
Exploring Tissue- and Sex-Specific DNA Methylation in Cattle Using a Pan-Mammalian Infinium Array

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Results
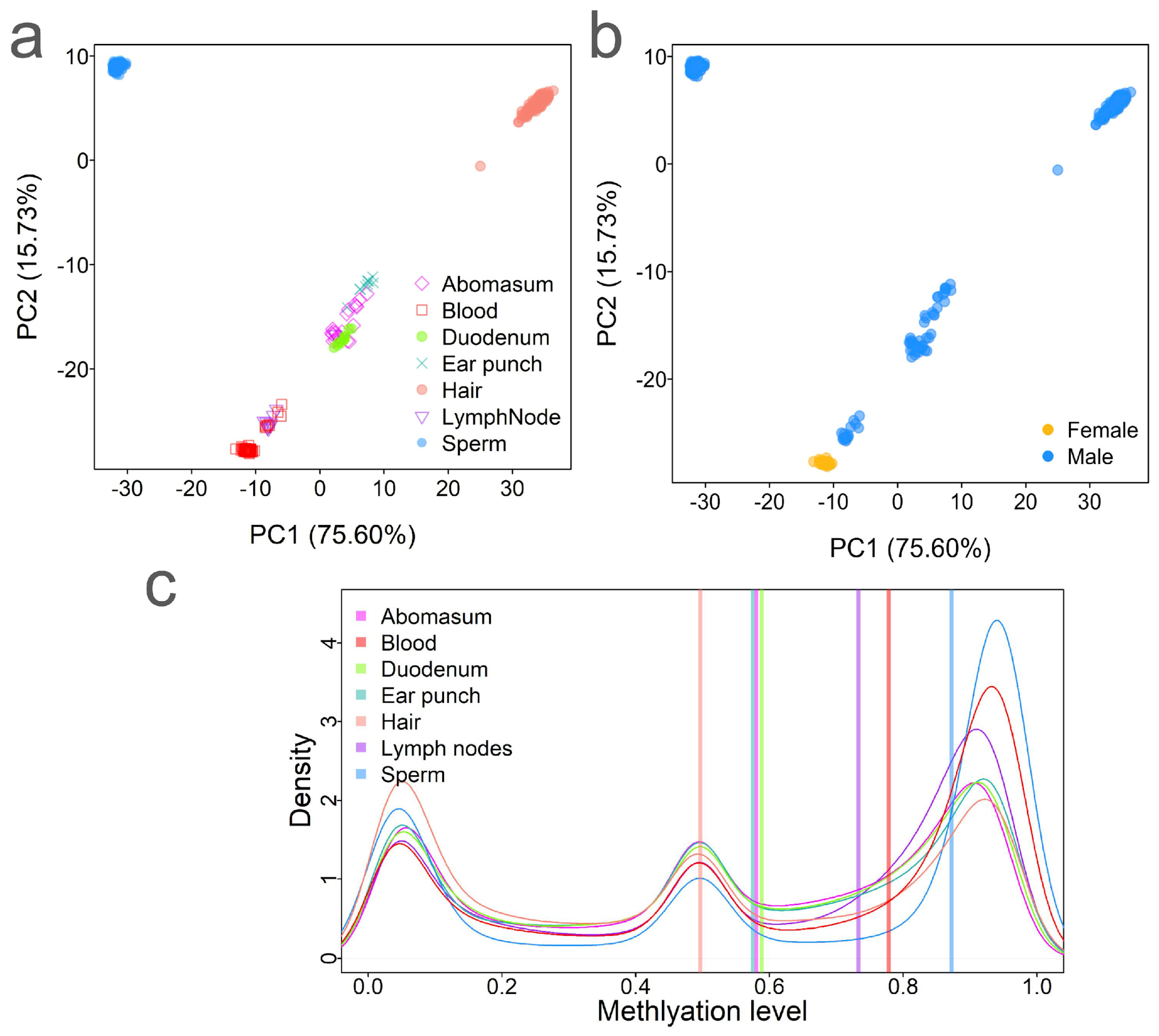
2.1. Application of the Infinium DNA Methylation Array in Cattle
2.2. Methylation Variations Among Tissues in Bos taurus
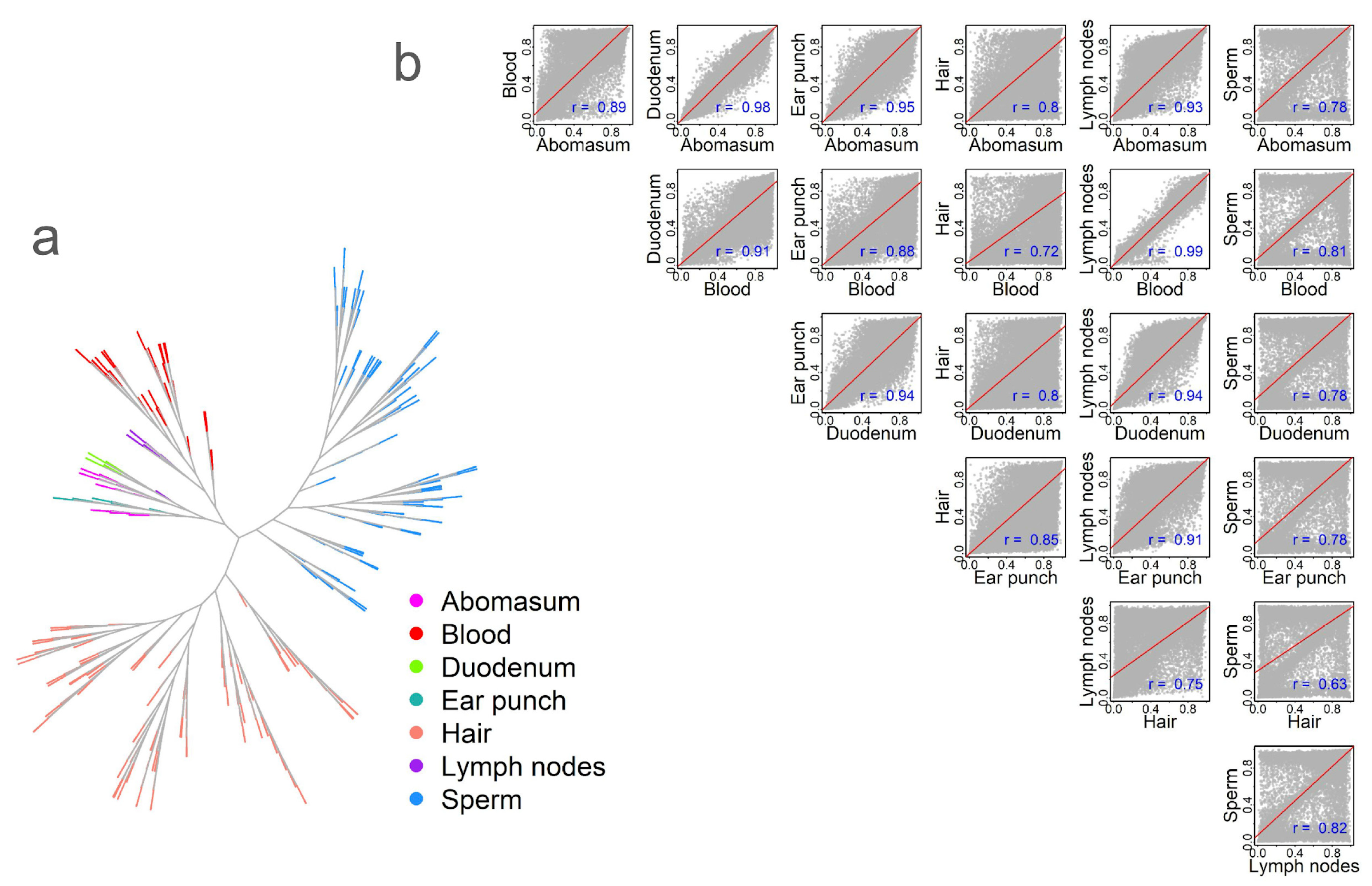
2.3. Methylation Relationships Among Tissues
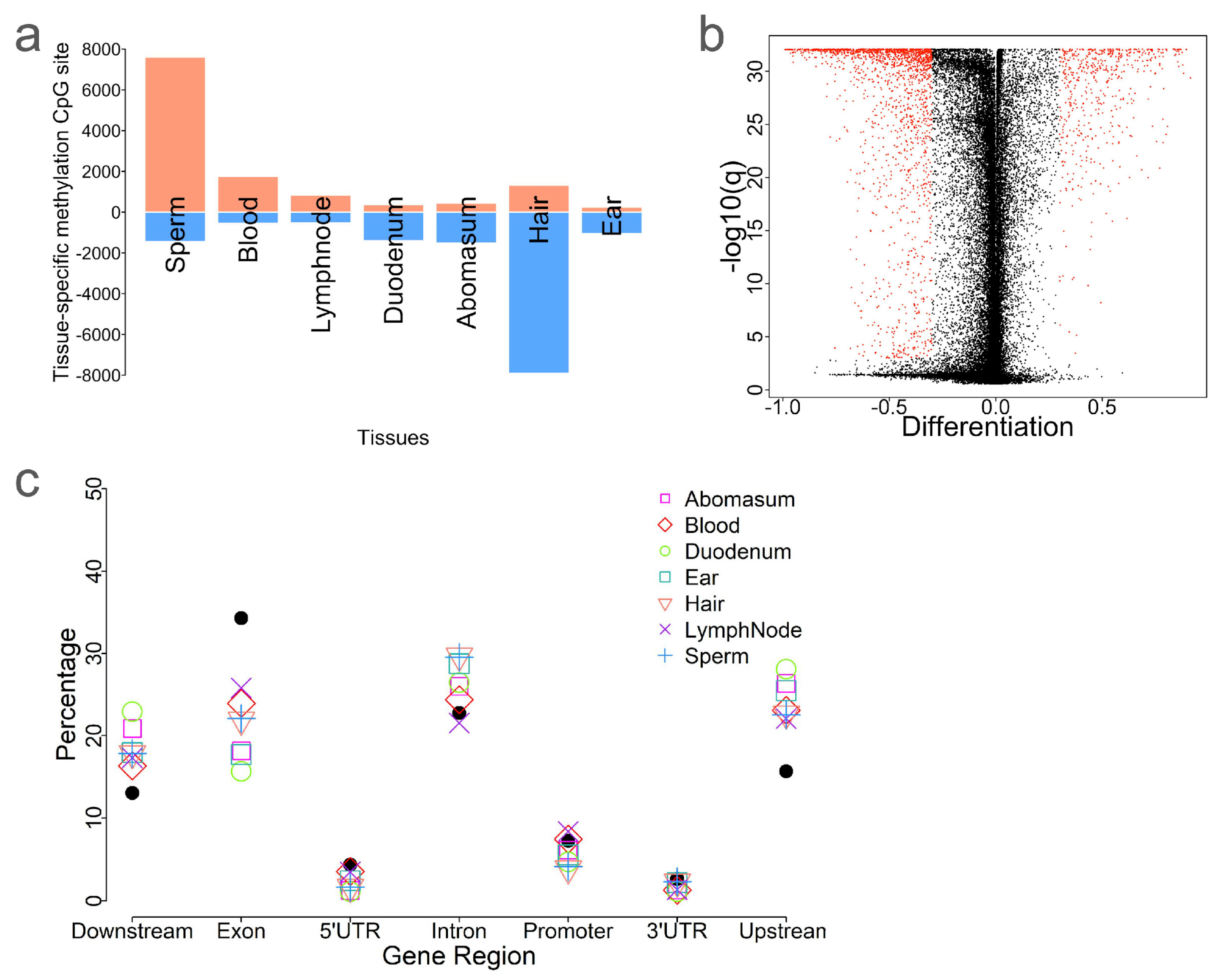
2.4. CpG Methylation Patterns Are Highly Tissue-Specific
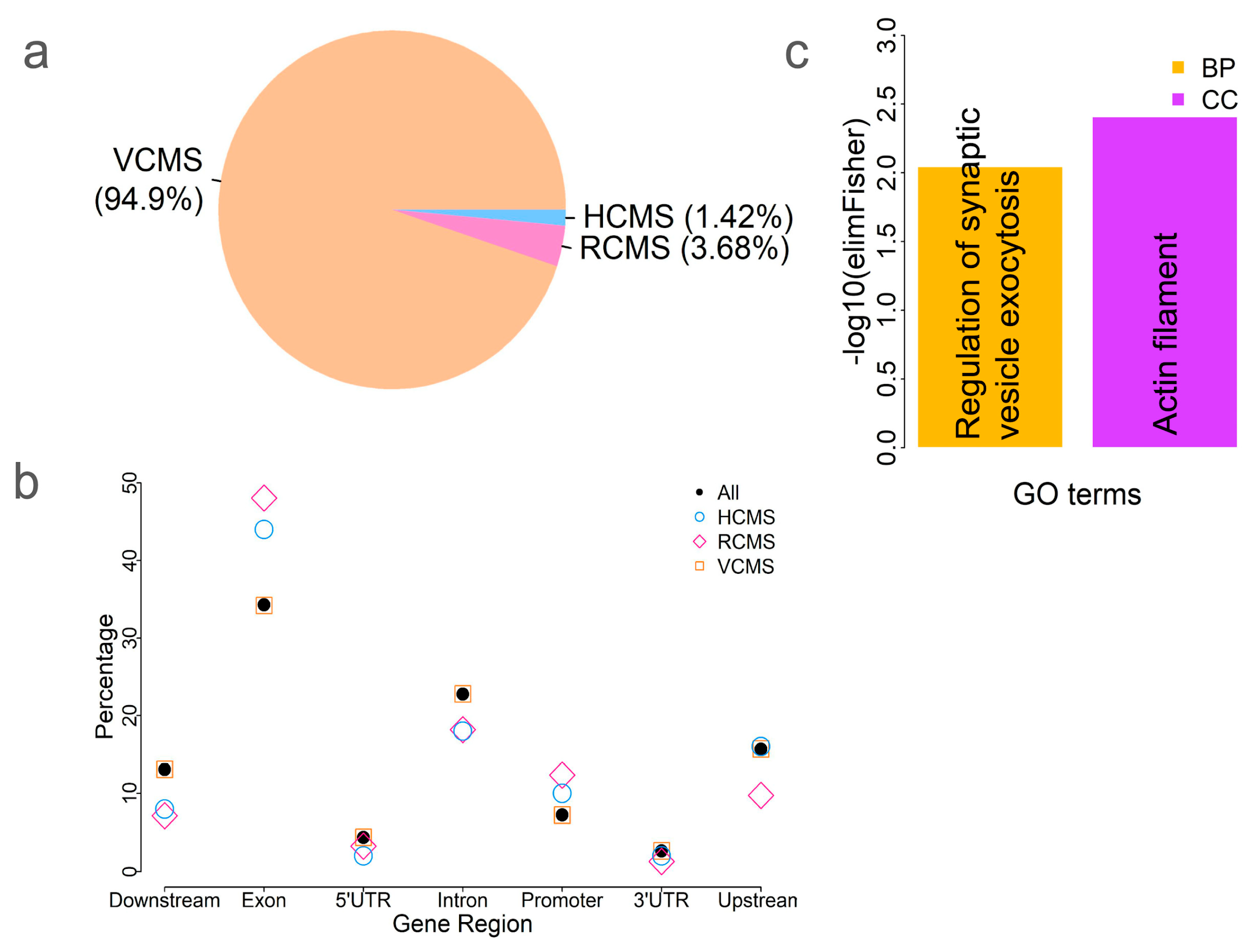
2.5. Identification of Tissue-Specific CpG Sites
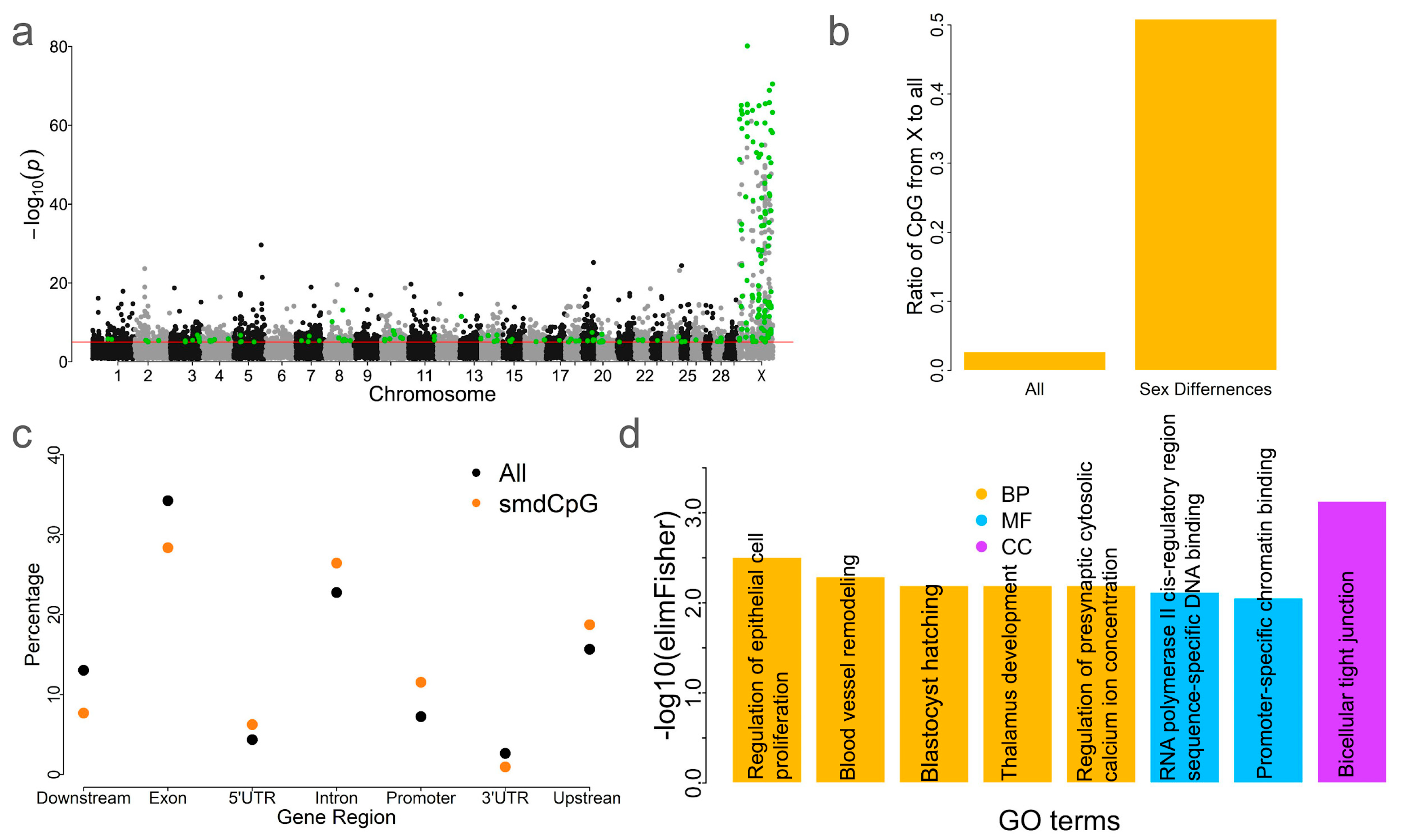
2.6. Sex Differences of DNA Methylation in Blood
3. Discussion
4. Materials and Methods
4.1. Methylation Data Generation
4.2. Principal Component Analysis
4.3. Phylogenetic Tree
4.4. Methylation Correlation Between Tissues
4.5. Identification of Highly Variable and Conserved Methylated CpG Sites
4.6. Tissue-Specific CpG Identification
4.7. GO Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, P.A. Functions of DNA Methylation: Islands, Start Sites, Gene Bodies and Beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of Epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, Maintaining and Modifying DNA Methylation Patterns in Plants and Animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Ribeiro, A.M.F.; Sanglard, L.P.; Wijesena, H.R.; Ciobanu, D.C.; Horvath, S.; Spangler, M.L. DNA Methylation Profile in Beef Cattle Is Influenced by Additive Genetics and Age. Sci. Rep. 2022, 12, 12016. [Google Scholar] [CrossRef] [PubMed]
