
Potential Therapeutic Exploitation of G Protein-Coupled Receptor 120 (GPR120/FFAR4) Signaling in Obesity-Related Metabolic Disorders


Abstract
1. Introduction
2. G Protein-Coupled Receptor (GPCR) Family
2.1. Classification of GPCRs
- -
- The classical A–F system, in which GPCRs are grouped into six classes based on sequence homology and functional similarity
- -
- A newer alternative classification proposed for vertebrates, known by its acronym GRAFS, which stands for glutamate, rhodopsin, adhesion, Frizzled/Taste2, and Secretin. The GRAFS system corresponds to classical classes C, A, B2 (Secretin receptor family, long N-terminal), F, and B1 + 3 (other secretins) [63,64,65].
2.2. G Protein-Coupled Receptor 120 (GPR120)
3. Inflammatory Background of Metabolic Diseases
3.1. Inflammatory Response in Obesity
3.2. Anti-Inflammatory and Metabolic Effects of GPR120 Signaling in the Context of Overweight and Obesity
3.2.1. GPR120 in Adipose Tissue
3.2.2. GPR120 and Gastrointestinal Hormones
3.2.3. GPR120 and the Endocrine Function of the Pancreas
4. Targeting GPR120 Signaling as a Promising Therapeutic Approach in Obesity: The Need for New Ligands
4.1. Non-LCFA GPR120 Agonists Derived from Compounds of Natural Origin
4.2. GPR120 Synthetic Agonists
5. In Summary: Challenges in the Therapeutic Use of GPR120 Agonists and Future Strategies
6. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 7TM receptors | seven transmembrane domain receptors, also known as G protein-coupled receptors (GPCRs) |
| 9-PAHSA | palmitic-acid-9-hydroxy-stearic-acid |
| Ω-3 FAs | omega-3-fatty acids |
| AD | Alzheimer’s disease |
| ALA | alpha-linolenic acid |
| ANGPTL2 | angiopoietin-like 2 protein |
| APCs | antigen-presenting cells |
| ARRβ2 | beta-arrestin 2 |
| AT | adipose tissue |
| ATP | adenosine triphosphate |
| BAT | brown adipose tissue |
| BMI | body mass index |
| cAMP | cyclic adenosine 3,5-monophosphate |
| CCK | cholecystokinin |
| CICR | calcium-induced calcium release |
| CNS | central nervous system |
| cpdA | compound A |
| CRF2 | corticotropin-releasing factor type-2 |
| CRF2R | corticotropin-releasing factor type-2 receptor |
| CTL | cytotoxic T lymphocyte |
| DAG | diacyl glycerol |
| DHA | docosahexaenoic acid |
| DIO | diet-induced obesity |
| DPP-IV | dipeptidyl peptidase-IV |
| EC50 | half maximal effective concentration |
| ECL1–ECL3 | three extracellular loops (from 1 to 3) forming the GPCR |
| eNAMPT | extracellular nicotinamide phosphoribosyl transferase |
| EPA | eicosapentaenoic acid |
| ERK1/2 | extracellular regulated protein kinases 1/2 |
| ERKs | extracellular signal-regulated kinases |
| FDA | U.S. Food and Drug Administration |
| FFAR1 | free fatty acid receptor 1, also known as GPR40 |
| FFAR4 | free fatty acid receptor 4, also known as GPR120 |
| FFAs | free fatty acids |
| FGF-21 | fibroblast growth factor-21 |
| GDP | guanosine diphosphate |
| GEF | guanine nucleotide exchange factor |
| GHIH | growth hormone-inhibiting hormone, also known as somatotropin release inhibiting factor (SRIF) |
| GIP | gastric inhibitory polypeptide, also known as glucose-dependent insulinotropic polypeptide |
| GLP-1 | glucagon-like peptide-1 |
| GLUT4 | glucose transporter type 4 |
| GPCRs | G protein-coupled receptors, also known as seven transmembrane domain (7TM) receptors |
| GPR40 | G protein-coupled receptor 40, also known as FFAR1 |
| GPR120 | G protein-coupled receptor 120, also known as FFAR4 |
| GPR120L | a long form of GPR120, |
| GPR120S | a short form of GPR120 |
| GRAFS | acronym defining classification of GPCRs that stands for glutamate, rhodopsin, adhesion, Frizzled/Taste2, and Secretin |
| GSIS | glucose-stimulated insulin secretion |
| GSSS | glucose-stimulated somatostatin secretion |
| GTP | guanosine triphosphate |
| HbA1c | hemoglobin A1C |
| HDL | high-density lipoprotein |
| HEC | human embryonic kidney |
| HFD | high-fat diet |
| HIF-1 | hypoxia-induced factor 1 |
| ICL1–ICL3 | three intracellular loops (from 1 to 3) forming the GPCR |
| IFN-γ | interferon gamma |
| IKKβ | nuclear factor kappa-B kinase |
| IL1R | interleukin 1 receptor |
| IL-1RA | interleukin 1 receptor antagonist |
| ILs | interleukins, including IL-1β, IL-6, IL-8), |
| iNAMPT | intracellular nicotinamide phosphoribosyl transferase |
| INSR | insulin receptor |
| IP3 | inositol 1,4,5-trisphosphate |
| IR | insulin resistance |
| IRSs | insulin receptor substrates |
| IVGTT | intravenous glucose tolerance test |
| JNK | c-Jun N-terminal kinase |
| KATP | ATP-sensitive potassium channels |
| LA | linolenic acid |
| LCFAs | long-chain fatty acids |
| LPS | lipopolysaccharide |
| MAP3K7 | TGF-β activated kinase 1 |
| M1, M2 macrophages | classically activated and alternatively activated macrophages, respectively |
| MARTs | mono ADP-ribosyl transferases |
| MCFAs | medium-chain fatty acids |
| MCP-1 | monocyte chemoattractant protein-1, also known as CCL2 |
| MMPs | matrix metalloproteinases |
| MiMP | mitochondrial membrane potential |
| NAc | nucleus accumben |
| N.A.D. | denotes no data or ambiguous data |
| NAFLD | nonalcoholic fatty liver disease |
| NAMPT | nicotinamide phosphoribosyl transferase |
| NCG | N-Carbamoyl-Beta-D-Glucopyranosylamine |
| NEMO | nuclear factor kappa-light-chain-enhancer of activated B cells (NF-кB) essential modulator |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 inflammasome | nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR)-containing protein (NLR) family member 3 inflammasome |
| PARPs | poly ADP-ribose polymerases |
| PC1/3 | proprotein convertase 1/3 |
| PDX1 | pancreatic duodenal homeobox-1 |
| pERK1/2 | phosphorylated extracellular regulated protein kinases 1/2 |
| PI3K | phosphoinositide 3-kinase |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| PKC | protein kinase C |
| PLC-β | phospholipase C-beta |
| PPARγ | peroxisome proliferator-activated receptor gamma |
| PUFAs | polyunsaturated fatty acids |
| RAW 264.7 | monocyte/macrophage-like cell line |
| ROS | reactive oxygen species |
| RXRα | retinoid X receptor alpha |
| SAAs | sulfur amino acids |
| S1P | sphingosine-1-phosphate |
| S1P1 | sphingosine-1-phosphate receptor-1 |
| SCFAs | short-chain fatty acids |
| SNP | single-nucleotide polymorphism |
| SPM | specialized pro-resolving mediator |
| SRIF | somatotropin release inhibiting factor, also known as growth hormone-inhibiting hormone (GHIH) |
| STC-1 | secretin tumor cell line 1 |
| S-V cells | stromal–vascular cells |
| T2D | type 2 diabetes mellitus |
| TAB1 | TAK1 binding protein 1 |
| TAK1 | TGFβ-activated kinase 1 |
| TGFβ | transforming growth factor-beta |
| Th1, Th2, Th17 | types of lymphocyte subpopulations |
| TLR4 | Toll-like receptor 4 |
| TM1–TM7 | seven transmembrane heptahelical structures (from 1 to 7) forming GPCR |
| TNFR | tumor necrosis factor receptor |
| UCN3 | urocortin 3 |
| UCP1 | uncoupling protein 1 |
| VEGF | vascular endothelial growth factor |
| WAT | white adipose tissue |
| ZDF | Zucker Diabetic Fatty (rats) |
References
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Tummolo, A.; Carella, R.; De Giovanni, D.; Paterno, G.; Simonetti, S.; Tolomeo, M.; Leone, P.; Barile, M. Micronutrient Deficiency in Inherited Metabolic Disorders Requiring Diet Regimen: A Brief Critical Review. Int. J. Mol. Sci. 2023, 24, 17024. [Google Scholar] [CrossRef]
- Nemer, M.; Osman, F.; Said, A. Dietary macro and micronutrients associated with MASLD: Analysis of a national US cohort database. Ann. Hepatol. 2024, 29, 101491. [Google Scholar] [CrossRef] [PubMed]
- Ahola, A.J.; Harjutsalo, V.; Thorn, L.M.; Freese, R.; Forsblom, C.; Mäkimattila, S.; Groop, P.H. The association between macronutrient intake and the metabolic syndrome and its components in type 1 diabetes. Br. J. Nutr. 2017, 117, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef]
- Safaei, M.; Sundararajan, E.A.; Driss, M.; Boulila, W.; Shapi’i, A. A systematic literature review on obesity: Understanding the causes & consequences of obesity and reviewing various machine learning approaches used to predict obesity. Comput. Biol. Med. 2021, 136, 104754. [Google Scholar] [CrossRef]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef]
- Boutari, C.; Mantzoros, C.S. A 2022 update on the epidemiology of obesity and a call to action: As its twin COVID-19 pandemic appears to be receding, the obesity and dysmetabolism pandemic continues to rage on. Metabolism 2022, 133, 155217. [Google Scholar] [CrossRef]
- World Obesity Federation. World Obesity Atlas 2024; World Obesity Federation: London, UK, 2024; Available online: https://data.worldobesity.org/publications/?cat=22 (accessed on 5 February 2025).
- Zhang, X.; Liu, J.; Ni, Y.; Yi, C.; Fang, Y.; Ning, Q.; Shen, B.; Zhang, K.; Liu, Y.; Yang, L.; et al. Global Prevalence of Overweight and Obesity in Children and Adolescents: A Systematic Review and Meta-Analysis. JAMA Pediatr. 2024, 178, 800–813. [Google Scholar] [CrossRef]
- Nuttall, F.Q. Body Mass Index: Obesity, BMI, and Health: A Critical Review. Nutr. Today 2015, 50, 117–128. [Google Scholar] [CrossRef]
- Tirthani, E.; Said, M.S.; Rehman, A. Genetics and Obesity. [Updated 31 July 2023]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024; Available online: https://www.ncbi.nlm.nih.gov/books/NBK573068/ (accessed on 5 February 2025).
