
Translating Molecular Psychiatry: From Biomarkers to Personalized Therapies—A Narrative Review



Abstract
1. Introduction
2. Genetic Markers
2.1. Major Depressive Disorder (MDD)
2.2. Generalized Anxiety Disorder (GAD)
2.3. Schizophrenia
2.4. Bipolar Disorder (BD)
- ANK3 influences the structural constituent of the cytoskeleton and protein binding and bridging.
- CACNA1C influences enzyme binding and ion channel activity.
- SYNE1 is located on chromosome 6q25.2 and is seen in the cerebellar hemisphere and cerebellum, where it encodes multiple proteins with roles in synaptic plasticity and function.
- ODZ4(TENM4) is another gene identified in BPD that belongs to the tenascin family (teneurin subfamily), and it is located on chromosome 11q14.1, with a role in protein homodimerization activity.
- TRANK1(LBA1) is located on chromosome 3p22.2, and its expression is influenced by valproic acid. Moreover, its dysregulation can disrupt neuronal development and differentiation and the synaptic plasticity of other genes.
3. Pharmacogenomics
3.1. CYP450 and Phenotypic Variations
3.1.1. Impact of CYP450 Genetic Variations on Antidepressant Response
3.1.2. Impact of CYP450 Genetic Variations on Antipsychotic Response
3.1.3. Impact of CYP450 Genetic Variations on Anxiolytic Response
3.2. Effects of CYP2D6 and CYP2C19 Variants on Neurotransmitter Regulation
3.3. Challenges
4. Metabolists
4.1. Insulin Resistance and Psychiatric Impact
4.2. Cholinesterases in Alzheimer’s Disease
4.3. Microbiota Gut–Brain Axis
4.4. Autophagy
4.5. Amyloid-Beta Peptides
5. Genetic Predisposition and Environmental Factors
5.1. Heritability of Psychiatric Disorders and Their Genetic Overlap
5.2. Fine Mapping Variants in GWAS Loci
5.3. Polygenic Risk Scores
6. Neuromodulation
6.1. Non-Invasive Brain Stimulation
6.1.1. Transcranial Magnetic Stimulation
6.1.2. Transcranial Direct Current Stimulation
6.2. Electroconvulsive Therapy
7. Neuroimaging
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Passos, I.C.; Ballester, P.; Rabelo-da-Ponte, F.D.; Kapczinski, F. Precision Psychiatry: The Future Is Now. Can. J. Psychiatry 2022, 67, 21–25. [Google Scholar] [CrossRef] [PubMed]
- State, M.W.; Lombroso, P.J.; Pauls, D.L.; Leckman, J.F. The genetics of childhood psychiatric disorders: A decade of progress. J. Am. Acad. Child Adolesc. Psychiatry 2000, 39, 946–962. [Google Scholar] [CrossRef]
- Belsky, J.; Bakermans-Kranenburg, M.J.; van IJzendoorn, M.H. For Better and For Worse: Differential Susceptibility to Environmental Influences. Curr. Dir. Psychol. Sci. 2007, 16, 300–304. [Google Scholar]
- Weissman, M.M. Is Depression Nature or Nurture? Yes. Am. J. Psychiatry 2020, 177, 376–377. [Google Scholar] [CrossRef] [PubMed]
- Levey, D.F.; Stein, M.B.; Wendt, F.R.; Pathak, G.A.; Zhou, H.; Aslan, M.; Quaden, R.; Harrington, K.M.; Nuñez, Y.Z.; Overstreet, C.; et al. Bi-ancestral depression GWAS in the Million Veteran Program and meta-analysis in >1.2 million individuals highlight new therapeutic directions. Nat. Neurosci. 2021, 24, 954–963. [Google Scholar] [CrossRef]
- Shadrina, M.; Bondarenko, E.A.; Slominsky, P.A. Genetics Factors in Major Depression Disease. Front. Psychiatry 2018, 9, 334. [Google Scholar] [CrossRef]
- Lacerda-Pinheiro, S.F.; Pinheiro Junior, R.F.; Pereira de Lima, M.A.; Lima da Silva, C.G.; Vieira dos Santos Mdo, S.; Teixeira Júnior, A.G.; Lima de Oliveira, P.N.; Ribeiro, K.D.; Rolim-Neto, M.L.; Bianco, B.A. Are there depression and anxiety genetic markers and mutations? A systematic review. J. Affect. Disord. 2014, 168, 387–398. [Google Scholar] [CrossRef]
- Power, R.A.; Tansey, K.E.; Buttenschøn, H.N.; Cohen-Woods, S.; Bigdeli, T.; Hall, L.S.; Kutalik, Z.; Lee, S.H.; Ripke, S.; Steinberg, S.; et al. Genome-wide Association for Major Depression Through Age at Onset Stratification: Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Biol. Psychiatry 2017, 81, 325–335. [Google Scholar] [CrossRef]
- Cai, N.; Revez, J.A.; Adams, M.J.; Andlauer, T.F.M.; Breen, G.; Byrne, E.M.; Clarke, T.K.; Forstner, A.J.; Grabe, H.J.; Hamilton, S.P.; et al. Minimal phenotyping yields genome-wide association signals of low specificity for major depression. Nat. Genet. 2020, 52, 437–447. [Google Scholar] [CrossRef]
- Hettema, J.M.; Neale, M.C.; Kendler, K.S. A review and meta-analysis of the genetic epidemiology of anxiety disorders. Am. J. Psychiatry 2001, 158, 1568–1578. [Google Scholar] [CrossRef]
- Kessler, R.C.; McGonagle, K.A.; Zhao, S.; Nelson, C.B.; Hughes, M.; Eshleman, S.; Wittchen, H.U.; Kendler, K.S. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch. Gen. Psychiatry 1994, 51, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Hettema, J.M.; Prescott, C.A.; Myers, J.M.; Neale, M.C.; Kendler, K.S. The structure of genetic and environmental risk factors for anxiety disorders in men and women. Arch. Gen. Psychiatry 2005, 62, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Waszczuk, M.A.; Zavos, H.M.; Gregory, A.M.; Eley, T.C. The phenotypic and genetic structure of depression and anxiety disorder symptoms in childhood, adolescence, and young adulthood. JAMA Psychiatry 2014, 71, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Dellava, J.E.; Kendler, K.S.; Neale, M.C. Generalized anxiety disorder and anorexia nervosa: Evidence of shared genetic variation. Depress. Anxiety 2011, 28, 728–733. [Google Scholar] [CrossRef]
- Hettema, J.M.; Prescott, C.A.; Kendler, K.S. Genetic and environmental sources of covariation between generalized anxiety disorder and neuroticism. Am. J. Psychiatry 2004, 161, 1581–1587. [Google Scholar] [CrossRef]
- You, J.S.; Hu, S.Y.; Chen, B.; Zhang, H.G. Serotonin transporter and tryptophan hydroxylase gene polymorphisms in Chinese patients with generalized anxiety disorder. Psychiatr. Genet. 2005, 15, 7–11. [Google Scholar] [CrossRef]
- Chang, C.C.; Chang, H.A.; Fang, W.H.; Chang, T.C.; Huang, S.Y. Gender-specific association between serotonin transporter polymorphisms (5-HTTLPR and rs25531) and neuroticism, anxiety and depression in well-defined healthy Han Chinese. J. Affect. Disord. 2017, 207, 422–428. [Google Scholar] [CrossRef]
- Molina, E.; Cervilla, J.; Rivera, M.; Torres, F.; Bellón, J.A.; Moreno, B.; King, M.; Nazareth, I.; Gutiérrez, B. Polymorphic variation at the serotonin 1-A receptor gene is associated with comorbid depression and generalized anxiety. Psychiatr. Genet. 2011, 21, 195–201. [Google Scholar] [CrossRef]
- Voltas, N.; Aparicio, E.; Arija, V.; Canals, J. Association study of monoamine oxidase-A gene promoter polymorphism (MAOA-uVNTR) with self-reported anxiety and other psychopathological symptoms in a community sample of early adolescents. J. Anxiety Disord. 2015, 31, 65–72. [Google Scholar] [CrossRef]
- Tadic, A.; Rujescu, D.; Szegedi, A.; Giegling, I.; Singer, P.; Möller, H.J.; Dahmen, N. Association of a MAOA gene variant with generalized anxiety disorder, but not with panic disorder or major depression. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2003, 117B, 1–6. [Google Scholar] [CrossRef]
- Klauke, B.; Deckert, J.; Reif, A.; Pauli, P.; Zwanzger, P.; Baumann, C.; Arolt, V.; Glöckner-Rist, A.; Domschke, K. Serotonin transporter gene and childhood trauma--a G × E effect on anxiety sensitivity. Depress. Anxiety 2011, 28, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Baumann, C.; Klauke, B.; Weber, H.; Domschke, K.; Zwanzger, P.; Pauli, P.; Deckert, J.; Reif, A. The interaction of early life experiences with COMT val158met affects anxiety sensitivity. Genes Brain Behav. 2013, 12, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Vendruscolo, L.F.; Izidio, G.S.; Takahashi, R.N.; Ramos, A. Chronic Methylphenidate Treatment during Adolescence Increases Anxiety-Related Behaviors and Ethanol Drinking in Adult Spontaneously Hypertensive Rats. Behav. Pharmacol. 2008, 19, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, W.; Okano, H.J.; Osumi, N.; Inoue, T.; Nakamura, S.; Sakakibara, S.I.; Miura, M.; Matsuo, N.; Darnell, R.B.; Okano, H. Mammalian ELAV-like neuronal RNA-binding proteins HuB and HuC promote neuronal development in both the central and the peripheral nervous systems. Proc. Natl. Acad. Sci. USA 1999, 96, 9885–9890. [Google Scholar] [CrossRef]
- Noureddine, M.A.; Qin, X.-J.; Oliveira, S.A.; Skelly, T.J.; van der Walt, J.; Hauser, M.A.; Pericak-Vance, M.A.; Vance, J.M.; Li, Y.-J. Association between the neuron-specific RNA-binding protein ELAVL4 and Parkinson disease. Hum. Genet. 2005, 117, 27–33. [Google Scholar] [CrossRef]
- Blennow, K.; Bogdanovic, N.; Gottfries, C.G.; Davidsson, P. The growth-associated protein GAP-43 is increased in the hippocampus and in the gyrus cinguli in schizophrenia. J. Mol. Neurosci. 1999, 13, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.Y.; Wang, J.-F.; Bezchlibnyk, Y.B.; Young, L.T. Immunoreactivity of 43kDa growth-associated protein is decreased in post mortem hippocampus of bipolar disorder and schizophrenia. Neurosci. Lett. 2007, 411, 123–127. [Google Scholar] [CrossRef]
- Yamada, K.; Iwayama, Y.; Hattori, E.; Iwamoto, K.; Toyota, T.; Ohnishi, T.; Ohba, H.; Maekawa, M.; Kato, T.; Yoshikawa, T. Genome-Wide Association Study of Schizophrenia in Japanese Population. PLoS ONE 2011, 6, e20468. [Google Scholar] [CrossRef]
- Vogel, F. Schizophrenia genesis: The origins of madness. Am. J. Hum. Genet. 1991, 48, 1218. [Google Scholar]
- Martinez-Moczygemba, M.; Huston, D.P. Biology of common beta receptor-signaling cytokines: IL-3, IL-5, and GM-CSF. J. Allergy Clin Immunol. 2003, 112, 653–665, quiz 666. [Google Scholar]
- Barreda, D.R.; Hanington, P.C.; Belosevic, M. Regulation of myeloid development and function by colony stimulating factors. Dev. Comp. Immunol. 2004, 28, 509–554. [Google Scholar] [CrossRef] [PubMed]
- Eaton, W.W.; Byrne, M.; Ewald, H.; Mors, O.; Chen, C.Y.; Agerbo, E.; Mortensen, P.B. Association of schizophrenia and autoimmune diseases: Linkage of Danish national registers. Am. J. Psychiatry 2006, 163, 521–528. [Google Scholar] [CrossRef]
- Need, A.C.; Ge, D.; Weale, M.E.; Maia, J.; Feng, S.; Heinzen, E.L.; Shianna, K.V.; Yoon, W.; Kasperaviciūte, D.; Gennarelli, M.; et al. A genome-wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet. 2009, 5, e1000373. [Google Scholar] [CrossRef]
- Gejman, P.V.; Sanders, A.R.; Kendler, K.S. Genetics of schizophrenia: New findings and challenges. Annu. Rev. Genom. Hum. Genet. 2011, 12, 121–144. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.H.; Smoller, J.W. The genetics of bipolar disorder. Neuroscience 2009, 164, 331–343. [Google Scholar] [CrossRef]
- Wehr, T.A.; Goodwin, F.K.; Wirz-Justice, A.; Breitmaier, J.; Craig, C. 48-hour sleep-wake cycles in manic-depressive illness: Naturalistic observations and sleep deprivation experiments. Arch. Gen. Psychiatry 1982, 39, 559–565. [Google Scholar] [CrossRef]
- Fähndrich, E. Effects of sleep deprivation on depressed patients of different nosological groups. Psychiatry Res. 1981, 5, 277–285. [Google Scholar] [CrossRef]
- Ko, C.H.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 2006, 15, R271–R277. [Google Scholar] [CrossRef]
- Cacabelos, R.; Cacabelos, P.; Gjumrakch, A. Genomics of schizophrenia and pharmacogenomics of antipsychotic drugs. Open J. Psychiatry 2013, 3, 46–139. [Google Scholar] [CrossRef]
- McQuillin, A.; Rizig, M.; Gurling, H.M. A microarray gene expression study of the molecular pharmacology of lithium carbonate on mouse brain mRNA to understand the neurobiology of mood stabilization and treatment of bipolar affective disorder. Pharmacogenet. Genom. 2007, 17, 605–617. [Google Scholar] [CrossRef]
- Smeland, O.B.; Frei, O.; Dale, A.M.; Andreassen, O.A. The polygenic architecture of schizophrenia—Rethinking pathogenesis and nosology. Nat. Rev. Neurol. 2020, 16, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Sadee, W.; Wang, D.; Hartmann, K.; Toland, A.E. Pharmacogenomics: Driving Personalized Medicine. Pharmacol. Rev. 2023, 75, 789–814. [Google Scholar] [CrossRef] [PubMed]
- Fiers, W.; Contreras, R.; Duerinck, F.; Haegeman, G.; Iserentant, D.; Merregaert, J.; Min Jou, W.; Molemans, F.; Raeymaekers, A.; Van den Berghe, A.; et al. Complete nucleotide sequence of bacteriophage MS2 RNA: Primary and secondary structure of the replicase gene. Nature 1976, 260, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef]
- Eichelbaum, M.; Ingelman-Sundberg, M.; Evans, W.E. Pharmacogenomics and individualized drug therapy. Annu. Rev. Med. 2006, 57, 119–137. [Google Scholar] [CrossRef]
- Lin, J.H.; Lu, A.Y. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 535–567. [Google Scholar] [CrossRef]
- de Leon, J.; Santoro, V.; D’Arrigo, C.; Spina, E. Interactions between antiepileptics and second-generation antipsychotics. Expert. Opin. Drug Metab. Toxicol. 2012, 8, 311–334. [Google Scholar] [CrossRef]
- Schoretsanitis, G.; Spina, E.; Hiemke, C.; de Leon, J. A systematic review and combined analysis of therapeutic drug monitoring studies for long-acting paliperidone. Expert. Rev. Clin. Pharmacol. 2018, 11, 1237–1253. [Google Scholar] [CrossRef]
- Schoretsanitis, G.; Kane, J.M.; de Leon, J. Adding Oral Contraceptives to Clozapine May Require Halving the Clozapine Dose: A New Case and a Literature Review. J. Clin. Psychopharmacol. 2020, 40, 308–310. [Google Scholar] [CrossRef]