- de Soutello, R.V.G.; Rodrigues, M.G.F.; Gonçalves, J.A.; Bello, H.J.S.; Pavan, B.E.; Ramos, E.S. Global Genomic Methylation Related to the Degree of Parasitism in Cattle. Sci. Rep. 2022, 12, 18135. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, L.; Bickhart, D.M.; Abdel Hay, E.H.; Schroeder, S.G.; Connor, E.E.; Alexander, L.J.; Sonstegard, T.S.; Van Tassell, C.P.; Chen, H.; et al. Reduced Representation Bisulphite Sequencing of Ten Bovine Somatic Tissues Reveals DNA Methylation Patterns and Their Impacts on Gene Expression. BMC Genom. 2016, 17, 779. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, S.; Hu, Y.; Fang, L.; Gao, Y.; Xia, H.; Schroeder, S.G.; Rosen, B.D.; Connor, E.E.; Li, C.; et al. Comparative Whole Genome DNA Methylation Profiling across Cattle Tissues Reveals Global and Tissue-Specific Methylation Patterns. BMC Biol. 2020, 18, 85. [Google Scholar] [CrossRef]
- Arneson, A.; Haghani, A.; Thompson, M.J.; Pellegrini, M.; Kwon, S.B.; Vu, H.; Maciejewski, E.; Yao, M.; Li, C.Z.; Lu, A.T.; et al. A Mammalian Methylation Array for Profiling Methylation Levels at Conserved Sequences. Nat. Commun. 2022, 13, 783. [Google Scholar] [CrossRef]
- Lu, A.T.; Fei, Z.; Haghani, A.; Robeck, T.R.; Zoller, J.A.; Li, C.Z.; Lowe, R.; Yan, Q.; Zhang, J.; Vu, H.; et al. Author Correction: Universal DNA methylation age across mammalian tissues. Nat. Aging 2023, 3, 1462. [Google Scholar] [CrossRef]
