

Recent Developments in the [1,2]-Phospha-Brook Rearrangement Reaction
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
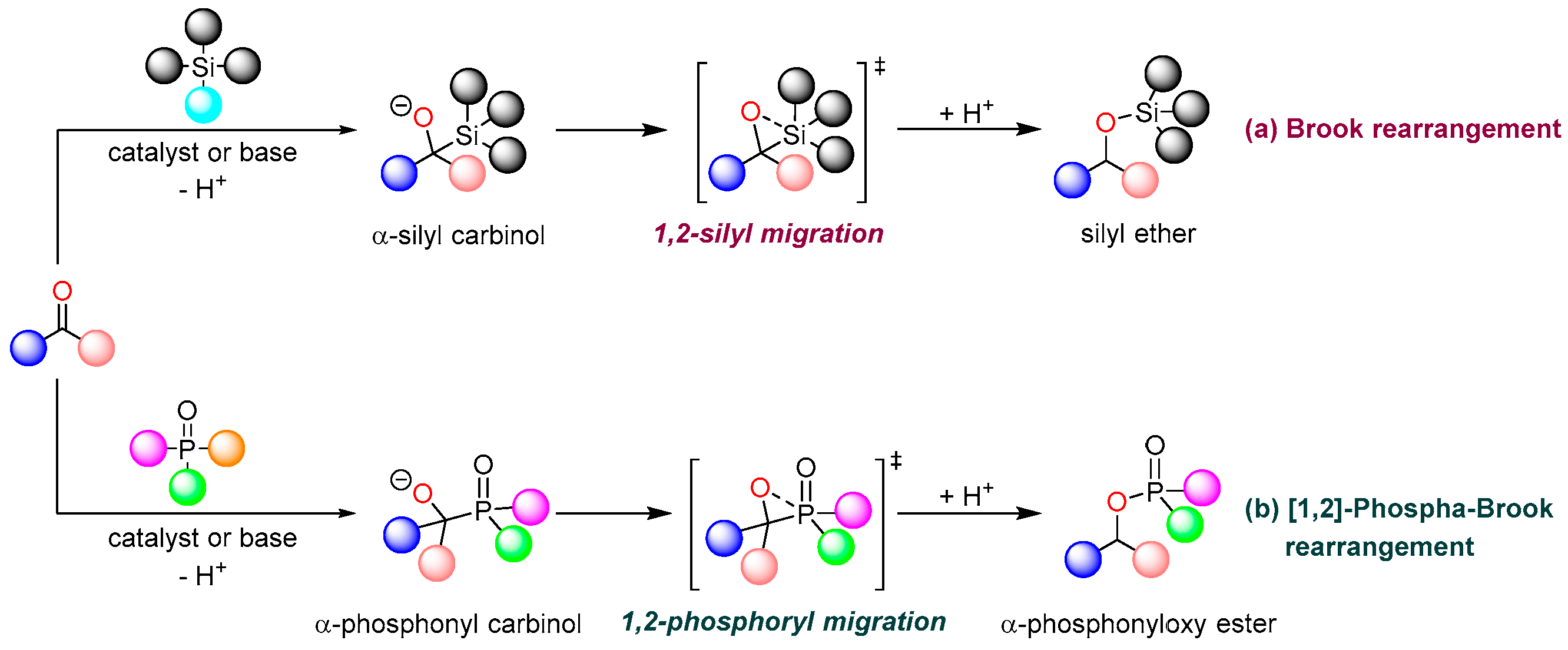
1. Introduction
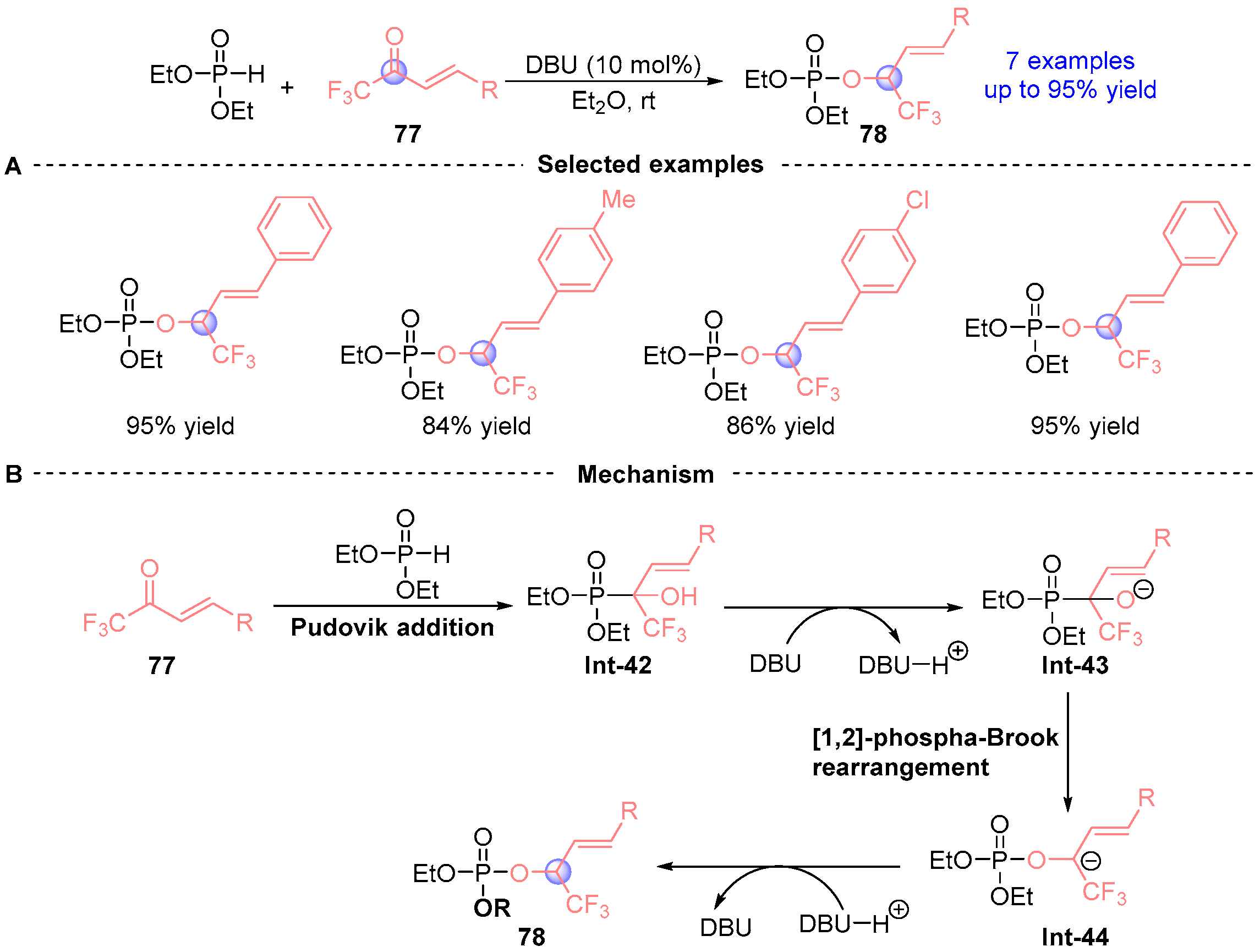
2. Aldehyde Rearrangement
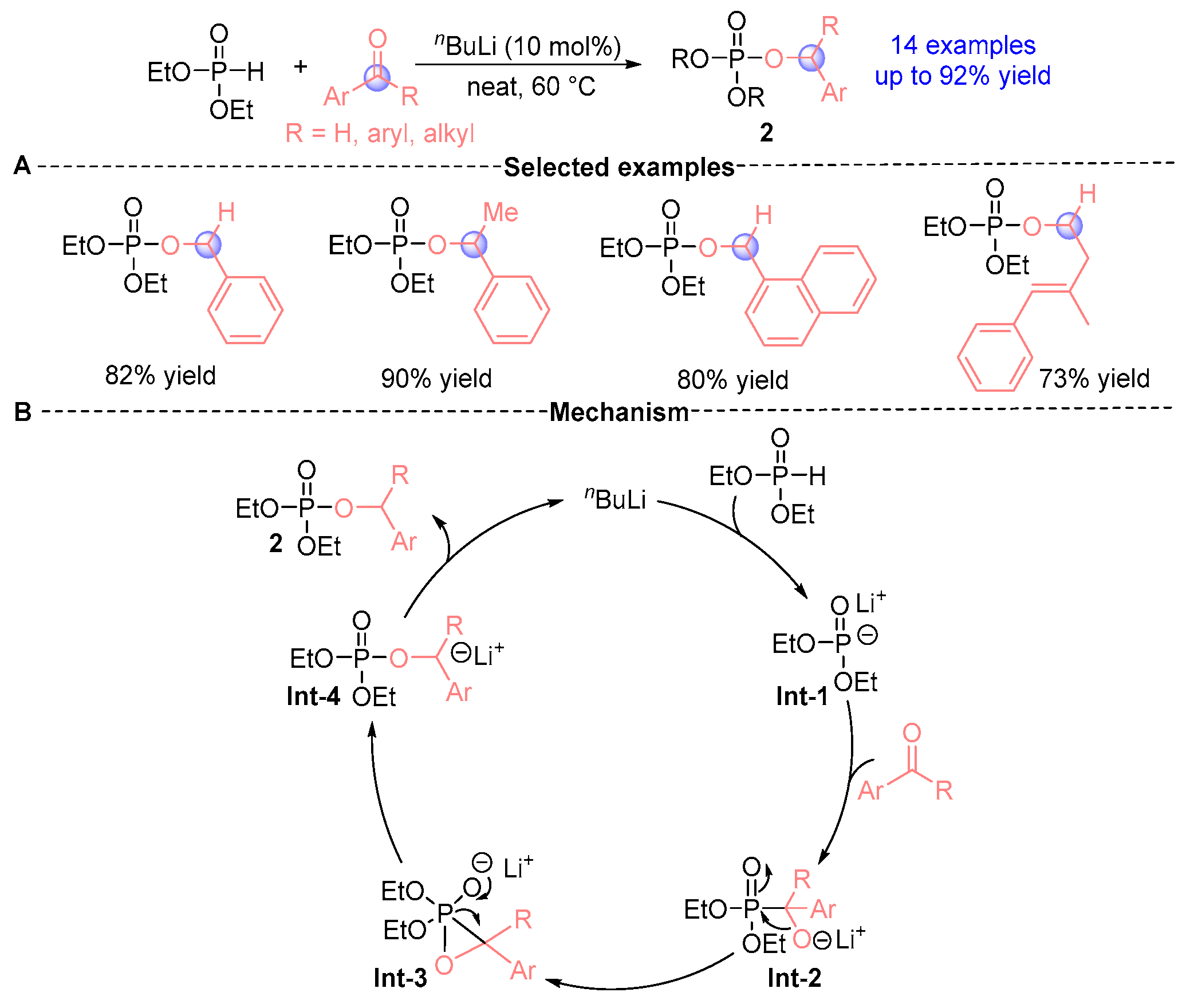
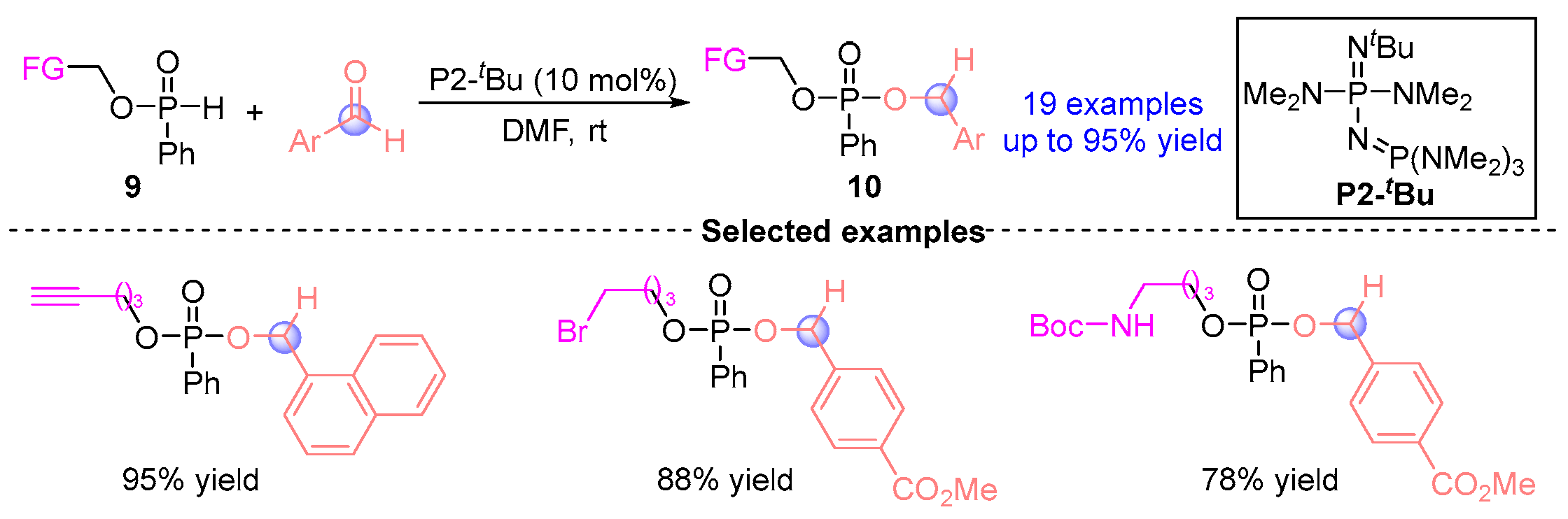
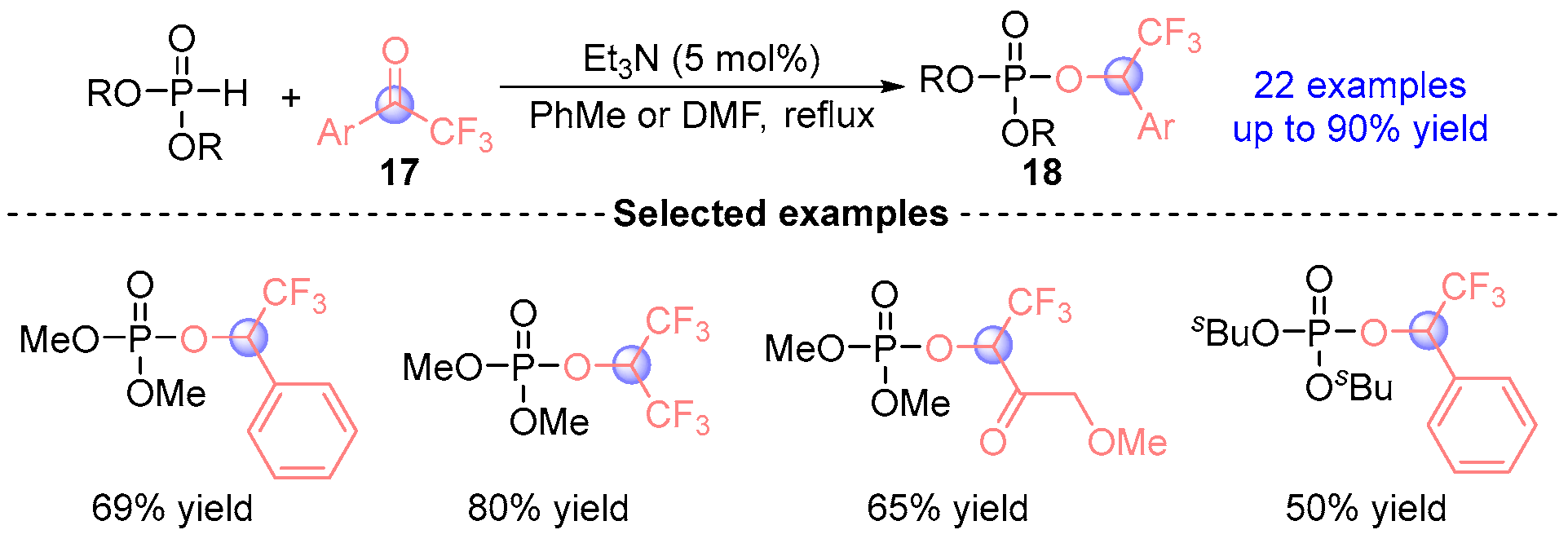
2.1. Aldehyde Rearrangement with Phosphodiesters
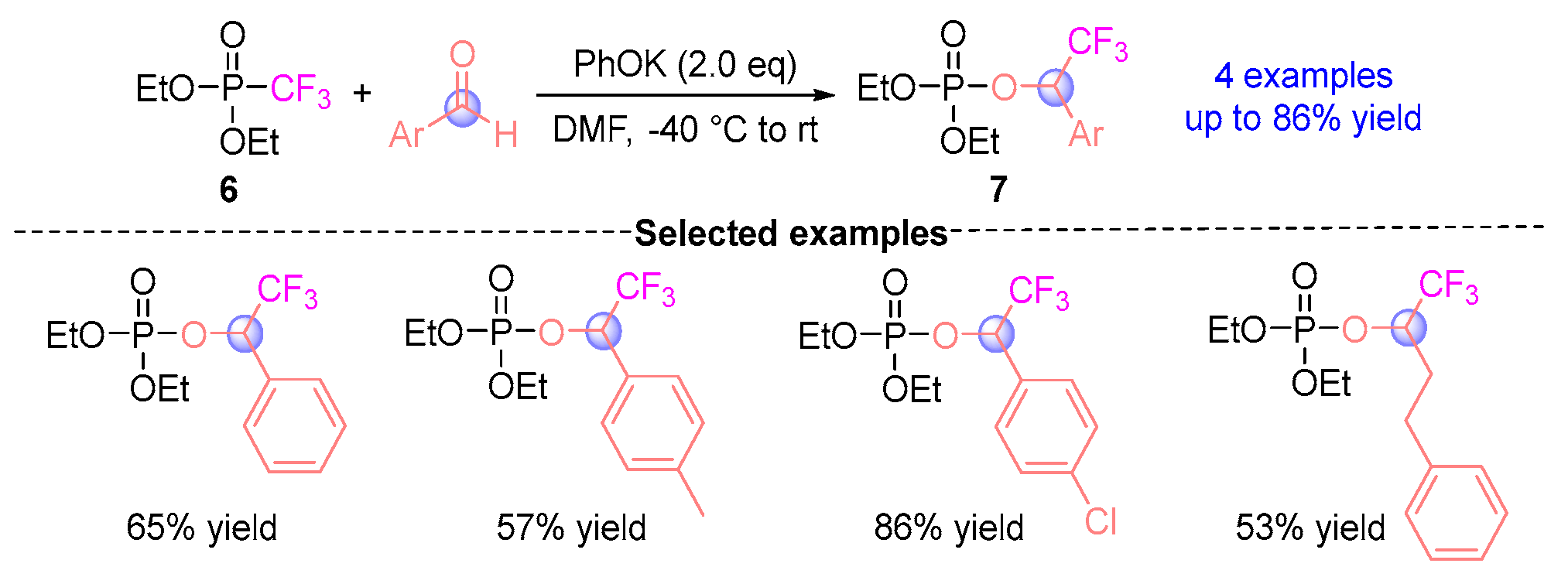
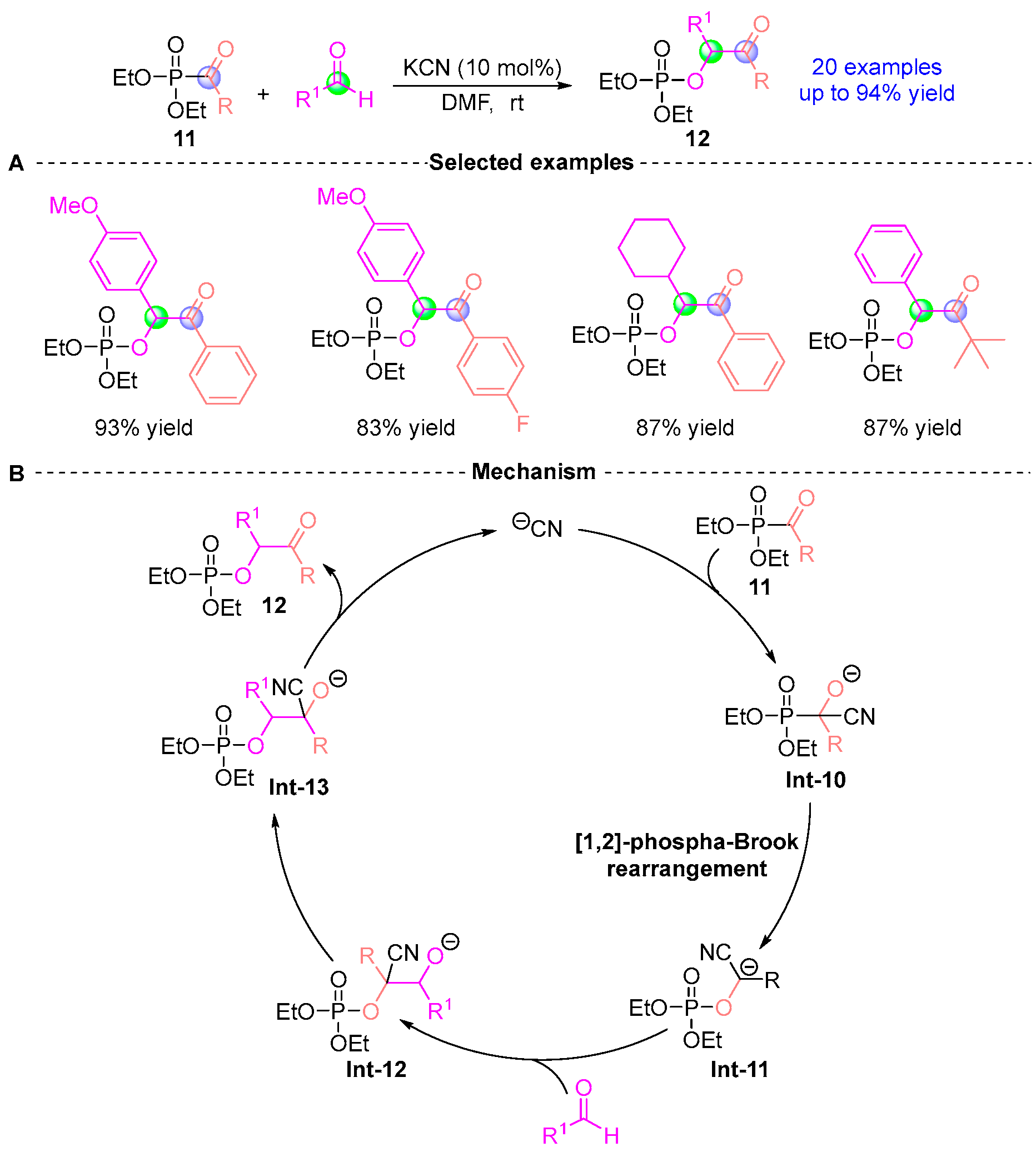
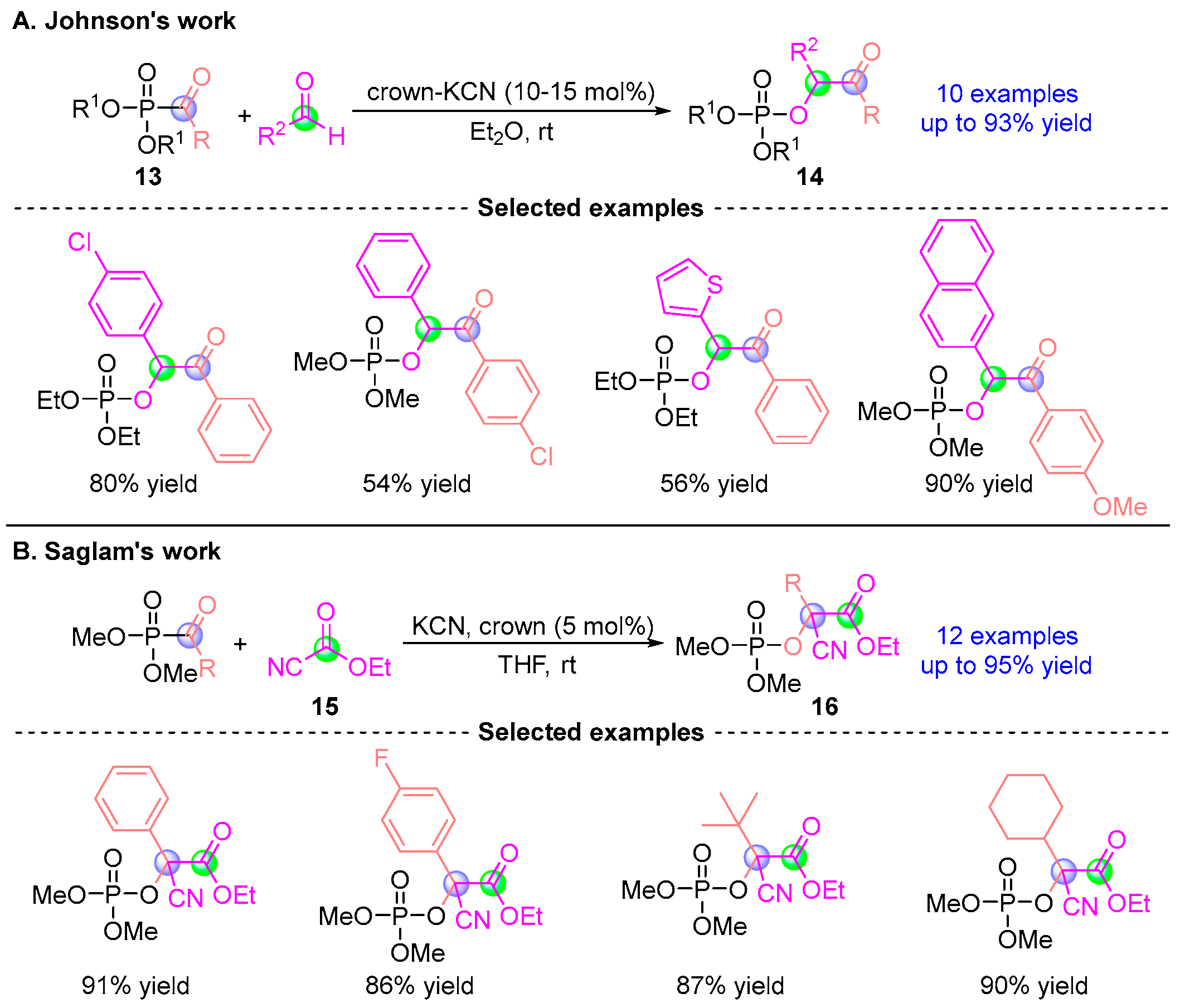
2.2. Aldehyde Rearrangement with Phosphodiester Precursors
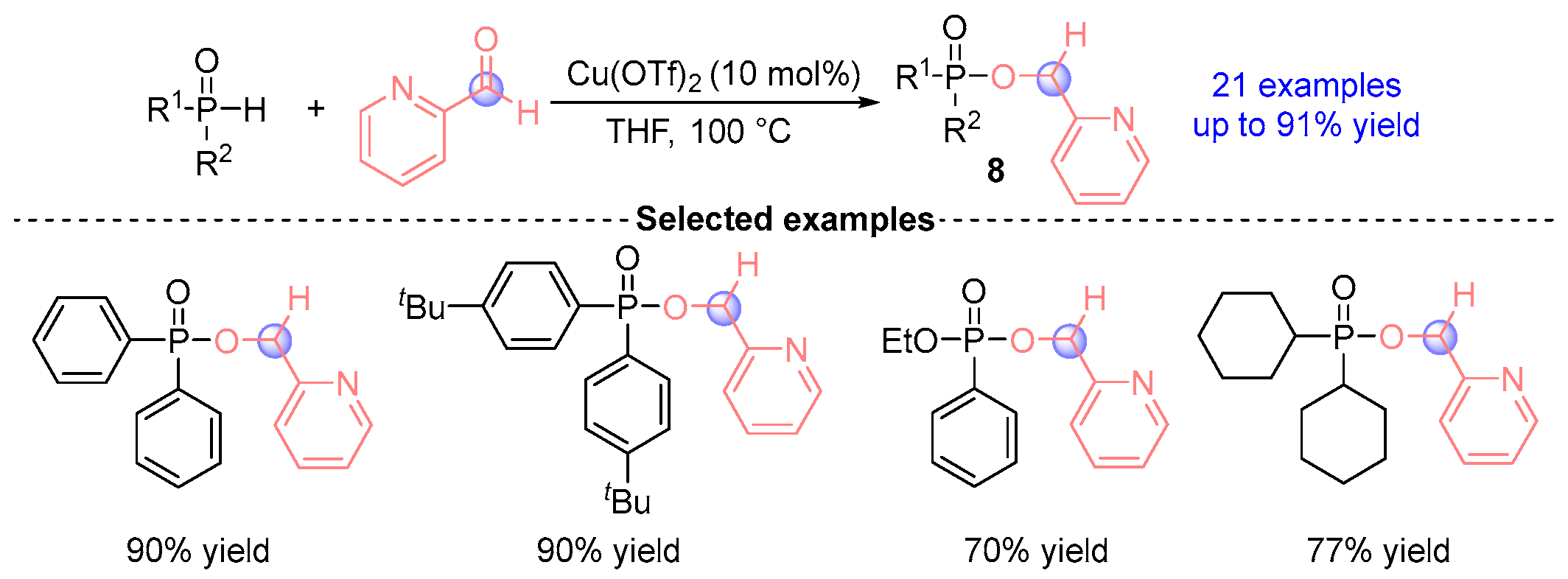
2.3. Aldehyde Rearrangement with Secondary Phosphine Oxides
2.4. Aldehyde Rearrangement with SPO-Modified Precursors
3. Ketone Rearrangement
3.1. Ketone Rearrangement with Phosphodiester Precursors
3.2. Ketone Rearrangement with Phosphodiesters
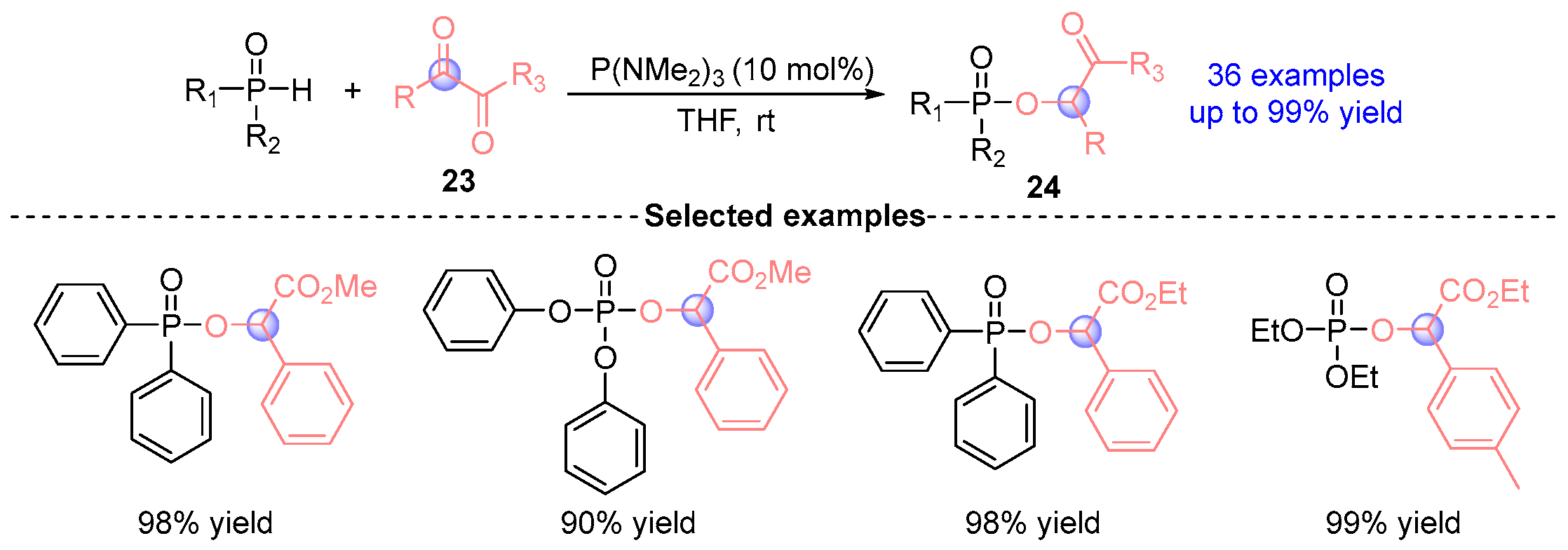
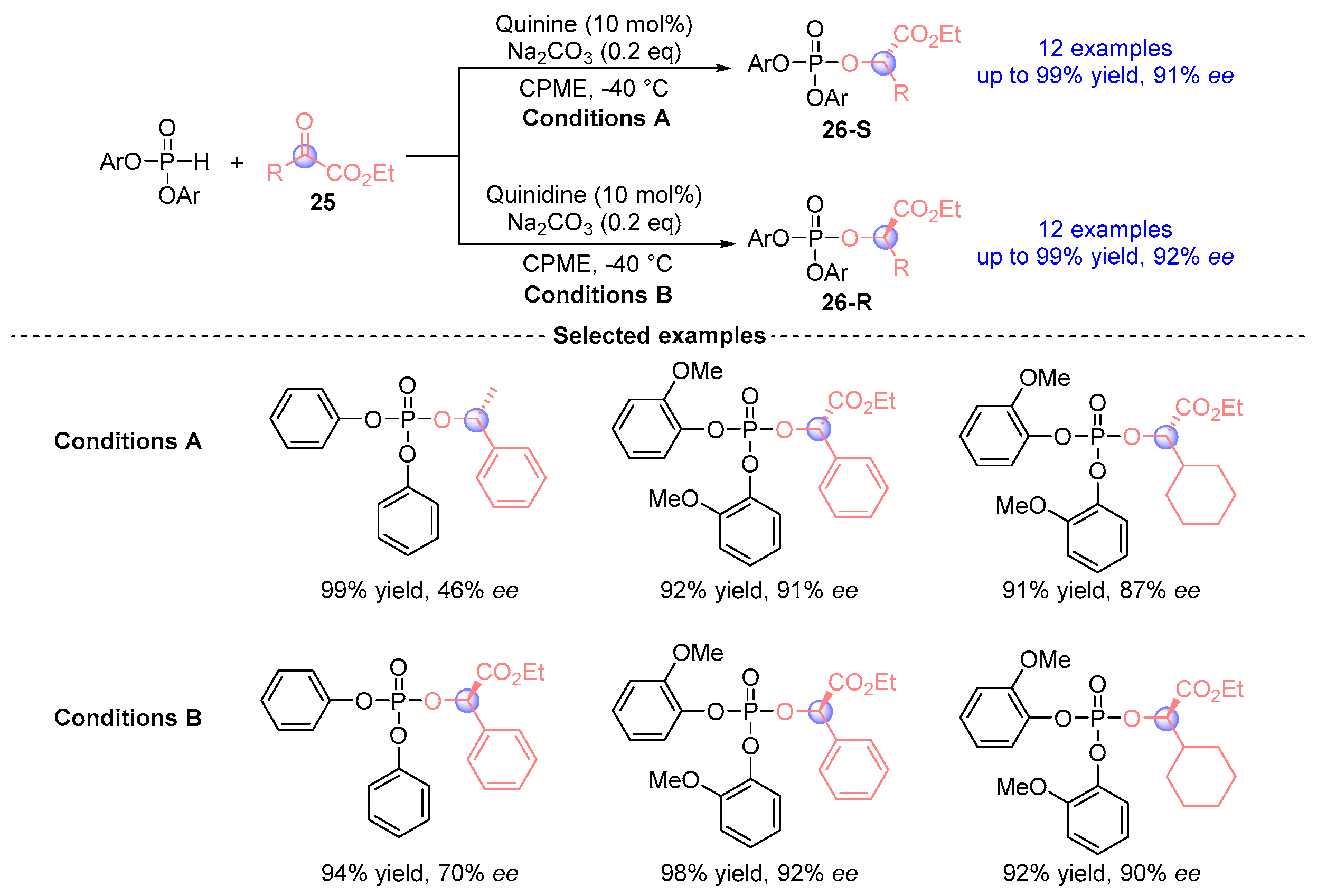
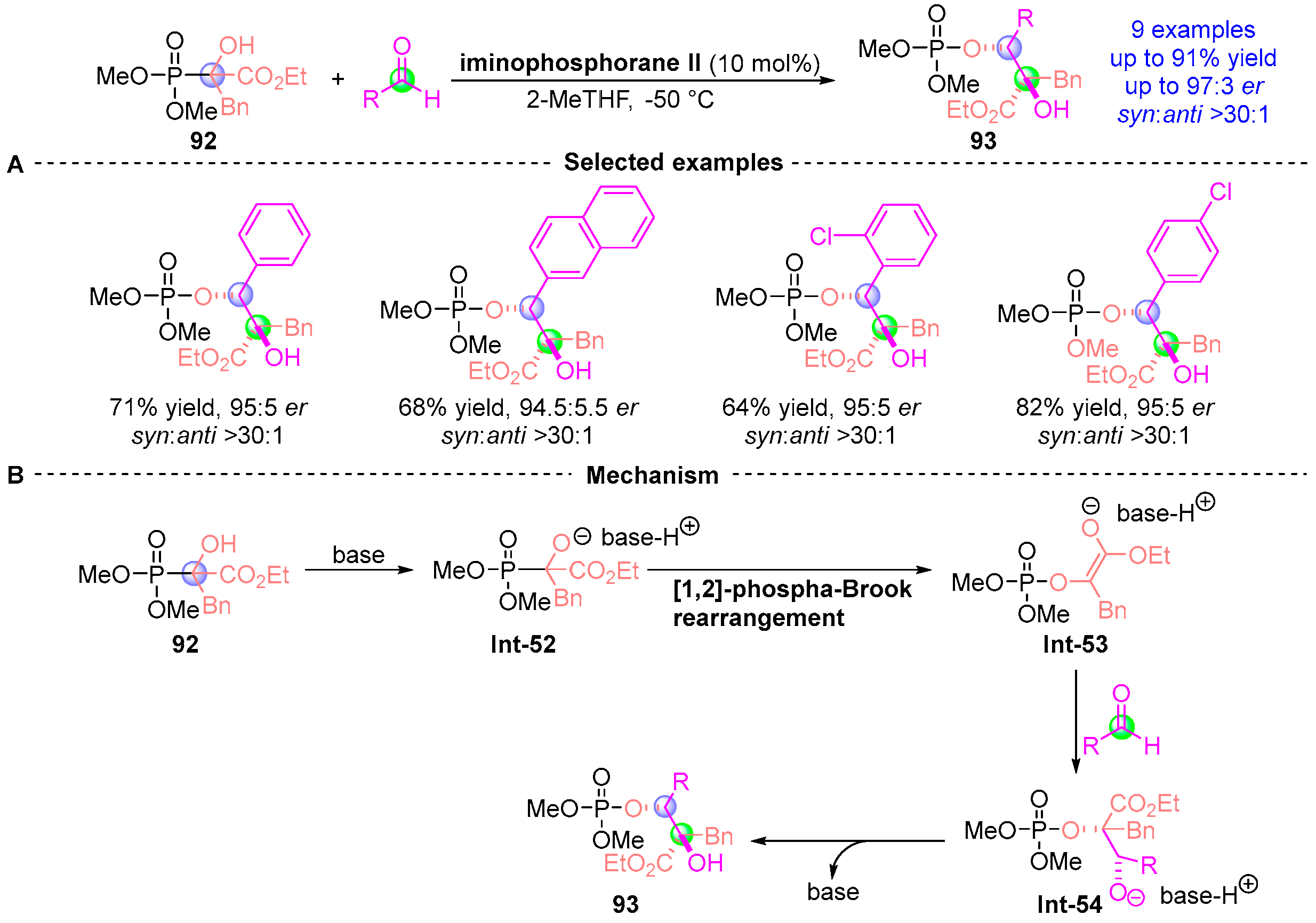
4. Ketoester Rearrangement
4.1. Ketoester Rearrangement with Phosphodiesters
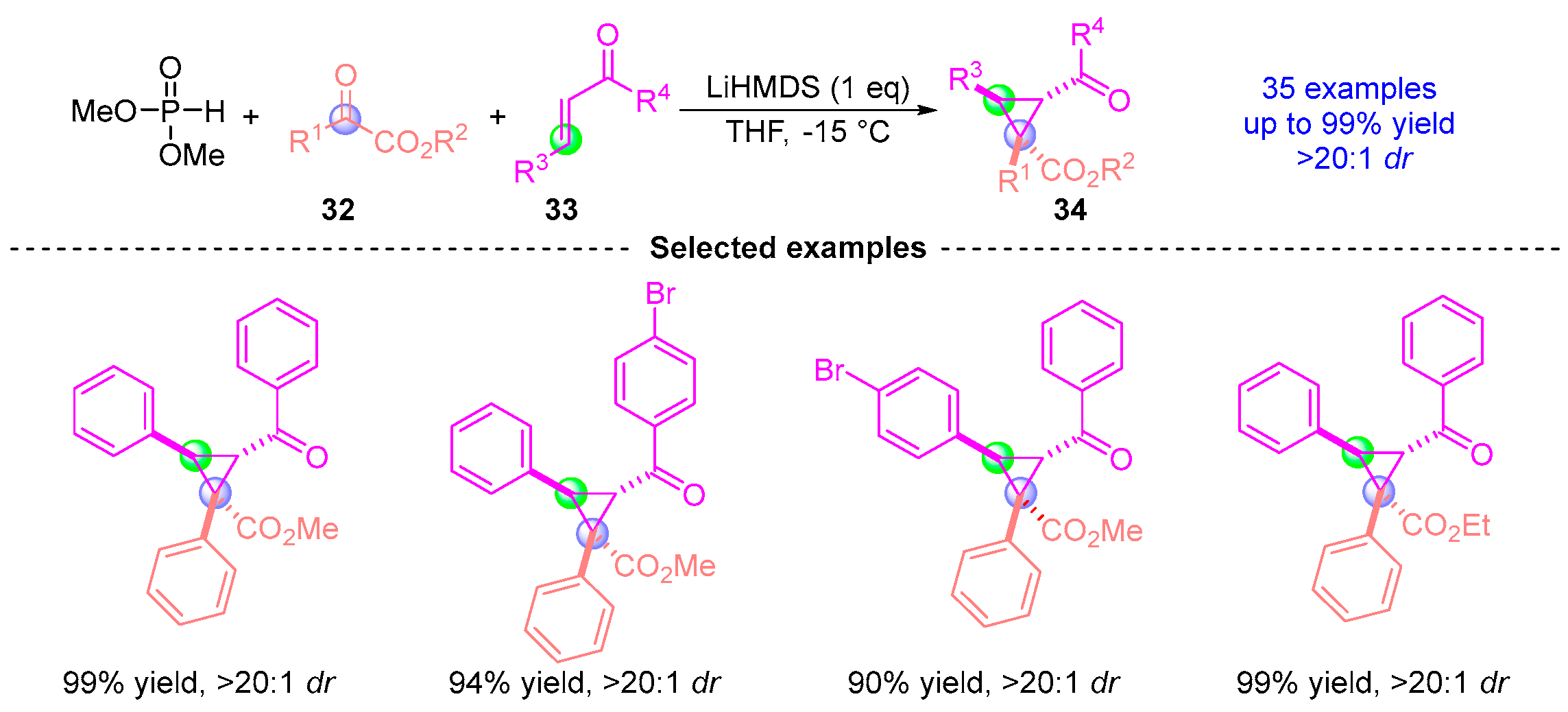
4.2. Ketoester Rearrangement with Three Components
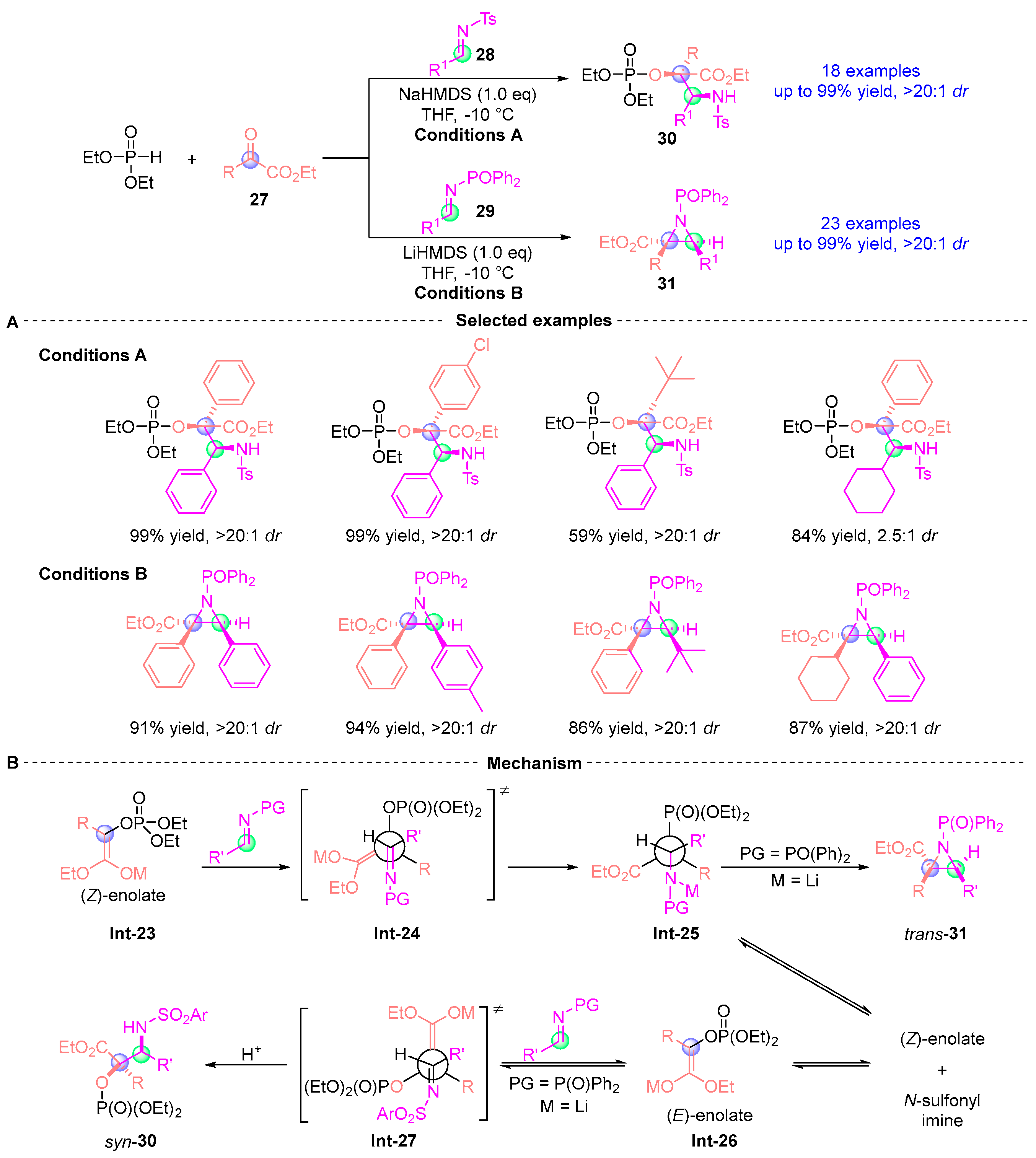
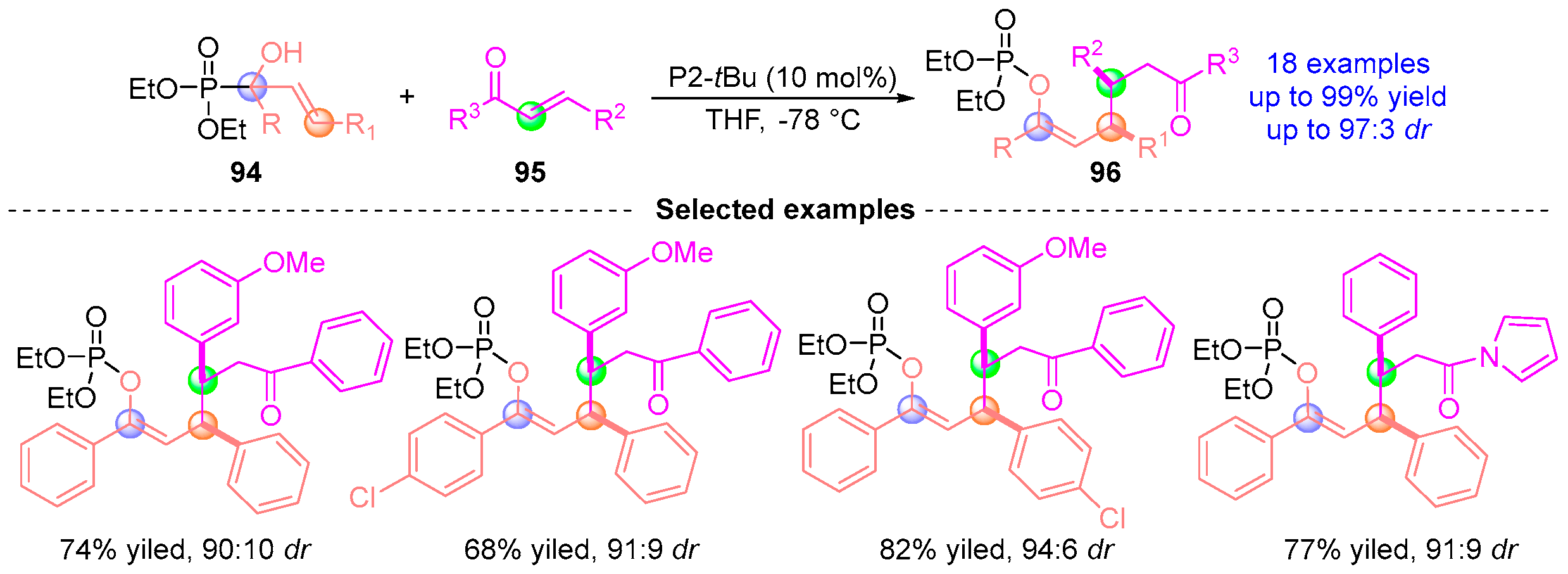
5. Ketoamide Rearrangement
5.1. Ketoamide Rearrangement with Phosphodiesters
5.2. Ketoamide Rearrangement with Three Components
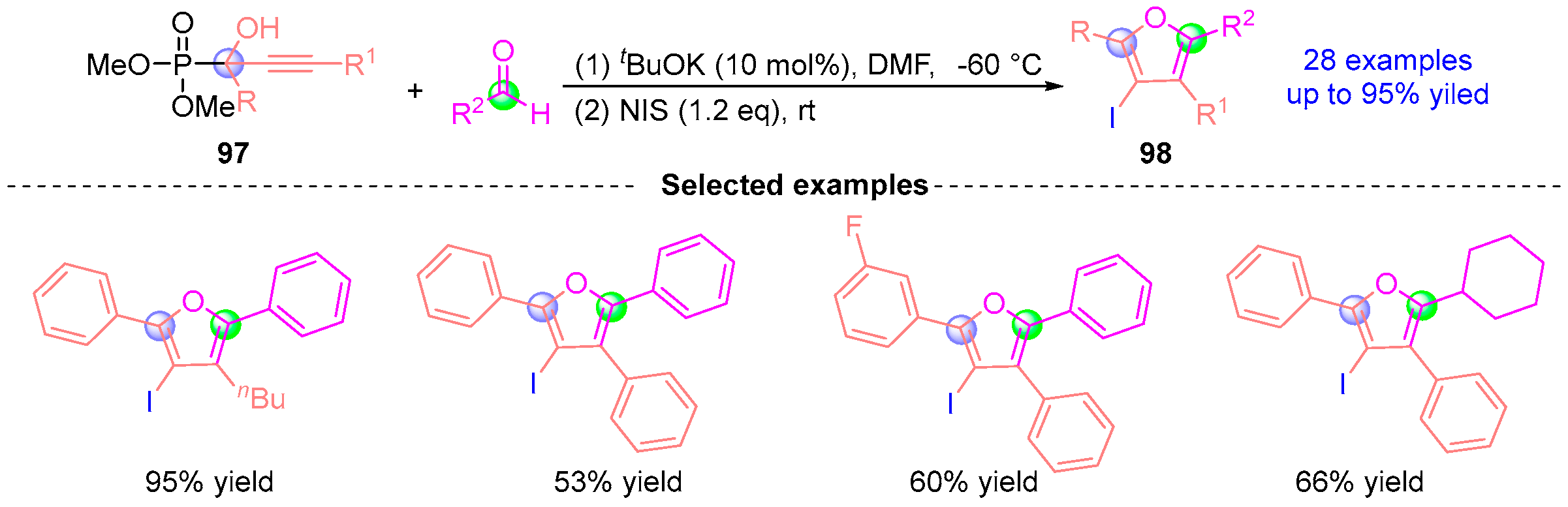
6. Ketene Rearrangement
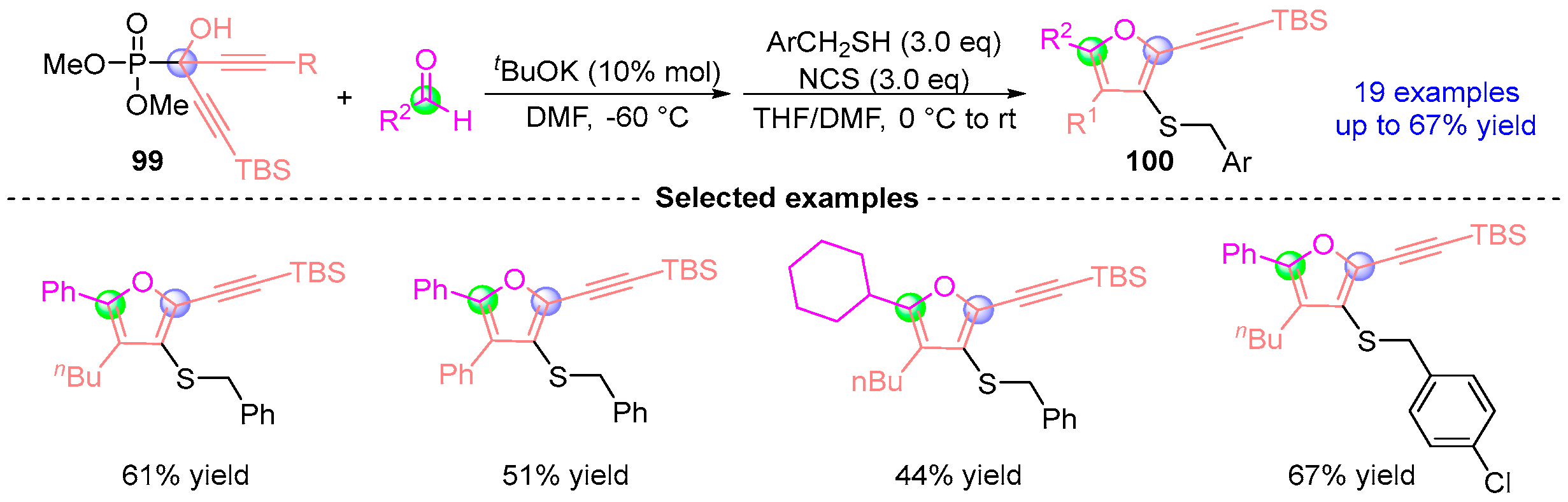
7. Alkyne Ketone Rearrangement
8. Multicomponent Rearrangement
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AChE | Acetylcholinesterase |
| BChE | Butyrylcholinesterase |
| BPR | Berry pseudorotation |
| CaE | Carboxylesterases |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DBU | 1,8-Diazabicyclo [5.4.0]undec-7-ene |
| DMAP | 4-Dimethylaminopyridine |
| DMF | N,N-Dimethylformamide |
| DFT | Density functional theory |
| KHMDS | Potassium Hexamethyldisilazide |
| LDA | Lithium Diisopropylamide |