- de Leon, J. The effects of antiepileptic inducers in neuropsychopharmacology, a neglected issue. Part II: Pharmacological issues and further understanding. Rev. Psiquiatr. Salud Ment. 2015, 8, 167–188. [Google Scholar] [CrossRef]
- Thorn, C.F.; Müller, D.J.; Altman, R.B.; Klein, T.E. PharmGKB summary: Clozapine pathway, pharmacokinetics. Pharmacogenet. Genom. 2018, 28, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Samer, C.F.; Lorenzini, K.I.; Rollason, V.; Daali, Y.; Desmeules, J.A. Applications of CYP450 testing in the clinical setting. Mol. Diagn. Ther. 2013, 17, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Alchakee, A.; Ahmed, M.; Eldohaji, L.; Alhaj, H.; Saber-Ayad, M. Pharmacogenomics in Psychiatry Practice: The Value and the Challenges. Int. J. Mol. Sci. 2022, 23, 13485. [Google Scholar] [CrossRef] [PubMed]
- Brousseau, D.C.; McCarver, D.G.; Drendel, A.L.; Divakaran, K.; Panepinto, J.A. The effect of CYP2D6 polymorphisms on the response to pain treatment for pediatric sickle cell pain crisis. J. Pediatr. 2007, 150, 623–626. [Google Scholar] [CrossRef]
- Stamer, U.M.; Lehnen, K.; Höthker, F.; Bayerer, B.; Wolf, S.; Hoeft, A.; Stuber, F. Impact of CYP2D6 genotype on postoperative tramadol analgesia. Pain 2003, 105, 231–238. [Google Scholar] [CrossRef]
- Cacabelos, R.; Torrellas, C. Pharmacogenomics of Antidepressants. HSOA J. Psychiatry Depress Anxiety 2015, 1, 001. [Google Scholar] [CrossRef]
- Fornaro, M.; Anastasia, A.; Valchera, A.; Carano, A.; Orsolini, L.; Vellante, F.; Rapini, G.; Olivieri, L.; Di Natale, S.; Perna, G.; et al. The FDA “Black Box” Warning on Antidepressant Suicide Risk in Young Adults: More Harm than Benefits? Front. Psychiatry 2019, 10, 294. [Google Scholar] [CrossRef]
- Pratt, V.M.; Del Tredici, A.L.; Hachad, H.; Ji, Y.; Kalman, L.V.; Scott, S.A.; Weck, K.E. Recommendations for Clinical CYP2C19 Genotyping Allele Selection: A Report of the Association for Molecular Pathology. J. Mol. Diagn. 2018, 20, 269–276. [Google Scholar] [CrossRef]
- Nassan, M.; Nicholson, W.T.; Elliott, M.A.; Rohrer Vitek, C.R.; Black, J.L.; Frye, M.A. Pharmacokinetic Pharmacogenetic Prescribing Guidelines for Antidepressants: A Template for Psychiatric Precision Medicine. Mayo Clin. Proc. 2016, 91, 897–907. [Google Scholar] [CrossRef]
- Sim, S.C.; Nordin, L.; Andersson, T.M.-L.; Virding, S.; Olsson, M.; Pedersen, N.L.; Ingelman-Sundberg, M. Association between CYP2C19 Polymorphism and Depressive Symptoms. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 1160–1166. [Google Scholar] [CrossRef]
- Jukić, M.M.; Opel, N.; Ström, J.; Carrillo-Roa, T.; Miksys, S.; Novalen, M.; Renblom, A.; Sim, S.C.; Peñas-Lledó, E.M.; Courtet, P.; et al. Elevated CYP2C19 Expression Is Associated with Depressive Symptoms and Hippocampal Homeostasis Impairment. Mol. Psychiatry 2017, 22, 1155–1163. [Google Scholar] [PubMed]
- Kuo, H.-W.; Liu, S.C.; Tsou, H.-H.; Liu, S.-W.; Lin, K.-M.; Lu, S.-C.; Hsiao, M.-C.; Hsiao, C.-F.; Liu, C.-Y.; Chen, C.-H.; et al. CYP1A2 Genetic Polymorphisms Are Associated with Early Antidepressant Escitalopram Metabolism and Adverse Reactions. Pharmacogenomics 2013, 14, 1191–1201. [Google Scholar] [PubMed]
- Lin, K.-M.; Tsou, H.-H.; Tsai, I.-J.; Hsiao, M.-C.; Hsiao, C.-F.; Liu, C.-Y.; Shen, W.W.; Tang, H.-S.; Fang, C.-K.; Wu, C.-S.; et al. CYP1A2 Genetic Polymorphisms Are Associated with Treatment Response to the Antidepressant Paroxetine. Pharmacogenomics 2010, 11, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, N.; Ren, D.; Bi, Y.; Xu, F.; Niu, W.; Sun, Q.; Guo, Z.; Yuan, R.; Yuan, F.; et al. CYP1A2 Genetic Polymorphism Is Associated with Treatment Remission to Antidepressant Venlafaxine in Han Chinese Population. Clin. Neuropharmacol. 2019, 42, 32–36. [Google Scholar]
- Cabaleiro, T.; Ochoa, D.; López-Rodríguez, R.; Román, M.; Novalbos, J.; Ayuso, C.; Abad-Santos, F. Effect of polymorphisms on the pharmacokinetics, pharmacodynamics, and safety of risperidone in healthy volunteers. Hum. Psychopharmacol. Clin. Exp. 2014, 29, 459–469. [Google Scholar] [CrossRef]
- Jose de Leon, M.D.; Susce, M.T.; Pan, R.M.; Fairchild, M.; Koch, W.H.; Wedlund, P.J. The CYP2D6 poor metabolizer phenotype may be associated with risperidone adverse drug reactions and discontinuation. J. Clin. Psychiatry 2005, 66, 15–27. [Google Scholar] [CrossRef]
- Schoretsanitis, G.; de Leon, J.; Diaz, F.J. Prolactin levels: Sex differences in the effects of risperidone, 9-hydroxyrisperidone levels, CYP2D6 and ABCB1 variants. Pharmacogenomics 2018, 19, 815–823. [Google Scholar] [CrossRef]
- Vandenberghe, F.; Guidi, M.; Choong, E.; von Gunten, A.; Conus, P.; Csajka, C.; Eap, C.B. Genetics-based population pharmacokinetics and pharmacodynamics of risperidone in a psychiatric cohort. Clin. Pharmacokinet. 2015, 54, 1259–1272. [Google Scholar] [CrossRef]
- Koller, D.; Belmonte, C.; Saiz-Rodríguez, M.; Zubiaur, P.; Román, M.; Ochoa, D.; Abad-Santos, F. Effects of aripiprazole on circadian prolactin secretion related to pharmacogenetics in healthy volunteers. Basic Clin. Pharmacol. Toxicol. 2020, 126, 236–246. [Google Scholar] [CrossRef]
- Llerena, A.; Berecz, R.; Dorado, P.; De la Rubia, A. QTc interval, CYP2D6 and CYP2C9 genotypes and risperidone plasma concentrations. J. Psychopharmacol. 2004, 18, 189–193. [Google Scholar]
- Doude van Troostwijk, L.J.; Koopmans, R.P.; Vermeulen, H.D.; Guchelaar, H.J. CYP1A2 activity is an important determinant of clozapine dosage in schizophrenic patients. Eur. J. Pharm. Sci. 2003, 20, 451–457. [Google Scholar] [PubMed]
- Olesen, O.V.; Linnet, K. Contributions of five human cytochrome P450 isoforms to the N-demethylation of clozapine in vitro at low and high concentrations. J. Clin. Pharmacol. 2001, 41, 823–832. [Google Scholar] [PubMed]
- Spina, E.; de Leon, J. Clinically relevant interactions between newer antidepressants and second-generation antipsychotics. Expert. Opin. Drug Metab. Toxicol. 2014, 10, 721–746. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R.; Smith, R.L. Inflammation-induced phenoconversion of polymorphic drug metabolizing enzymes: Hypothesis with implications for personalized medicine. Drug Metab. Dispos. 2015, 43, 400–410. [Google Scholar] [CrossRef]
- Spina, E.; Hiemke, C.; de Leon, J. Assessing drug-drug interactions through therapeutic drug monitoring when administering oral second-generation antipsychotics. Expert. Opin. Drug Metab. Toxicol. 2016, 12, 407–422. [Google Scholar]
- Eggert, A.E.; Crismon, M.L.; Dorson, P.G. Lack of effect of fluoxetine on plasma clozapine concentrations. J. Clin. Psychiatry 1994, 55, 454–455. [Google Scholar]
- Joos, A.A.; König, F.; Frank, U.G.; Kaschka, W.P.; Mörike, K.E.; Ewald, R. Dose-dependent pharmacokinetic interaction of clozapine and paroxetine in an extensive metabolizer. Pharmacopsychiatry 1997, 30, 266–270. [Google Scholar]
- Centorrino, F.; Baldessarini, R.J.; Frankenburg, F.R.; Kando, J.; Volpicelli, S.A.; Flood, J.G. Serum levels of clozapine and norclozapine in patients treated with selective serotonin reuptake inhibitors. Am. J. Psychiatry 1996, 153, 820–822. [Google Scholar]
- Zubiaur, P.; Figueiredo-Tor, L.; Villapalos-García, G.; Soria-Chacartegui, P.; Navares-Gómez, M.; Novalbos, J.; Matas, M.; Calleja, S.; Mejía-Abril, G.; Román, M.; et al. Association between CYP2C19 and CYP2B6 phenotypes and the pharmacokinetics and safety of diazepam. Biomed. Pharmacother. 2022, 155, 113747. [Google Scholar] [CrossRef]
- Raaska, K.; Raitasuo, V.; Arstila, M.; Neuvonen, P.J. Bacterial pneumonia can increase serum concentration of clozapine. Eur. J. Clin. Pharmacol. 2002, 58, 321–322. [Google Scholar]
- de Leon, J.; Diaz, F.J. Serious respiratory infections can increase clozapine levels and contribute to side effects: A case report. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- de Leon, J.; Ruan, C.J.; Verdoux, H.; Wang, C. Clozapine is strongly associated with the risk of pneumonia and inflammation. Gen. Psychiatr. 2020, 33, e100183. [Google Scholar] [CrossRef] [PubMed]
- Spina, E.; Barbieri, M.A.; Cicala, G.; de Leon, J. Clinically Relevant Interactions between Atypical Antipsychotics and Anti-Infective Agents. Pharmaceuticals 2020, 13, 439. [Google Scholar] [CrossRef] [PubMed]
- Sachse, C.; Brockmöller, J.; Bauer, S.; Roots, I. Functional significance of a C-->A polymorphism in intron 1 of the cytochrome P450 CYP1A2 gene tested with caffeine. Br. J. Clin. Pharmacol. 1999, 47, 445–449. [Google Scholar] [CrossRef]
- van der Weide, J.; Steijns, L.S.; van Weelden, M.J. The effect of smoking and cytochrome P450 CYP1A2 genetic polymorphism on clozapine clearance and dose requirement. Pharmacogenetics 2003, 13, 169–172. [Google Scholar]
- Markowitz, J.S.; DeVane, C.L. Suspected ciprofloxacin inhibition of olanzapine resulting in increased plasma concentration. J. Clin. Psychopharmacol. 1999, 19, 289–291. [Google Scholar]
- Callaghan, J.T.; Bergstrom, R.F.; Ptak, L.R.; Beasley, C.M. Olanzapine. Pharmacokinetic and pharmacodynamic profile. Clin. Pharmacokinet. 1999, 37, 177–193. [Google Scholar]
- Dev, V.; Raniwalla, J. Quetiapine: A review of its safety in the management of schizophrenia. Drug Saf. 2000, 23, 295–307. [Google Scholar]
- Wong, Y.W.; Yeh, C.; Thyrum, P.T. The effects of concomitant phenytoin administration on the steady-state pharmacokinetics of quetiapine. J. Clin. Psychopharmacol. 2001, 21, 89–93. [Google Scholar]
- Chan, P.K.; Lam, R.W.; Perry, K.F. Mania precipitated by light therapy for patients with SAD. J. Clin. Psychiatry 1994, 55, 454. [Google Scholar]
- Skryabin, V.Y.; Zastrozhin, M.S.; Torrado, M.V.; Grishina, E.A.; Ryzhikova, K.A.; Shipitsyn, V.V.; Galaktionova, T.E.; Sorokin, A.S.; Bryun, E.A.; Sychev, D.A. How Do CYP2C19*2 and CYP2C19*17 Genetic Polymorphisms Affect the Efficacy and Safety of Diazepam in Patients with Alcohol Withdrawal Syndrome? Drug Metab. Pers. Ther. 2020, 35, 20190026. [Google Scholar] [CrossRef] [PubMed]
- Skryabin, V.Y.; Zastrozhin, M.; Torrado, M.; Grishina, E.; Ryzhikova, K.; Shipitsyn, V.; Galaktionova, T.; Bryun, E.; Sychev, D. Effects of CYP2C19*17 Genetic Polymorphisms on Plasma and Saliva Concentrations of Diazepam in Patients with Alcohol Withdrawal Syndrome. Psychiatr. Genet. 2022, 32, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Tolle-Sander, S. Midazolam Exhibits Characteristics of a Highly Permeable P-Glycoprotein Substrate. Pharm. Res. 2003, 20, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, K.; Gibbs, M.A. CYP3A5 Polymorphism and Alprazolam Pharmacokinetics/Pharmacodynamics. Clin. Pharmacol. Ther. 2006, 80, 719–720. [Google Scholar] [CrossRef]
- Park, J.; Kim, K.; Park, P.; Lee, O.; Kang, D.; Shon, J.; Liu, K.; Shin, J. Effect of CYP3A5*3 Genotype on the Pharmacokinetics and Pharmacodynamics of Alprazolam in Healthy Subjects. Clin. Pharmacol. Ther. 2006, 79, 590–599. [Google Scholar] [CrossRef]
- Mulder, T.A.M.; van Eerden, R.A.G.; de With, M.; Elens, L.; Hesselink, D.A.; Matic, M.; Bins, S.; Mathijssen, R.H.J.; van Schaik, R.H.N. CYP3A4*22 Genotyping in Clinical Practice: Ready for Implementation? Front. Genet. 2021, 12, 711943. [Google Scholar] [CrossRef]
- Zubiaur, P.; Abad-Santos, F. Use of Pharmacogenetics for Benzodiazepine Prescription: State of the Art and Expectations. Pharmacogenomics 2022, 23, 949–952. [Google Scholar] [CrossRef]
- Niwa, T.; Yasuda, S.; Yamamoto, Y.; Murakami, M.; Ishii, R. Contribution of the human cytochrome P450 2C subfamily to the metabolism of and the interactions with endogenous compounds including steroid hormones. Die Pharm.-Int. J. Pharm. Sci. 2021, 76, 611–613. [Google Scholar]
- Haduch, A.; Bromek, E.; Daniel, W.A. Role of brain cytochrome P450 (CYP2D) in the metabolism of monoaminergic neurotransmitters. Pharmacol. Rep. 2013, 65, 1519–1528. [Google Scholar] [CrossRef]
- Kirchheiner, J.; Seeringer, A.; Godoy, A.L.; Ohmle, B.; Maier, C.; Beschoner, P.; Sim, E.J.; Viviani, R. CYP2D6 in the brain: Genotype effects on resting brain perfusion. Mol. Psychiatry 2011, 16, 333–341. [Google Scholar] [CrossRef]
- Hicks, J.K.; Bishop, J.R.; Sangkuhl, K.; Müller, D.J.; Ji, Y.; Leckband, S.G.; Leeder, J.S.; Graham, R.L.; Chiulli, D.L.; LLerena, A.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin. Pharmacol. Ther. 2015, 98, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.; Bang, M.; Kim, A.; Cho, D.Y.; Lee, S.H. Influence of cytochrome P450 2D6 polymorphism on hippocampal white matter and treatment response in schizophrenia. NPJ Schizophr. 2021, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhen, Y.; Miksys, S.; Beyoğlu, D.; Krausz, K.W.; Tyndale, R.F.; Yu, A.; Idle, J.R.; Gonzalez, F.J. Potential role of CYP2D6 in the central nervous system. Xenobiotica 2013, 43, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Haduch, A.; Daniel, W.A. The engagement of brain cytochrome P450 in the metabolism of endogenous neuroactive substrates: A possible role in mental disorders. Drug Metab. Rev. 2018, 50, 415–429. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Table of Pharmacogenetic Associations. Available online: https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations (accessed on 1 February 2025).