- Zhang, J.; Sheng, H.; Hu, C.; Li, F.; Cai, B.; Ma, Y.; Wang, Y.; Ma, Y. Effects of DNA Methylation on Gene Expression and Phenotypic Traits in Cattle: A Review. Int. J. Mol. Sci. 2023, 24, 11882. [Google Scholar] [CrossRef]
- Wang, X.; Li, W.; Feng, X.; Li, J.; Liu, G.E.; Fang, L.; Yu, Y. Correction: Harnessing Male Germline Epigenomics for the Genetic Improvement in Cattle. J. Anim. Sci. Biotechnol. 2023, 14, 110. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Zhou, Y.; Liu, S.; Jiang, J.; Bickhart, D.M.; Null, D.J.; Li, B.; Schroeder, S.G.; Rosen, B.D.; Cole, J.B.; et al. Comparative Analyses of Sperm DNA Methylomes among Human, Mouse and Cattle Provide Insights into Epigenomic Evolution and Complex Traits. Epigenetics 2019, 14, 260–276. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Fang, L.; Zhou, Y.; Santos, D.J.A.; Xiang, R.; Daetwyler, H.D.; Chamberlain, A.J.; Cole, J.B.; Li, C.; Yu, Y.; et al. Analyses of Inter-Individual Variations of Sperm DNA Methylation and Their Potential Implications in Cattle. BMC Genom. 2019, 20, 888. [Google Scholar] [CrossRef]
- Zhou, Y.; Connor, E.E.; Bickhart, D.M.; Li, C.; Baldwin, R.L.; Schroeder, S.G.; Rosen, B.D.; Yang, L.; Van Tassell, C.P.; Liu, G.E. Comparative Whole Genome DNA Methylation Profiling of Cattle Sperm and Somatic Tissues Reveals Striking Hypomethylated Patterns in Sperm. GigaScience 2018, 7, giy039. [Google Scholar] [CrossRef] [PubMed]
- Caulton, A.; Dodds, K.G.; McRae, K.M.; Couldrey, C.; Horvath, S.; Clarke, S.M. Development of Epigenetic Clocks for Key Ruminant Species. Genes 2022, 13, 96. [Google Scholar] [CrossRef]