- Smith, E.N.L.; Chandanathil, M.; Millis, R.M. Epigenetic Mechanisms in Obesity: Broadening Our Understanding of the Disease. Cureus 2023, 15, e47875. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Svensson, S.I.A.; Rohde-Zimmermann, K.; Kovacs, P.; Böttcher, Y. Genetics and Epigenetics in Obesity: What Do We Know so Far? Curr. Obes. Rep. 2023, 12, 482–501. [Google Scholar] [CrossRef]
- Lee, A.; Cardel, M.; Donahoo, W.T. Social and Environmental Factors Influencing Obesity. [Updated 12 October 2019]. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000; Available online: https://www.ncbi.nlm.nih.gov/books/NBK278977/ (accessed on 10 February 2025).
- Yang, M.; Liu, S.; Zhang, C. The Related Metabolic Diseases and Treatments of Obesity. Healthcare 2022, 10, 1616. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Haboubi, H.; Haboubi, N. Adult obesity complications: Challenges and clinical impact. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820934955. [Google Scholar] [CrossRef]
- Jin, X.; Qiu, T.; Li, L.; Yu, R.; Chen, X.; Li, C.; Proud, C.G.; Jiang, T. Pathophysiology of obesity and its associated diseases. Acta Pharm. Sin. B 2023, 13, 2403–2424. [Google Scholar] [CrossRef] [PubMed]
- Pinto, K.R.D.; Feckinghaus, C.M.; Hirakata, V.N. Obesity as a predictive factor for chronic kidney disease in adults: Systematic review and meta-analysis. Braz. J. Med. Biol. Res. 2021, 54, e10022. [Google Scholar] [CrossRef]
- Rajesh, Y.; Sarkar, D. Association of Adipose Tissue and Adipokines with Development of Obesity-Induced Liver Cancer. Int. J. Mol. Sci. 2021, 22, 2163. [Google Scholar] [CrossRef]
- Tzenios, N.; Tazanios, M.E.; Chahine, M. The impact of BMI on breast cancer—An updated systematic review and meta-analysis. Medicine 2024, 103, e36831. [Google Scholar] [CrossRef]
- Tzenios, N.; Tazanios, M.E.; Chahine, M. The impact of body mass index on prostate cancer: An updated systematic review and meta-analysis. Medicine 2022, 101, e30191. [Google Scholar] [CrossRef]
- Opio, J.; Wynne, K.; Attia, J.; Oldmeadow, C.; Hancock, S.; Kelly, B.; Inder, K.; McEvoy, M. Metabolic Health, Overweight or Obesity, and Depressive Symptoms among Older Australian Adults. Nutrients 2024, 16, 928. [Google Scholar] [CrossRef]
- Maurizi, G.; Della Guardia, L.; Maurizi, A.; Poloni, A. Adipocytes properties and crosstalk with immune system in obesity-related inflammation. J. Cell. Physiol. 2018, 233, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Savulescu-Fiedler, I.; Mihalcea, R.; Dragosloveanu, S.; Scheau, C.; Baz, R.O.; Caruntu, A.; Scheau, A.E.; Caruntu, C.; Benea, S.N. The Interplay between Obesity and Inflammation. Life 2024, 14, 856. [Google Scholar] [CrossRef]
- Guria, S.; Hoory, A.; Das, S.; Chattopadhyay, D.; Mukherjee, S. Adipose tissue macrophages and their role in obesity-associated insulin resistance: An overview of the complex dynamics at play. Biosci. Rep. 2023, 43, BSR20220200. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef]
- Szukiewicz, D. Molecular Mechanisms for the Vicious Cycle between Insulin Resistance and the Inflammatory Response in Obesity. Int. J. Mol. Sci. 2023, 24, 9818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, T.; Lu, X.; Lan, X.; Chen, Z.; Lu, S. G protein-coupled receptors (GPCRs): Advances in structures, mechanisms, and drug discovery. Signal Transduct. Target. Ther. 2024, 9, 88. [Google Scholar] [CrossRef]
- Liu, S.; Anderson, P.J.; Rajagopal, S.; Lefkowitz, R.J.; Rockman, H.A. G Protein-Coupled Receptors: A Century of Research and Discovery. Circ. Res. 2024, 135, 174–197. [Google Scholar] [CrossRef]
- Liu, H.D.; Wang, W.B.; Xu, Z.G.; Liu, C.H.; He, D.F.; Du, L.P.; Li, M.Y.; Yu, X.; Sun, J.P. FFA4 receptor (GPR120): A hot target for the development of anti-diabetic therapies. Eur. J. Pharmacol. 2015, 763 Pt B, 160–168. [Google Scholar] [CrossRef]
- Al Mahri, S.; Malik, S.S.; Al Ibrahim, M.; Haji, E.; Dairi, G.; Mohammad, S. Free Fatty Acid Receptors (FFARs) in Adipose: Physiological Role and Therapeutic Outlook. Cells 2022, 11, 750. [Google Scholar] [CrossRef]
- Lefkowitz, R.; Kobilka, B. Royal Swedish Academy of Sciences. The Nobel Prize in Chemistry 2012. Retrieved 10 October 2012. Available online: https://www.nobelprize.org/prizes/chemistry/2012/summary/ (accessed on 10 February 2025).
- Vithani, N.; Todd, T.D.; Singh, S.; Trent, T.; Blumer, K.J.; Bowman, G.R. G Protein Activation Occurs via a Largely Universal Mechanism. J. Phys. Chem. B 2024, 128, 3554–3562. [Google Scholar] [CrossRef]
- Afzal, M.S. G proteins: Binary switches in health and disease. Cent. Eur. J. Immunol. 2020, 45, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Kamato, D.; Thach, L.; Bernard, R.; Chan, V.; Zheng, W.; Kaur, H.; Brimble, M.; Osman, N.; Little, P.J. Structure, Function, Pharmacology, and Therapeutic Potential of the G Protein, Gα/q,11. Front. Cardiovasc. Med. 2015, 2, 14. [Google Scholar] [CrossRef]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Dong, D.; Guo, L.; Dong, X.; Leng, J.; Zhao, B.; Guo, Y.D.; Zhang, N. Research Advances in Heterotrimeric G-Protein α Subunits and Uncanonical G-Protein Coupled Receptors in Plants. Int. J. Mol. Sci. 2021, 22, 8678. [Google Scholar] [CrossRef]
- Rehman, S.; Rahimi, N.; Dimri, M. Biochemistry, G Protein Coupled Receptors. [Updated 30 July 2023]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024; Available online: https://www.ncbi.nlm.nih.gov/books/NBK518966/ (accessed on 10 February 2025).
- Odoemelam, C.S.; Percival, B.; Wallis, H.; Chang, M.W.; Ahmad, Z.; Scholey, D.; Burton, E.; Williams, I.H.; Kamerlin, C.L.; Wilson, P.B. G-Protein coupled receptors: Structure and function in drug discovery. RSC Adv. 2020, 10, 36337–36348. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G protein-coupled receptors: Structure and function-based drug discovery. Signal Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef]
- O’Hayre, M.; Vázquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 2013, 13, 412–424. [Google Scholar] [CrossRef]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef]
- Wess, J. Designer GPCRs as Novel Tools to Identify Metabolically Important Signaling Pathways. Front. Endocrinol. 2021, 12, 706957. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef]
- Edward Zhou, X.; Melcher, K.; Eric Xu, H. Structural biology of G protein-coupled receptor signaling complexes. Protein Sci. 2019, 28, 487–501. [Google Scholar] [CrossRef]
- Cheng, L.; Xia, F.; Li, Z.; Shen, C.; Yang, Z.; Hou, H.; Sun, S.; Feng, Y.; Yong, X.; Tian, X.; et al. Structure, function and drug discovery of GPCR signaling. Mol. Biomed. 2023, 4, 46. [Google Scholar] [CrossRef]
- Laschet, C.; Dupuis, N.; Hanson, J. The G protein-coupled receptors deorphanization landscape. Biochem. Pharmacol. 2018, 153, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20, G protein-coupled receptors. Br. J. Pharmacol. 2019, 176 (Suppl. S1), S21–S141. [Google Scholar] [CrossRef]
- Santhanam, B.; Sluter, M.; Babu, M.M. Exploring GPCR signaling pathway networks as cancer therapeutic targets. Cell Genom. 2024, 4, 100560. [Google Scholar] [CrossRef] [PubMed]
- Arora, C.; Matic, M.; Bisceglia, L.; Di Chiaro, P.; De Oliveira Rosa, N.; Carli, F.; Clubb, L.; Nemati Fard, L.A.; Kargas, G.; Diaferia, G.R.; et al. The landscape of cancer-rewired GPCR signaling axes. Cell Genom. 2024, 4, 100557. [Google Scholar] [CrossRef]
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735, Erratum in Circ. Res. 2018, 123, e34. https://doi.org/10.1161/RES.0000000000000235. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Deng, Y.; Jiang, Z.; Qing, H. G Protein-Coupled Receptors (GPCRs) in Alzheimer’s Disease: A Focus on BACE1 Related GPCRs. Front. Aging Neurosci. 2016, 8, 58. [Google Scholar] [CrossRef]
- Wong, T.S.; Li, G.; Li, S.; Gao, W.; Chen, G.; Gan, S.; Zhang, M.; Li, H.; Wu, S.; Du, Y. G protein-coupled receptors in neurodegenerative diseases and psychiatric disorders. Signal Transduct. Target. Ther. 2023, 8, 177. [Google Scholar] [CrossRef]
- Schöneberg, T.; Liebscher, I. Mutations in G Protein-Coupled Receptors: Mechanisms, Pathophysiology and Potential Therapeutic Approaches. Pharmacol. Rev. 2021, 73, 89–119. [Google Scholar] [CrossRef]
- Thompson, M.D.; Percy, M.E.; Cole, D.E.C.; Bichet, D.G.; Hauser, A.S.; Gorvin, C.M. G protein-coupled receptor (GPCR) gene variants and human genetic disease. Crit. Rev. Clin. Lab. Sci. 2024, 61, 317–346. [Google Scholar] [CrossRef] [PubMed]
- Secundo, L.; Snitz, K.; Weissler, K.; Pinchover, L.; Shoenfeld, Y.; Loewenthal, R.; Agmon-Levin, N.; Frumin, I.; Bar-Zvi, D.; Shushan, S.; et al. Individual olfactory perception reveals meaningful nonolfactory genetic information. Proc. Natl. Acad. Sci. USA 2015, 112, 8750–8755. [Google Scholar] [CrossRef]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR drug discovery: Integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 2019, 18, 59–82. [Google Scholar] [CrossRef] [PubMed]
