Abstract
3-(1H-Indol-3-yl)-4-(morpholin-4-yl)cyclobut-3-ene-1,2-dione was obtained in good yields (72–82%) by nucleophilic substitution of 3-chloro-4-(1H-indol-3-yl)cyclobut-3-ene-1,2-dione with morpholine.
The indole ring system is a widespread structural motif in a variety of biologically active species []. While it is obvious to use such a privileged structure as a template for further drug development, it has been suggested that medicinal chemists should direct their attention to “unusual scaffolds” for the sake of novelty and originality []. Along these lines, a number of studies report novel bioactive compounds comprising a combination of the indole core with less common motifs such as 2,5-diazabicyclo[2.2.1]heptanes [], benzazepinones [,,], epoxides [], and aroylhydrazides []. The squaric acid diamide structure was recently highlighted as an unusual medicinal chemistry scaffold of high potential [,]. Surprisingly there are only scattered examples in the literature in which the indole and the squaramide scaffold are combined [,,]. A survey of the literature revealed only a single study describing the synthesis of indolylsquarylamides []. We here report the preparation of the title compound as the first example of an indolylsquarylamide unsubstituted at the indole nitrogen. A 1,2-disubstituted analog of the compound is mentioned in the literature without reporting analytical data [].
In order to establish suitable reaction conditions various ratios of 1 and 2 were stirred in different solvents under reflux (scheme 1, Table 1). The best yields were obtained with ethanol as solvent and a 1:2.2 ratio of 1 and 2. Exchanging ethanol for acetonitrile did not lead to better results. Furthermore, the replacement of one equivalent of morpholine (2) by triethylamine also failed to provide higher yields. The reaction with a 1:1 ratio of 1 and 2 afforded lower yields and unreacted educt 1. The product precipitated from the reaction mixture and was collected by simple filtration. The work-up of the filtrate increased the yield only by 3–6%. The optimized procedure furnished satisfactory yields of a crystalline product that proved to be stable under ambient storage conditions.
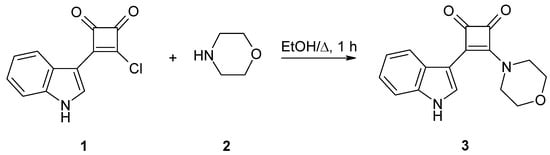
Scheme 1.
Synthesis of the title compound 3.

Table 1.
Conditons and reagents for optimization of synthesis procedure.
Experimental
General
Melting points were determined in open-glass capillaries on an electric variable heater (Electrothermal IA 9100). FT-IR absorption spectra were recorded on a Thermo Nicolet FT-IR 200 spectrometer using KBr pellets. 1HNMR and 13CNMR spectra were recorded on a Bruker Avance DRX-400 (NMR laboratories of the Chemical Institutes of the Technische Universität Braunschweig) using DMSO-d6 as solvent. Chemical shifts are reported as parts per million (ppm) downfield from TMS used as an internal standard. The elemental analysis was recorded on a CE Instruments FlashEA® 1112 Elemental Analyzer. The reaction was monitored by TLC (Macherey-Nagel Polygram SIL G/UV254) using ethyl acetate as eluent. The mass spectrum was recorded on a ThermoFisher Scientific LTQ-Orbitrap Velos (department of mass spectrometry of the Chemical Institutes of the Technische Universität Braunschweig). HPLC was performed on a Merck Hitachi LaChrom Elite system (pump: L-2130, DAD detector: L-2450; autosampler: L-2200; column: Merck LiChroCART 125-4, LiChrospher 100 RP-18 (5 μm); eluent: acetonitrile/water (30:70), elution rate 1.000 mL/min; detection wavelength: 254 nm and 280 nm; overall run time: 15 min); tms = total retention time, ts = dead time.
3-Chloro-4-(1H-indol-3-yl)cyclobut-3-ene-1,2-dione (1) was synthesized by chlorosquarylation of indole according to the procedure described in lit. []. Morpholine was purchased from a commercial supplier and distilled at atmospheric pressure prior to use.