- Kordowitzki, P.; Haghani, A.; Zoller, J.A.; Li, C.Z.; Raj, K.; Spangler, M.L.; Horvath, S. Epigenetic Clock and Methylation Study of Oocytes from a Bovine Model of Reproductive Aging. Aging Cell 2021, 20, e13349. [Google Scholar] [CrossRef]
- Sugrue, V.J.; Zoller, J.A.; Narayan, P.; Lu, A.T.; Ortega-Recalde, O.J.; Grant, M.J.; Bawden, C.S.; Rudiger, S.R.; Haghani, A.; Bond, D.M.; et al. Correction: Castration Delays Epigenetic Aging and Feminizes DNA Methylation at Androgen-Regulated Loci. eLife 2023, 12, e92968. [Google Scholar] [CrossRef]
- Kropp, J.; Carrillo, J.A.; Namous, H.; Daniels, A.; Salih, S.M.; Song, J.; Khatib, H. Male Fertility Status Is Associated with DNA Methylation Signatures in Sperm and Transcriptomic Profiles of Bovine Preimplantation Embryos. BMC Genom. 2017, 18, 280. [Google Scholar] [CrossRef]
- Reik, W.; Dean, W.; Walter, J. Epigenetic Reprogramming in Mammalian Development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef]
- Boschiero, C.; Beshah, E.; Zhu, X.; Tuo, W.; Liu, G.E. Profiling Genome-Wide Methylation Patterns in Cattle Infected with Ostertagia Ostertagi. Int. J. Mol. Sci. 2025, 26, 89. [Google Scholar] [CrossRef]
- Hu, Z.; Boschiero, C.; Li, C.-J.; Connor, E.E.; Baldwin, R.L.; Liu, G.E. Unraveling the Genetic Basis of Feed Efficiency in Cattle through Integrated DNA Methylation and CattleGTEx Analysis. Genes 2023, 14, 2121. [Google Scholar] [CrossRef]