| LiHMDS | Lithium Hexamethyldisilazide |
| NaHMDS | Sodium Hexamethyldisilazide |
| NIS | N-Iodosuccinimide |
| PhOK | Potassium phenoxide |
| SPO | Secondary phosphine oxides |
| TMG | Tetramethylguanidine |
| TMSOTf | Trimethylsilyl triflate |
| nBuLi | n-butyllithium |
| tBuOK | potassium tert-butoxide |
References
- Brook, A.G. Isomerism of Some α-Hydroxysilanes to Silyl Ethers. J. Am. Chem. Soc. 1958, 80, 1886–1889. [Google Scholar] [CrossRef]
- Yang, F.; Wang, J.; Dong, Y.; Zhang, N.; Zhang, C. Transition Metal Promoted Brook Rearrangement and Its Related Reactions. Tetrahedron 2024, 168, 134351. [Google Scholar] [CrossRef]
- Kondoh, A.; Terada, M. Development of Molecular Transformations on the Basis of Catalytic Generation of Anionic Species by Organosuperbase. Bull. Chem. Soc. Jpn. 2021, 94, 339–356. [Google Scholar] [CrossRef]
- Kondoh, A.; Terada, M. [1,2]-Phospha-Brook Rearrangement as Tool for Generation of Anionic Nucleophiles in Addition Reactions under Bronsted Base Catalysis. Asian. J. Org. Chem. 2023, 12, e202300003. [Google Scholar]
- Kaur, R.; Singh, R.P. Stereoselective Reductive Coupling Reactions Utilizing 1,2-Phospha-Brook Rearrangement: A Powerful Umpolung Approach. J. Org. Chem. 2023, 88, 10325–10338. [Google Scholar]
- Kaïm, L.E.; Gaultier, L.; Grimaud, L.; Santos, D.A. Formation of New Phosphates from Aldehydes by a DBU-Catalysed Phospha-Brook Rearrangement in a Polar Solvent. Synlett 2005, 15, 2335–2336. [Google Scholar]
- Pallikonda, G.; Santosh, R.; Ghosal, S.; Chakravarty, M. n-BuLi-Triggered Phospha-Brook Rearrangement: Efficient Synthesis of Organophosphates from Ketones and Aldehydes. Tetrahedron Lett. 2015, 56, 3796–3798. [Google Scholar]
- Ranga, S.; Chakravarty, M.; Chatterjee, T.; Ghosal, S. Mechanistic Insights into n-BuLi Mediated Phospha-Brook Rearrangement. New J. Chem. 2019, 43, 9886–9890. [Google Scholar]
- Wang, B.; Gao, Q.; Jiang, H.; Wu, W. [1,2]-Phospha-Brook Rearrangement-Initiated Palladium-Catalyzed Cyclization Reaction of Isocyanides and o-Bromobenzaldehydes: Access to 2H-Isoindole-1-carboxamides and 2H-Isoindole-1-carbonitriles. Chin. J. Chem. 2025, 43, 620–626. [Google Scholar]