- Cicali, E.J.; Weitzel, K.W.; Elsey, A.R.; Orlando, F.A.; Vinson, M.; Mosley, S.; Smith, D.M.; Davis, R.; Drum, L.; Estores, D.; et al. Challenges and lessons learned from clinical pharmacogenetic implementation of multiple gene–drug pairs across ambulatory care settings. Genet. Med. 2019, 21, 2264–2274. [Google Scholar] [CrossRef]
- Luzum, J.A.; Luzum, M.J. Physicians’ attitudes toward pharmacogenetic testing before and after pharmacogenetic education. Pers. Med. 2016, 13, 119–127. [Google Scholar] [CrossRef]
- Mikat-Stevens, N.A.; Larson, I.A.; Tarini, B.A. Primary-care providers’ perceived barriers to integration of genetics services: A systematic review of the literature. Genet. Med. 2015, 17, 169–176. [Google Scholar] [CrossRef]
- Shad, M.U. Genetic testing for antipsychotic pharmacotherapy: Bench to bedside. Behav. Sci. 2021, 11, 97. [Google Scholar] [CrossRef]
- Cuéllar-Barboza, A.B.; McElroy, S.L.; Veldic, M.; Singh, B.; Kung, S.; Romo-Nava, F.; Nunez, N.A.; Cabello-Arreola, A.; Coombes, B.J.; Prieto, M.; et al. Potential pharmacogenomic targets in bipolar disorder: Considerations for current testing and the development of decision support tools to individualize treatment selection. Int. J. Bipolar Disord. 2020, 8, 1–7. [Google Scholar] [CrossRef]
- Padberg, F.; Bulubas, L.; Mizutani-Tiebel, Y.; Burkhardt, G.; Kranz, G.S.; Koutsouleris, N.; Kambeitz, J.; Hasan, A.; Takahashi, S.; Keeser, D.; et al. The intervention, the patient and the illness—Personalizing non-invasive brain stimulation in psychiatry. Exp. Neurol. 2021, 341, 113713. [Google Scholar] [CrossRef]
- Hallett, M. Transcranial magnetic stimulation: A primer. Neuron 2007, 55, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Watson, K.; Nasca, C.; Aasly, L.; McEwen, B.; Rasgon, N. Insulin resistance, an unmasked culprit in depressive disorders: Promises for interventions. Neuropharmacology 2018, 136 Pt B, 327–334. [Google Scholar] [CrossRef]
- Fernandes, B.S.; Salagre, E.; Enduru, N.; Grande, I.; Vieta, E.; Zhao, Z. Insulin resistance in depression: A large meta-analysis of metabolic parameters and variation. Neurosci. Biobehav. Rev. 2022, 139, 104758. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- van der Heide, L.P.; Kamal, A.; Artola, A.; Gispen, W.H.; Ramakers, G.M. Insulin modulates hippocampal activity-dependent synaptic plasticity in a N-methyl-d-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J. Neurochem. 2005, 94, 1158–1166. [Google Scholar] [CrossRef]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef]
- Lee, C.C.; Huang, C.C.; Hsu, K.S. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology 2011, 61, 867–879. [Google Scholar] [CrossRef]
- Jurcovicova, J. Glucose transport in brain—Effect of inflammation. Endocr. Regul. 2014, 48, 35–48. [Google Scholar] [CrossRef]
- Pearson-Leary, J.; McNay, E.C. Novel roles for the insulin-regulated glucose transporter-4 in hippocampally dependent memory. J. Neurosci. 2016, 36, 11851–11864. [Google Scholar] [CrossRef]
- Garwood, C.J.; Ratcliffe, L.E.; Morgan, S.V.; Simpson, J.E.; Owens, H.; Vazquez-Villaseñor, I.; Heath, P.R.; Romero, I.A.; Ince, P.G.; Wharton, S.B. Insulin and IGF1 signalling pathways in human astrocytes in vitro and in vivo; characterisation, subcellular localisation and modulation of the receptors. Mol. Brain 2015, 8, 51. [Google Scholar] [CrossRef]
- Heni, M.; Hennige, A.M.; Peter, A.; Siegel-Axel, D.; Ordelheide, A.M.; Krebs, N.; Machicao, F.; Fritsche, A.; Häring, H.U.; Staiger, H. Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS ONE 2011, 6, e21594. [Google Scholar] [CrossRef] [PubMed]
- Cairns, K.; McCarvill, T.; Ruzickova, M.; Calkin, C.V. Course of bipolar illness worsens after onset of insulin resistance. J. Psychiatr. Res. 2018, 102, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Calkin, C.V.; Ruzickova, M.; Uher, R.; Hajek, T.; Slaney, C.M.; Garnham, J.S.; O’Donovan, M.C.; Alda, M. Insulin resistance and outcome in bipolar disorder. Br. J. Psychiatry 2015, 206, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Guha, P.; Bhowmick, K.; Mazumder, P.; Ghosal, M.; Chakraborty, I.; Burman, P. Assessment of insulin resistance and metabolic syndrome in drug naive patients of bipolar disorder. Indian. J. Clin. Biochem. 2014, 29, 51–56. [Google Scholar] [CrossRef]
- Fanelli, G.; Franke, B.; De Witte, W.; Ruisch, I.H.; Haavik, J.; van Gils, V.; Jansen, W.J.; Vos, S.J.B.; Lind, L.; Buitelaar, J.K.; et al. Insulinopathies of the brain? Genetic overlap between somatic insulin-related and neuropsychiatric disorders. Transl. Psychiatry 2022, 12, 59. [Google Scholar] [CrossRef]
- Mittal, K.; Katare, D.P. Shared links between type 2 diabetes mellitus and Alzheimer’s disease: A review. Diabetes Metab. Syndr. 2016, 10 (Suppl. S1), S144–S149. [Google Scholar] [CrossRef]
- Grajales, D.; Ferreira, V.; Valverde, Á.M. Second-Generation Antipsychotics and Dysregulation of Glucose Metabolism: Beyond Weight Gain. Cells 2019, 8, 1336. [Google Scholar] [CrossRef]
- Mazereel, V.; Detraux, J.; Vancampfort, D.; van Winkel, R.; De Hert, M. Impact of Psychotropic Medication Effects on Obesity and the Metabolic Syndrome in People With Serious Mental Illness. Front. Endocrinol. 2020, 11, 573479. [Google Scholar] [CrossRef]
- Yang, W.; Zheng, L.; Zheng, B.; Zeng, S.; Li, J.; Liang, B.; Zhu, J.; Zhang, M. A Meta-Analysis of Abnormal Glucose Metabolism in First-Episode Drug-Naive Schizophrenia. Psychiatr. Danub. 2020, 32, 46–54. [Google Scholar] [CrossRef]
- Fernandez-Egea, E.; Miller, B.; Bernardo, M.; Donner, T.; Kirkpatrick, B. Parental history of type 2 diabetes in patients with nonaffective psychosis. Schizophr. Res. 2008, 98, 302–306. [Google Scholar] [CrossRef]
- Pillinger, T.; Beck, K.; Gobjila, C.; Donocik, J.G.; Jauhar, S.; Howes, O.D. Impaired glucose homeostasis in first-episode schizophrenia: A systematic review and meta-analysis. JAMA Psychiatry 2017, 74, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Holt, R.I. Association between antipsychotic medication use and diabetes. Curr. Diabetes Rep. 2019, 19, 96. [Google Scholar] [CrossRef] [PubMed]
- Chadha, R.; Meador-Woodruff, J. S192. AKT-MTOR signaling pathway is downregulated in schizophrenia. Schizophr. Bull. 2018, 44, S400. [Google Scholar] [CrossRef]
- Zhao, Z.; Ksiezak-Reding, H.; Riggio, S.; Haroutunian, V.; Pasinetti, G.M. Insulin receptor deficits in schizophrenia and in cellular and animal models of insulin receptor dysfunction. Schizophr. Res. 2006, 84, 1–14. [Google Scholar] [CrossRef]
- Emamian, E.S.; Hall, D.; Birnbaum, M.J.; Karayiorgou, M.; Gogos, J.A. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat. Genet. 2004, 36, 131–137. [Google Scholar] [CrossRef]
- Wijtenburg, S.A.; Kapogiannis, D.; Korenic, S.A.; Mullins, R.J.; Tran, J.; Gaston, F.E.; Chen, S.; Mustapic, M.; Hong, L.E.; Rowland, L.M. Brain insulin resistance and altered brain glucose are related to memory impairments in schizophrenia. Schizophr. Res. 2019, 208, 324–330. [Google Scholar] [CrossRef]
- Meyer, J.M.; Correll, C.U. Increased metabolic potential, efficacy, and safety of emerging treatments in schizophrenia. CNS Drugs 2023, 37, 545–570. [Google Scholar] [CrossRef]
- Corrao, M.M.; Nelson, L.A. Olanzapine/samidorphan: A new combination treatment for schizophrenia and bipolar i disorder intended to reduce weight gain. CNS Drugs 2022, 36, 605–616. [Google Scholar] [CrossRef]
- Correll, C.U.; Stein, E.; Graham, C.; DiPetrillo, L.; Akerman, S.; Stanford, A.D.; Jiang, Y.; Yagoda, S.; McDonnell, D.; Hopkinson, C. Reduction in multiple cardiometabolic risk factors with combined olanzapine/samidorphan compared with olanzapine: Post hoc analyses from a 24-week phase 3 study. Schizophr. Bull. 2023, 49, 454–463. [Google Scholar] [CrossRef]
- Svensson, C.K.; Larsen, J.R.; Vedtofte, L.; Jakobsen, M.S.L.; Jespersen, H.R.; Jakobsen, M.I.; Koyuncu, K.; Schjerning, O.; Nielsen, J.; Ekstrøm, C.T.; et al. One-year follow-up on liraglutide treatment for prediabetes and overweight/obesity in clozapine- or olanzapine-treated patients. Acta Psychiatr. Scand. 2019, 139, 26–36. [Google Scholar] [CrossRef]
- Benedict, C.; Kern, W.; Schultes, B.; Born, J.; Hallschmid, M. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J. Clin. Endocrinol. Metab. 2008, 93, 1339–1344. [Google Scholar] [PubMed]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long Acting Intranasal Insulin Detemir Improves Cognition for Adults with Mild Cognitive Impairment or Early-Stage Alzheimer’s Disease Dementia. J. Alzheimers Dis. 2015, 45, 1269–1270, Erratum in: J. Alzheimers Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Lotta, B. Targeting acetylcholinesterase: Identification of chemical leads by high throughput screening, structure determination and molecular modeling. PLoS ONE 2011, 6, e26039. [Google Scholar]
- Silman, I.; Sussman, J.L. Acetylcholinesterase: How is structure related to function? Chem. Biol. Interact. 2008, 175, 3–10. [Google Scholar] [CrossRef]
- Tripathi, A. Acetylcholinsterase: A versatile enzyme of nervous system. Ann. Neurosci. 2008, 15, 106–111. [Google Scholar]
- Schröder, H.; Zilles, K.; Maelicke, A.; Hajós, F. Immunohistoand cytochemical localization of cortical cholinoceptors in rat and man. Brain Res. 1989, 502, 287–295. [Google Scholar]
- Schröder, H.; Zilles, K.; Luiten, P.G.M.; Strosberg, A.D. Immunocytochemical visualization of muscarinic cholinoceptors in the human cerebral cortex. Brain Res. 1990, 514, 249–258. [Google Scholar] [CrossRef]
- McCormick, D.A.; Prince, D.A. Two types of muscarinic responses to acetylcholine in mammalian cortical neurons. Proc. Natl. Acad. Sci. USA 1985, 82, 6344–6348. [Google Scholar] [CrossRef]
- Umbriaco, D.; Watkins, K.C.; Descarries, L.; Cozzari, C.; Hartman, B.K. Ultrastructural and morphometric features of the acetylcholine innervation in adult rat parietal cortex: An electron microscopic study in serial sections. J. Comp. Neurol. 1994, 348, 351–373. [Google Scholar] [CrossRef]
- Yamasaki, M.; Matsui, M.; Watanabi, M. Preferential localization of muscarininc M1 receptor on dendritic shaft and spine of cortical pyramidal cells and its anatomical evidence for volume transmission. J. Neurosci. 2010, 30, 4408–4418. [Google Scholar] [PubMed]
- Ashford, J.W. Treatment of Alzheimer’s disease: The legacy of the cholinergic hypothesis, neuroplasticity, and future directions. J. Alzheimers Dis. 2015, 47, 149–156. [Google Scholar] [CrossRef]
- Groner, E.; Ashani, Y.; Schorer-Apelbaum, D.; Sterling, J.; Herzig, Y.; Weinstock, M. The kinetics of inhibition of human acetylcholinesterase and butyrylcholinesterase by two series of novel carbamates. Mol. Pharmacol. 2007, 71, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P. Pharmacodynamic-tolerability relationships of cholinesterase inhibitors for Alzheimer’s disease. CNS Drugs 2001, 15, 375–390. [Google Scholar] [PubMed]
- Jouvet, M. The role of monoamines acetylcholine-containing neurons in the regulation of the sleep-waking cycle. In Ergebnisse der Physiologie; Springer: Berlin/Heidelberg, Germany, 1972; pp. 166–307. [Google Scholar]
- Baghdoyan, H.A.; Rodrigo-Angulo, M.L.; McCarley, R.W.; Hobson, J.A. Site-specific enhancement and suppression of desynchronized sleep signs following cholinergic stimulation of three brainstem regions. Brain Res. 1984, 306, 39–52. [Google Scholar] [CrossRef]
- Webster, H.H.; Jones, B.E. Neurotoxic lesions of the dorsolateral pontomesencephalic tegmentum-cholinergic cell area in the cat. II Effects upon sleep-waking states. Brain Res. 1988, 458, 285–302. [Google Scholar] [CrossRef]
- Shouse, M.N.; Siegel, J.M. Pontine regulation of REM sleep components in cats: Integrity of the pedunculopontine tegmentum (PPT) is important for phasic events but unnecessary for atonia during REM sleep. Brain Res. 1992, 571, 50–63. [Google Scholar]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70, S38–S44. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar]
- Lin, P.; Ding, B.; Feng, C.; Yin, S.; Zhang, T.; Qi, X.; Lv, H.; Guo, X.; Dong, K.; Zhu, Y.; et al. Prevotella and Klebsiella proportions in fecal microbial communities are potential characteristic parameters for patients with major depressive disorder. J. Affect. Disord. 2017, 207, 300–304. [Google Scholar]
- Adeshirlarijaney, A.; Gewirtz, A.T. Considering gut microbiota in treatment of type 2 diabetes mellitus. Gut Microbes 2020, 11, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Socała, K.; Doboszewska, U.; Szopa, A.; Serefko, A.; Włodarczyk, M.; Zielińska, A.; Poleszak, E.; Fichna, J.; Wlaź, P. The role of microbiota-gut-brain axis in neuropsychiatric and neurological disorders. Pharm. Res. 2021, 172, 105840. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.N.M.; Kumar, J.; Nawawi, K.N.M.; Ali, R.A.R.; Mokhtar, N.M. Irritable Bowel Syndrome, Depression, and Neurodegeneration: A Bidirectional Communication from Gut to Brain. Nutrients 2021, 13, 3061. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.G.; Goldenthal, A.R.; Uhlemann, A.C.; Mann, J.J.; Miller, J.M.; Sublette, M.E. Systematic Review of Gut Microbiota and Major Depression. Front. Psychiatry 2019, 10, 34. [Google Scholar] [CrossRef]
- Erny, D.; Hrabe de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- McCusker, R.H.; Kelley, K.W. Immune-neural connections: How the immune system’s response to infectious agents influences behavior. J. Exp. Biol. 2013, 216 Pt 1, 84–98. [Google Scholar] [CrossRef]
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J.M. Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef]