- Hall, E.; Volkov, P.; Dayeh, T.; Esguerra, J.L.S.; Salö, S.; Eliasson, L.; Rönn, T.; Bacos, K.; Ling, C. Sex Differences in the Genome-Wide DNA Methylation Pattern and Impact on Gene Expression, microRNA Levels and Insulin Secretion in Human Pancreatic Islets. Genome Biol. 2014, 15, 522. [Google Scholar] [CrossRef]
- Åsenius, F.; Gorrie-Stone, T.J.; Brew, A.; Panchbhaya, Y.; Williamson, E.; Schalkwyk, L.C.; Rakyan, V.K.; Holland, M.L.; Marzi, S.J.; Williams, D.J. The DNA Methylome of Human Sperm Is Distinct from Blood with Little Evidence for Tissue-Consistent Obesity Associations. PLoS Genet. 2020, 16, e1009035. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Baum, M.; Carbon, J. DNA Methylation Regulates Phenotype-Dependent Transcriptional Activity in Candida Albicans. Proc. Natl. Acad. Sci. USA 2011, 108, 11965–11970. [Google Scholar] [CrossRef]
- Dominguez, R.; Holmes, K.C. Actin Structure and Function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef]
- Maschietto, M.; Bastos, L.C.; Tahira, A.C.; Bastos, E.P.; Euclydes, V.L.V.; Brentani, A.; Fink, G.; de Baumont, A.; Felipe-Silva, A.; Francisco, R.P.V.; et al. Sex Differences in DNA Methylation of the Cord Blood Are Related to Sex-Bias Psychiatric Diseases. Sci. Rep. 2017, 7, 44547. [Google Scholar] [CrossRef] [PubMed]
- Noguerol, J.; Roustan, P.-J.; N’Taye, M.; Delcombel, L.; Rolland, C.; Guiraud, L.; Sagnat, D.; Edir, A.; Bonnart, C.; Denadai-Souza, A.; et al. Sexual Dimorphism in PAR2-Dependent Regulation of Primitive Colonic Cells. Biol. Sex Differ. 2019, 10, 47. [Google Scholar] [CrossRef]