- Saikia, S.; Bordoloi, M.; Sarmah, R. Established and In-trial GPCR Families in Clinical Trials: A Review for Target Selection. Curr. Drug Targets 2019, 20, 522–539. [Google Scholar] [CrossRef]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54.e19. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Scholz, N.; Langenhan, T.; Schöneberg, T. Revisiting the classification of adhesion GPCRs. Ann. N. Y. Acad. Sci. 2019, 1456, 80–95. [Google Scholar] [CrossRef]
- Krishnan, A.; Nijmeijer, S.; de Graaf, C.; Schiöth, H.B. Classification, Nomenclature, and Structural Aspects of Adhesion GPCRs. Handb. Exp. Pharmacol. 2016, 234, 15–41. [Google Scholar] [CrossRef]
- Wittlake, A.; Prömel, S.; Schöneberg, T. The Evolutionary History of Vertebrate Adhesion GPCRs and Its Implication on Their Classification. Int. J. Mol. Sci. 2021, 22, 11803. [Google Scholar] [CrossRef]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Common activation mechanism of class A GPCRs. eLife 2019, 8, e50279. [Google Scholar] [CrossRef]
- Stuttgen, G.M.; Sahoo, D. FFAR4, A New Player in Cardiometabolic Disease? Endocrinology 2021, 162, bqab111. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Gu, Z.; Heid, B.; Akers, R.M.; Jiang, H. Identification and characterization of the bovine G protein-coupled receptor GPR41 and GPR43 genes. J. Dairy Sci. 2009, 92, 2696–2705. [Google Scholar] [CrossRef]
- Ang, Z.; Ding, J.L. GPR41 and GPR43 in Obesity and Inflammation - Protective or Causative? Front. Immunol. 2016, 7, 28. [Google Scholar] [CrossRef]
- Luscombe, V.B.; Lucy, D.; Bataille, C.J.R.; Russell, A.J.; Greaves, D.R. 20 Years an Orphan: Is GPR84 a Plausible Medium-Chain Fatty Acid-Sensing Receptor? DNA Cell Biol. 2020, 39, 1926–1937. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G.; Alvarez-Curto, E.; Watterson, K.R.; Ulven, T.; Hudson, B.D. Characterizing pharmacological ligands to study the long-chain fatty acid receptors GPR40/FFA1 and GPR120/FFA4. Br. J. Pharmacol. 2015, 172, 3254–3265. [Google Scholar] [CrossRef] [PubMed]
- Son, S.E.; Kim, N.J.; Im, D.S. Development of Free Fatty Acid Receptor 4 (FFA4/GPR120) Agonists in Health Science. Biomol. Ther. 2021, 29, 22–30. [Google Scholar] [CrossRef]
- Galindo, M.M.; Voigt, N.; Stein, J.; van Lengerich, J.; Raguse, J.D.; Hofmann, T.; Meyerhof, W.; Behrens, M. G protein-coupled receptors in human fat taste perception. Chem. Senses 2012, 37, 123–139. [Google Scholar] [CrossRef]
- Carullo, G.; Mazzotta, S.; Vega-Holm, M.; Iglesias-Guerra, F.; Vega-Pérez, J.M.; Aiello, F.; Brizzi, A. GPR120/FFAR4 Pharmacology: Focus on Agonists in Type 2 Diabetes Mellitus Drug Discovery. J. Med. Chem. 2021, 64, 4312–4332. [Google Scholar] [CrossRef]
- Moore, K.; Zhang, Q.; Murgolo, N.; Hosted, T.; Duffy, R. Cloning, expression, and pharmacological characterization of the GPR120 free fatty acid receptor from cynomolgus monkey: Comparison with human GPR120 splice variants. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2009, 154, 419–426. [Google Scholar] [CrossRef]
- Watson, S.J.; Brown, A.J.; Holliday, N.D. Differential signaling by splice variants of the human free fatty acid receptor GPR120. Mol. Pharmacol. 2012, 81, 631–642. [Google Scholar] [CrossRef]
- Zhang, X.; Guseinov, A.A.; Jenkins, L.; Li, K.; Tikhonova, I.G.; Milligan, G.; Zhang, C. Structural basis for the ligand recognition and signaling of free fatty acid receptors. Sci. Adv. 2024, 10, eadj2384. [Google Scholar] [CrossRef] [PubMed]
- Sadler, F.; Ma, N.; Ritt, M.; Sharma, Y.; Vaidehi, N.; Sivaramakrishnan, S. Autoregulation of GPCR signalling through the third intracellular loop. Nature 2023, 615, 734–741. [Google Scholar] [CrossRef]
- Burns, R.N.; Moniri, N.H. Agonism with the omega-3 fatty acids alpha-linolenic acid and docosahexaenoic acid mediates phosphorylation of both the short and long isoforms of the human GPR120 receptor. Biochem. Biophys. Res. Commun. 2010, 396, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free Fatty Acid Receptors in Health and Disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef] [PubMed]
- Vestmar, M.A.; Andersson, E.A.; Christensen, C.R.; Hauge, M.; Glümer, C.; Linneberg, A.; Witte, D.R.; Jørgensen, M.E.; Christensen, C.; Brandslund, I.; et al. Functional and genetic epidemiological characterisation of the FFAR4 (GPR120) p.R270H variant in the Danish population. J. Med. Genet. 2016, 53, 616–623. [Google Scholar] [CrossRef]
- Oh, D.Y.; Walenta, E. Omega-3 Fatty Acids and FFAR4. Front. Endocrinol. 2014, 5, 115. [Google Scholar] [CrossRef]
- Cheshmehkani, A.; Senatorov, I.S.; Kandi, P.; Singh, M.; Britt, A.; Hayslett, R.; Moniri, N.H. Fish oil and flax seed oil supplemented diets increase FFAR4 expression in the rat colon. Inflamm. Res. 2015, 64, 809–815. [Google Scholar] [CrossRef]
- Yin, J.; Li, H.; Meng, C.; Chen, D.; Chen, Z.; Wang, Y.; Wang, Z.; Chen, G. Inhibitory effects of omega-3 fatty acids on early brain injury after subarachnoid hemorrhage in rats: Possible involvement of G protein-coupled receptor 120/β-arrestin2/TGF-β activated kinase-1 binding protein-1 signaling pathway. Int. J. Biochem. Cell Biol. 2016, 75, 11–22. [Google Scholar] [CrossRef]
- Cornall, L.M.; Mathai, M.L.; Hryciw, D.H.; McAinch, A.J. Diet-induced obesity up-regulates the abundance of GPR43 and GPR120 in a tissue specific manner. Cell. Physiol. Biochem. 2011, 28, 949–958. [Google Scholar] [CrossRef]
- Karakuła-Juchnowicz, H.; Róg, J.; Juchnowicz, D.; Morylowska-Topolska, J. GPR120, Mechanism of action, role and potential for medical applications. Postępy Hig. Med. Doświadczalnej 2017, 71, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, C.; Hong, Y.H.; Iga, T.; Hishikawa, D.; Suzuki, Y.; Song, S.H.; Choi, K.C.; Adachi, T.; Hirasawa, A.; Tsujimoto, G.; et al. The regulation of adipogenesis through GPR120. Biochem. Biophys. Res. Commun. 2007, 354, 591–597. [Google Scholar] [CrossRef]
- Moriyama, R.; Deura, C.; Imoto, S.; Nose, K.; Fukushima, N. Expression of the long-chain fatty acid receptor GPR120 in the gonadotropes of the mouse anterior pituitary gland. Histochem. Cell Biol. 2015, 143, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Noda, K.; Miwa, M.; Minabe, S.; Hagiwara, T.; Hirasawa, A.; Matsuyama, S.; Moriyama, R. Colocalization of GPR120 and anterior pituitary hormone-producing cells in female Japanese Black cattle. J. Reprod. Dev. 2020, 66, 135–141. [Google Scholar] [CrossRef]
- Deura, C.; Kimura, Y.; Nonoyama, T.; Moriyama, R. Gpr120 mRNA expression in gonadotropes in the mouse pituitary gland is regulated by free fatty acids. J. Reprod. Dev. 2020, 66, 249–254. [Google Scholar] [CrossRef]
- Duah, M.; Zhang, K.; Liang, Y.; Ayarick, V.A.; Xu, K.; Pan, B. Immune regulation of poly unsaturated fatty acids and free fatty acid receptor 4. J. Nutr. Biochem. 2023, 112, 109222. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Lee, K.P.; Park, S.J.; Kang, S.; Huang, J.; Lee, J.M.; Sato, K.; Chung, H.Y.; Okajima, F.; Im, D.S. Omega-3 fatty acids induce Ca(2+) mobilization responses in human colon epithelial cell lines endogenously expressing FFA4. Acta Pharmacol. Sin. 2015, 36, 813–820. [Google Scholar] [CrossRef]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef]
- Kasonga, A.E.; Kruger, M.C.; Coetzee, M. Free fatty acid receptor 4-β-arrestin 2 pathway mediates the effects of different classes of unsaturated fatty acids in osteoclasts and osteoblasts. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 281–289. [Google Scholar] [CrossRef]
- Alharbi, A.G.; Tobin, A.B.; Milligan, G. How Arrestins and GRKs Regulate the Function of Long Chain Fatty Acid Receptors. Int. J. Mol. Sci. 2022, 23, 12237. [Google Scholar] [CrossRef]
- Schulte, G.; Fredholm, B.B. Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal 2003, 15, 813–827. [Google Scholar] [CrossRef]
- Anbazhagan, A.N.; Priyamvada, S.; Gujral, T.; Bhattacharyya, S.; Alrefai, W.A.; Dudeja, P.K.; Borthakur, A. A novel anti-inflammatory role of GPR120 in intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2016, 310, C612–C621. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Y.; Coin, I. Biochemical insights into structure and function of arrestins. FEBS J. 2021, 288, 2529–2549. [Google Scholar] [CrossRef]
- Zhao, Y.F. Free fatty acid receptors in the endocrine regulation of glucose metabolism: Insight from gastrointestinal-pancreatic-adipose interactions. Front. Endocrinol. 2022, 13, 956277. [Google Scholar] [CrossRef]
- Sidhu, S.S.; Thompson, D.G.; Warhurst, G.; Case, R.M.; Benson, R.S. Fatty acid-induced cholecystokinin secretion and changes in intracellular Ca2+ in two enteroendocrine cell lines, STC-1 and GLUTag. J. Physiol. 2000, 528 Pt 1, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 2001, 70, 281–312. [Google Scholar] [CrossRef] [PubMed]
- Danciu, T.E.; Adam, R.M.; Naruse, K.; Freeman, M.R.; Hauschka, P.V. Calcium regulates the PI3K-Akt pathway in stretched osteoblasts. FEBS Lett. 2003, 536, 193–197. [Google Scholar] [CrossRef]