- Kielian, T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J. Neurosci. Res. 2006, 83, 711–730. [Google Scholar] [CrossRef]
- Zhu, F.; Ju, Y.; Wang, W.; Wang, Q.; Guo, R.; Ma, Q.; Sun, Q.; Fan, Y.; Xie, Y.; Yang, Z.; et al. Metagenome-wide association of gut microbiome features for schizophrenia. Nat. Commun. 2020, 11, 1612. [Google Scholar] [CrossRef]
- Coyle, J.T. NMDA receptor and schizophrenia: A brief history. Schizophr. Bull. 2012, 38, 920–926. [Google Scholar] [CrossRef]
- Nieto, R.; Kukuljan, M.; Silva, H. BDNF and schizophrenia: From neurodevelopment to neuronal plasticity, learning, and memory. Front. Psychiatry 2013, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Zeng, B.; Zhou, C.; Liu, M.; Fang, Z.; Xu, X.; Zeng, L.; Chen, J.; Fan, S.; Du, X.; et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol. Psychiatry 2016, 21, 786–796. [Google Scholar] [CrossRef]
- Li, N.; Wang, Q.; Wang, Y.; Sun, A.; Lin, Y.; Jin, Y.; Li, X. Fecal microbiota transplantation from chronic unpredictable mild stress mice donors affects anxiety-like and depression-like behavior in recipient mice via the gut microbiota-inflammation-brain axis. Stress 2019, 22, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 10. [Google Scholar] [CrossRef]
- Vulevic, J.; Juric, A.; Walton, G.E.; Claus, S.P.; Tzortzis, G.; Toward, R.E.; Gibson, G.R. Influence of galacto-oligosaccharide mixture (B-GOS) on gut microbiota, immune parameters and metabonomics in elderly persons. Br. J. Nutr. 2015, 114, 586–595. [Google Scholar] [CrossRef]
- Akbari, E.; Asemi, Z.; Kakhaki, R.D.; Bahmani, F.; Kouchaki, E.; Tamtaji, O.R.; Hamidi, G.A.; Salami, M. Effect of Probiotic Supplementation on Cognitive Function and Metabolic Status in Alzheimer’s Disease: A Randomized, Double-Blind and Controlled Trial. Front. Aging Neurosci. 2016, 8, 256. [Google Scholar] [CrossRef]
- Tarr, A.J.; Galley, J.D.; Fisher, S.E.; Chichlowski, M.; Berg, B.M.; Bailey, M.T. The prebiotics 3′Sialyllactose and 6′Sialyllactose diminish stressor-induced anxiety-like behavior and colonic microbiota alterations: Evidence for effects on the gut-brain axis. Brain Behav. Immun. 2015, 50, 166–177. [Google Scholar] [CrossRef]
- Liu, R.T.; Walsh, R.F.L.; Sheehan, A.E. Prebiotics and probiotics for depression and anxiety: A systematic review and meta-analysis of controlled clinical trials. Neurosci. Biobehav. Rev. 2019, 102, 13–23. [Google Scholar] [CrossRef]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef]
- Buffington, S.A.; di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 2016, 165, 1762–1775. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Bercik, P.; Park, A.J.; Sinclair, D.; Khoshdel, A.; Lu, J.; Huang, X.; Deng, Y.; Blennerhassett, P.A.; Fahnestock, M.; Moine, D.; et al. The anxiolytic effect of Bifidobacterium longum NCC3001 involves vagal pathways for gut-brain communication. Neurogastroenterol. Motil. 2011, 23, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef]
- Guo, M.; Li, R.; Wang, Y.; Ma, S.; Zhang, Y.; Li, S.; Zhang, H.; Liu, Z.; You, C.; Zheng, H. Lactobacillus plantarum ST-III modulates abnormal behavior and gut microbiota in a mouse model of autism spectrum disorder. Physiol. Behav. 2022, 257, 113965. [Google Scholar] [CrossRef]
- Abdellatif, B.; McVeigh, C.; Bendriss, G.; Chaari, A. The Promising Role of Probiotics in Managing the Altered Gut in Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 4159. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Horesh, Y.; Katsel, P.; Haroutunian, V.; Domany, E. Gene expression signature is shared by patients with Alzheimer’s disease and schizophrenia at the superior temporal gyrus. Eur. J. Neurol. 2011, 18, 410–424. [Google Scholar] [CrossRef]
- Rapoport, J.L.; Giedd, J.N.; Blumenthal, J.; Hamburger, S.; Jeffries, N.; Fernandez, T.; Nicolson, R.; Bedwell, J.; Lenane, M.; Zijdenbos, A.; et al. Progressive cortical change during adolescence in childhood-onset schizophrenia. A longitudinal magnetic resonance imaging study. Arch. Gen. Psychiatry 1999, 56, 649–654. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Merenlender-Wagner, A.; Malishkevich, A.; Shemer, Z.; Udawela, M.; Gibbons, A.; Scarr, E.; Gozes, I. Autophagy has a key role in the pathophysiology of schizophrenia. Mol. Psychiatry 2015, 20, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Panda, S.; Lin, J.D. Temporal orchestration of circadian autophagy rhythm by C/EBPbeta. EMBO J. 2011, 30, 4642–4651. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Dang, F.; Li, P.; Wang, P.; Xu, Q.; Liu, Z.; Li, Y.; Wu, Y.; Chen, Y.; Liu, Y. The circadian protein Period2 suppresses mTORC1 activity via recruiting Tsc1 to mTORC1 complex. Cell Metab. 2019, 29, 653–667.e656. [Google Scholar]
- Pastore, N.; Vainshtein, A.; Herz, N.J.; Huynh, T.; Brunetti, L.; Klisch, T.J.; Mutarelli, M.; Annunziata, P.; Kinouchi, K.; Brunetti-Pierri, N.; et al. Nutrient-sensitive transcription factors TFEB and TFE3 couple autophagy and metabolism to the peripheral clock. EMBO J. 2019, 38, e101347. [Google Scholar]
- Barakat, D.J.; Mendonca, J.; Barberi, T.; Zhang, J.; Kachhap, S.K.; Paz-Priel, I.; Friedman, A.D. C/EBPbeta regulates sensitivity to bortezomib in prostate cancer cells by inducing REDD1 and autophagosome-lysosome fusion. Cancer Lett. 2016, 375, 152–161. [Google Scholar]
- Vasconcelos-Ferreira, A.; Carmo-Silva, S.; Codesso, J.M.; Silva, P.; Martinez, A.R.M.; França, M.C., Jr.; Nóbrega, C.; Pereira de Almeida, L. The autophagy-enhancing drug carbamazepine improves neuropathology and motor impairment in mouse models of Machado-Joseph disease. Neuropathol. Appl. Neurobiol. 2022, 48, e12763. [Google Scholar]
- Sarkar, S.; Rubinsztein, D.C. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol. Biosyst. 2008, 4, 895–901. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, J.; Pan, H.; Hu, P.; Hao, Y.; Cai, W.; Yuan, J. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc. Natl. Acad. Sci. USA 2007, 104, 19023–19028. [Google Scholar]
- Zhang, H.; Shang, Y.; Xiao, X.; Yu, M.; Zhang, T. Prenatal stress-induced impairments of cognitive flexibility and bidirectional synaptic plasticity are possibly associated with autophagy in adolescent male-offspring. Exp Neurol. 2017, 298 Pt A, 68–78. [Google Scholar]
- Shih, J.H.; Chiu, C.H.; Ma, K.H.; Huang, Y.S.; Shiue, C.Y.; Yeh, T.Y.; Kao, L.T.; Lin, Y.Y.; Li, I.H. Autophagy inhibition plays a protective role against 3, 4-methylenedioxymethamphetamine (MDMA)-induced loss of serotonin transporters and depressive-like behaviors in rats. Pharmacol. Res. 2019, 142, 283–293. [Google Scholar]
- Daley, E.; Wilkie, D.; Loesch, A.; Hargreaves, I.P.; Kendall, D.A.; Pilkington, G.J.; Bates, T.E. Chlorimipramine: A novel anticancer agent with a mitochondrial target. Biochem. Biophys. Res. Commun. 2005, 328, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 443. [Google Scholar]
- Rossi, M.; Munarriz, E.R.; Bartesaghi, S.; Milanese, M.; Dinsdale, D.; Guerra-Martin, M.A.; Bampton, E.T.; Glynn, P.; Bonanno, G.; Knight, R.A.; et al. Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. J. Cell Sci. 2009, 122 Pt 18, 3330–3339. [Google Scholar]
- Zschocke, J.; Zimmermann, N.; Berning, B.; Ganal, V.; Holsboer, F.; Rein, T. Antidepressant drugs diversely affect autophagy pathways in astrocytes and neurons--dissociation from cholesterol homeostasis. Neuropsychopharmacology 2011, 36, 1754–1768. [Google Scholar]
- Zschocke, J.; Rein, T. Antidepressants encounter autophagy in neural cells. Autophagy 2011, 7, 1247–1248. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar]
- van Oostveen, W.M.; de Lange, E.C. Imaging techniques in Alzheimer’s disease: A review of applications in early diagnosis and longitudinal monitoring. Int. J. Mol. Sci. 2021, 22, 2110. [Google Scholar] [CrossRef]
- Samuelsson, J.; Kern, S.; Zetterberg, H.; Blennow, K.; Rothenberg, E.; Wallengren, O.; Skoog, I.; Zettergren, A. A Western-style dietary pattern is associated with cerebrospinal fluid biomarker levels for preclinical Alzheimer’s disease—A population-based cross-sectional study among 70-year-olds. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12183. [Google Scholar]
- González Olmo, B.M.; Butler, M.J.; Barrientos, R.M. Evolution of the Human Diet and Its Impact on Gut Microbiota, Immune Responses, and Brain Health. Nutrients 2021, 13, 196. [Google Scholar] [CrossRef]
- Hill, E.; Goodwill, A.M.; Gorelik, A.; Szoeke, C. Diet and biomarkers of Alzheimer’s disease: A systematic review and meta-analysis. Neurobiol. Aging 2019, 76, 45–52. [Google Scholar] [CrossRef]
- Vinciguerra, F.; Graziano, M.; Hagnäs, M.; Frittitta, L.; Tumminia, A. Influence of the Mediterranean and Ketogenic Diets on Cognitive Status and Decline: A Narrative Review. Nutrients 2020, 12, 1019. [Google Scholar] [CrossRef] [PubMed]
- Więckowska-Gacek, A.; Mietelska-Porowska, A.; Wydrych, M.; Wojda, U. Western diet as a trigger of Alzheimer’s disease: From metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration. Ageing Res. Rev. 2021, 70, 101397. [Google Scholar] [CrossRef] [PubMed]
- Hersant, H.; Grossberg, G. The ketogenic diet and Alzheimer’s disease. J. Nutr. Health aging 2022, 26, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.Q.; Yu, L.L.; Qi, G.Y.; Mi, Y.S.; Wu, W.Q.; Lee, Y.K.; Zhai, Q.X.; Tian, F.W.; Chen, W. Can dietary patterns prevent cognitive impairment and reduce Alzheimer’s disease risk: Exploring the underlying mechanisms of effects. Neurosci. Biobehav. Rev. 2022, 135, 104556. [Google Scholar] [CrossRef]
- Craft, S.; Neth, B.J.; Mintz, A.; Sai, K.; Shively, N.; Dahl, D.; Baker, L.D.; Cunnane, S.; Register, T.C.; Gage, H.D. O4-05-03: Ketogenic diet effects on brain ketone metabolism and Alzheimer’s disease Csf biomarkers. Alzheimer’s Dement. 2016, 12, P342–P343. [Google Scholar] [CrossRef]
- Neth, B.J.; Mintz, A.; Whitlow, C.; Jung, Y.; Sai, K.S.; Register, T.C.; Kellar, D.; Lockhart, S.N.; Hoscheidt, S.; Maldjian, J.; et al. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: A pilot study. Neurobiol. Aging 2020, 86, 54–63. [Google Scholar] [CrossRef]
- Muñoz, G.A.; Castañeda-Gómez, Á.M.; Monsalve, M.P.; Salazar, J.P.; Velandia, M.C.; Arbeláez, F.B. Efecto de la dieta cetogénica baja en calorías sobre la composición corporal en adultos con sobrepeso y obesidad: Revisión sistemática y metaanálisis. Rev. Nutr. Clín. Metab. 2021, 4, 98–113. [Google Scholar] [CrossRef]
- Vassilaki, M.; Aakre, J.A.; Syrjanen, J.A.; Mielke, M.M.; Geda, Y.E.; Kremers, W.K.; Machulda, M.M.; Alhurani, R.E.; Staubo, S.C.; Knopman, D.S.; et al. Mediterranean diet, its components, and amyloid imaging biomarkers. J. Alzheimer’s Dis. 2018, 64, 281–290. [Google Scholar] [CrossRef]
- Kim, J.W.; Byun, M.S.; Yi, D.; Lee, J.H.; Ko, K.; Jeon, S.Y.; Sohn, B.K.; Lee, J.Y.; Kim, Y.K.; Shin, S.A.; et al. Association of moderate alcohol intake with in vivo amyloid-beta deposition in human brain: A cross-sectional study. PLoS Med. 2020, 17, e1003022. [Google Scholar] [CrossRef]
- Gong, Y.S.; Hou, F.L.; Guo, J.; Lin, L.; Zhu, F.Y. Effects of alcohol intake on cognitive function and β-amyloid protein in APP/PS1 transgenic mice. Food Chem. Toxicol. 2021, 151, 112105. [Google Scholar] [CrossRef]
- Ramos-Lopez, O.; Milagro, F.I.; Riezu-Boj, J.I.; Martinez, J.A. Epigenetic signatures underlying inflammation: An interplay of nutrition, physical activity, metabolic diseases, and environmental factors for personalized nutrition. Inflamm. Res. 2021, 70, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, A.; Kalk, N.; Sewell, G.; Ritchie, C.W.; Lingford-Hughes, A. Alcohol and Alzheimer’s disease—Does alcohol dependence contribute to beta-amyloid deposition, neuroinflammation and neurodegeneration in Alzheimer’s disease? Alcohol Alcohol. 2017, 52, 151–158. [Google Scholar] [PubMed]
- Doorduijn, A.S.; De Van Der Schueren, M.A.; Van De Rest, O.; De Leeuw, F.A.; Hendriksen, H.M.; Teunissen, C.E.; Scheltens, P.; van der Flier, W.M.; Visser, M. Energy intake and expenditure in patients with Alzheimer’s disease and mild cognitive impairment: The NUDAD project. Alzheimer’s Res. Ther. 2020, 12, 1–8. [Google Scholar]
- Yeh, S.H.; Shie, F.S.; Liu, H.K.; Yao, H.H.; Kao, P.C.; Lee, Y.H.; Chen, L.M.; Hsu, S.M.; Chao, L.J.; Wu, K.W.; et al. A high-sucrose diet aggravates Alzheimer’s disease pathology, attenuates hypothalamic leptin signaling, and impairs food-anticipatory activity in APPswe/PS1dE9 mice. Neurobiol. Aging 2020, 90, 60–74. [Google Scholar] [CrossRef]
- Luo, D.; Hou, X.; Hou, L.; Wang, M.; Xu, S.; Dong, C.; Liu, X. Effect of pioglitazone on altered expression of Aβ metabolism-associated molecules in the brain of fructose-drinking rats, a rodent model of insulin resistance. Eur. J. Pharmacol. 2011, 664, 14–19. [Google Scholar] [CrossRef]
- Sanderlin, A.H.; Hoscheidt, S.M.; Hanson, A.J.; Baker, L.D.; Sink, K.M.; Wittmer, P.; Craft, S. P3-019: The effects of diet intervention on metabolic health and cerebral spinal fluid Alzheimer’s disease biomarkers: A randomized trial. Alzheimer’s Dement. 2018, 14, P1069–P1070. [Google Scholar] [CrossRef]
- Iqbal, G.; Braidy, N.; Ahmed, T. Blood-based biomarkers for predictive diagnosis of cognitive impairment in a Pakistani population. Front. Aging Neurosci. 2020, 12, 223. [Google Scholar] [CrossRef]
- Jia, J.; Hu, J.; Huo, X.; Miao, R.; Zhang, Y.; Ma, F. Effects of vitamin D supplementation on cognitive function and blood Aβ-related biomarkers in older adults with Alzheimer’s disease: A randomised, double-blind, placebo-controlled trial. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1347–1352. [Google Scholar] [CrossRef]
