 1H and 125.73 MHz for 13C at room temperature. For the 1H spectrum, a 1.3 mm double resonance MAS probe head was used with a sample spinning frequency of 60 kHz and no homonuclear decoupling. This spectrum was referenced to a single resonance observed in adamantane for protons at 1.8 ppm with respect to neat tetramethylsilane (TMS). For the 13C spectrum, a 4 mm double resonance (1H/X) MAS probe was used with 14 kHz frequency, and a standard 13C CP-MAS pulse sequence with a contact pulse time of 2 ms. The spectrum was recorded under high-power proton decoupling (100 kHz) with a TPPM (Two-Pulse Phase Modulation) sequence by averaging 100 transients, with a recycle delay of 5 s. The recorded 13C NMR spectrum is calibrated with respect to the CH3 line in TMS (tetramethylsilane) through an indirect procedure that uses α-Glycine (176.5 ppm for the 13COOH line) as an external reference.
4. Conclusions
Starting from amorphous pravastatin, we successfully prepared a crystalline form of pravastatin tert-butylamine salt whose crystal structure was determined by single-crystal X-ray diffraction and further analyzed by ss-NMR.
Supplementary Materials
The data can be seen in the Supplementary Materials.
Author Contributions
Conceptualization: I.-G.G. and M.-O.M.; methodology: M.-O.M.; X-ray crystal structure: A.T.; ss-NMR: X.F.; investigation: I.-G.G., M.-O.M., A.T. and X.F.; writing—original draft preparation: I.-G.G., M.-O.M., A.T. and X.F; writing—review and editing; I.-G.G., M.-O.M., A.T. and X.F. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge the financial support from the MCID through the “Nucleu” Programme within the National Plan for Research, Development and Innovation 2022–2027, project PN23 24 01 05.
Data Availability Statement
The CIF file of the compound was deposited via the Cambridge Crystallographic Data Centre with the deposit number of 2382089. It can be obtained free of charge following written application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (Fax: +44-1223-336033); upon request via e-mail to maria.miclaus@itim-cj.ro or by access to http://www.ccdc.cam.ac.uk (accessed on 18 September 2024).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Taylor, F.; Huffman, M.D.; Macedo, A.F.; Moore, T.H.M.; Burke, M.; Smith, G.D.; Ward, K.; Ebrahim, S.; Gay, H.C. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2013, 2013, CD004816. [Google Scholar] [CrossRef] [PubMed]
- Nanovskaya, T.N.; Patrikeeva, S.L.; Paul, J.; Constantine, M.; Hakins, G.D.V.; Ahmed, M.S. Transplacentar Transfer and Distribution of Pravastatin. Am. J. Obstet. Gynecol. 2013, 209, 373.e1–373.e5. [Google Scholar] [CrossRef] [PubMed]
- Del Sol, A.I.; Nanayakkara, P.W.B. Pravastatin: An evidence-based statin? Expert Opin. Drug Metab. Toxicol. 2008, 4, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Yamaoka, O.; Uchida, K.; Morigami, N.; Sugimoto, Y.; Fujita, T.; Inoue, T.; Fuchi, T.; Hachisuka, M.; Ueshima, H.; et al. Pravastatin reduces restenosis after coronary angioplasty of high grade stenotic lesions: Results of SHIPS (SHIga Pravastatin Study). Cardiovasc. Drug Ther. 1996, 10, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Valenti, S.; Barrio, M.; Negrier, P.; Romanini, M.; Macovez, R.; Tamarit, J.L. Comparative physical Study of Three pharmaceutically Active Benzodiazepine Derivatives: Crystalline versus Amorphous State and Crystallization Tendency. Mol. Pharm. 2021, 18, 1819–1832. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.G.; Kumar, A.; Gide, P.S. Formulation of solid self-nanoemulsifying drug delivery systems using N-methyl pyrrolidone as cosolvent. Drug Dev. Ind. Pharm. 2015, 41, 1460–1463. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins. 1. Drug Solubilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef]
- Turza, A.; Borodi, G.; Muresan-Pop, M.; Ulici, A. Polymorphism and β-cyclodextrin complexation of methyldrostanolone. J. Mol. Struct. 2022, 1250, 131852. [Google Scholar] [CrossRef]
- Turza, A.; Ulici, A.; Muresan-Pop, M.; Borodi, G. Solid forms and β-cyclodextrin complexation of turinabol. Acta Cryst. C 2022, C78, 305–313. [Google Scholar] [CrossRef]
- Borodi, G.; Miclaus, M.O.; Muresan-Pop, M.; Turza, A. Solid Forms and β-cyclodextrin complexation of Oxymetholone and Crystal Structure of Metribolone. Crystals 2024, 14, 483. [Google Scholar] [CrossRef]
- Porter, C.J.; Trevaskis, N.L.; Charman, W.N. Lipid and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Borodi, G.; Turza, A.; Onija, O.; Bende, A. Succinic, fumaric, adipic and oxalic acid cocrystals of promethazine hydrochloride. Acta Cryst. Sect. C Struct. Chem. 2019, 75, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Kanaujia, P.; Poovizhi, P.; Ng, W.K.; Tan, R.B.H. Amorphous formulations for dissolution and bioavailability enhancement of poorly soluble APIs. Powder Technol. 2015, 285, 2–15. [Google Scholar] [CrossRef]
- Sato, S.; Furukawa, Y. X-ray crystal structure of the tert-octyamine salt (RMS-431) of pravastatin. J. Antibiot. 1988, 41, 1265. [Google Scholar] [CrossRef] [PubMed]
- Bis, J.A.; Zaworotko, M.J. The 2-Aminopyridinium-carboxylate Supramolecular Heterosynthon: A Robust Motif for Generation of Multiple-Component Crystals. Cryst. Growth Des. 2005, 5, 1169–1179. [Google Scholar] [CrossRef]
- CrysAlis PRO. Rigaku Oxford Diffraction; CrysAlis PRO: Yarnton, UK, 2015. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP-a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).