- Cherkupally, P.; Beier, P. Alkoxide-Induced Nucleophilic Trifluoromethylation using Diethyl Trifluoromethylphosphonate. Tetrahedron Lett. 2010, 51, 252–255. [Google Scholar]
- Yang, J.; Qian, D.-W.; Yang, S.-D. Lewis acid-Catalyzed Pudovik Reaction–Phospha-Brook Rearrangement Sequence to Access Phosphoric Esters. Beilstein. J. Org. Chem. 2022, 18, 1188–1194. [Google Scholar] [CrossRef]
- Kondoh, A.; Terada, M. Brønsted Base-Catalyzed 1,2-Addition/[1,2]-Phospha-Brook Rearrangement Sequence Providing Functionalized Phosphonates. Org. Biomol. Chem. 2022, 20, 2863–2866. [Google Scholar] [CrossRef] [PubMed]
- Demir, A.S.; Reis, Ö.; İǧdir, A.Ç.; Esiringü, İ.; Eymur, S. Generation of Acyl anion Equivalents from Acylphosphonates via Phosphonate-Phosphate Rearrangement: A highly Practical Method for Cross-Benzoin Reaction. J. Org. Chem. 2005, 70, 10584–10587. [Google Scholar] [PubMed]
- Bausch, C.C.; Johnson, J.S. Cyanide-Catalyzed Additions of Acyl Phosphonates to Aldehydes: A New Acyl Donor for Benzoin-Type Reactions. Adv. Synth. Catal. 2005, 347, 1207–1211. [Google Scholar] [CrossRef]
- Demir, A.S.; Reis, B.; Reis, Ö.; Eymuer, S.; Goellue, M.; Tural, S.; Saglam, G. Cyanide Ion Promoted Addition of Acyl Phosphonates to Ethyl Cyanoformate: Synthesis of Tertiary Carbinols via Tandem Carbon−Carbon Bond Formations. J. Org. Chem. 2007, 72, 7439–7442. [Google Scholar] [CrossRef]
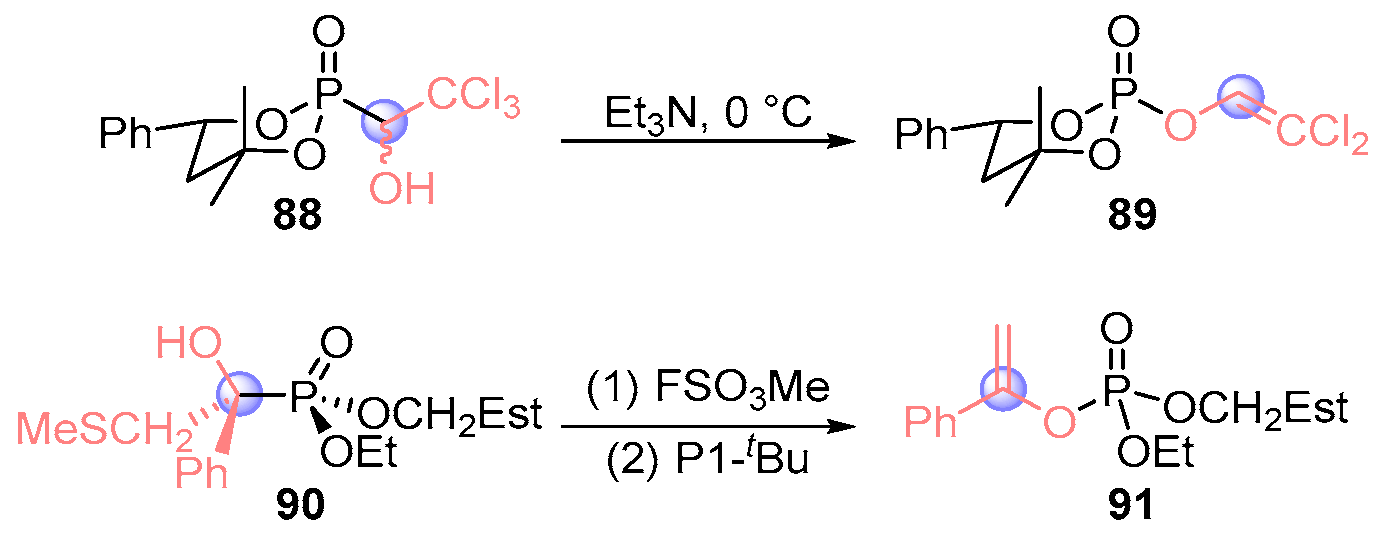
- Makhaeva, G.F.; Aksinenko, A.Y.; Sokolov, V.B.; Serebryakova, O.G.; Richardson, R.J. Synthesis of Organophosphates with Fluorine-Containing Leaving Groups as Serine Esterase Inhibitors with Potential for Alzheimer Disease Therapeutics. Bioorg. Med. Chem. Lett. 2009, 19, 5528–5530. [Google Scholar] [CrossRef] [PubMed]
- Aksinenko, A.Y.; Sokolov, V.B.; Goreva, T.V.; Makhaeva, G.F. Synthesis of O-phosphorylated 1-Substituted 2,2,2-Trifluoroethanols, Serine Hydrolase Inhibitors. Russ. Chem. Bull. 2010, 59, 102–106. [Google Scholar] [CrossRef]