- Nourhashemi, F.; Hooper, C.; Cantet, C.; Féart, C.; Gennero, I.; Payoux, P.; Salabert, A.S.; Guyonnet, S.; De Souto Barreto, P.; Vellas, B.; et al. Cross-sectional associations of plasma vitamin D with cerebral β-amyloid in older adults at risk of dementia. Alzheimer’s Res. Ther. 2018, 10, 1–7. [Google Scholar] [CrossRef]
- Ma, F.; Li, Q.; Zhou, X.; Zhao, J.; Song, A.; Li, W.; Liu, H.; Xu, W.; Huang, G. Effects of folic acid supplementation on cognitive function and Aβ-related biomarkers in mild cognitive impairment: A randomized controlled trial. Eur. J. Nutr. 2019, 58, 345–356. [Google Scholar] [CrossRef]
- Oikonomidi, A.; Lewczuk, P.; Kornhuber, J.; Smulders, Y.; Linnebank, M.; Semmler, A.; Popp, J. Homocysteine metabolism is associated with cerebrospinal fluid levels of soluble amyloid precursor protein and amyloid beta. J. Neurochem. 2016, 139, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Polderman, T.J.; Benyamin, B.; De Leeuw, C.A.; Sullivan, P.F.; Van Bochoven, A.; Visscher, P.M.; Posthuma, D. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet. 2015, 47, 702–709. [Google Scholar] [CrossRef]
- Pettersson, E.; Larsson, H.; Lichtenstein, P. Common psychiatric disorders share the same genetic origin: A multivariate sibling study of the Swedish population. Mol. Psychiatry 2016, 21, 717–721. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.K.; Loh, P.R.; Finucane, H.K.; Ripke, S.; Yang, J.; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef]
- van der Meer, D.; Hindley, G.; Shadrin, A.A.; Smeland, O.B.; Parker, N.; Dale, A.M.; Frei, O.; Andreassen, O.A. Mapping the Genetic Landscape of Psychiatric Disorders With the MiXeR Toolset. Biol. Psychiatry 2025. advance online publication. [Google Scholar] [CrossRef]
- Lee, P.H.; Anttila, V.; Won, H.; Feng, Y.-C.A.; Rosenthal, J.; Zhu, Z.; Tucker-Drob, E.M.; Nivard, M.G.; Grotzinger, A.D.; Posthuma, D.; et al. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 2019, 179, 1469–1482. [Google Scholar] [CrossRef]
- Bahrami, S.; Shadrin, A.; Frei, O.; O’Connell, K.S.; Bettella, F.; Krull, F.; Fan, C.C.; Røssberg, J.I.; Hindley, G.; Ueland, T.; et al. Genetic loci shared between major depression and intelligence with mixed directions of effect. Nat. Hum. Behav. 2021, 18, 1–7. [Google Scholar] [CrossRef]
- Szmulewicz, A.G.; Valerio, M.P.; Smith, J.M.; Samamé, C.; Martino, D.J.; Strejilevich, S.A. Neuropsychological profiles of major depressive disorder and bipolar disorder during euthymia. A systematic literature review of comparative studies. Psychiatry Res. 2017, 248, 127–133. [Google Scholar] [CrossRef]
- Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium. A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry 2012, 18, 10–38. [Google Scholar]
- Savage, J.E.; Jansen, P.R.; Stringer, S.; Watanabe, K.; Bryois, J.; De Leeuw, C.A.; Nagel, M.; Awasthi, S.; Barr, P.B.; Coleman, J.R.; et al. Genome-wide association meta-analysis in 269, 867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 2018, 50, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Smeland, O.B.; Bahrami, S.; Frei, O.; Shadrin, A.; O’Connell, K.; Savage, J.; Watanabe, K.; Krull, F.; Bettella, F.; Steen, N.E.; et al. Genome-wide analysis reveals extensive genetic overlap between schizophrenia, bipolar disorder, and intelligence. Mol. Psychiatry 2020, 25, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Barch, D.M. Neuropsychological abnormalities in schizophrenia and major mood disorders: Similarities and differences. Curr. Psychiatry Rep. 2009, 11, 313–319. [Google Scholar] [CrossRef]
- Hill, W.D.; Harris, S.E.; Deary, I.J. What genome-wide association studies reveal about the association between intelligence and mental health. Curr. Opin. Psychol. 2019, 27, 25–30. [Google Scholar] [CrossRef]
- Beck, A.T. The evolution of the cognitive model of depression and its neurobiological correlates. Am. J. Psychiatry 2008, 165, 969–977. [Google Scholar] [CrossRef]
- Werme, J.; Tissink, E.P.; de Lange, S.C.; van den Heuvel, M.P.; Posthuma, D.; de Leeuw, C.A. Local genetic correlation analysis links depression with molecular and brain imaging endophenotypes. medRxiv 2023. [Google Scholar] [CrossRef]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef]
- Demontis, D.; Walters, R.K.; Martin, J.; Mattheisen, M.; Als, T.D.; Agerbo, E.; Baldursson, G.; Belliveau, R.; Bybjerg-Grauholm, J.; Bækvad-Hansen, M.; et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 2019, 51, 63–75. [Google Scholar] [CrossRef]
- Yu, D.; Sul, J.H.; Tsetsos, F.; Nawaz, M.S.; Huang, A.Y.; Zelaya, I.; Illmann, C.; Osiecki, L.; Darrow, S.M.; Hirschtritt, M.E.; et al. Interrogating the genetic determinants of Tourette’s syndrome and other tic disorders through genome-wide association studies. Am. J. Psychiatry 2019, 176, 217–227. [Google Scholar] [CrossRef]
- Watson, H.J.; Yilmaz, Z.; Thornton, L.M.; Hübel, C.; Coleman, J.R.; Gaspar, H.A.; Bryois, J.; Hinney, A.; Leppä, V.M.; Mattheisen, M.; et al. Genome-wide association study identifies eight risk loci and implicates metabo-psychiatric origins for anorexia nervosa. Nat. Genet. 2019, 51, 1207–1214. [Google Scholar]
- Sallis, H.; Evans, J.; Wootton, R.; Krapohl, E.; Oldehinkel, A.J.; Davey Smith, G.; Paternoster, L. Genetics of depressive symptoms in adolescence. BMC Psychiatry 2017, 17, 1–8. [Google Scholar]
- Cantor, R.M.; Navarro, L.; Won, H.; Walker, R.L.; Lowe, J.K.; Geschwind, D.H. ASD restricted and repetitive behaviors associated at 17q21. 33: Genes prioritized by expression in fetal brains. Mol. Psychiatry 2018, 23, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- St Pourcain, B.; Whitehouse, A.O.; Ang, W.Q.; Warrington, N.M.; Glessner, J.T.; Wang, K.; Timpson, N.J.; Evans, D.M.; Kemp, J.P.; Ring, S.M.; et al. Common variation contributes to the genetic architecture of social communication traits. Mol. Autism 2013, 4, 1–2. [Google Scholar]
- Olff, M.; Langeland, W.; Draijer, N.; Gersons, B.P.R. Gender differences in posttraumatic stress disorder. Psychol. Bull. 2007, 133, 183–204. [Google Scholar] [CrossRef]
- Craddock, N.; Sklar, P. Genetics of bipolar disorder. Lancet 2013, 381, 1654–1662. [Google Scholar] [CrossRef]
- Song, J.; Bergen, S.E.; Kuja-Halkola, R.; Larsson, H.; Landén, M.; Lichtenstein, P. Bipolar disorder and its relation to major psychiatric disorders: A family-based study in the S wedish population. Bipolar Disord. 2015, 17, 184–193. [Google Scholar] [CrossRef]
- Kendler, K.S.; Ohlsson, H.; Sundquist, J.; Sundquist, K. An extended Swedish national adoption study of bipolar disorder illness and cross-generational familial association with schizophrenia and major depression. JAMA Psychiatry 2020, 77, 814–822. [Google Scholar]
- Yapici Eser, H.; Kacar, A.S.; Kilciksiz, C.M.; Yalcinay-Inan, M.; Ongur, D. Prevalence and Associated Features of Anxiety Disorder Comorbidity in Bipolar Disorder: A Meta-Analysis and Meta-Regression Study. Front. Psychiatry 2018, 9, 229. [Google Scholar]
- Achim, A.M.; Maziade, M.; Raymond, E.; Olivier, D.; Merette, C.; Roy, M.A. How prevalent are anxiety disorders in schizophrenia? A meta-analysis and critical review on a significant association. Schizophr. Bull. 2011, 37, 811–821. [Google Scholar] [CrossRef]
- Fuller-Thomson, E.; Carrique, L.; MacNeil, A. Generalized anxiety disorder among adults with attention deficit hyperactivity disorder. J. Affect. Disord. 2022, 299, 707–714. [Google Scholar]
- Wetherell, J.L.; Palmer, B.W.; Thorp, S.R.; Patterson, T.L.; Golshan, S.; Jeste, D.V. Anxiety symptoms and quality of life in middle-aged and older outpatients with schizophrenia and schizoaffective disorder. J. Clin. Psychiatry 2003, 64, 1476–1482. [Google Scholar] [PubMed]
- Spoorthy, M.S.; Chakrabarti, S.; Grover, S. Comorbidity of bipolar and anxiety disorders: An overview of trends in research. World J. Psychiatry 2019, 9, 7–29. [Google Scholar] [PubMed]
- Penninx, B.W.; Pine, D.S.; Holmes, E.A.; Reif, A. Anxiety disorders. Lancet 2021, 397, 914–927. [Google Scholar]
- Purves, K.L.; Coleman, J.R.I.; Meier, S.M.; Rayner, C.; Davis, K.A.S.; Cheesman, R.; Bækvad-Hansen, M.; Børglum, A.D.; Wan Cho, S.; Jürgen Deckert, J.; et al. A major role for common genetic variation in anxiety disorders. Mol. Psychiatry 2020, 25, 3292–3303. [Google Scholar]
- Ohi, K.; Otowa, T.; Shimada, M.; Sasaki, T.; Tanii, H. Shared genetic etiology between anxiety disorders and psychiatric and related intermediate phenotypes. Psychol. Med. 2020, 50, 692–704. [Google Scholar]
- Frei, O.; Holland, D.; Smeland, O.B.; Shadrin, A.A.; Fan, C.C.; Maeland, S.; O’Connell, K.S.; Wang, Y.; Djurovic, S.; Thompson, W.K.; et al. Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat. Commun. 2019, 10, 2417. [Google Scholar]
- Werme, J.; van der Sluis, S.; Posthuma, D.; de Leeuw, C.A. An integrated framework for local genetic correlation analysis. Nat. Genet. 2022, 54, 274–282. [Google Scholar]
- Andreassen, O.A.; Djurovic, S.; Thompson, W.K.; Schork, A.J.; Kendler, K.S.; O’Donovan, M.C.; Rujescu, D.; Werge, T.; van de Bunt, M.; Morris, A.P.; et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am. J. Hum. Genet. 2013, 92, 197–209. [Google Scholar]
- Smeland, O.B.; Frei, O.; Shadrin, A.; O’Connell, K.; Fan, C.C.; Bahrami, S.; Holland, D.; Djurovic, S.; Thompson, W.K.; Dale, A.M.; et al. Discovery of shared genomic loci using the conditional false discovery rate approach. Hum. Genet. 2020, 139, 85–94. [Google Scholar]
- Zhou, X.; Keitner, G.I.; Qin, B.; Ravindran, A.V.; Bauer, M.; Del Giovane, C.; Zhao, J.; Liu, Y.; Fang, Y.; Zhang, Y.; et al. Atypical antipsychotic augmentation for treatment-resistant depression: A systematic review and network meta-analysis. Int. J. Neuropsychopharmacol. 2015, 18, pyv060. [Google Scholar]
- Pain, O.; Hodgson, K.; Trubetskoy, V.; Ripke, S.; Marshe, V.S.; Adams, M.J.; Byrne, E.M.; Campos, A.I.; Carrillo-Roa, T.; Cattaneo, A.; et al. Identifying the Common Genetic Basis of Antidepressant Response. Biol. Psychiatry Glob. Open Sci. 2022, 2, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.; Crowley, J.J.; Lindefors, N.; Ljótsson, B.; Hedman-Lagerlöf, E.; Boberg, J.; El Alaoui, S.; Karlsson, R.; Lu, Y.; Mattheisen, M.; et al. Genetics of response to cognitive behavior therapy in adults with major depression: A preliminary report. Mol. Psychiatry 2019, 24, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Gilman, S.E.; Houck, P.R.; Szanto, K.; Reynolds, C.F. Socioeconomic status and anxiety as predictors of antidepressant treatment response and suicidal ideation in older adults. Soc. Psychiatry Psychiatr. Epidemiol. 2009, 44, 272–277. [Google Scholar] [CrossRef]
- Wellcome Trust Case Control, C. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef]
- Broekema, R.V.; Bakker, O.B.; Jonkers, I.H. A practical view of fine-mapping and gene prioritization in the post-genome-wide association era. Open Biol. 2020, 10, 190221. [Google Scholar] [CrossRef]
- Seifuddin, F.; Mahon, P.B.; Judy, J.; Pirooznia, M.; Jancic, D.; Taylor, J.; Goes, F.S.; Potash, J.B.; Zandi, P.P. Meta-analysis of genetic association studies on bipolar disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 508–518. [Google Scholar] [CrossRef]
- Farrell, M.S.; Werge, T.; Sklar, P.; Owen, M.J.; Ophoff, R.A.; O’Donovan, M.C.; Corvin, A.; Cichon, S.; Sullivan, P.F. Evaluating historical candidate genes for schizophrenia. Mol. Psychiatry 2015, 20, 555–562. [Google Scholar] [CrossRef]
- Lee, P.H.; Feng, Y.C.A.; Smoller, J.W. Pleiotropy and cross-disorder genetics among psychiatric disorders. Biol. Psychiatry 2021, 89, 20–31. [Google Scholar] [CrossRef]
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Hindley, G.F.L.; Frei, O.; Smeland, O.B. New insights from the last decade of research in psychiatric genetics: Discoveries, challenges and clinical implications. World Psychiatry Off. J. World Psychiatr. Assoc. (WPA) 2023, 22, 4–24. [Google Scholar] [CrossRef]
- Nagel, M.; Jansen, P.R.; Stringer, S.; Watanabe, K.; de Leeuw, C.A.; Bryois, J.; Savage, J.E.; Hammerschlag, A.R.; Skene, N.G.; Muñoz-Manchado, A.B.; et al. Meta-analysis of genome-wide association studies for neuroticism in 449, 484 individuals identifies novel genetic loci and pathways. Nat. Genet. 2018, 50, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.A.; Kumar, K.; Harvey, A.; Huguet, G.; Urchs, S.G.W.; Schultz, L.M.; Sharmarke, H.; Jizi, K.; Martin, C.O.; Younis, N.; et al. Brain functional connectivity mirrors genetic pleiotropy in psychiatric conditions. Brain A J. Neurol. 2023, 146, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Chawner, S.J.; Owen, M.J.; Holmans, P.; Raymond, F.L.; Skuse, D.; Hall, J.; van den Bree, M.B. Genotype–phenotype associations in children with copy number variants associated with high neuropsychiatric risk in the UK (IMAGINE-ID): A case-control cohort study. Lancet Psychiatry 2019, 6, 493–505. [Google Scholar] [CrossRef]
- Sha, Z.; Wager, T.D.; Mechelli, A.; He, Y. Common dysfunction of large-scale neurocognitive networks across psychiatric disorders. Biol. Psychiatry 2019, 85, 375–388. [Google Scholar] [CrossRef]
- Biswal, B.; Yetkin, F.Z.; Haughton, V.M.; Hyde, J.S. Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn. Reson. Med. 1995, 34, 537–541. [Google Scholar] [CrossRef]
- Cao, H.; Zhou, H.; Cannon, T.D. Functional connectome-wide associations of schizophrenia polygenic risk. Mol. Psychiatry 2021, 26, 2553–2561. [Google Scholar] [CrossRef]