- Bermejo-Alvarez, P.; Rizos, D.; Rath, D.; Lonergan, P.; Gutierrez-Adan, A. Sex Determines the Expression Level of One Third of the Actively Expressed Genes in Bovine Blastocysts. Proc. Natl. Acad. Sci. USA 2010, 107, 3394–3399. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative Analysis of 111 Reference Human Epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- MacPhillamy, C.; Chen, T.; Hiendleder, S.; Williams, J.L.; Alinejad-Rokny, H.; Low, W.Y. DNA Methylation Analysis to Differentiate Reference, Breed, and Parent-of-Origin Effects in the Bovine Pangenome Era. GigaScience 2024, 13, giae061. [Google Scholar] [CrossRef]
- Lokk, K.; Modhukur, V.; Rajashekar, B.; Märtens, K.; Mägi, R.; Kolde, R.; Koltšina, M.; Nilsson, T.K.; Vilo, J.; Salumets, A.; et al. Erratum to: DNA Methylome Profiling of Human Tissues Identifies Global and Tissue-Specific Methylation Patterns. Genome Biol. 2016, 17, 224. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Bilmez, Y.; Ozturk, S. Dynamic Changes of Histone Methylation in Male Germ Cells during Spermatogenesis. FS Rev. 2023, 4, 187–205. [Google Scholar] [CrossRef]
- Siebert-Kuss, L.M.; Dietrich, V.; Di Persio, S.; Bhaskaran, J.; Stehling, M.; Cremers, J.-F.; Sandmann, S.; Varghese, J.; Kliesch, S.; Schlatt, S.; et al. Genome-Wide DNA Methylation Changes in Human Spermatogenesis. Am. J. Hum. Genet. 2024, 111, 1125–1139. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wang, Y.; Gu, H.; Wang, X. Technologies and Applications of Single-Cell DNA Methylation Sequencing. Theranostics 2023, 13, 2439–2454. [Google Scholar] [CrossRef] [PubMed]