3-(1H-Indol-3-yl)-4-(morpholin-4-yl)cyclobut-3-ene-1,2-dione (3)
3-Chloro-4-(1H-indol-3-yl)cyclobut-3-ene-1,2-dione (1) (0.231 g; 1.00 mmol) was added to a solution of morpholine (2) (0.192 g; 2.20 mmol) in ethanol (15 mL). This suspension was stirred for 1 h under reflux. After storage at room temperature for 24 h the mixture was filtered through a paper filter. The residue was crystallized from ethanol (99.6%) to yield yellow crystals (0.20–0.23 g; 72–82%; n = 3).
Mp: 264–265 °C (dec.).
MS (EI, rel. intensity) m/z: 282.1 [M+] (30), 226.1 (100).
IR (KBr) (cm−1): 3428 (N-H), 3125 (C-H, arom.), 2916 (C-H, aliph.), 1777 and 1708 (C=O), 1590 (C=C, arom.).
1H-NMR (400 MHz, DMSO-d6) δ (ppm): 3.7–3.8 (m, 4H, CH2), 3.86 (br. s, 4H, CH2), 7.14 (ddd, 1H, J = 1.1, 7.1, 8.1 Hz, CH arom.), 7.21 (ddd, 1H, J = 1.2, 7.1, 8.2 Hz, CH arom.), 7.47 (dt, 1H, J = 8.0, 0.9 Hz, CH arom.), 7.73 (d, 1H, J = 2.8 Hz, CH arom.), 7.91 (dd, 1H, J = 0.5, 7.8 Hz, CH arom.), 12.00 (s, 1H, NH).
13C-NMR (100 MHz, DMSO-d6) δ (ppm): 47.5 (2 C, CH2), 65.9 (2 C, CH2), 105.0 (C quat.), 112.0 (CH), 120.3 (CH), 121.6 (CH), 122.5 (CH), 125.6 (C quat.), 127.5 (CH), 136.3 (C quat.), 161.1 (C quat.), 176.3 (C quat.), 188.1 (C quat.), 191.2 (C quat.).
HPLC (AUC %): 99.4% at 254 nm, 99.9% at 280 nm; tms = 4.03 min; ts (DMSO) = 1.00 min.
Anal. calculated for C16H14N2O3: C, 68.08; H, 5.00; N, 9.92. Found: C, 68.10; H, 4.87; N, 9.79.
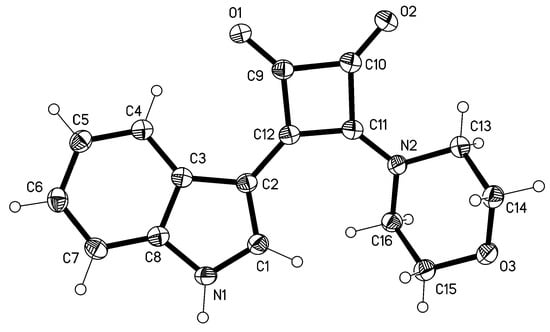
X-Ray Structure Determination of 3
Crystal data: Triclinic, space group P, a = 6.8488(5), b = 8.1491(6), c = 12.2500(8) Å, α = 73.888(7), β = 75.955(6), γ = 89.166(6), V = 636.18(8) Å3, Z = 2, Dx = 1.474 Mg m–3. Data collection: A yellow lath 0.30 × 0.08 × 0.04 mm was mounted on an Oxford Diffraction diffractometer. A total of 19988 intensities were registered using Cu Kα radiation to 2θ 152°. The crystal was twinned by 180° rotation about the x axis. Only non-overlapped reflections were used, resulting in a completeness of 92%. Structure refinement: The structure was refined anisotropically on F2 using the program SHELXL-97 []. The NH hydrogen was refined freely; other H were included using a riding model starting from calculated positions. The final wR2 was 0.103 for 194 parameters and 2427 unique reflections, with a conventional R1 of 0.038; S = 1.06, max. Δρ = 0.2 e Å–3. Complete data have been deposited at the Cambridge Crystallographic Data Centre under the number CCDC-882817. These data can be obtained free of charge from www.ccdc.cam.ac.uk/data_request/cif.
Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3Acknowledgments
The authors are grateful to H.-O. Burmeister, S. Meyer, and P. Reich for recording the IR spectra and performing the elemental analysis.
References
- Sharma, V.; Kumar, P.; Pathak, D. Biological importance of the indole nucleus in recent years: A comprehensive review. J. Heterocycl. Chem. 2010, 47, 491–502. [Google Scholar] [CrossRef]
- Marson, C.M. New and unusual scaffolds in medicinal chemistry. Chem. Soc. Rev. 2011, 40, 5514–5533. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Bunnelle, W.H.; Ryther, K.B.; Anderson, D.J.; Malysz, J.; Helfrich, R.; Grønlien, J.H.; Håkerud, M.; Peters, D.; Schrimpf, M.R.; et al. Syntheses and structure-activity relationship (SAR) studies of 2,5-diazabicyclo[2.2.1]heptanes as novel alpha7 neuronal nicotinic receptor (NNR) ligands. Bioorg. Med. Chem. Lett. 2010, 20, 3636–3639. [Google Scholar] [CrossRef] [PubMed]
- Kunick, C.; Zeng, Z.; Gussio, R.; Zaharevitz, D.; Leost, M.; Totzke, F.; Schächtele, C.; Kubbutat, M.H.G.; Meijer, L.; Lemcke, T. Structure-aided optimization of kinase inhibitors derived from alsterpaullone. ChemBioChem 2005, 6, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Reichwald, C.; Shimony, O.; Dunkel, U.; Sacerdoti-Sierra, N.; Jaffe, C.L.; Kunick, C. 2-(3-Aryl-3-oxopropen-1-yl)-9-tert-butyl-paullones: A new antileishmanial chemotype. J. Med. Chem. 2008, 51, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Wieking, K.; Knockaert, M.; Leost, M.; Zaharevitz, D.W.; Meijer, L.; Kunick, C. Synthesis of paullones with aminoalkyl side chains. Arch. Pharm. Pharm. Med. Chem. 2002, 335, 311–317. [Google Scholar] [CrossRef]
- Xie, X.; Lemcke, T.; Gussio, R.; Zaharevitz, D.W.; Leost, M.; Meijer, L.; Kunick, C. Epoxide-containing side chains enhance antiproliferative activity of paullones. Eur. J. Med. Chem. 2005, 40, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Preu, L.; Lemcke, T.; Totzke, F.; Schächtele, C.; Kubbutat, M.H.G.; Kunick, C. Dual IGF-1R/SRC inhibitors based on a N'-aroyl-2-(1H-indol-3-yl)-2-oxoacetohydrazide structure. Eur. J. Med. Chem. 2011, 46, 2759–2769. [Google Scholar] [CrossRef] [PubMed]
- Storer, R.I.; Aciroa, C.; Jones, L.H. Squaramides: Physical properties, synthesis and applications. Chem. Soc. Rev. 2011, 40, 2330–2346. [Google Scholar] [CrossRef] [PubMed]
- Shinada, T.; Nakagawa, Y.; Hayashi, K.; Corzo, G.; Nakajima, T.; Ohfune, Y. Synthesis and paralytic activities of squaryl amino acid-containing polyamine toxins. Amino Acids 2003, 24, 293–301. [Google Scholar] [PubMed]
- Charton, J.; Déprez, B.P.; Déprez-Poulain, R.F. Synthesis of a 200-member library of squaric acid N-hydroxylamide amides. Bioorg. Med. Chem. Lett. 2008, 18, 4968–4971. [Google Scholar] [CrossRef] [PubMed]
- Arrington, K.L.; Dudkin, V.Y. Novel inhibitors of checkpoint kinase 1. ChemMedChem 2007, 2, 1571–1585. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.H.; Thiel, S.H.; Gaschler, O. Oxocarbons and related compounds. Part 24. Chlorosquarylation of indoles. J. Chem. Soc. Perkin Trans. 1 1996, 495–496. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).