- Vassos, E.; Di Forti, M.; Coleman, J.; Iyegbe, C.; Prata, D.; Euesden, J.; O’Reilly, P.; Curtis, C.; Kolliakou, A.; Patel, H.; et al. An examination of polygenic score risk prediction in individuals with first-episode psychosis. Biol. Psychiatry 2017, 81, 470–477. [Google Scholar] [CrossRef]
- Zilhão, N.R.; Abdellaoui, A.; Smit, D.J.A.; Cath, D.C.; Hottenga, J.J.; Boomsma, D.I. Polygenic prediction of obsessive compulsive symptoms. Mol. Psychiatry 2018, 23, 168–169. [Google Scholar] [CrossRef]
- Peyrot, W.J.; Milaneschi, Y.; Abdellaoui, A.; Sullivan, P.F.; Hottenga, J.J.; Boomsma, D.I.; Penninx, B.W. Effect of polygenic risk scores on depression in childhood trauma. Br. J. Psychiatry 2014, 205, 113–119. [Google Scholar] [CrossRef]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet 2013, 381, 1371–1379. [Google Scholar] [CrossRef]
- Amare, A.T.; Schubert, K.O.; Hou, L.; Clark, S.R.; Papiol, S.; Heilbronner, U.; Degenhardt, F.; Tekola-Ayele, F.; Hsu, Y.H.; Shekhtman, T.; et al. Association of polygenic score for schizophrenia and HLA antigen and inflammation genes with response to lithium in bipolar affective disorder: A genome-wide association study. JAMA Psychiatry 2018, 75, 65–74. [Google Scholar] [PubMed]
- Legge, S.E.; Hamshere, M.L.; Ripke, S.; Pardinas, A.F.; Goldstein, J.I.; Rees, E.; Richards, A.L.; Leonenko, G.; Jorskog, L.F.; Clozapine-Induced Agranulocytosis Consortium; et al. Genome-wide common and rare variant analysis provides novel insights into clozapine-associated neutropenia. Mol. Psychiatry 2017, 22, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Pardiñas, A.F.; Smart, S.E.; Willcocks, I.R.; Holmans, P.A.; Dennison, C.A.; Lynham, A.J.; Legge, S.E.; Baune, B.T.; Bigdeli, T.B.; Cairns, M.J.; et al. Interaction Testing and Polygenic Risk Scoring to Estimate the Association of Common Genetic Variants With Treatment Resistance in Schizophrenia. JAMA Psychiatry 2022, 79, 260–269. [Google Scholar] [CrossRef]
- Meerman, J.J.; Ter Hark, S.E.; Janzing, J.G.E.; Coenen, M.J.H. The Potential of Polygenic Risk Scores to Predict Antidepressant Treatment Response in Major Depression: A Systematic Review. J. Affect. Disord. 2022, 304, 1–11. [Google Scholar] [CrossRef]
- García-González, J.; Tansey, K.E.; Hauser, J.; Henigsberg, N.; Maier, W.; Mors, O.; Placentino, A.; Rietschel, M.; Souery, D.; Žagar, T.; et al. Pharmacogenetics of antidepressant response: A polygenic approach. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 75, 128–134. [Google Scholar] [CrossRef]
- Fabbri, C.; Hagenaars, S.P.; John, C.; Williams, A.T.; Shrine, N.; Moles, L.; Hanscombe, K.B.; Serretti, A.; Shepherd, D.J.; Free, R.C.; et al. Genetic and clinical characteristics of treatment-resistant depression using primary care records in two UK cohorts. Mol. Psychiatry 2021, 26, 3363–3373. [Google Scholar] [CrossRef]
- Li, Q.S.; Wajs, E.; Ochs-Ross, R.; Singh, J.; Drevets, W.C. Genome-wide association study and polygenic risk score analysis of esketamine treatment response. Sci. Rep. 2020, 10, 12649. [Google Scholar] [CrossRef]
- Foo, J.C.; Streit, F.; Frank, J.; Witt, S.H.; Treutlein, J.; Baune, B.T.; Moebus, S.; Jöckel, K.H.; Forstner, A.J.; et al.; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium Evidence for increased genetic risk load for major depression in patients assigned to electroconvulsive therapy. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2019, 180, 35–45. [Google Scholar] [CrossRef]
- Luykx, J.J.; Loef, D.; Lin, B.; van Diermen, L.; Nuninga, J.O.; van Exel, E.; Oudega, M.L.; Rhebergen, D.; Schouws, S.N.T.M.; van Eijndhoven, P.; et al. Interrogating Associations Between Polygenic Liabilities and Electroconvulsive Therapy Effectiveness. Biol. Psychiatry 2022, 91, 531–539. [Google Scholar] [CrossRef]
- Amare, A.T.; Schubert, K.O.; Hou, L.; Clark, S.R.; Papiol, S.; Cearns, M.; Heilbronner, U.; Degenhardt, F.; Tekola-Ayele, F.; Hsu, Y.H.; et al. Association of polygenic score for major depression with response to lithium in patients with bipolar disorder. Mol. Psychiatry 2021, 26, 2457–2470. [Google Scholar] [CrossRef]
- Schubert, K.O.; Thalamuthu, A.; Amare, A.T.; Frank, J.; Streit, F.; Adl, M.; Akula, N.; Akiyama, K.; Ardau, R.; Arias, B.; et al. Combining schizophrenia and depression polygenic risk scores improves the genetic prediction of lithium response in bipolar disorder patients. Transl. Psychiatry 2021, 11, 606. [Google Scholar] [CrossRef] [PubMed]
- Mistry, S.; Harrison, J.R.; Smith, D.J.; Escott-Price, V.; Zammit, S. The use of polygenic risk scores to identify phenotypes associated with genetic risk of bipolar disorder and depression: A systematic review. J. Affect. Disord. 2018, 234, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Rosa, M.A.; Lisanby, S.H. Somatic treatments for mood disorders. Neuropsychopharmacology 2012, 37, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Hallett, M.; Rossini, P.M.; Pascual-Leone, A. Safety of TMS Consensus Group Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clin. Neurophysiol. 2009, 120, 2008–2039. [Google Scholar] [CrossRef]
- Fertonani, A.; Rosini, S.; Cotelli, M.; Rossini, P.M.; Miniussi, C. Naming facilitation induced by transcranial direct current stimulation. Behav. Brain Res. 2010, 208, 311–318. [Google Scholar] [CrossRef]
- Loo, C.K.; Sachdev, P.; Mitchell, P.B.; Gandevia, S.C.; Malhi, G.S.; Todd, G.; Taylor, J.L. A study using transcranial magnetic stimulation to investigate motor mechanisms in psychomotor retardation in depression. Int. J. Neuropsychopharmacol. 2008, 11, 935–946. [Google Scholar] [CrossRef]
- Xia, G.; Gajwani, P.; Muzina, D.J.; Kemp, D.E.; Gao, K.; Ganocy, S.J.; Calabrese, J.R. Treatment-emergent mania in unipolar and bipolar depression: Focus on repetitive transcranial magnetic stimulation. Int. J. Neuropsychopharmacol. 2008, 11, 119–130. [Google Scholar] [CrossRef]
- Zangiabadi, N.; Ladino, L.D.; Sina, F.; Orozco-Hernández, J.P.; Carter, A.; Téllez-Zenteno, J.F. Deep Brain Stimulation and Drug-Resistant Epilepsy: A Review of the Literature. Front. Neurol. 2019, 10, 601. [Google Scholar] [CrossRef]
- Milev, R.V.; Giacobbe, P.; Kennedy, S.H.; Blumberger, D.M.; Daskalakis, Z.J.; Downar, J.; Modirrousta, M.; Patry, S.; Vila-Rodriguez, F.; Lam, R.W.; et al. Canadian network for mood and anxiety treatments (CANMAT) 2016 clinical guidelines for the management of adults with major depressive disorder: Section 4: Neurostimulation treatments. Can. J. Psychiatry 2016, 61, 561–575. [Google Scholar] [CrossRef]
- Brunoni, A.R.; Chaimani, A.; Moffa, A.H.; Razza, L.B.; Gattaz, W.F.; Daskalakis, Z.J.; Carvalho, A.F. Repetitive transcranial magnetic stimulation for the acute treatment of major depressive episodes: A systematic review with network meta-analysis. JAMA Psychiatry 2017, 74, 143–152. [Google Scholar] [CrossRef]
- Bares, M.; Kopecek, M.; Novak, T.; Stopkova, P.; Sos, P.; Kozeny, J.; Brunovsky, M.; Höschl, C. Low frequency (1-Hz), right prefrontal repetitive transcranial magnetic stimulation (rTMS) compared with venlafaxine ER in the treatment of resistant depression: A double-blind, single-centre, randomized study. J. Affect. Disord. 2009, 118, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Brunelin, J.; Jalenques, I.; Trojak, B.; Attal, J.; Szekely, D.; Gay, A.; Januel, D.; Haffen, E.; Schott-Pethelaz, A.M.; Brault, C.; et al. The efficacy and safety of low frequency repetitive transcranial magnetic stimulation for treatment-resistant depression: The results from a large multicenter French RCT. Brain Stimul. 2014, 7, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Berlim, M.T.; Van den Eynde, F.; Daskalakis, Z.J. High-frequency repetitive transcranial magnetic stimulation accelerates and enhances the clinical response to antidepressants in major depression: A meta-analysis of randomized, double-blind, and sham-controlled trials. J. Clin. Psychiatry 2013, 74, e122-9. [Google Scholar] [CrossRef] [PubMed]
- McGirr, A.; Karmani, S.; Arsappa, R.; Berlim, M.T.; Thirthalli, J.; Muralidharan, K.; Yatham, L.N. Clinical efficacy and safety of repetitive transcranial magnetic stimulation in acute bipolar depression. World Psychiatry 2016, 15, 85–86. [Google Scholar] [CrossRef]
- Luber, B.M.; Davis, S.; Bernhardt, E.; Neacsiu, A.; Kwapil, L.; Lisanby, S.H.; Strauman, T.J. Using neuroimaging to individualize TMS treatment for depression: Toward a new paradigm for imaging-guided intervention. Neuroimage 2017, 148, 1–7. [Google Scholar] [CrossRef]
- Martin, D.M.; McClintock, S.M.; Forster, J.J.; Lo, T.Y.; Loo, C.K. Cognitive enhancing effects of rTMS administered to the prefrontal cortex in patients with depression: A systematic review and meta-analysis of individual task effects. Depress. Anxiety 2017, 34, 1029–1039. [Google Scholar] [CrossRef]
- Cao, X.; Deng, C.; Su, X.; Guo, Y. Response and Remission Rates Following High-Frequency vs. Low-Frequency Repetitive Transcranial Magnetic Stimulation (rTMS) Over Right DLPFC for Treating Major Depressive Disorder (MDD): A Meta-Analysis of Randomized, Double-Blind Trials. Front. Psychiatry 2018, 9, 413. [Google Scholar] [CrossRef]
- Shi, C.; Yu, X.; Cheung, E.F.; Shum, D.H.; Chan, R.C. Revisiting the therapeutic effect of rTMS on negative symptoms in schizophrenia: A meta-analysis. Psychiatry Res. 2014, 215, 505–513. [Google Scholar] [CrossRef]
- Slotema, C.W.; Blom, J.D.; van Lutterveld, R.; Hoek, H.W.; Sommer, I.E. Review of the efficacy of transcranial magnetic stimulation for auditory verbal hallucinations. Biol. Psychiatry 2014, 76, 101–110. [Google Scholar] [CrossRef]
- Wobrock, T.; Guse, B.; Cordes, J.; Wolwer, W.; Winterer, G.; Gaebel, W.; Langguth, B.; Landgrebe, M.; Eichhammer, P.; Frank, E.; et al. Left prefrontal high-frequency repetitive transcranial magnetic stimulation for the treatment of schizophrenia with predominant negative symptoms: A sham-controlled, randomized multicenter trial. Biol. Psychiatry 2015, 77, 979–988. [Google Scholar] [CrossRef]
- Dilkov, D.; Hawken, E.R.; Kaludiev, E.; Milev, R. Repetitive transcranial magnetic stimulation of the right dorsal lateral prefrontal cortex in the treatment of generalized anxiety disorder: A randomized, double-blind sham controlled clinical trial. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 78, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Aly, M.; Dagan, Y.; Allart, A.; Lisanby, S.H. Randomized sham controlled trial of repetitive transcranial magnetic stimulation to the dorsolateral prefrontal cortex for the treatment of panic disorder with comorbid major depression. J. Affect. Disord. 2013, 144, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Boggio, P.S.; Rocha, M.; Oliveira, M.O.; Fecteau, S.; Cohen, R.B.; Campanha, C.; Ferreira-Santos, E.; Meleiro, A.; Corchs, F.; Zaghi, S.; et al. Noninvasive brain stimulation with high-frequency and low-intensity repetitive transcranial magnetic stimulation treatment for posttraumatic stress disorder. J. Clin. Psychiatry 2010, 71, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.J.; Antal, A.; Bikson, M.; Boggio, P.S.; Brunoni, A.R.; Celnik, P.; Cohen, L.G.; Fregni, F.; Herrmann, C.S.; Kappenman, E.S.; et al. A technical guide to tDCS, and related non-invasive brain stimulation tools. Clin. Neurophysiol. 2016, 127, 1031–1048. [Google Scholar] [CrossRef]
- Brunoni, A.R.; Nitsche, M.A.; Bolognini, N.; Bikson, M.; Wagner, T.; Merabet, L.; Edwards, D.J.; Valero-Cabre, A.; Rotenberg, A.; Pascual-Leone, A.; et al. Clinical research with transcranial direct current stimulation (tDCS): Challenges and future directions. Brain Stimul. 2012, 5, 175–195. [Google Scholar] [CrossRef]
- Batsikadze, G.; Moliadze, V.; Paulus, W.; Kuo, M.F.; Nitsche, M.A. Partially non-linear stimulation intensity-dependent effects of direct current stimulation on motor cortex excitability in humans. J. Physiol. 2013, 591, 1987–2000. [Google Scholar] [CrossRef]
- Monte-Silva, K.; Kuo, M.F.; Hessenthaler, S.; Fresnoza, S.; Liebetanz, D.; Paulus, W.; Nitsche, M.A. Induction of late LTP-like plasticity in the human motor cortex by repeated non-invasive brain stimulation. Brain Stimul. 2013, 6, 424–432. [Google Scholar] [CrossRef]
- Nitsche, M.A.; Paulus, W. Excitability changes induced in the human motor cortex by weak transcranial direct current stimulation. J. Physiol. 2000, 527, 633–639. [Google Scholar] [CrossRef]
- Nitsche, M.A.; Paulus, W. Sustained excitability elevations induced by transcranial DC motor cortex stimulation in humans. Neurology 2001, 57, 1899–1901. [Google Scholar] [CrossRef]
- Brunoni, A.R.; Amadera, J.; Berbel, B.; Volz, M.S.; Rizzerio, B.G.; Fregni, F. A systematic review on reporting and assessment of adverse effects associated with transcranial direct current stimulation. Int. J. Neuropsychopharmacol. 2011, 14, 1133–1145. [Google Scholar] [CrossRef]
- Ezquerro, F.; Moffa, A.H.; Bikson, M.; Khadka, N.; Aparicio, L.V.; de Sampaio-Junior, B.; Fregni, F.; Bensenor, I.M.; Lotufo, P.A.; Pereira, A.C.; et al. The influence of skin redness on blinding in transcranial direct current stimulation studies: A crossover trial. Neuromodulation 2017, 20, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Brunoni, A.R.; Valiengo, L.; Baccaro, A.; Zanao, T.A.; de Oliveira, J.F.; Goulart, A.; Boggio, P.S.; Lotufo, P.A.; Benseñor, I.M.; Fregni, F.; et al. The sertraline vs. electrical current therapy for treating depression clinical study: Results from a factorial, randomized, controlled trial. JAMA Psychiatry 2013, 70, 383–391. [Google Scholar] [PubMed]