- Liang, Y.; Yin, W.; Yin, Y.; Zhang, W. Ghrelin Based Therapy of Metabolic Diseases. Curr. Med. Chem. 2021, 28, 2565–2576. [Google Scholar] [CrossRef]
- Bang, A.S.; Soule, S.G.; Yandle, T.G.; Richards, A.M.; Pemberton, C.J. Characterisation of proghrelin peptides in mammalian tissue and plasma. J. Endocrinol. 2007, 192, 313–323. [Google Scholar] [CrossRef]
- Simonds, W.F. G protein regulation of adenylate cyclase. Trends Pharmacol. Sci. 1999, 20, 66–73. [Google Scholar] [CrossRef]
- Fredriksson, R.; Höglund, P.J.; Gloriam, D.E.; Lagerström, M.C.; Schiöth, H.B. Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett. 2003, 554, 381–388. [Google Scholar] [CrossRef] [PubMed]
- da Silva Batista, E.; Nakandakari, S.C.B.R.; Ramos da Silva, A.S.; Pauli, J.R.; Pereira de Moura, L.; Ropelle, E.R.; Camargo, E.A.; Cintra, D.E. Omega-3 pleiad: The multipoint anti-inflammatory strategy. Crit. Rev. Food Sci. Nutr. 2024, 64, 4817–4832. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lei, X.T.; Huang, Q.; Wang, T.; Sun, H.B.; Wang, H.Y. A novel GPR120-selective agonist promotes insulin secretion and improves chronic inflammation. Life Sci. 2021, 269, 119029. [Google Scholar] [CrossRef]
- Suckow, A.T.; Polidori, D.; Yan, W.; Chon, S.; Ma, J.Y.; Leonard, J.; Briscoe, C.P. Alteration of the glucagon axis in GPR120 (FFAR4) knockout mice: A role for GPR120 in glucagon secretion. J. Biol. Chem. 2014, 289, 15751–15763. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; Zhang, Z. Recent Advance in Regulatory Effect of GRP120 on Bone Metabolism. Aging Dis. 2023, 14, 1714–1727. [Google Scholar] [CrossRef]
- Zhang, X.; Macielag, M.J. GPR120 agonists for the treatment of diabetes: A patent review (2014 present). Expert. Opin. Ther. Pat. 2020, 30, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Qian, Y.; Wu, M.S.; Petrov, A.; Dai, G.; Wang, S.P.; Zhu, Y.; Shen, X.; Muise, E.S.; Chen, Y.; et al. GPR120 suppresses adipose tissue lipolysis and synergizes with GPR40 in antidiabetic efficacy. J. Lipid Res. 2017, 58, 1561–1578. [Google Scholar] [CrossRef]
- Sun, Q.; Li, J.; Gao, F. New insights into insulin: The anti-inflammatory effect and its clinical relevance. World J. Diabetes 2014, 5, 89–96. [Google Scholar] [CrossRef]
- Chang, Y.W.; Hung, L.C.; Chen, Y.C.; Wang, W.H.; Lin, C.Y.; Tzeng, H.H.; Suen, J.L.; Chen, Y.H. Insulin Reduces Inflammation by Regulating the Activation of the NLRP3 Inflammasome. Front. Immunol. 2021, 11, 587229. [Google Scholar] [CrossRef]
- Chen, Q.; Yu, W.; Shi, J.; Shen, J.; Gao, T.; Zhang, J.; Xi, F.; Li, J.; Li, N. Insulin alleviates the inflammatory response and oxidative stress injury in cerebral tissues in septic rats. J. Inflamm. 2014, 11, 18. [Google Scholar] [CrossRef]
- Huang, C.T.; Lue, J.H.; Cheng, T.H.; Tsai, Y.J. Glycemic control with insulin attenuates sepsis-associated encephalopathy by inhibiting glial activation via the suppression of the nuclear factor kappa B and mitogen-activated protein kinase signaling pathways in septic rats. Brain Res. 2020, 1738, 146822. [Google Scholar] [CrossRef]
- Khanna, D.; Khanna, S.; Khanna, P.; Kahar, P.; Patel, B.M. Obesity: A Chronic Low-Grade Inflammation and Its Markers. Cureus 2022, 14, e22711. [Google Scholar] [CrossRef] [PubMed]
- Matuschik, L.; Riabov, V.; Schmuttermaier, C.; Sevastyanova, T.; Weiss, C.; Klüter, H.; Kzhyshkowska, J. Hyperglycemia Induces Inflammatory Response of Human Macrophages to CD163-Mediated Scavenging of Hemoglobin-Haptoglobin Complexes. Int. J. Mol. Sci. 2022, 23, 1385. [Google Scholar] [CrossRef]
- Moganti, K.; Li, F.; Schmuttermaier, C.; Riemann, S.; Klüter, H.; Gratchev, A.; Harmsen, M.C.; Kzhyshkowska, J. Hyperglycemia induces mixed M1/M2 cytokine profile in primary human monocyte-derived macrophages. Immunobiology 2017, 222, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lan, C.; Benlagha, K.; Camara, N.O.S.; Miller, H.; Kubo, M.; Heegaard, S.; Lee, P.; Yang, L.; Forsman, H.; et al. The interaction of innate immune and adaptive immune system. MedComm (2020) 2024, 5, e714. [Google Scholar] [CrossRef]
- Engin, A.B. Adipocyte-Macrophage Cross-Talk in Obesity. Adv. Exp. Med. Biol. 2017, 960, 327–343. [Google Scholar] [CrossRef]
- Hu, T.; Liu, C.H.; Lei, M.; Zeng, Q.; Li, L.; Tang, H.; Zhang, N. Metabolic regulation of the immune system in health and diseases: Mechanisms and interventions. Signal Transduct. Target. Ther. 2024, 9, 268. [Google Scholar] [CrossRef] [PubMed]
- Ignacio, R.M.; Kim, C.S.; Kim, S.K. Immunological Profiling of Obesity. J. Lifestyle Med. 2014, 4, 1–7. [Google Scholar] [CrossRef]
- Li, X.; Ren, Y.; Chang, K.; Wu, W.; Griffiths, H.R.; Lu, S.; Gao, D. Adipose tissue macrophages as potential targets for obesity and metabolic diseases. Front. Immunol. 2023, 14, 1153915. [Google Scholar] [CrossRef]
- Rowan, C.R.; McManus, J.; Boland, K.; O’Toole, A. Visceral adiposity and inflammatory bowel disease. Int. J. Colorectal Dis. 2021, 36, 2305–2319. [Google Scholar] [CrossRef]
- Santillana, N.; Astudillo-Guerrero, C.; D’Espessailles, A.; Cruz, G. White Adipose Tissue Dysfunction: Pathophysiology and Emergent Measurements. Nutrients 2023, 15, 1722. [Google Scholar] [CrossRef]
- Lee, Y.H.; Park, J.; Min, S.; Kang, O.; Kwon, H.; Oh, S.W. Impact of Visceral Obesity on the Risk of Incident Metabolic Syndrome in Metabolically Healthy Normal Weight and Overweight Groups: A Longitudinal Cohort Study in Korea. Korean J. Fam. Med. 2020, 41, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Mongraw-Chaffin, M.; Hairston, K.G.; Hanley, A.J.G.; Tooze, J.A.; Norris, J.M.; Palmer, N.D.; Bowden, D.W.; Lorenzo, C.; Chen, Y.I.; Wagenknecht, L.E. Association of Visceral Adipose Tissue and Insulin Resistance with Incident Metabolic Syndrome Independent of Obesity Status: The IRAS Family Study. Obesity 2021, 29, 1195–1202. [Google Scholar] [CrossRef]
- Navaneeth, G.C.; Hiremath, R.; Poojary, S.R.; Kini, D.V.; Chittaragi, K.B. Computed tomographic abdominal fat volume estimation—A handy tool to predict the risk of metabolic syndrome. Pol. J. Radiol. 2023, 88, e379–e388. [Google Scholar] [CrossRef] [PubMed]
- Daryabor, G.; Kabelitz, D.; Kalantar, K. An update on immune dysregulation in obesity-related insulin resistance. Scand. J. Immunol. 2019, 89, e12747. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Yuan, M.; Wang, Y.; Wang, Y.; Bao, W.; Zeng, S.; Zhang, D.; Liu, P.; Sun, L. Association Between Visceral Obesity and Glycemic Control in Patients with Type 2 Diabetes Mellitus: A Retrospective Study. Diabetes Metab. Syndr. Obes. 2024, 17, 2869–2880. [Google Scholar] [CrossRef]
- Doyle, S.L.; Donohoe, C.L.; Lysaght, J.; Reynolds, J.V. Visceral obesity, metabolic syndrome, insulin resistance and cancer. Proc. Nutr. Soc. 2012, 71, 181–189. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, Y.; Chen, Y.; Liu, T.; Liu, C.; Lin, S.; Xie, H.; Ma, X.; Wang, Z.; Shi, J.; et al. Metabolic obesity phenotypes and the risk of cancer: A prospective study of the Kailuan cohort. Front. Endocrinol. 2024, 15, 1333488. [Google Scholar] [CrossRef]
- Rumyantsev, K.A.; Polyakova, V.V.; Sorokina, I.V.; Feoktistova, P.S.; Khatkov, I.E.; Bodunova, N.A.; Zhukova, L.G. The Gut Microbiota Impacts Gastrointestinal Cancers through Obesity, Diabetes, and Chronic Inflammation. Life 2024, 14, 1219. [Google Scholar] [CrossRef]
- Kolb, H. Obese visceral fat tissue inflammation: From protective to detrimental? BMC Med. 2022, 20, 494. [Google Scholar] [CrossRef]
- Yu, J.Y.; Choi, W.J.; Lee, H.S.; Lee, J.W. Relationship between inflammatory markers and visceral obesity in obese and overweight Korean adults: An observational study. Medicine 2019, 98, e14740. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meng, Y.; He, S.; Tan, X.; Zhang, Y.; Zhang, X.; Wang, L.; Zheng, W. Macrophages, Chronic Inflammation, and Insulin Resistance. Cells 2022, 11, 3001. [Google Scholar] [CrossRef]
- Graßmann, S.; Wirsching, J.; Eichelmann, F.; Aleksandrova, K. Association Between Peripheral Adipokines and Inflammation Markers: A Systematic Review and Meta-Analysis. Obesity 2017, 25, 1776–1785. [Google Scholar] [CrossRef] [PubMed]
- Wnuk, A.; Stangret, A.; Wątroba, M.; Płatek, A.E.; Skoda, M.; Cendrowski, K.; Sawicki, W.; Szukiewicz, D. Can adipokine visfatin be a novel marker of pregnancy-related disorders in women with obesity? Obes. Rev. 2020, 21, e13022. [Google Scholar] [CrossRef]
- Kirichenko, T.V.; Markina, Y.V.; Bogatyreva, A.I.; Tolstik, T.V.; Varaeva, Y.R.; Starodubova, A.V. The Role of Adipokines in Inflammatory Mechanisms of Obesity. Int. J. Mol. Sci. 2022, 23, 14982. [Google Scholar] [CrossRef] [PubMed]
- Saddi-Rosa, P.; Oliveira, C.S.; Giuffrida, F.M.; Reis, A.F. Visfatin, glucose metabolism and vascular disease: A review of evidence. Diabetol. Metab. Syndr. 2010, 2, 21. [Google Scholar] [CrossRef]