- Li, Q.; Sun, Y.-M.; Yao, L.; Ji, S.-Y.; Zheng, H.-X.; Wen, J.-H.; Xu, Q.; Zhao, C.-Q. Stereoselective O-Phosphorylation of Aldehydes and Ketones via Phospha-Brook Rearrangement: The Stereochemistry and Intermolecular Mechanism. New J. Chem. 2024, 48, 18520–18525. [Google Scholar] [CrossRef]
- Kondoh, A.; Aoki, T.; Terada, M. Synthesis of Phenanthrene Derivatives by Intramolecular Cyclization Utilizing the [1,2]-Phospha-Brook Rearrangement Catalyzed by a Brønsted Base. Chem. Eur. J. 2015, 21, 12577–12580. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, N.; Wu, Z.-G.; Wu, X.; Wang, M.; Huang, W.; Zi, Y. Sequential In Situ-Formed Kukhtin−Ramirez Adduct and P(NMe2)3-Catalyzed O-Phosphination of α-Dicarbonyls with P(O)−H. Org. Lett. 2023, 25, 7595–7600. [Google Scholar] [CrossRef]
- Hayashi, M.; Nakamura, S. Catalytic Enantioselective Protonation of α-Oxygenated Ester Enolates Prepared through Phospha-Brook Rearrangement. Angew. Chem. Int. Ed. 2011, 50, 2249–2252. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Liu, H.; Lu, C.-D.; Xu, Y.-J. Diethyl Phosphite Initiated Coupling of α-Ketoesters with Imines for Synthesis of α-Phosphonyloxy-β-Amino Acid Derivatives and Aziridine-2-Carboxylates. Org. Lett. 2016, 18, 880–883. [Google Scholar]
- Yin, D.; Liu, H.; Lu, C.-D.; Xu, Y.-J. Dialkyl Phosphite-Initiated Cyclopropanation of α,β-Unsaturated Ketones using α-Ketoesters or Isatin Derivatives. J. Org. Chem. 2017, 82, 3252–3261. [Google Scholar] [CrossRef]
- Kondoh, A.; Terada, M. Brønsted Base-Catalyzed Three-Component Coupling Reaction of α-Ketoesters, Imines, and Diethyl Phosphite Utilizing [1,2]-Phospha-Brook Rearrangement. Org. Biomol. Chem. 2016, 14, 4704–4711. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, A.; Aoki, T.; Terada, M. Organocatalytic Arylation of α-Ketoesters Based on Umpolung Strategy: Phosphazene-Catalyzed SNAr Reaction Utilizing [1,2]-Phospha-Brook Rearrangement. Chem.-Eur. J. 2018, 24, 13110–13113. [Google Scholar]