- Brunoni, A.R.; Moffa, A.H.; Sampaio-Junior, B.; Borrione, L.; Moreno, M.L.; Fernandes, R.A.; Veronezi, B.P.; Nogueira, B.S.; Aparicio, L.V.M.; Razza, L.B.; et al. Trial of electrical direct-current therapy versus escitalopram for depression. N. Engl. J. Med. 2017, 376, 2523–2533. [Google Scholar] [PubMed]
- Brunelin, J.; Mondino, M.; Gassab, L.; Haesebaert, F.; Gaha, L.; Suaud-Chagny, M.F.; Saoud, M.; Mechri, A.; Poulet, E. Examining transcranial direct-current stimulation (tDCS) as a treatment for hallucinations in schizophrenia. Am. J. Psychiatry 2012, 169, 719–724. [Google Scholar] [CrossRef]
- Mondino, M.; Jardri, R.; Suaud-Chagny, M.F.; Saoud, M.; Poulet, E.; Brunelin, J. Effects of fronto-temporal transcranial direct current stimulation on auditory verbal hallucinations and resting-state functional connectivity of the left temporo-parietal junction in patients with schizophrenia. Schizophr. Bull. 2016, 42, 318–326. [Google Scholar] [CrossRef]
- Boggio, P.S.; Liguori, P.; Sultani, N.; Rezende, L.; Fecteau, S.; Fregni, F. Cumulative priming effects of cortical stimulation on smoking cue-induced craving. Neurosci. Lett. 2009, 463, 82–86. [Google Scholar]
- Fecteau, S.; Agosta, S.; Hone-Blanchet, A.; Fregni, F.; Boggio, P.; Ciraulo, D.; Pascual-Leone, A. Modulation of smoking and decision-making behaviors with transcranial direct current stimulation in tobacco smokers: A preliminary study. Drug Alcohol. Depend. 2014, 140, 78–84. [Google Scholar]
- Batista, E.K.; Klauss, J.; Fregni, F.; Nitsche, M.A.; Nakamura-Palacios, E.M. A randomized placebo-controlled trial of targeted prefrontal cortex modulation with bilateral tDCS in patients with crack-cocaine dependence. Int J Neuropsychopharmacol. 2015, 18, pyv066. [Google Scholar] [CrossRef]
- Klauss, J.; Penido Pinheiro, L.C.; Silva Merlo, B.L.; de Almeida Correia Santos, G.; Fregni, F.; Nitsche, M.A.; Miyuki Nakamura-Palacios, E. A randomized controlled trial of targeted prefrontal cortex modulation with tDCS in patients with alcohol dependence. Int. J. Neuropsychopharmacol. 2014, 17, 1793–1803. [Google Scholar] [CrossRef]
- Kritzer, M.D.; Peterchev, A.V.; Camprodon, J.A. Electroconvulsive Therapy: Mechanisms of Action, Clinical Considerations, and Future Directions. Harv. Rev. Psychiatry 2023, 31, 101–113. [Google Scholar] [CrossRef]
- Bauer, M.; Pfennig, A.; Severus, E.; Whybrow, P.C.; Angst, J.; Möller, H.J. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of unipolar depressive disorders part 1: Update 2013 on the acute continuation treatment of unipolar depressive disorders. World J. Biol. Psychiatry 2013, 14, 334–385. [Google Scholar] [PubMed]
- Zielinski, R.J.; Roose, S.P.; Devanand, D.P.; Woodring, S.; Sackeim, H.A. Cardiovascular complications of ECT in depressed patients with cardiac disease. Am. J. Psychiatry 1993, 150, 904–909. [Google Scholar] [PubMed]
- Dominiak, M.; Antosik-Wójcińska, A.Z.; Goetz, Z.; Sikorska, O.; Stefanowski, B.; Gorostiza, D.; Święcicki, Ł. Efficacy, safety and tolerability of formula-based unilateral vs bilateral electroconvulsive therapy in the treatment of major depression: A randomized open label controlled trial. J. Psychiatr. Res. 2021, 133, 52–59. [Google Scholar] [PubMed]
- Dong, M.; Zhu, X.M.; Zheng, W.; Li, X.H.; Ng, C.H.; Ungvari, G.S.; Xiang, Y.T. Electroconvulsive therapy for older adult patients with major depressive disorder: A systematic review of randomized controlled trials. Psychogeriatrics 2018, 18, 468–475. [Google Scholar]
- Sienaert, P.; Vansteelandt, K.; Demyttenaere, K.; Peuskens, J. Randomized comparison of ultra-brief bifrontal and unilateral electroconvulsive therapy for major depression: Clinical efficacy. J. Affect. Disord. 2009, 116, 106–112. [Google Scholar]
- Sienaert, P.; Vansteelandt, K.; Demyttenaere, K.; Peuskens, J. Randomized comparison of ultra-brief bifrontal and unilateral electroconvulsive therapy for major depression: Cognitive side-effects. J. Affect. Disord. 2010, 122, 60–67. [Google Scholar]
- Bodnar, A.; Krzywotulski, M.; Lewandowska, A.; Chlopocka-Wozniak, M.; Bartkowska-Sniatkowska, A.; Michalak, M.; Rybakowski, J.K. Electroconvulsive therapy and cognitive functions in drug-resistant depression. World J. Biol. Psychiatry 2016, 17, 159–164. [Google Scholar]
- Trifu, S.; Sevcenco, A.; Stănescu, M.; Drăgoi, A.M.; Cristea, M.B. Efficacy of electroconvulsive therapy as a potential first-choice treatment in treatment-resistant depression (Review). Exp. Ther. Med. 2021, 22, 1281. [Google Scholar]
- van Duist, M.; Spaans, H.P.; Verwijk, E.; Kok, R.M. ECT nonremitters: Prognosis and treatment after 12 unilateral electroconvulsive therapy sessions for major depression. J. Affect. Disord. 2020, 272, 501–507. [Google Scholar]
- Petrides, G.; Malur, C.; Braga, R.J.; Bailine, S.H.; Schooler, N.R.; Malhotra, A.K.; Kane, J.M.; Sanghani, S.; Goldberg, T.E.; John, M. Electroconvulsive therapy augmentation in clozapine-resistant schizophrenia: A prospective, randomized study. Am. J. Psychiatry 2015, 172, 52–58. [Google Scholar] [CrossRef]
- Shelef, A.; Mazeh, D.; Berger, U.; Baruch, Y.; Barak, Y. Acute electroconvulsive therapy followed by maintenance electroconvulsive therapy decreases hospital re-admission rates of older patients with severe mental illness. J. ECT 2015, 31, 125–128. [Google Scholar] [CrossRef]
- Kristensen, D.; Bauer, J.; Hageman, I.; Jørgensen, M.B. Electroconvulsive therapy for treating schizophrenia: A chart review of patients from two catchment areas. Eur. Arch. Psychiatry Clin. Neurosci. 2011, 261, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Chanpattana, W.; Chakrabhand, M.L.; Sackeim, H.A.; Kitaroonchai, W.; Kongsakon, R.; Techakasem, P.; Buppanharun, W.; Tuntirungsee, Y.; Kirdcharoen, N. Continuation ECT in treatment-resistant schizophrenia: A controlled study. J. ECT 1999, 15, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Henderson, T.A.; van Lierop, M.J.; McLean, M.; Uszler, J.M.; Thornton, J.F.; Siow, Y.H.; Pavel, D.G.; Cardaci, J.; Cohen, P. Functional Neuroimaging in Psychiatry-Aiding in Diagnosis and Guiding Treatment. What the American Psychiatric Association Does Not Know. Front. Psychiatry 2020, 11, 276. [Google Scholar] [CrossRef] [PubMed]
- Cumming, P.; Abi-Dargham, A.; Gründer, G. Molecular imaging of schizophrenia: Neurochemical findings in a heterogeneous and evolving disorder. Behav. Brain Res. 2021, 398, 113004. [Google Scholar] [CrossRef]
- Clark, C.M.; Kessler, R.; Buchsbaum, M.S.; Margolin, R.A.; Holcomb, H.H. Correlational methods for determining regional coupling of cerebral glucose metabolism: A pilot study. Biol. Psychiatry 1984, 19, 663–678. [Google Scholar]
- Katz, M.; Buchsbaum, M.S.; Siegel, B.V., Jr.; Wu, J.; Haier, R.J.; Bunney, W.E., Jr. Correlational patterns of cerebral glucose metabolism in never-medicated schizophrenics. Neuropsychobiology 1996, 33, 1–11. [Google Scholar] [CrossRef]
- Siegel, B.V., Jr.; Buchsbaum, M.S.; Najafi, A.; Wu, M. Activity in 70 Unmedicated Male Schizophrenic Patients. Am. J. Psychiatry 1993, 1, 1325. [Google Scholar]
- Potkin, S.G.; Alva, G.; Fleming, K.; Anand, R.; Keator, D.; Carreon, D.; Doo, M.; Jin, Y.; Wu, J.C.; Fallon, J.H.; et al. A PET study of the pathophysiology of negative symptoms in schizophrenia. Am. J. Psychiatry 2002, 159, 227–237. [Google Scholar] [CrossRef]
- Kumakura, Y.; Cumming, P.; Vernaleken, I.; Buchholz, H.G.; Siessmeier, T.; Heinz, A.; Kienast, T.; Bartenstein, P.; Gründer, G. Elevated [18F] fluorodopamine turnover in brain of patients with schizophrenia: An [18F] fluorodopa/positron emission tomography study. J. Neurosci. 2007, 27, 8080–8087. [Google Scholar] [CrossRef]
- Vernaleken, I.; Kumakura, Y.; Cumming, P.; Buchholz, H.G.; Siessmeier, T.; Stoeter, P.; Müller, M.J.; Bartenstein, P.; Gründer, G. Modulation of [18F] fluorodopa (FDOPA) kinetics in the brain of healthy volunteers after acute haloperidol challenge. Neuroimage 2006, 30, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Gründer, G.; Vernaleken, I.; Müller, M.J.; Davids, E.; Heydari, N.; Buchholz, H.G.; Bartenstein, P.; Munk, O.L.; Stoeter, P.; Wong, D.F.; et al. Subchronic haloperidol downregulates dopamine synthesis capacity in the brain of schizophrenic patients in vivo. Neuropsychopharmacology 2003, 28, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Grace, A.A.; Bunney, B.S.; Moore, H.; Todd, C.L. Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci. 1997, 20, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Avram, M.; Brandl, F.; Cabello, J.; Leucht, C.; Scherr, M.; Mustafa, M.; Leucht, S.; Ziegler, S.; Sorg, C. Reduced striatal dopamine synthesis capacity in patients with schizophrenia during remission of positive symptoms. Brain 2019, 142, 1813–1826. [Google Scholar] [CrossRef]
- Demjaha, A.; Murray, R.M.; McGuire, P.K.; Kapur, S.; Howes, O.D. Dopamine synthesis capacity in patients with treatment-resistant schizophrenia. Am. J. Psychiatry 2012, 169, 1203–1210. [Google Scholar] [CrossRef]
- Kim, E.; Howes, O.D.; Veronese, M.; Beck, K.; Seo, S.; Park, J.W.; Lee, J.S.; Lee, Y.S.; Kwon, J.S. Presynaptic dopamine capacity in patients with treatment-resistant schizophrenia taking clozapine: An [18F] DOPA PET study. Neuropsychopharmacology 2017, 42, 941–950. [Google Scholar] [CrossRef]
- Veronese, M.; Santangelo, B.; Jauhar, S.; D’Ambrosio, E.; Demjaha, A.; Salimbeni, H.; Huajie, J.; McCrone, P.; Turkheimer, F.; Howes, O. A potential biomarker for treatment stratification in psychosis: Evaluation of an [18F] FDOPA PET imaging approach. Neuropsychopharmacology 2021, 46, 1122–1132. [Google Scholar] [CrossRef]
- Marotta, G.; Delvecchio, G.; Pigoni, A.; Mandolini, G.; Ciappolino, V.; Oldani, L.; Madonna, D.; Grottaroli, M.; Altamura, A.C.; Brambilla, P. The metabolic basis of psychosis in bipolar disorder: A positron emission tomography study. Bipolar Disord. 2019, 21, 151–158. [Google Scholar] [CrossRef]
- Bench, C.J.; Friston, K.J.; Brown, R.G.; Scott, L.C.; Frackowiak, R.S.; Dolan, R.J. The anatomy of melancholia–focal abnormalities of cerebral blood flow in major depression. Psychol. Med. 1992, 22, 607–615. [Google Scholar] [CrossRef]
- Dolan, R.J.; Bench, C.J.; Brown, R.G.; Scott, L.C.; Friston, K.J.; Frackowiak, R.S. Regional cerebral blood flow abnormalities in depressed patients with cognitive impairment. J. Neurol. Neurosurg. Psychiatry 1992, 55, 768–773. [Google Scholar] [CrossRef]
- Kumar, A.; Newberg, A.; Alavi, A.; Berlin, J.; Smith, R.; Reivich, M. Regional cerebral glucose metabolism in late-life depression and Alzheimer disease: A preliminary positron emission tomography study. Proc. Natl. Acad. Sci. USA 1993, 90, 7019–7023. [Google Scholar] [CrossRef] [PubMed]
- Newberg, A.B.; Moss, A.S.; Monti, D.A.; Alavi, A. Positron emission tomography in psychiatric disorders. Ann. N. Y. Acad. Sci. 2011, 1228, E13–E25. [Google Scholar] [CrossRef] [PubMed]
- Deschwanden, A.; Karolewicz, B.; Feyissa, A.M.; Treyer, V.; Ametamey, S.M.; Johayem, A.; Burger, C.; Auberson, Y.P.; Sovago, J.; Stockmeier, C.A.; et al. Reduced metabotropic glutamate receptor 5 density in major depression determined by [11C] ABP688 PET and postmortem study. Am. J. Psychiatry 2011, 168, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Martinot, M.L.; Bragulat, V.; Artiges, E.; Dollé, F.; Hinnen, F.; Jouvent, R.; Martinot, J. Decreased presynaptic dopamine function in the left caudate of depressed patients with affective flattening and psychomotor retardation. Am. J. Psychiatry 2001, 158, 314–316. [Google Scholar] [CrossRef]
- Meyer, J.H.; Kapur, S.; Eisfeld, B.; Brown, G.M.; Houle, S.; DaSilva, J.; Wilson, A.A.; Rafi-Tari, S.; Mayberg, H.S.; Kennedy, S.H.; et al. The effect of paroxetine on 5-HT2A receptors in depression: An [18F] setoperone PET imaging study. Am. J. Psychiatry 2001, 158, 78–85. [Google Scholar] [CrossRef]
- Zanardi, R.; Artigas, F.; Moresco, R.; Colombo, C.; Messa, C.; Gobbo, C.; Smeraldi, E.; Fazio, F. Increased 5-hydroxytryptamine-2 receptor binding in the frontal cortex of depressed patients responding to paroxetine treatment: A positron emission tomography scan study. J. Clin. Psychopharmacol. 2001, 21, 53–58. [Google Scholar] [CrossRef]
- Pardo, J.V.; Sheikh, S.A.; Schwindt, G.; Lee, J.T.; Adson, D.E.; Rittberg, B.; Abuzzahab, F.S., Sr. A preliminary study of resting brain metabolism in treatment-resistant depression before and after treatment with olanzapine-fluoxetine combination. PLoS ONE 2020, 15, e0226486. [Google Scholar] [CrossRef]
- Tiger, M.; Veldman, E.R.; Ekman, C.J.; Halldin, C.; Svenningsson, P.; Lundberg, J. A randomized placebo-controlled PET study of ketamine’ s effect on serotonin1B receptor binding in patients with SSRI-resistant depression. Transl. Psychiatry 2020, 10, 159. [Google Scholar] [CrossRef]
- Saggar, M.; Uddin, L.Q. Pushing the boundaries of psychiatric neuroimaging to ground diagnosis in biology. ENeuro 2019, 6, 1–8. [Google Scholar] [CrossRef]
- Collin, G.; Scholtens, L.H.; Kahn, R.S.; Hillegers, M.H.; van den Heuvel, M.P. Affected anatomical rich club and structural–functional coupling in young offspring of schizophrenia and bipolar disorder patients. Biol. Psychiatry 2017, 82, 746–755. [Google Scholar] [CrossRef]
- Albrecht, D.S.; Kim, M.; Akeju, O.; Torrado-Carvajal, A.; Edwards, R.R.; Zhang, Y.; Bergan, C.; Protsenko, E.; Kucyi, A.; Wasan, A.D.; et al. The neuroinflammatory component of negative affect in patients with chronic pain. Mol. Psychiatry 2021, 26, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gong, L.; He, C.; Wang, Q.; Ren, Q.; Xie, C. Alzheimer’s Disease Neuroimaging Initiative. Default mode network connectivity moderates the relationship between the APOE genotype and cognition and individualizes identification across the Alzheimer’s disease spectrum. J. Alzheimer’s Dis. 2019, 70, 843–860. [Google Scholar] [CrossRef] [PubMed]