- Dakroub, A.; ANasser, S.; Younis, N.; Bhagani, H.; Al-Dhaheri, Y.; Pintus, G.; Eid, A.A.; El-Yazbi, A.F.; Eid, A.H. Visfatin: A Possible Role in Cardiovasculo-Metabolic Disorders. Cells 2020, 9, 2444. [Google Scholar] [CrossRef]
- Abdalla, M.M.I. Role of visfatin in obesity-induced insulin resistance. World J. Clin. Cases 2022, 10, 10840–10851. [Google Scholar] [CrossRef]
- McLaughlin, T.; Ackerman, S.E.; Shen, L.; Engleman, E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J. Clin. Investig. 2017, 127, 5–13. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hotamisligil, G.S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Investig. 2003, 112, 1785–1788. [Google Scholar] [CrossRef] [PubMed]
- Pitharouli, M.C.; Hagenaars, S.P.; Glanville, K.P.; Coleman, J.R.I.; Hotopf, M.; Lewis, C.M.; Pariante, C.M. Elevated C-Reactive Protein in Patients With Depression, Independent of Genetic, Health, and Psychosocial Factors: Results From the UK Biobank. Am. J. Psychiatry 2021, 178, 522–529. [Google Scholar] [CrossRef]
- Parkitny, L.; McAuley, J.H.; Di Pietro, F.; Stanton, T.R.; O’Connell, N.E.; Marinus, J.; van Hilten, J.J.; Moseley, G.L. Inflammation in complex regional pain syndrome: A systematic review and meta-analysis. Neurology 2013, 80, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Rodelo, C.; Arellano-Plancarte, A.; Hernandez-Aranda, J.; Landa-Galvan, H.V.; Parra-Mercado, G.K.; Moreno-Licona, N.J.; Hernandez-Gonzalez, K.D.; Catt, K.J.; Villalobos-Molina, R.; Olivares-Reyes, J.A. Angiotensin II Inhibits Insulin Receptor Signaling in Adipose Cells. Int. J. Mol. Sci. 2022, 23, 6048. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.K.; Jang, Y.J.; Park, H.S.; Kim, J.H.; Hong, J.P.; Lee, Y.J.; Heo, Y.S. Enhanced ANGPTL2 expression in adipose tissues and its association with insulin resistance in obese women. Sci. Rep. 2018, 8, 13976. [Google Scholar] [CrossRef]
- Corbin, J.A.; Bhaskar, V.; Goldfine, I.D.; Issafras, H.; Bedinger, D.H.; Lau, A.; Michelson, K.; Gross, L.M.; Maddux, B.A.; Kuan, H.F.; et al. Inhibition of insulin receptor function by a human, allosteric monoclonal antibody: A potential new approach for the treatment of hyperinsulinemic hypoglycemia. MAbs 2014, 6, 262–272. [Google Scholar] [CrossRef]
- Willard, D.L.; Stevenson, M.; Steenkamp, D. Type B insulin resistance syndrome. Curr. Opin. Endocrinol. Diabetes Obes. 2016, 23, 318–323. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Pei, J.; Wang, B.; Wang, D. Current Studies on Molecular Mechanisms of Insulin Resistance. J. Diabetes Res. 2022, 2022, 1863429. [Google Scholar] [CrossRef]
- Lorenzo, M.; Fernández-Veledo, S.; Vila-Bedmar, R.; Garcia-Guerra, L.; De Alvaro, C.; Nieto-Vazquez, I. Insulin resistance induced by tumor necrosis factor-alpha in myocytes and brown adipocytes. J. Anim. Sci. 2008, 86 (Suppl. S14), E94–E104. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Mechanistic Insight into Oxidative Stress-Triggered Signaling Pathways and Type 2 Diabetes. Molecules 2022, 27, 950. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.M.; Pennings, N. Insulin Resistance. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; An, X.; Yang, C.; Sun, W.; Ji, H.; Lian, F. The crucial role and mechanism of insulin resistance in metabolic disease. Front. Endocrinol. 2023, 14, 1149239. [Google Scholar] [CrossRef] [PubMed]
- Auguet, T.; Guiu-Jurado, E.; Berlanga, A.; Terra, X.; Martinez, S.; Porras, J.A.; Ceausu, A.; Sabench, F.; Hernandez, M.; Aguilar, C.; et al. Downregulation of lipogenesis and fatty acid oxidation in the subcutaneous adipose tissue of morbidly obese women. Obesity 2014, 22, 2032–2038. [Google Scholar] [CrossRef]
- Zhou, W.; Ramachandran, D.; Mansouri, A.; Dailey, M.J. Glucose stimulates intestinal epithelial crypt proliferation by modulating cellular energy metabolism. J. Cell Physiol. 2018, 233, 3465–3475. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Duca, F.A.; Swartz, T.D.; Sakar, Y.; Covasa, M. Decreased intestinal nutrient response in diet-induced obese rats: Role of gut peptides and nutrient receptors. Int. J. Obes. 2013, 37, 375–381. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, T.; Zhang, D.; Leung, P.S. GPR120 protects lipotoxicity-induced pancreatic β-cell dysfunction through regulation of PDX1 expression and inhibition of islet inflammation. Clin. Sci. 2019, 133, 101–116. [Google Scholar] [CrossRef]
- Miyabe, M.; Gin, A.; Onozawa, E.; Daimon, M.; Yamada, H.; Oda, H.; Mori, A.; Momota, Y.; Azakami, D.; Yamamoto, I.; et al. Genetic variants of the unsaturated fatty acid receptor GPR120 relating to obesity in dogs. J. Vet. Med. Sci. 2015, 77, 1201–1206. [Google Scholar] [CrossRef]
- Luo, L.; Liu, M. Adipose tissue in control of metabolism. J. Endocrinol. 2016, 231, R77–R99. [Google Scholar] [CrossRef]
- Stojko, M.; Spychał, A.; Nikel, K.; Kołodziej, R.; Zalejska-Fiolka, J. The Impact of Diet on Lipoprotein(a) Levels. Life 2024, 14, 1403. [Google Scholar] [CrossRef]
- Wallace, T.C. Health Effects of Coconut Oil-A Narrative Review of Current Evidence. J. Am. Coll. Nutr. 2019, 38, 97–107. [Google Scholar] [CrossRef]
- Panchal, S.K.; Carnahan, S.; Brown, L. Coconut Products Improve Signs of Diet-Induced Metabolic Syndrome in Rats. Plant Foods Hum. Nutr. 2017, 72, 418–424. [Google Scholar] [CrossRef]
- Araújo de Vasconcelos, M.H.; Tavares, R.L.; Dutra, M.L.D.V.; Batista, K.S.; D’Oliveira, A.B.; Pinheiro, R.O.; Pereira, R.A.; Lima, M.D.S.; Salvadori, M.G.D.S.S.; de Souza, E.L.; et al. Extra virgin coconut oil (Cocos nucifera L.) intake shows neurobehavioural and intestinal health effects in obesity-induced rats. Food Funct. 2023, 14, 6455–6469. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Qadri, S.; Luukkonen, P.K.; Ragnarsdottir, O.; McGlinchey, A.; Jäntti, S.; Juuti, A.; Arola, J.; Schlezinger, J.J.; Webster, T.F.; et al. Exposure to environmental contaminants is associated with altered hepatic lipid metabolism in non-alcoholic fatty liver disease. J. Hepatol. 2022, 76, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Fang, R.; Wang, H.; Xu, D.X.; Yang, J.; Huang, X.; Cozzolino, D.; Fang, M.; Huang, Y. A review of environmental metabolism disrupting chemicals and effect biomarkers associating disease risks: Where exposomics meets metabolomics. Environ. Int. 2022, 158, 106941. [Google Scholar] [CrossRef]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef] [PubMed]
- An, S.M.; Cho, S.H.; Yoon, J.C. Adipose Tissue and Metabolic Health. Diabetes Metab. J. 2023, 47, 595–611. [Google Scholar] [CrossRef]
- Teixeira, L.; Whitmire, J.K.; Bourgeois, C. Editorial: The role of adipose tissue and resident immune cells in infections. Front. Immunol. 2024, 15, 1360262. [Google Scholar] [CrossRef]
- Rosenwald, M.; Wolfrum, C. The origin and definition of brite versus white and classical brown adipocytes. Adipocyte 2014, 3, 4–9. [Google Scholar] [CrossRef]
- Chen, Y.; Pan, R.; Pfeifer, A. Fat tissues, the brite and the dark sides. Pflugers Arch. 2016, 468, 1803–1807. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Yamada, T. UCP1 Dependent and Independent Thermogenesis in Brown and Beige Adipocytes. Front. Endocrinol. 2020, 11, 498. [Google Scholar] [CrossRef]
- Kazak, L.; Chouchani, E.T.; Jedrychowski, M.P.; Erickson, B.K.; Shinoda, K.; Cohen, P.; Vetrivelan, R.; Lu, G.Z.; Laznik-Bogoslavski, D.; Hasenfuss, S.C.; et al. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell 2015, 163, 643–655. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Kazak, L.; Chouchani, E.T.; Bogaczyńska, M.G.; Paranjpe, I.; Wainwright, G.L.; Bétourné, A.; Kajimura, S.; Spiegelman, B.M.; Kirichok, Y. Mitochondrial Patch Clamp of Beige Adipocytes Reveals UCP1-Positive and UCP1-Negative Cells Both Exhibiting Futile Creatine Cycling. Cell Metab. 2017, 25, 811–822.e4. [Google Scholar] [CrossRef]
- van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508, Erratum in N. Engl. J. Med. 2009, 360, 1917. https://doi.org/10.1056/NEJMoa0808718. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, D.; Xiang, J.; Zhou, J.; Cao, H.; Che, Q.; Bai, Y.; Guo, J.; Su, Z. Non-shivering Thermogenesis Signalling Regulation and Potential Therapeutic Applications of Brown Adipose Tissue. Int. J. Biol. Sci. 2021, 17, 2853–2870. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Delgado, G.; Martinez-Tellez, B.; Acosta, F.M.; Virtue, S.; Vidal-Puig, A.; Gil, A.; Llamas-Elvira, J.M.; Ruiz, J.R. Brown Adipose Tissue Volume and Fat Content Are Positively Associated With Whole-Body Adiposity in Young Men-Not in Women. Diabetes 2021, 70, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Chiadak, J.D.; Arsenijevic, T.; Verstrepen, K.; Gregoire, F.; Bolaky, N.; Delforge, V.; Flamand, V.; Perret, J.; Delporte, C. Forskolin Inhibits Lipopolysaccharide-Induced Modulation of MCP-1 and GPR120 in 3T3-L1 Adipocytes through an Inhibition of NFκB. Mediat. Inflamm. 2016, 2016, 1431789. [Google Scholar] [CrossRef]