- Horwitz, M.A.; Zavesky, B.P.; Martinez-Alvarado, J.I.; Johnson, J.S. Asymmetric Organocatalytic Reductive Coupling Reactions between Benzylidene Pyruvates and Aldehydes. Org. Lett. 2016, 18, 36–39. [Google Scholar] [PubMed]
- Kaur, R.; Singh, D.; Singh, R.P. Stereoselective Synthesis of Dihydrocoumarins via [1,2]-PhosphaBrook Rearrangement in Three-Component Coupling Reaction of α-Ketoesters, o-Quinone Methides, and Dialkyl Phosphites. J. Org. Chem. 2021, 86, 15702–15711. [Google Scholar] [CrossRef]
- Zhang, J.; Su, J.-Y.; Liu, Y.-Z.; Li, H.; Wang, Q.; Deng, W.-P. Palladium-Catalyzed Allylic Alkylation Enabled by Ketone Umpolung via Pudovik Addition/[1,2]-Phospha-Brook Rearrangement. Sci. China Chem. 2023, 66, 2810–2816. [Google Scholar] [CrossRef]
- Sakihara, M.; Shimoyama, S.; Kurosawa, M.B.; Yamaguchi, J. Deoxygenative Functionalizations of Aromatic Dicarbonyls and Aldehydes. Bull. Chem. Soc. Jpn. 2024, 97, uoae078. [Google Scholar]
- Sakihara, M.; Kurosawa, M.B.; Watanabe, M.; Shimoyama, S.; Yamaguchi, J. Deoxygenative Hetero- and Carbofunctionalizations of Diarylketones. J. Org. Chem. 2024, 89, 8157–8167. [Google Scholar]
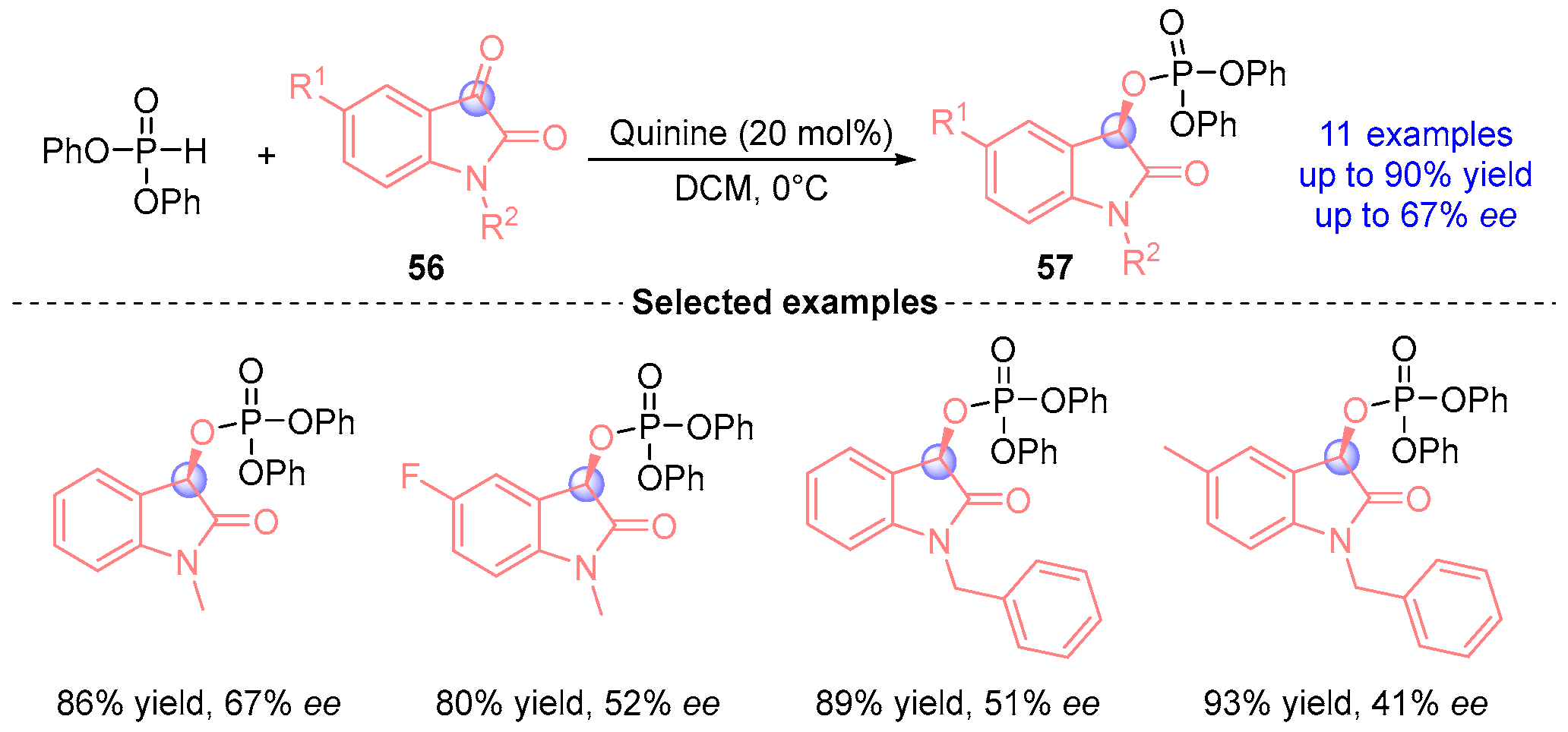
- Peng, L.; Wang, L.-L.; Bai, J.-F.; Jia, L.-N.; Yang, Q.-C.; Huang, Q.-C.; Xu, X.-Y.; Wang, L.-X. Highly Effective and Enantioselective Phospho-Aldol Reaction of Diphenyl Phosphite with N-Alkylated Isatins Catalyzed by Quinine. Tetrahedron Lett. 2011, 52, 1157–1160, Erratum in Tetrahedron Lett. 2011, 52, 6207–6209. [Google Scholar] [CrossRef]
- Kondoh, A.; Aoki, T.; Terada, M. Intramolecular Cyclization of Alkynyl α-Ketoanilide Utilizing 1,2 -Phospha-Brook Rearrangement Catalyzed by Phosphazene Base. Org. Lett. 2014, 16, 3528–3531. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Guo, S.; He, F.; Zhang, Y.; Wu, Z.; Wu, J. DBU Catalyzed Phospho-Aldol-Brook Rearrangement for Rapid Preparation of α-Phosphates Amide in Solvent-Free Conditions. Catalysts 2020, 10, 1445. [Google Scholar] [CrossRef]