- Ercan, S. Mechanisms of X Chromosome Dosage Compensation. J. Genom. 2015, 3, 1–19. [Google Scholar] [CrossRef]
- Meyer, B.J. Mechanisms of Sex Determination and X-Chromosome Dosage Compensation. Genetics 2022, 220, iyab197. [Google Scholar] [CrossRef] [PubMed]
- Demanelis, K.; Jasmine, F.; Chen, L.S.; Chernoff, M.; Tong, L.; Delgado, D.; Zhang, C.; Shinkle, J.; Sabarinathan, M.; Lin, H.; et al. Determinants of Telomere Length across Human Tissues. Science 2020, 369, eaaz6876. [Google Scholar] [CrossRef]
- Estermann, M.A.; Williams, S.; Hirst, C.E.; Roly, Z.Y.; Serralbo, O.; Adhikari, D.; Powell, D.; Major, A.T.; Smith, C.A. Insights into Gonadal Sex Differentiation Provided by Single-Cell Transcriptomics in the Chicken Embryo. Cell Rep. 2020, 31, 107491. [Google Scholar] [CrossRef]
- Andersson, L.; Archibald, A.L.; Bottema, C.D.; Brauning, R.; Burgess, S.C.; Burt, D.W.; Casas, E.; Cheng, H.H.; Clarke, L.; Couldrey, C.; et al. Coordinated International Action to Accelerate Genome-to-Phenome with FAANG, the Functional Annotation of Animal Genomes Project. Genome Biol. 2015, 16, 57. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A Flexible and Comprehensive Bioconductor Package for the Analysis of Infinium DNA Methylation Microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R Language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Total | Hypermethylated | Hypomethylated |
|---|---|---|---|
| Sperm | 9039 | 7609 | 1430 |
| Lymph Node | 1359 | 839 | 520 |
| Hair | 9227 | 1323 | 7904 |
| Ear Punch | 1300 | 247 | 1053 |
| Duodenum | 1761 | 365 | 1396 |