- Song, T.; Yang, Y.; Zhou, Y.; Wei, H.; Peng, J. GPR120, a critical role in adipogenesis, inflammation, and energy metabolism in adipose tissue. Cell Mol. Life Sci. 2017, 74, 2723–2733. [Google Scholar] [CrossRef]
- Otto, T.C.; Lane, M.D. Adipose development: From stem cell to adipocyte. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 229–242. [Google Scholar] [CrossRef]
- Ichimura, A.; Hirasawa, A.; Poulain-Godefroy, O.; Bonnefond, A.; Hara, T.; Yengo, L.; Kimura, I.; Leloire, A.; Liu, N.; Iida, K.; et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature 2012, 483, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, A.; Hara, T.; Hirasawa, A. Regulation of Energy Homeostasis via GPR120. Front. Endocrinol. 2014, 5, 111. [Google Scholar] [CrossRef]
- Song, T.; Zhou, Y.; Peng, J.; Tao, Y.X.; Yang, Y.; Xu, T.; Peng, J.; Ren, J.; Xiang, Q.; Wei, H. GPR120 promotes adipogenesis through intracellular calcium and extracellular signal-regulated kinase 1/2 signal pathway. Mol. Cell Endocrinol. 2016, 434, 1–13. [Google Scholar] [CrossRef]
- Christian, M. Elucidation of the roles of brown and brite fat genes: GPR120 is a modulator of brown adipose tissue function. Exp. Physiol. 2020, 105, 1201–1205. [Google Scholar] [CrossRef]
- Quesada-López, T.; Cereijo, R.; Turatsinze, J.V.; Planavila, A.; Cairó, M.; Gavaldà-Navarro, A.; Peyrou, M.; Moure, R.; Iglesias, R.; Giralt, M.; et al. The lipid sensor GPR120 promotes brown fat activation and FGF21 release from adipocytes. Nat. Commun. 2016, 7, 13479. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Umemoto, T.; Kakei, M.; Momomura, S.I.; Kawakami, M.; Ishikawa, S.E.; Hara, K. Eicosapentaenoic acid shows anti-inflammatory effect via GPR120 in 3T3-L1 adipocytes and attenuates adipose tissue inflammation in diet-induced obese mice. Nutr. Metab. 2017, 14, 33. [Google Scholar] [CrossRef]
- Murano, I.; Severi, I.; Venema, W.; Cecchini, M.P.; Kershaw, E.E.; Barbatelli, G.; Haemmerle, G.; Zechner, R.; Cinti, S. Brown adipose tissue whitening leads to brown adipocyte death and adipose tissue inflammation. J. Lipid Res. 2018, 59, 784–794. [Google Scholar] [CrossRef]
- Pærregaard, S.I.; Agerholm, M.; Serup, A.K.; Ma, T.; Kiens, B.; Madsen, L.; Kristiansen, K.; Jensen, B.A. FFAR4 (GPR120) Signaling Is Not Required for Anti-Inflammatory and Insulin-Sensitizing Effects of Omega-3 Fatty Acids. Mediat. Inflamm. 2016, 2016, 1536047. [Google Scholar] [CrossRef] [PubMed]
- Sveiven, S.N.; Anesko, K.; Morgan, J.; Nair, M.G.; Nordgren, T.M. Lipid-Sensing Receptor FFAR4 Modulates Pulmonary Epithelial Homeostasis following Immunogenic Exposures Independently of the FFAR4 Ligand Docosahexaenoic Acid (DHA). Int. J. Mol. Sci. 2023, 24, 7072. [Google Scholar] [CrossRef]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef]
- Katsuma, S.; Hatae, N.; Yano, T.; Ruike, Y.; Kimura, M.; Hirasawa, A.; Tsujimoto, G. Free fatty acids inhibit serum deprivation-induced apoptosis through GPR120 in a murine enteroendocrine cell line STC-1. J. Biol. Chem. 2005, 280, 19507–19515. [Google Scholar] [CrossRef] [PubMed]
- Cawthon, C.R.; de La Serre, C.B. The critical role of CCK in the regulation of food intake and diet-induced obesity. Peptides 2021, 138, 170492. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Katsuma, S.; Adachi, T.; Koshimizu, T.A.; Hirasawa, A.; Tsujimoto, G. Free fatty acids induce cholecystokinin secretion through GPR120. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 377, 523–527. [Google Scholar] [CrossRef]
- Popoviciu, M.S.; Păduraru, L.; Yahya, G.; Metwally, K.; Cavalu, S. Emerging Role of GLP-1 Agonists in Obesity: A Comprehensive Review of Randomised Controlled Trials. Int. J. Mol. Sci. 2023, 24, 10449. [Google Scholar] [CrossRef]
- McKillop, A.M.; Miskelly, M.G.; Moran, B.M.; Flatt, P.R. Incretins play an important role in FFA4/GPR120 regulation of glucose metabolism by GW-9508. Life Sci. 2023, 318, 121475. [Google Scholar] [CrossRef] [PubMed]
- Loona, D.P.S.; Das, B.; Kaur, R.; Kumar, R.; Yadav, A.K. Free Fatty Acid Receptors (FFARs): Emerging Therapeutic Targets for the Management of Diabetes Mellitus. Curr. Med. Chem. 2023, 30, 3404–3440. [Google Scholar] [CrossRef]
- Patil, M.; Casari, I.; Warne, L.N.; Falasca, M. G protein-coupled receptors driven intestinal glucagon-like peptide-1 reprogramming for obesity: Hope or hype? Biomed. Pharmacother. 2024, 172, 116245. [Google Scholar] [CrossRef]
- Sankoda, A.; Harada, N.; Iwasaki, K.; Yamane, S.; Murata, Y.; Shibue, K.; Thewjitcharoen, Y.; Suzuki, K.; Harada, T.; Kanemaru, Y.; et al. Long-Chain Free Fatty Acid Receptor GPR120 Mediates Oil-Induced GIP Secretion Through CCK in Male Mice. Endocrinology 2017, 158, 1172–1180. [Google Scholar] [CrossRef]
- Sankoda, A.; Harada, N.; Kato, T.; Ikeguchi, E.; Iwasaki, K.; Yamane, S.; Murata, Y.; Hirasawa, A.; Inagaki, N. Free fatty acid receptors, G protein-coupled receptor 120 and G protein-coupled receptor 40, are essential for oil-induced gastric inhibitory polypeptide secretion. J. Diabetes Investig. 2019, 10, 1430–1437. [Google Scholar] [CrossRef]
- Iwasaki, K.; Harada, N.; Sasaki, K.; Yamane, S.; Iida, K.; Suzuki, K.; Hamasaki, A.; Nasteska, D.; Shibue, K.; Joo, E.; et al. Free fatty acid receptor GPR120 is highly expressed in enteroendocrine K cells of the upper small intestine and has a critical role in GIP secretion after fat ingestion. Endocrinology 2015, 156, 837–846. [Google Scholar] [CrossRef]
- Yamane, S.; Harada, N.; Inagaki, N. Mechanisms of fat-induced gastric inhibitory polypeptide/glucose-dependent insulinotropic polypeptide secretion from K cells. J. Diabetes Investig. 2016, 7 (Suppl. S1), 20–26. [Google Scholar] [CrossRef]
- Yasuda, T.; Harada, N.; Hatoko, T.; Ichimura, A.; Ikeguchi-Ogura, E.; Murata, Y.; Wada, N.; Kiyobayashi, S.; Yamane, S.; Hirasawa, A.; et al. Inhibition of GPR120 signaling in intestine ameliorates insulin resistance and fatty liver under high-fat diet feeding. Am. J. Physiol. Endocrinol. Metab. 2023, 324, E449–E460. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 2018, 20 (Suppl. S1), 5–21. [Google Scholar] [CrossRef] [PubMed]
- Codoñer-Alejos, A.; Carrasco-Luna, J.; Carrasco-García, Á.; Codoñer-Franch, P. Reduced Free Fatty Acid Receptor 4 Gene Expression is Associated With Extreme Obesity and Insulin Resistance in Children. J. Pediatr. Gastroenterol. Nutr. 2022, 74, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Stone, V.M.; Dhayal, S.; Brocklehurst, K.J.; Lenaghan, C.; Sörhede Winzell, M.; Hammar, M.; Xu, X.; Smith, D.M.; Morgan, N.G. GPR120 (FFAR4) is preferentially expressed in pancreatic delta cells and regulates somatostatin secretion from murine islets of Langerhans. Diabetologia 2014, 57, 1182–1191. [Google Scholar] [CrossRef]
- Croze, M.L.; Flisher, M.F.; Guillaume, A.; Tremblay, C.; Noguchi, G.M.; Granziera, S.; Vivot, K.; Castillo, V.C.; Campbell, S.A.; Ghislain, J.; et al. Free fatty acid receptor 4 inhibitory signaling in delta cells regulates islet hormone secretion in mice. Mol. Metab. 2021, 45, 101166. [Google Scholar] [CrossRef]
- Rorsman, P.; Huising, M.O. The somatostatin-secreting pancreatic δ-cell in health and disease. Nat. Rev. Endocrinol. 2018, 14, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Moran, B.M.; Abdel-Wahab, Y.H.; Flatt, P.R.; McKillop, A.M. Evaluation of the insulin-releasing and glucose-lowering effects of GPR120 activation in pancreatic β-cells. Diabetes Obes. Metab. 2014, 16, 1128–1139. [Google Scholar] [CrossRef]
- Vergari, E.; Denwood, G.; Salehi, A.; Zhang, Q.; Adam, J.; Alrifaiy, A.; Wernstedt Asterholm, I.; Benrick, A.; Chibalina, M.V.; Eliasson, L.; et al. Somatostatin secretion by Na+-dependent Ca2+-induced Ca2+ release in pancreatic delta-cells. Nat. Metab. 2020, 2, 32–40. [Google Scholar] [CrossRef]
- Denwood, G.; Tarasov, A.; Salehi, A.; Vergari, E.; Ramracheya, R.; Takahashi, H.; Nikolaev, V.O.; Seino, S.; Gribble, F.; Reimann, F.; et al. Glucose stimulates somatostatin secretion in pancreatic δ-cells by cAMP-dependent intracellular Ca2+ release. J. Gen. Physiol. 2019, 151, 1094–1115. [Google Scholar] [CrossRef]
- Greenhill, C. Urocortin 3 function in glucose metabolism. Nat. Rev. Endocrinol. 2022, 18, 333. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.Y.; Gao, M.X.; Qi, H.B.; An, W.T.; Lin, J.Y.; Ning, S.L.; Yang, F.; Xiao, P.; Cheng, J.; Pan, W.; et al. Differential contributions of G protein- or arrestin subtype-mediated signalling underlie urocortin 3-induced somatostatin secretion in pancreatic δ cells. Br. J. Pharmacol. 2024, 181, 2600–2621. [Google Scholar] [CrossRef]
- Du, Y.Q.; Sha, X.Y.; Cheng, J.; Wang, J.; Lin, J.Y.; An, W.T.; Pan, W.; Zhang, L.J.; Tao, X.N.; Xu, Y.F.; et al. Endogenous Lipid-GPR120 Signaling Modulates Pancreatic Islet Homeostasis to Different Extents. Diabetes 2022, 71, 1454–1471. [Google Scholar] [CrossRef]