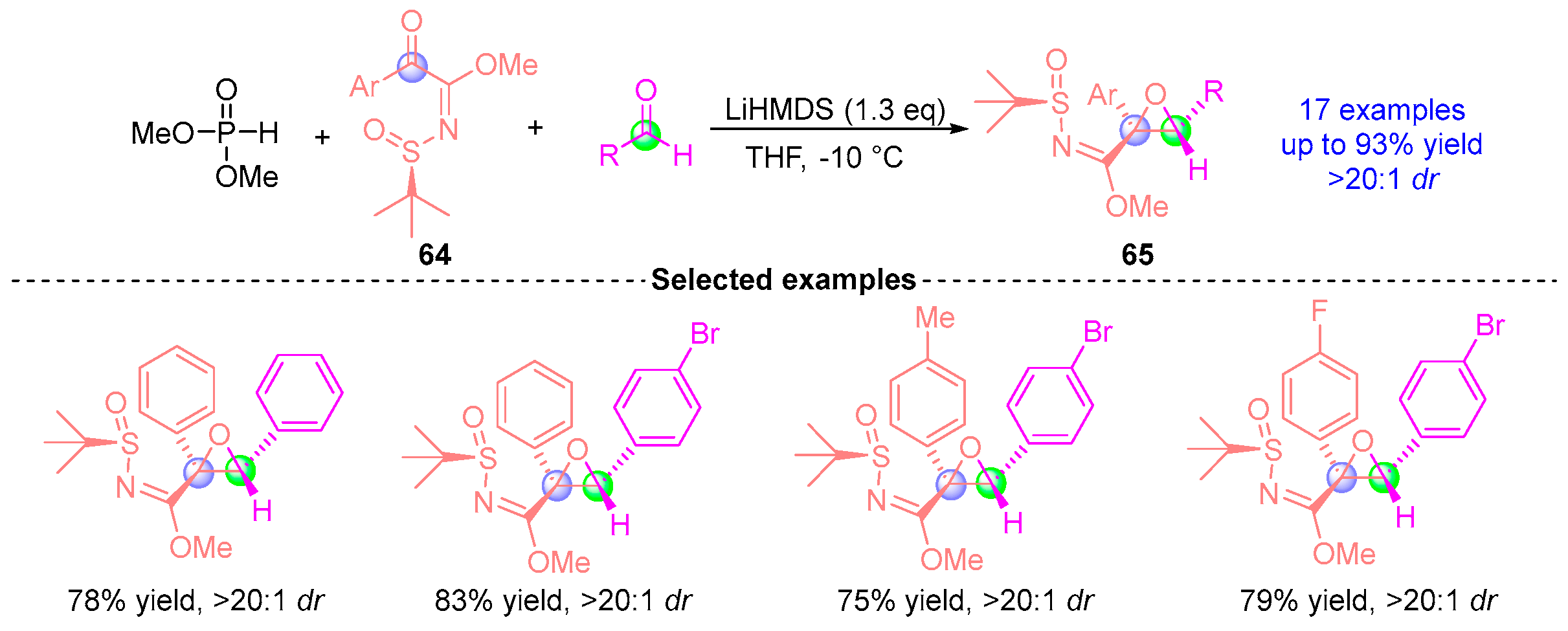
- Huang, W.; Liu, H.; Lu, C.-D.; Xu, Y.-J. Diastereoselective Synthesis of 2-Methoxyimidoyloxiranes Via Dimethyl Phosphite-Mediated Coupling of α-Keto N-Sulfinyl Imidates with Aldehydes. Chem. Commun. 2016, 52, 13592–13595. [Google Scholar] [CrossRef] [PubMed]
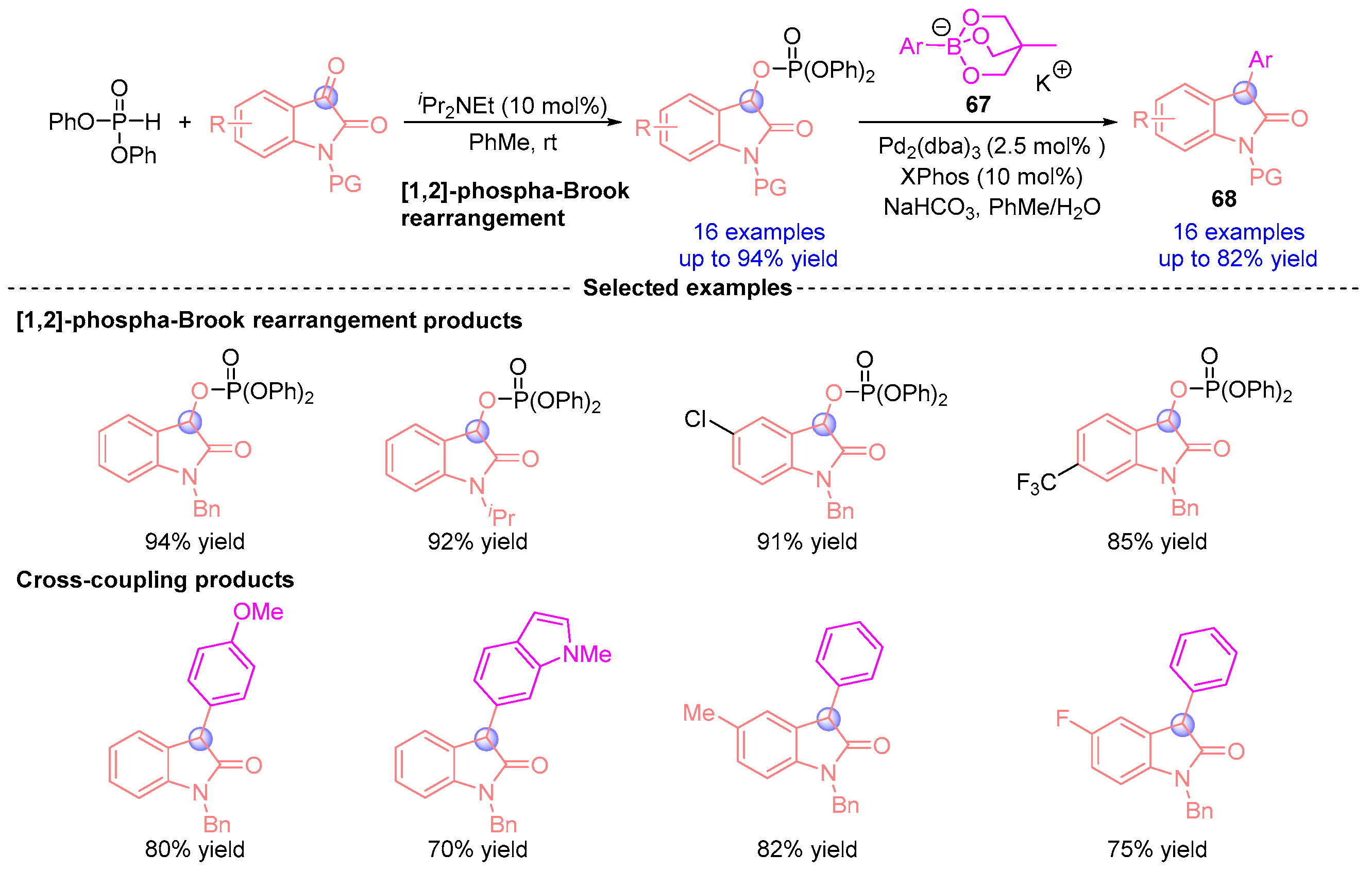
- Kondoh, A.; Takei, A.; Terada, M. Novel Methodology for the Efficient Synthesis of 3-Aryloxindoles: [1,2]-Phospha-Brook Rearrangement–Palladium-Catalyzed Cross-Coupling Sequence. Synlett 2016, 27, 1848–1853. [Google Scholar] [CrossRef]
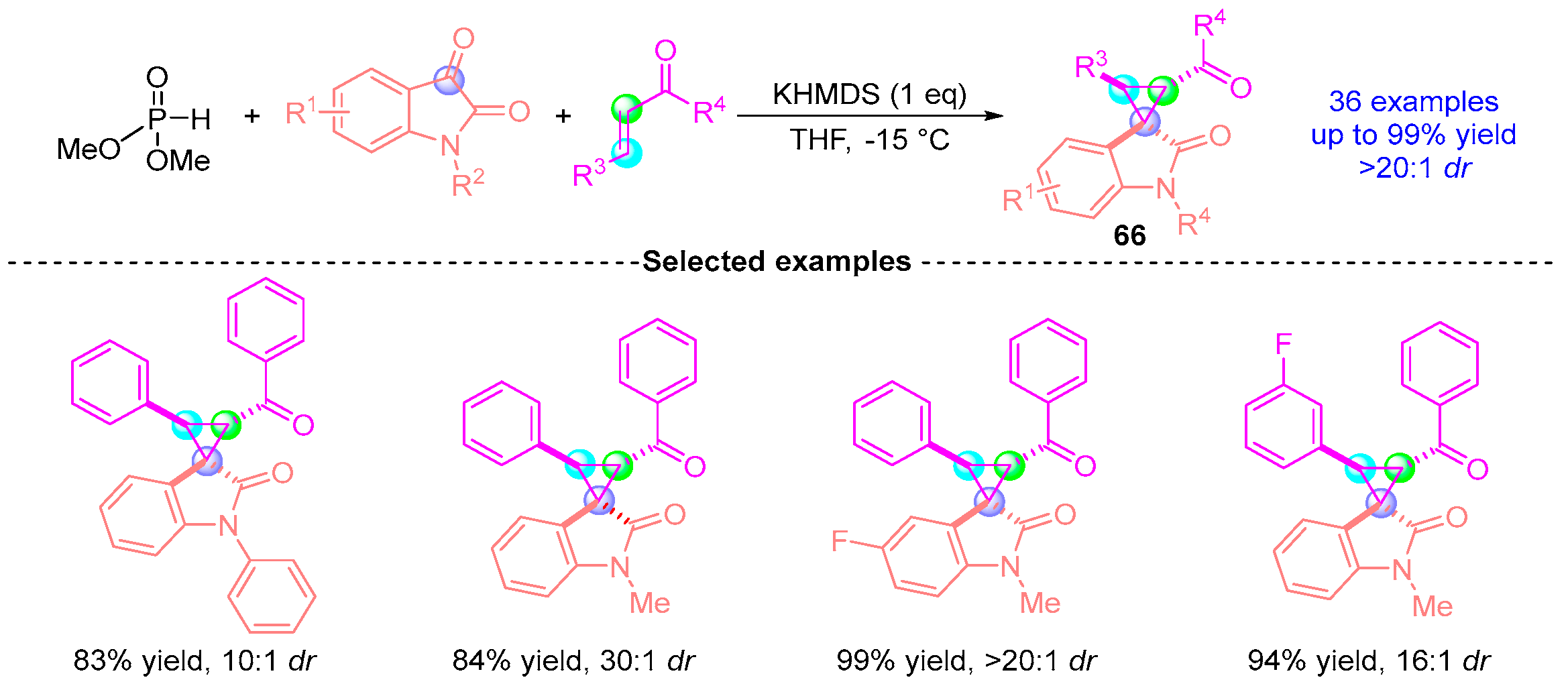
- Horwitz, M.A.; Tanaka, N.; Yokosaka, T.; Uraguchi, D.; Johnson, J.S.; Ooi, T. Enantioselective Reductive Multicomponent Coupling Reactions between Isatins and Aldehydes. Chem. Sci. 2015, 6, 6086–6090. [Google Scholar] [CrossRef]
- Motevalli, S.; Johnson, J.S. Phosphite-Mediated Reductive Cross-Coupling of Isatins and Nitrostyrenes. Synthesis 2017, 49, 2663–2676. [Google Scholar]
- Lin, Q.; Wang, S.; Weng, R.; Cao, W.; Feng, X. Chiral Lewis Acid-Catalyzed Asymmetric Multicomponent Michael Reaction through [1,2]-Phospha-Brook Rearrangement. Org. Lett. 2023, 25, 6262–6266. [Google Scholar] [CrossRef]
- Kaur, R.; Rautela, S.S.; Ali, M.; Singh, R.P. Deciphering Asymmetric Brønsted Base-Aminocatalytic Mode in Pudovik/1,2-Phospha-Brook Rearrangement/Michael Cascade Reaction. J. Org. Chem. 2024, 89, 14177–14182. [Google Scholar] [CrossRef]
- Zhu, X.-Y.; Chen, J.-R.; Lu, L.-Q.; Xiao, W.-J. An Efficient Synthesis of Enol Phosphates via Organic Base-Promoted Addition of Phosphites to 4-Oxo-Enoates. Tetrahedron 2012, 68, 6032–6037. [Google Scholar] [CrossRef]
- Zhang, F.; Jiang, M.; Liu, J.-T. Base-Controlled Reaction of α,β-Unsaturated Trifluoromethyl Ketone and Dialkyl Phosphite. Tetrahedron Lett. 2017, 58, 1871–1874. [Google Scholar]
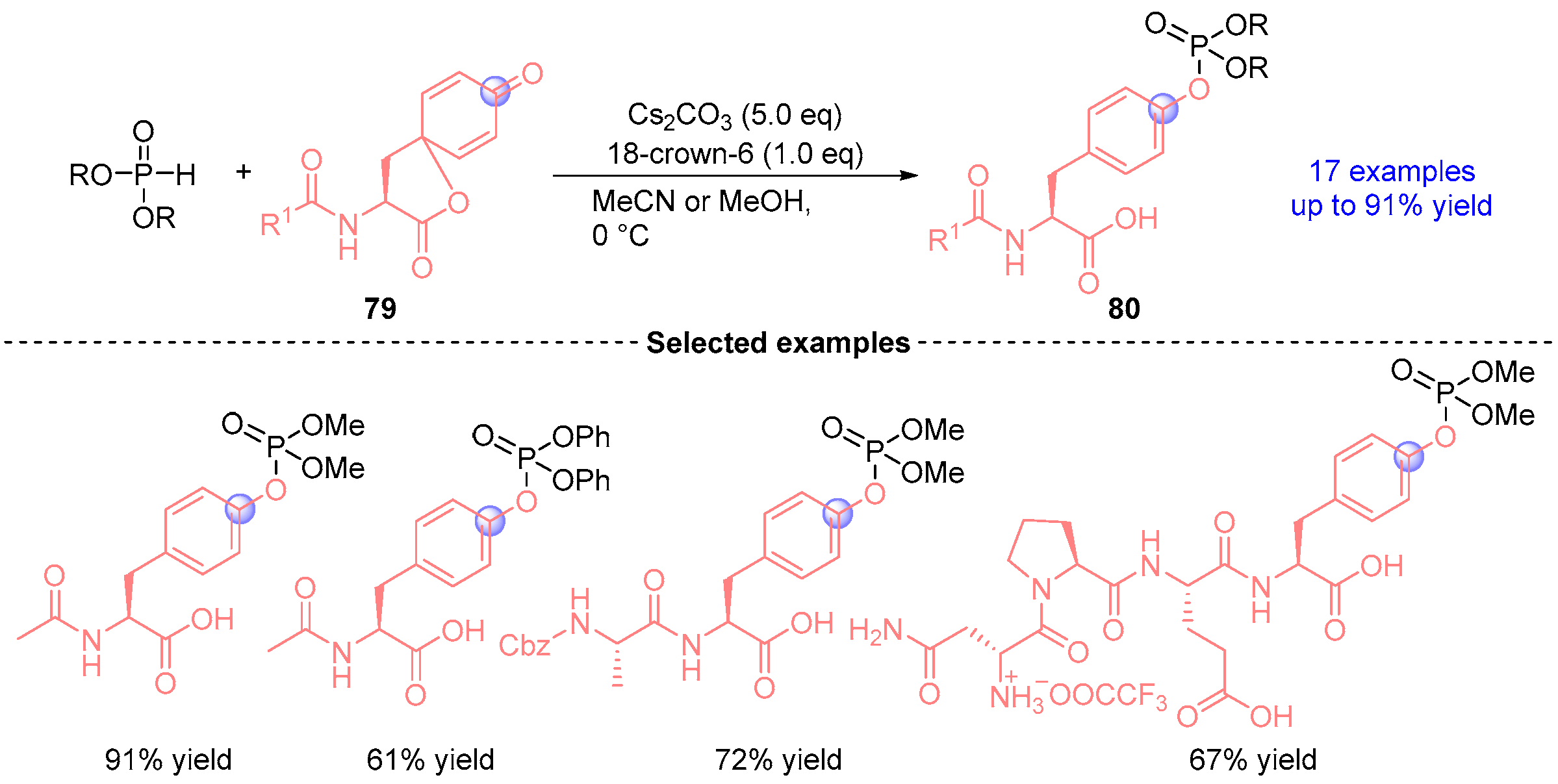