| Blood | 2305 | 1758 | 547 |
| Abomasum | 1956 | 434 | 1522 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Z.; Boschiero, C.; Neupane, M.; Bhowmik, N.; Yang, L.; Kilian, L.; DeJarnette, J.M.; Sargolzaei, M.; Harstine, B.; Li, C.-J.; et al. Exploring Tissue- and Sex-Specific DNA Methylation in Cattle Using a Pan-Mammalian Infinium Array. Int. J. Mol. Sci. 2025, 26, 4284. https://doi.org/10.3390/ijms26094284
Hu Z, Boschiero C, Neupane M, Bhowmik N, Yang L, Kilian L, DeJarnette JM, Sargolzaei M, Harstine B, Li C-J, et al. Exploring Tissue- and Sex-Specific DNA Methylation in Cattle Using a Pan-Mammalian Infinium Array. International Journal of Molecular Sciences. 2025; 26(9):4284. https://doi.org/10.3390/ijms26094284
Chicago/Turabian StyleHu, Zhenbin, Clarissa Boschiero, Mahesh Neupane, Nayan Bhowmik, Liu Yang, Levi Kilian, James Mel DeJarnette, Mehdi Sargolzaei, Bo Harstine, Cong-Jun Li, and et al. 2025. "Exploring Tissue- and Sex-Specific DNA Methylation in Cattle Using a Pan-Mammalian Infinium Array" International Journal of Molecular Sciences 26, no. 9: 4284. https://doi.org/10.3390/ijms26094284
APA StyleHu, Z., Boschiero, C., Neupane, M., Bhowmik, N., Yang, L., Kilian, L., DeJarnette, J. M., Sargolzaei, M., Harstine, B., Li, C.-J., Tuo, W., Baldwin, R. L., VI, Van Tassell, C. P., Sattler, C. G., & Liu, G. E. (2025). Exploring Tissue- and Sex-Specific DNA Methylation in Cattle Using a Pan-Mammalian Infinium Array. International Journal of Molecular Sciences, 26(9), 4284. https://doi.org/10.3390/ijms26094284