- Whitfield-Gabrieli, S.; Ghosh, S.S.; Nieto-Castanon, A.; Saygin, Z.; Doehrmann, O.; Chai, X.J.; Reynolds, G.O.; Hofmann, S.G.; Pollack, M.H.; Gabrieli, J.D. Brain connectomics predict response to treatment in social anxiety disorder. Mol. Psychiatry 2016, 21, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Hojjati, S.H.; Ebrahimzadeh, A.; Babajani-Feremi, A. Identification of the early stage of Alzheimer’s disease using structural MRI and resting-state fMRI. Front. Neurol. 2019, 10, 904. [Google Scholar] [CrossRef]
- Meyer-Lindenberg, A. Neural connectivity as an intermediate phenotype: Brain networks under genetic control. Hum. Brain Mapp. 2009, 30, 1938–1946. [Google Scholar] [CrossRef]
- Shenton, M.E.; Dickey, C.C.; Frumin, M.; McCarley, R.W. A review of MRI findings in schizophrenia. Schizophr. Res. 2001, 49, 1–52. [Google Scholar] [CrossRef]
- McCarley, R.W.; Wible, C.G.; Frumin, M.; Hirayasu, Y.; Levitt, J.J.; Fischer, I.A.; Shenton, M.E. MRI anatomy of schizophrenia. Biol. Psychiatry 1999, 45, 1099–1119. [Google Scholar] [CrossRef]
- Duarte, J.M.; Xin, L. Magnetic resonance spectroscopy in schizophrenia: Evidence for glutamatergic dysfunction and impaired energy metabolism. Neurochem. Res. 2019, 44, 102–116. [Google Scholar] [CrossRef]
- Bora, E.; Harrison, B.J.; Davey, C.G.; Yücel, M.; Pantelis, C. Meta-analysis of volumetric abnormalities in cortico-striatal-pallidal-thalamic circuits in major depressive disorder. Psychol. Med. 2012, 42, 671–681. [Google Scholar] [CrossRef]
- Kim, M.J.; Hamilton, J.P.; Gotlib, I.H. Reduced caudate gray matter volume in women with major depressive disorder. Psychiatry Res. Neuroimaging 2008, 164, 114–122. [Google Scholar] [CrossRef]
- Khundakar, A.; Morris, C.; Oakley, A.; Thomas, A.J. Morphometric analysis of neuronal and glial cell pathology in the caudate nucleus in late-life depression. Am. J. Geriatr. Psychiatry 2011, 19, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Pillay, S.S.; Renshaw, P.F.; Bonello, C.M.; Lafer, B.; Fava, M.; Yurgelun-Todd, D. A quantitative magnetic resonance imaging study of caudate and lenticular nucleus gray matter volume in primary unipolar major depression: Relationship to treatment response and clinical severity. Psychiatry Res. Neuroimaging 1998, 84, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Pizzagalli, D.A.; Holmes, A.J.; Dillon, D.G.; Goetz, E.L.; Birk, J.L.; Bogdan, R.; Dougherty, D.D.; Iosifescu, D.V.; Rauch, S.L.; Fava, M. Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am. J. Psychiatry 2009, 166, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Perlman, K.; Benrimoh, D.; Israel, S.; Rollins, C.; Brown, E.; Tunteng, J.F.; You, R.; You, E.; Tanguay-Sela, M.; Snook, E.; et al. A systematic meta-review of predictors of antidepressant treatment outcome in major depressive disorder. J. Affect. Disord. 2019, 243, 503–515. [Google Scholar] [CrossRef]
- Willner, P.; Scheel-Krüger, J.; Belzung, C. The neurobiology of depression and antidepressant action. Neurosci. Biobehav. Rev. 2013, 37, 2331–2371. [Google Scholar] [CrossRef]
- Doi, T.; Fan, Y.; Gold, J.I.; Ding, L. The caudate nucleus contributes causally to decisions that balance reward and uncertain visual information. Elife 2020, 9, e56694. [Google Scholar] [CrossRef]
- Satterthwaite, T.D.; Kable, J.W.; Vandekar, L.; Katchmar, N.; Bassett, D.S.; Baldassano, C.F.; Ruparel, K.; Elliott, M.A.; Sheline, Y.I.; Gur, R.C.; et al. Common and dissociable dysfunction of the reward system in bipolar and unipolar depression. Neuropsychopharmacology 2015, 40, 2258–2268. [Google Scholar] [CrossRef]
- Peciña, M.; Sikora, M.; Avery, E.T.; Heffernan, J.; Peciña, S.; Mickey, B.J.; Zubieta, J.K. Striatal dopamine D2/3 receptor-mediated neurotransmission in major depression: Implications for anhedonia, anxiety and treatment response. Eur. Neuropsychopharmacol. 2017, 27, 977–986. [Google Scholar] [CrossRef]
- Whitfield-Gabrieli, S.; Ford, J.M. Default mode network activity and connectivity in psychopathology. Annu. Rev. Clin. Psychol. 2012, 8, 49–76. [Google Scholar] [CrossRef]
- Menon, V. Large-scale brain networks and psychopathology: A unifying triple network model. Trends Cogn. Sci. 2011, 15, 483–506. [Google Scholar] [CrossRef]
- Shulman, G.L.; Fiez, J.A.; Corbetta, M.; Buckner, R.L.; Miezin, F.M.; Raichle, M.E.; Peterson, S.E. Common blood flow changes across visual tasks: II. Decreases in cerebral cortex. J. Cogn. Neurosci. 1997, 9, 648–663. [Google Scholar] [CrossRef] [PubMed]
- Raichle, M.E.; MacLeod, A.M.; Snyder, A.Z.; Powers, W.J.; Gusnard, D.A.; Shulman, G.L. A default mode of brain function. Proc. Natl. Acad. Sci. USA 2001, 98, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Weissman, D.H.; Roberts, K.C.; Visscher, K.M.; Woldorff, M.G. The neural bases of momentary lapses in attention. Nat. Neurosci. 2006, 9, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.; Deckersbach, T.; Dougherty, D.D.; Hooley, J.M. The default mode network and rumination in individuals at risk for depression. Soc. Cogn. Affect. Neurosci. 2023, 18, nsad032. [Google Scholar] [CrossRef]
- Buckner, R.L.; Snyder, A.Z.; Shannon, B.J.; LaRossa, G.; Sachs, R.; Fotenos, A.F.; Sheline, Y.I.; Klunk, W.E.; Mathis, C.A.; Morris, J.C.; et al. Molecular, structural, and functional characterization of Alzheimer’s disease: Evidence for a relationship between default activity, amyloid, and memory. J. Neurosci. 2005, 25, 7709–7717. [Google Scholar] [CrossRef]
- Greicius, M.D.; Srivastava, G.; Reiss, A.L.; Menon, V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: Evidence from functional MRI. Proc. Natl. Acad. Sci. USA 2004, 101, 4637–4642. [Google Scholar] [CrossRef]
- Spreng, R.N.; Mar, R.A.; Kim, A.S. The common neural basis of autobiographical memory, prospection, navigation, theory of mind, and the default mode: A quantitative meta-analysis. J. Cogn. Neurosci. 2009, 21, 489–510. [Google Scholar]
- Cohen, R.M.; Weingartner, H.; Smallberg, S.A.; Pickar, D.; Murphy, D.L. Effort and cognition in depression. Arch. Gen. Psychiatry 1982, 39, 593–597. [Google Scholar] [CrossRef]
- Frith, C.D. The Cognitive Neuropsychology of Schizophrenia; Psychology Press: East Sussex, UK, 2014. [Google Scholar]
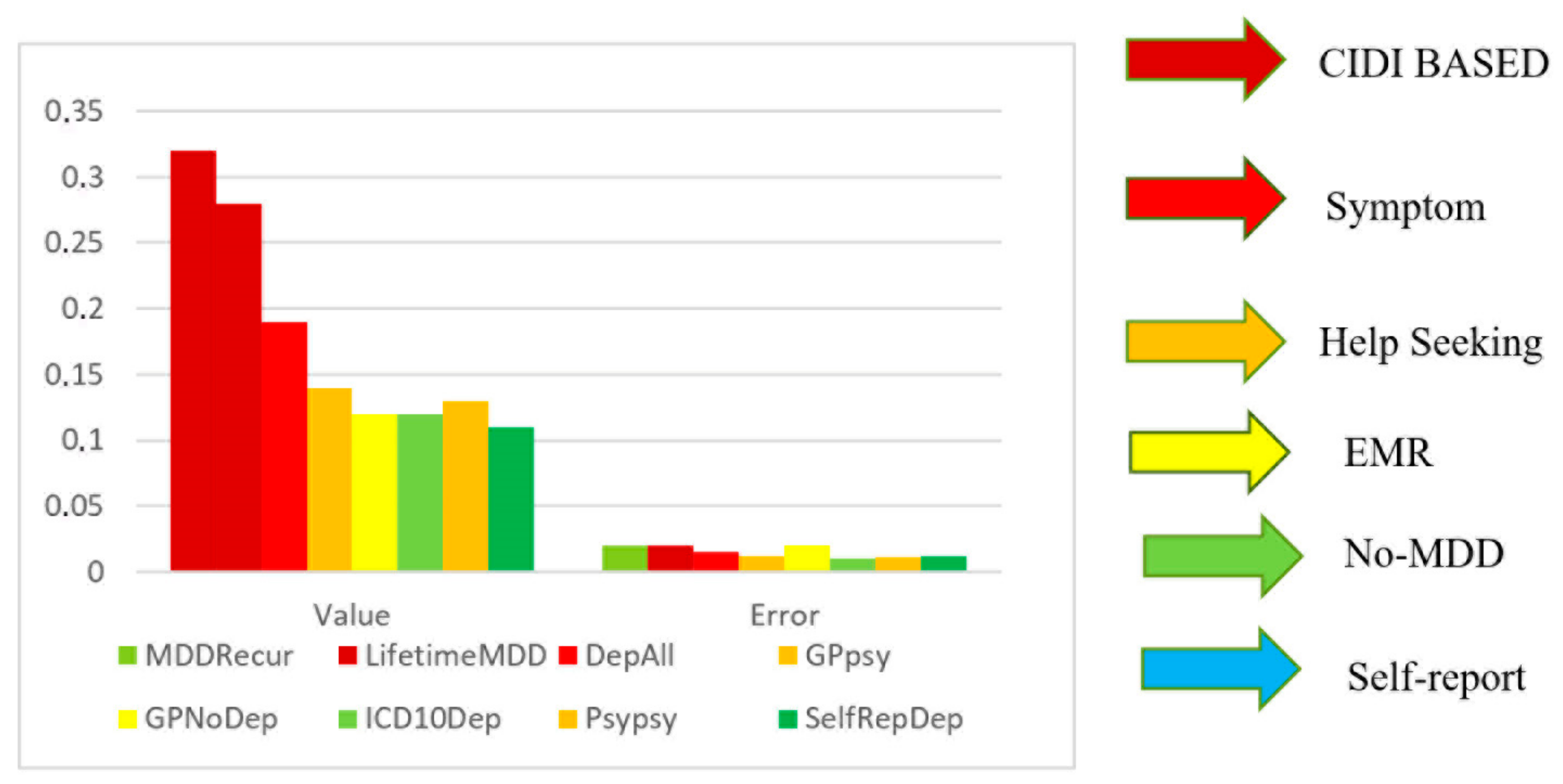



{kind=link}
{kind=link}
{kind=link}
| Twin Pair | Twin 1 Neuroticism | Twin 1 Generalized Anxiety Disorder | Twin 2 Neuroticism | Twin 2 Generalized Anxiety Disorder |
|---|---|---|---|---|
| Male–male | ||||
| Twin 1: neuroticism | 0.318 | 0.358 | 0.206 | |
| Twin 1: generalized anxiety disorder | 0.394 | 0.174 | 0.128 | |
| Twin 2: neuroticism | 0.207 | 0.074 | 0.394 | |
| Twin 2: generalized anxiety disorder | 0.064 | 0.093 | 0.316 | |
| Female–female | ||||
| Twin 1: neuroticism | 0.569 | 0.303 | 0.327 | |
| Twin 1: generalized anxiety disorder | 0.195 | 0.214 | 0.150 | |
| Twin 2: neuroticism | 0.119 | 0.142 | 0.462 | |
| Twin 2: generalized anxiety disorder | 0.229 | 0.186 | 0.468 | |
| Opposite-sex | ||||
| Twin 1: neuroticism | ||||
| Twin 1: generalized anxiety disorder | 0.339 | |||
| Twin 2: neuroticism | 0.113 | 0.095 | ||
| Twin 2: generalized anxiety disorder | 0.038 | 0.059 | 0.211 | |
| Gene Symbol | Gene Description | Chromosome | Functions |
|---|---|---|---|
| APOE | Apolipoprotein E | 19 | A main apoprotein of the chylomicron that binds to a specific receptor on liver cells and peripheral cells; essential for the normal catabolism of triglyceride-rich lipoprotein constituents |
| CFTR | Cystic fibrosis transmembrane conductance regulator (ATP-binding cassette subfamily C, member 7) | 7 | Involved in multidrug resistance; functions as a chloride channel and controls the regulation of other transport pathways |
| CNTNAP2 | Contactin-associated protein-like 2 | 7 | A member of the neurexin family; functions in the vertebrate nervous system as a cell adhesion molecule and receptor |
| COMT | Catechol-O-methyltransferase | 22 | Catecholamine neurotransmitter metabolism; VCFS region (22q deletion syndrome) |
| DISC1 | Disrupted in schizophrenia 1 | 1 | Neurite outgrowth and cortical development; disrupted in t(1;11)(q42.1;q14.3) |
| DRD2 | Dopamine receptor D2 | 11 | G protein-coupled receptor for dopamine; inhibits adenylyl cyclase |
| DTNBP1 | Dystrobrevin-binding protein 1 | 6 | A component of the biogenesis of lysosome-related organelles complex 1 |
| ERBB4 | V-erb-a erythroblastic leukemia viral oncogene | 2 | Receptor for neuregulins; cell differentiation |
| FMR1 | Fragile X mental retardation 1 | 23 | May be involved in nucleus → cytoplasm mRNA trafficking |
| HTR2A | 5-hydroxytryptamine (serotonin) receptor 2A | 13 | G protein-coupled receptor for serotonin; activates phosphoinositide hydrolysis |
| NRG1 | Neuregulin 1 | 8 | Signaling protein that mediates cell–cell interactions; has roles in growth and development |
| NRGN | Neurogranin | 11 | Postsynaptic protein kinase substrate; learning and memory; glutamate signaling |
| NRXN1 | Neurexin 1 | 2 | Functions in the nervous system as a cell adhesion molecule and receptor |
| PARK7 | Parkinson’s disease (autosomal recessive, early onset) 7 | 1 | Positive regulator of androgen receptor-dependent transcription; apparently protects neurons against oxidative stress and cell death |
| SCNA | Synuclein, alpha (non-A4 component of amyloid precursor) | 4 | May serve to integrate presynaptic signaling and membrane trafficking |
| TCF4 | Transcription factor 4 | 18 | Neuronal transcriptional factor; neurogenesis |
| VIPR2 | Vasoactive intestinal peptide receptor 2 | 7 | Peptide that functions as a neurotransmitter and a neuroendocrine hormone |
| ZNF804A | Zinc finger protein 804A | 2 | Transcription factor; neuronal connectivity in the dorsolateral prefrontal cortex |
| Genotype | Enzymatic Activity | Impact of Diazepam | Clinical Consequences |
|---|---|---|---|
| CYP2C19 PM (Poor Metabolizer) | Low/Absent | Slow metabolism, drug accumulation | Increased risk of adverse reactions (dependence, tolerance) |
| CYP2C19*2 (Allele) | Reduced activity | Slow metabolism, drug accumulation | Higher risk of side effects |
| CYP2C19*17 (Allele) | Increased activity | Rapid metabolism, faster drug clearance | Reduced efficacy, possible higher dose needed |
| CYP2B6 PM (Poor Metabolizer) | Low | Slow metabolism, drug accumulation | Possible need for dose reduction |
| Medication | Type | Effect on Autophagy | Possible Mechanism/Notes |
|---|---|---|---|
| Lithium | Mood stabilizer | Promotes autophagy | Enhances autophagic activity in the brain |
| Fluspirilene | Antipsychotic | Stimulates autophagy | Identified in a small-molecule screen |
| Trifluoperazine | Antipsychotic | Stimulates autophagy | May help reverse the downregulation of autophagic genes in the BA22 region |
| Pimozide | Antipsychotic | Stimulates autophagy | Could enhance the expression of autophagy-related proteins |
| Clomipramine | Antidepressant (TCA) | Presence of autophagy-related structures, but unclear effect | May induce or inhibit autophagic flux; further experiments are required |
| Desmethylclomipramine | Metabolite of clomipramine | Inhibits functional autophagy | Disrupts autophagic flux |
| Amitriptyline | Antidepressant (TCA) | Enhances autophagy | Observed in primary neurons and astrocytes |
| Citalopram | Antidepressant (SSRI) | Enhances autophagy | Similar effect to amitriptyline |
| Venlafaxine | Antidepressant (SNRI) | No significant effect on autophagy | Does not appear to alter autophagic processes |
| Vitamin | Effect on Aβ Levels | Notes |
|---|---|---|
| Vitamin D | ↓ Plasma Aβ42 in AD patients after supplementation | Positive correlation with Aβ42 in CSF; no association in older adults without dementia |
| Vitamin B12 | Low levels correlate with ↑ Aβ42 | Deficiency may contribute to higher Aβ accumulation |
| Folic Acid | Supplementation may ↓ Aβ42 levels | Potential protective effect |
| Vitamin E | No significant change in Aβ42 after supplementation in AD patients | Antioxidant properties but no direct effect on Aβ levels observed |
| Vitamin C | No significant change in Aβ42 after supplementation in AD patients | Similar to vitamin E; no observed reduction in Aβ |
| α-Lipoic Acid | No significant change in Aβ42 after supplementation in AD patients | Antioxidant effects but no impact on Aβ levels |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capatina, T.-F.; Oatu, A.; Babasan, C.; Trifu, S. Translating Molecular Psychiatry: From Biomarkers to Personalized Therapies—A Narrative Review. Int. J. Mol. Sci. 2025, 26, 4285. https://doi.org/10.3390/ijms26094285
Capatina T-F, Oatu A, Babasan C, Trifu S. Translating Molecular Psychiatry: From Biomarkers to Personalized Therapies—A Narrative Review. International Journal of Molecular Sciences. 2025; 26(9):4285. https://doi.org/10.3390/ijms26094285
Chicago/Turabian StyleCapatina, Tudor-Florentin, Anamaria Oatu, Casandra Babasan, and Simona Trifu. 2025. "Translating Molecular Psychiatry: From Biomarkers to Personalized Therapies—A Narrative Review" International Journal of Molecular Sciences 26, no. 9: 4285. https://doi.org/10.3390/ijms26094285
APA StyleCapatina, T.-F., Oatu, A., Babasan, C., & Trifu, S. (2025). Translating Molecular Psychiatry: From Biomarkers to Personalized Therapies—A Narrative Review. International Journal of Molecular Sciences, 26(9), 4285. https://doi.org/10.3390/ijms26094285