- Fukuta, T.; Tatsumu, T.; Fujiyoshi, K.; Koyama, T.; Kawashima, S.A.; Mitsunuma, H.; Yamatsugu, K.; Kanai, M. Umpolung Phosphorylation of Tyrosine via 1,2-Phospha-Brook Rearrangement. Org. Lett. 2024, 26, 8827–8831. [Google Scholar] [CrossRef]
- Kondoh, A.; Ishikawa, S.; Aoki, T.; Terada, M. Synthesis of 2,3-Allenylamides Utilizing 1,2 -Phospha-Brook Rearrangement and Their Application to Gold-catalyzed Cycloisomerization Providing 2-Aminofuran Derivatives. Chem. Commun. 2016, 52, 12513–12516. [Google Scholar] [CrossRef]
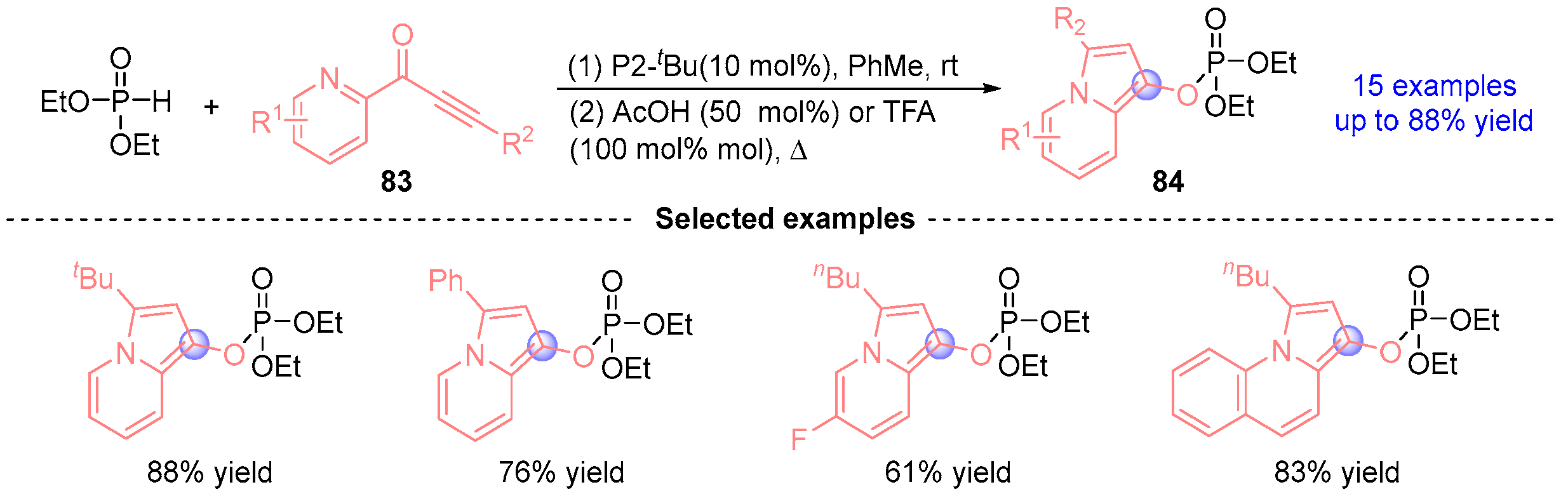
- Kondoh, A.; Koda, K.; Kamata, Y.; Terada, M. Synthesis of Indolizine Derivatives Utilizing 1,2-Phospha-Brook Rearrangement/Cycloisomerization Sequence. Chem. Lett. 2017, 46, 1020–1023. [Google Scholar]
- Lin, Q.; Zheng, S.; Chen, L.; Wu, J.; Li, J.; Liu, P.; Dong, S.; Liu, X.; Peng, Q.; Feng, X. Catalytic Regio-and Enantioselective Protonation for the Synthesis of Chiral Allenes: Synergistic Effect of the Counterion and Water. Angew. Chem. Int. Ed. 2022, 61, e202203650. [Google Scholar] [CrossRef]
- Zheng, J.Y.; Wang, F.; Zhang, Y.; Zheng, Z.; Wu, J.-H.; Ren, X.; Su, Z.; Chen, W.; Wang, T. Novel Stereo-Induction Pattern in Pudovik Addition/Phospha-Brook Rearrangement Towards Chiral Trisubstituted Allenes. Angew. Chem. Int. Ed. 2024, 63, e202403707. [Google Scholar]
- Pallitsch, K.; Roller, A.; Hammerschmidt, F. The Stereochemical Course of the α-Hydroxyphosphonate-Phosphate Rearrangement. Chem.-Eur. J. 2015, 21, 10200–10206. [Google Scholar]
- Prechelmacher, S.; Mereiter, K.; Hammerschmidt, F. The α-Hydroxyphosphonate-Phosphate Rearrangement of a Noncyclic Substrate-Some New Observations. Org. Biomol. Chem. 2018, 16, 3672–3680. [Google Scholar]
- Corbett, M.T.; Uraguchi, D.; Ooi, T.; Johnson, J.S. Base-Catalyzed Direct Aldolization of α-Alkyl-α-Hydroxy Trialkyl Phosphonoacetates. Angew. Chem. Int. Ed. 2012, 51, 4865–4869. [Google Scholar]