- Hong, S.W.; Lee, J.; Moon, S.J.; Kwon, H.; Park, S.E.; Rhee, E.J.; Lee, W.Y. Docosahexanoic Acid Attenuates Palmitate-Induced Apoptosis by Autophagy Upregulation via GPR120/mTOR Axis in Insulin-Secreting Cells. Endocrinol. Metab. 2024, 39, 353–363. [Google Scholar] [CrossRef]
- Zhang, D.; So, W.Y.; Wang, Y.; Wu, S.Y.; Cheng, Q.; Leung, P.S. Insulinotropic effects of GPR120 agonists are altered in obese diabetic and obese non-diabetic states. Clin. Sci. 2017, 131, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Owolabi, A.I.; Corbett, R.C.; Flatt, P.R.; McKillop, A.M. Positive interplay between FFAR4/GPR120, DPP-IV inhibition and GLP-1 in beta cell proliferation and glucose homeostasis in obese high fat fed mice. Peptides 2024, 177, 171218. [Google Scholar] [CrossRef]
- Ichimura, A.; Hasegawa, S.; Kasubuchi, M.; Kimura, I. Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front. Pharmacol. 2014, 5, 236. [Google Scholar] [CrossRef] [PubMed]
- Pedroni, L.; Perugino, F.; Magnaghi, F.; Dall’Asta, C.; Galaverna, G.; Dellafiora, L. Free fatty acid receptors beyond fatty acids: A computational journey to explore peptides as possible binders of GPR120. Curr. Res. Food Sci. 2024, 8, 100710. [Google Scholar] [CrossRef]
- Suzuki, T.; Igari, S.; Hirasawa, A.; Hata, M.; Ishiguro, M.; Fujieda, H.; Itoh, Y.; Hirano, T.; Nakagawa, H.; Ogura, M.; et al. Identification of G protein-coupled receptor 120-selective agonists derived from PPARgamma agonists. J. Med. Chem. 2008, 51, 7640–7644. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, W.; Spinetti, T.; Tardivel, A.; Castillo, R.; Bourquin, C.; Guarda, G.; Tian, Z.; Tschopp, J.; Zhou, R. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 2013, 38, 1154–1163. [Google Scholar] [CrossRef]
- Li, X.; Yu, Y.; Funk, C.D. Cyclooxygenase-2 induction in macrophages is modulated by docosahexaenoic acid via interactions with free fatty acid receptor 4 (FFA4). FASEB J. 2013, 27, 4987–4997. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Leung, P.S. Potential roles of GPR120 and its agonists in the management of diabetes. Drug Des. Dev. Ther. 2014, 8, 1013–1027. [Google Scholar] [CrossRef]
- Pal, A.; Curtin, J.F.; Kinsella, G.K. In silico and in vitro screening for potential anticancer candidates targeting GPR120. Bioorg. Med. Chem. Lett. 2021, 31, 127672. [Google Scholar] [CrossRef]
- Rubbino, F.; Garlatti, V.; Garzarelli, V.; Massimino, L.; Spanò, S.; Iadarola, P.; Cagnone, M.; Giera, M.; Heijink, M.; Guglielmetti, S.; et al. GPR120 prevents colorectal adenocarcinoma progression by sustaining the mucosal barrier integrity. Sci. Rep. 2022, 12, 381. [Google Scholar] [CrossRef]
- He, Q.; Zhu, S.; Lin, M.; Yang, Q.; Wei, L.; Zhang, J.; Jiang, X.; Zhu, D.; Lu, X.; Chen, Y.Q. Increased GPR120 level is associated with gestational diabetes mellitus. Biochem. Biophys. Res. Commun. 2019, 512, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Pompili, S.; Cappariello, A.; Vetuschi, A.; Sferra, R. G-Protein-Coupled Receptor 120 Agonist Mitigates Steatotic and Fibrotic Features Triggered in Obese Mice by the Administration of a High-Fat and High-Carbohydrate Diet. ACS Omega 2024, 9, 31899–31909. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G.; Alvarez-Curto, E.; Hudson, B.D.; Prihandoko, R.; Tobin, A.B. FFA4/GPR120, Pharmacology and Therapeutic Opportunities. Trends Pharmacol. Sci. 2017, 38, 809–821. [Google Scholar] [CrossRef]
- Abedi, E.; Sahari, M.A. Long-chain polyunsaturated fatty acid sources and evaluation of their nutritional and functional properties. Food Sci. Nutr. 2014, 2, 443–463. [Google Scholar] [CrossRef]
- Murff, H.J.; Edwards, T.L. Endogenous Production of Long-Chain Polyunsaturated Fatty Acids and Metabolic Disease Risk. Curr. Cardiovasc. Risk Rep. 2014, 8, 418. [Google Scholar] [CrossRef]
- Rodriguez-Pacheco, F.; Garcia-Serrano, S.; Garcia-Escobar, E.; Gutierrez-Repiso, C.; Garcia-Arnes, J.; Valdes, S.; Gonzalo, M.; Soriguer, F.; Moreno-Ruiz, F.J.; Rodriguez-Cañete, A.; et al. Effects of obesity/fatty acids on the expression of GPR120. Mol. Nutr. Food Res. 2014, 58, 1852–1860, Erratum in Mol. Nutr. Food Res. 2015, 59, 2338. https://doi.org/10.1002/mnfr.201570115. [Google Scholar] [CrossRef]
- Holliday, N.D.; Watson, S.J.; Brown, A.J. Drug discovery opportunities and challenges at g protein coupled receptors for long chain free Fatty acids. Front. Endocrinol. 2012, 2, 112. [Google Scholar] [CrossRef]
- Hansen, S.V.; Ulven, T. Pharmacological Tool Compounds for the Free Fatty Acid Receptor 4 (FFA4/GPR120). Handb. Exp. Pharmacol. 2017, 236, 33–56. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Dalli, J. New pro-resolving n-3 mediators bridge resolution of infectious inflammation to tissue regeneration. Mol. Aspects Med. 2018, 64, 1–17. [Google Scholar] [CrossRef]
- Yore, M.M.; Syed, I.; Moraes-Vieira, P.M.; Zhang, T.; Herman, M.A.; Homan, E.A.; Patel, R.T.; Lee, J.; Chen, S.; Peroni, O.D.; et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 2014, 159, 318–332. [Google Scholar] [CrossRef]
- Wang, Y.M.; Liu, H.X.; Fang, N.Y. 9-PAHSA promotes browning of white fat via activating G-protein-coupled receptor 120 and inhibiting lipopolysaccharide/NF-kappa B pathway. Biochem. Biophys. Res. Commun. 2018, 506, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Hirasawa, A.; Sun, Q.; Sadakane, K.; Itsubo, C.; Iga, T.; Adachi, T.; Koshimizu, T.A.; Hashimoto, T.; Asakawa, Y.; et al. Novel selective ligands for free fatty acid receptors GPR120 and GPR40. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 380, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Hirasawa, A.; Hara, T.; Kimura, I.; Adachi, T.; Awaji, T.; Ishiguro, M.; Suzuki, T.; Miyata, N.; Tsujimoto, G. Structure-activity relationships of GPR120 agonists based on a docking simulation. Mol. Pharmacol. 2010, 78, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, A.; Hara, T.; Katsuma, S.; Adachi, T.; Tsujimoto, G. Free fatty acid receptors and drug discovery. Biol. Pharm. Bull. 2008, 31, 1847–1851. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, H.; Yan, A.; Zhu, J.; Liu, K.; Chen, D.; Xie, R.; Xu, X.; Su, X. Grifolic acid induces mitochondrial membrane potential loss and cell death of RAW264.7 macrophages. Mol. Med. Rep. 2018, 17, 3281–3287. [Google Scholar] [CrossRef]
- Bouyahya, A.; El Allam, A.; Zeouk, I.; Taha, D.; Zengin, G.; Goh, B.H.; Catauro, M.; Montesano, D.; El Omari, N. Pharmacological Effects of Grifolin: Focusing on Anticancer Mechanisms. Molecules 2022, 27, 284. [Google Scholar] [CrossRef]
- Wilson, C.H.; Kumar, S. Caspases in metabolic disease and their therapeutic potential. Cell Death Differ. 2018, 25, 1010–1024. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Riley, J.S. When cell death goes wrong: Inflammatory outcomes of failed apoptosis and mitotic cell death. Cell Death Differ. 2023, 30, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Konda, V.R.; Desai, A.; Darland, G.; Grayson, N.; Bland, J.S. KDT501, a derivative from hops, normalizes glucose metabolism and body weight in rodent models of diabetes. PLoS ONE 2014, 9, e87848. [Google Scholar] [CrossRef]
- Kern, P.A.; Finlin, B.S.; Ross, D.; Boyechko, T.; Zhu, B.; Grayson, N.; Sims, R.; Bland, J.S. Effects of KDT501 on Metabolic Parameters in Insulin-Resistant Prediabetic Humans. J. Endocr. Soc. 2017, 1, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Finlin, B.S.; Zhu, B.; Kok, B.P.; Godio, C.; Westgate, P.M.; Grayson, N.; Sims, R.; Bland, J.S.; Saez, E.; Kern, P.A. The Influence of a KDT501, a Novel Isohumulone, on Adipocyte Function in Humans. Front. Endocrinol. 2017, 8, 255. [Google Scholar] [CrossRef]
- Nagasawa, T.; Ishimaru, K.; Higashiyama, S.; Hama, Y.; Mitsutake, S. Teadenol A in microbial fermented tea acts as a novel ligand on GPR120 to increase GLP-1 secretion. Food Funct. 2020, 11, 10534–10541. [Google Scholar] [CrossRef]
- Wilches, I.; Jiménez-Castillo, P.; Cuzco, N.; Clos, M.V.; Jiménez-Altayó, F.; Peñaherrera, E.; Jerves-Andrade, L.; Tobar, V.; Vander Heyden, Y.; Leon-Tamariz, F.; et al. Anti-inflammatory and sedative activities of Peperomia galioides: In vivo studies in mice. Nat. Prod. Res. 2021, 35, 1657–1661. [Google Scholar] [CrossRef]
- Ye, C.; Jin, M.; Li, R.; Sun, J.; Wang, R.; Wang, J.; Li, S.; Zhou, W.; Li, G. Phytochemical and chemotaxonomic study on the leaves of Rhododendron dauricum L. Biochem. Syst. Ecol. 2020, 90, 104038. [Google Scholar] [CrossRef]
- Wang, Y.; Alkhalidy, H.; Liu, D. The Emerging Role of Polyphenols in the Management of Type 2 Diabetes. Molecules 2021, 26, 703. [Google Scholar] [CrossRef]
- Nukata, M.; Hashimoto, T.; Yamamoto, I.; Iwasaki, N.; Tanaka, M.; Asakawa, Y. Neogrifolin derivatives possessing anti-oxidative activity from the mushroom Albatrellus ovinus. Phytochemistry 2002, 59, 731–737. [Google Scholar] [CrossRef]
- Grabovyi, G.A.; Mohr, J.T. Total Synthesis of Grifolin, Grifolic Acid, LL-Z1272α, LL-Z1272β, and Ilicicolinic Acid A. Org. Lett. 2016, 18, 5010–5013. [Google Scholar] [CrossRef]