- Kondoh, A.; Aoki, T.; Terada, M. Generation and Application of Homoenolate Equivalents Utilizing [1,2]-Phospha-Brook Rearrangement under Brønsted Base Catalysis. Chem.-Eur. J. 2017, 23, 2769–2773. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, A.; Aita, K.; Ishikawa, S.; Terada, M. Synthesis of Tetrasubstituted Furans through One-Pot Formal [3+2] Cycloaddition Utilizing [1,2]-Phospha-Brook Rearrangement. Org. Lett. 2020, 22, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, A.; Aita, K.; Terada, M. Synthesis of 2,3,5,6-Tetrasubstituted Thieno [3,2-b]furans through Formal [3+2] Cycloaddition Utilizing [1,2]-Phospha-Brook Rearrangement/Brønsted Base-Mediated Cyclization Sequence. Adv. Synth. Catal. 2024, 366, 4723–4728. [Google Scholar]
- Kondoh, A.; Suzuki, H.; Hirozane, T.; Terada, M. Catalytic Generation of Benzyl Anions from Aryl Ketones Utilizing [1,2]-Phospha-Brook Rearrangement and Their Application to Synthesis of Tertiary Benzylic Alcohols. Chem. Eur. J. 2024, 30, e202402967. [Google Scholar]
- Kondoh, A.; Terada, M. Catalytic Generation and Intermolecular Addition of Diarylmethyl Anions Utilizing [1,2]-Phospha-Brook Rearrangement Under Brønsted Base Catalysis. Adv. Synth. Catal. 2024, 366, 1857–1862. [Google Scholar] [CrossRef]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Wu, Q.; Huang, Y.; Shi, E.; Li, J. Recent Developments in the [1,2]-Phospha-Brook Rearrangement Reaction. Int. J. Mol. Sci. 2025, 26, 3065. https://doi.org/10.3390/ijms26073065
Li N, Wu Q, Huang Y, Shi E, Li J. Recent Developments in the [1,2]-Phospha-Brook Rearrangement Reaction. International Journal of Molecular Sciences. 2025; 26(7):3065. https://doi.org/10.3390/ijms26073065
Chicago/Turabian StyleLi, Ning, Qian Wu, Yu Huang, Enxue Shi, and Junchen Li. 2025. "Recent Developments in the [1,2]-Phospha-Brook Rearrangement Reaction" International Journal of Molecular Sciences 26, no. 7: 3065. https://doi.org/10.3390/ijms26073065
APA StyleLi, N., Wu, Q., Huang, Y., Shi, E., & Li, J. (2025). Recent Developments in the [1,2]-Phospha-Brook Rearrangement Reaction. International Journal of Molecular Sciences, 26(7), 3065. https://doi.org/10.3390/ijms26073065