- Elsayed, E.A.; El Enshasy, H.; Wadaan, M.A.; Aziz, R. Mushrooms: A potential natural source of anti-inflammatory compounds for medical applications. Mediat. Inflamm. 2014, 2014, 805841. [Google Scholar] [CrossRef] [PubMed]
- Muszyńska, B.; Grzywacz-Kisielewska, A.; Kała, K.; Gdula-Argasińska, J. Anti-inflammatory properties of edible mushrooms: A review. Food Chem. 2018, 243, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Janssen, S.; Laermans, J.; Iwakura, H.; Tack, J.; Depoortere, I. Sensing of fatty acids for octanoylation of ghrelin involves a gustatory G-protein. PLoS ONE 2012, 7, e40168. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.N.A. An insight into the multifunctional role of ghrelin and structure activity relationship studies of ghrelin receptor ligands with clinical trials. Eur. J. Med. Chem. 2022, 235, 114308. [Google Scholar] [CrossRef]
- Usui, A.; Nakamura, A.; Era, M.; Matsuo, Y.; Tanaka, T.; Ishimaru, K. A New Flavonoid from Camellia sinensis Fermented Tea. Nat. Prod. Commun. 2016, 11, 1281–1282. [Google Scholar] [CrossRef]
- Wulandari, R.A.; Amano, M.; Yanagita, T.; Tanaka, T.; Kouno, I.; Kawamura, D.; Ishimaru, K. New phenolic compounds from Camellia sinensis L. leaves fermented with Aspergillus sp. J. Nat. Med. 2011, 65, 594–597. [Google Scholar] [CrossRef]
- Katanasaka, Y.; Sunagawa, Y.; Miyazaki, Y.; Funamoto, M.; Shimizu, S.; Shimizu, K.; Yamakage, H.; Satoh-Asahara, N.; Toyama, K.; Sabashi, T.; et al. Ameliorating prediabetic subject status via fermented tea supplementation: A randomized, double-blind, parallel-group comparison study. J. Funct. Foods. 2022, 97, 105257. [Google Scholar] [CrossRef]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef]
- Klobučar, I.; Habisch, H.; Klobučar, L.; Trbušić, M.; Pregartner, G.; Berghold, A.; Kostner, G.M.; Scharnagl, H.; Madl, T.; Frank, S.; et al. Serum Levels of Adiponectin Are Strongly Associated with Lipoprotein Subclasses in Healthy Volunteers but Not in Patients with Metabolic Syndrome. Int. J. Mol. Sci. 2024, 25, 5050. [Google Scholar] [CrossRef]
- Ma, S.; Li, Z.; Yang, Y.; Zhang, L.; Li, M.; Du, L. Fluorescent Ligand-Based Discovery of Small-Molecule Sulfonamide Agonists for GPR120. Front. Chem. 2022, 10, 816014. [Google Scholar] [CrossRef] [PubMed]
- Shimpukade, B.; Hudson, B.D.; Hovgaard, C.K.; Milligan, G.; Ulven, T. Discovery of a potent and selective GPR120 agonist. J. Med. Chem. 2012, 55, 4511–4515. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, C.P.; Peat, A.J.; McKeown, S.C.; Corbett, D.F.; Goetz, A.S.; Littleton, T.R.; McCoy, D.C.; Kenakin, T.P.; Andrews, J.L.; Ammala, C.; et al. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40, identification of agonist and antagonist small molecules. Br. J. Pharmacol. 2006, 148, 619–628. [Google Scholar] [CrossRef]
- Li, Y.; Yu, H.; Lopes-Virella, M.F.; Huang, Y. GPR40/GPR120 Agonist GW9508 Improves Metabolic Syndrome-Exacerbated Periodontitis in Mice. Int. J. Mol. Sci. 2024, 25, 9622. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G.; Shimpukade, B.; Ulven, T.; Hudson, B.D. Complex Pharmacology of Free Fatty Acid Receptors. Chem. Rev. 2017, 117, 67–110. [Google Scholar] [CrossRef]
- Hudson, B.D.; Shimpukade, B.; Mackenzie, A.E.; Butcher, A.J.; Pediani, J.D.; Christiansen, E.; Heathcote, H.; Tobin, A.B.; Ulven, T.; Milligan, G. The pharmacology of TUG-891, a potent and selective agonist of the free fatty acid receptor 4 (FFA4/GPR120), demonstrates both potential opportunity and possible challenges to therapeutic agonism. Mol. Pharmacol. 2013, 84, 710–725. [Google Scholar] [CrossRef]
- Ji, G.; Guo, Q.; Xue, Q.; Kong, R.; Wang, S.; Lei, K.; Liu, R.; Wang, X. Novel GPR120 Agonists with Improved Pharmacokinetic Profiles for the Treatment of Type 2 Diabetes. Molecules 2021, 26, 6907. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Wei, S.; Wang, M.; Xu, Y.; Hu, W.; Gao, Z.; Liu, R.; Wang, S.; Ji, G. Discovery of Novel and Selective G-Protein Coupled Receptor 120 (GPR120) Agonists for the Treatment of Type 2 Diabetes Mellitus. Molecules 2022, 27, 9018. [Google Scholar] [CrossRef]
- Adams, G.L.; Velazquez, F.; Jayne, C.; Shah, U.; Miao, S.; Ashley, E.R.; Madeira, M.; Akiyama, T.E.; Di Salvo, J.; Suzuki, T.; et al. Discovery of Chromane Propionic Acid Analogues as Selective Agonists of GPR120 with in Vivo Activity in Rodents. ACS Med. Chem. Lett. 2016, 8, 96–101. [Google Scholar] [CrossRef]
- Sparks, S.M.; Aquino, C.; Banker, P.; Collins, J.L.; Cowan, D.; Diaz, C.; Dock, S.T.; Hertzog, D.L.; Liang, X.; Swiger, E.D.; et al. Exploration of phenylpropanoic acids as agonists of the free fatty acid receptor 4 (FFA4): Identification of an orally efficacious FFA4 agonist. Bioorganic Med. Chem. Lett. 2017, 27, 1278–1283. [Google Scholar] [CrossRef]
- Oh, D.Y.; Walenta, E.; Akiyama, T.E.; Lagakos, W.S.; Lackey, D.; Pessentheiner, A.R.; Sasik, R.; Hah, N.; Chi, T.J.; Cox, J.M.; et al. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat. Med. 2014, 20, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Son, S.E.; Park, S.J.; Koh, J.M.; Im, D.S. Free fatty acid receptor 4 (FFA4) activation ameliorates 2,4-dinitrochlorobenzene-induced atopic dermatitis by increasing regulatory T cells in mice. Acta Pharmacol. Sin. 2020, 41, 1337–1347. [Google Scholar] [CrossRef]
- Sundström, L.; Myhre, S.; Sundqvist, M.; Ahnmark, A.; McCoull, W.; Raubo, P.; Groombridge, S.D.; Polla, M.; Nyström, A.C.; Kristensson, L.; et al. The acute glucose lowering effect of specific GPR120 activation in mice is mainly driven by glucagon-like peptide 1. PLoS ONE 2017, 12, e0189060. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, A.G.; Miskelly, M.G.; Flatt, P.R.; McKillop, A.M. Pharmacological potential of novel agonists for FFAR4 on islet and enteroendocrine cell function and glucose homeostasis. Eur. J. Pharm. Sci. 2020, 142, 105104. [Google Scholar] [CrossRef]
- Cox, J.M.; Chu, H.D.; Chelliah, M.V.; Debenham, J.S.; Eagen, K.; Lan, P.; Lombardo, M.; London, C.; Plotkin, M.A.; Shah, U.; et al. Design, Synthesis, and Evaluation of Novel and Selective G-protein Coupled Receptor 120 (GPR120) Spirocyclic Agonists. ACS Med. Chem. Lett. 2016, 8, 49–54. [Google Scholar] [CrossRef]
- Chen, X.; Liu, C.; Ruan, L. G-Protein-Coupled Receptors 120 Agonist III Improves Hepatic Inflammation and ER Stress in Steatohepatitis. Dig. Dis. Sci. 2021, 66, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Watterson, K.R.; Hansen, S.V.F.; Hudson, B.D.; Alvarez-Curto, E.; Raihan, S.Z.; Azevedo, C.M.G.; Martin, G.; Dunlop, J.; Yarwood, S.J.; Ulven, T.; et al. Probe-Dependent Negative Allosteric Modulators of the Long-Chain Free Fatty Acid Receptor FFA4. Mol. Pharmacol. 2017, 91, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.M.; Watterson, K.R.; Wargent, E.T.; Hansen, S.V.; Hudson, B.D.; Kępczyńska, M.A.; Dunlop, J.; Shimpukade, B.; Christiansen, E.; Milligan, G.; et al. Non-Acidic Free Fatty Acid Receptor 4 Agonists with Antidiabetic Activity. J. Med. Chem. 2016, 59, 8868–8878. [Google Scholar] [CrossRef]
- Dragano, N.R.V.; Solon, C.; Ramalho, A.F.; de Moura, R.F.; Razolli, D.S.; Christiansen, E.; Azevedo, C.; Ulven, T.; Velloso, L.A. Polyunsaturated fatty acid receptors, GPR40 and GPR120, are expressed in the hypothalamus and control energy homeostasis and inflammation. J. Neuroinflamm. 2017, 14, 91. [Google Scholar] [CrossRef]
- Tian, M.; Wu, Z.; Heng, J.; Chen, F.; Guan, W.; Zhang, S. Novel advances in understanding fatty acid-binding G protein-coupled receptors and their roles in controlling energy balance. Nutr. Rev. 2022, 80, 187–199. [Google Scholar] [CrossRef]
- Gong, Z.; Yoshimura, M.; Aizawa, S.; Kurotani, R.; Zigman, J.M.; Sakai, T.; Sakata, I. G protein-coupled receptor 120 signaling regulates ghrelin secretion in vivo and in vitro. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E28–E35. [Google Scholar] [CrossRef] [PubMed]
- Graciano, M.F.; Valle, M.M.; Curi, R.; Carpinelli, A.R. Evidence for the involvement of GPR40 and NADPH oxidase in palmitic acid-induced superoxide production and insulin secretion. Islets 2013, 5, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Raptis, D.A.; Limani, P.; Jang, J.H.; Ungethüm, U.; Tschuor, C.; Graf, R.; Humar, B.; Clavien, P.A. GPR120 on Kupffer cells mediates hepatoprotective effects of ω3-fatty acids. J. Hepatol. 2014, 60, 625–632. [Google Scholar] [CrossRef]
- Chen, Y.L.; Lin, Y.P.; Sun, C.K.; Huang, T.H.; Yip, H.K.; Chen, Y.T. Extracorporeal shockwave against inflammation mediated by GPR120 receptor in cyclophosphamide-induced rat cystitis model. Mol. Med. 2018, 24, 60. [Google Scholar] [CrossRef] [PubMed]
- Olmo, I.; Teuber, S.; Larrazabal, C.; Alarcon, P.; Raipane, F.; Burgos, R.A.; Hidalgo, M.A. Docosahexaenoic acid and TUG-891 activate free fatty acid-4 receptor in bovine neutrophils. Vet. Immunol. Immunopathol. 2019, 209, 53–60. [Google Scholar] [CrossRef]
- Gozal, D.; Qiao, Z.; Almendros, I.; Zheng, J.; Khalyfa, A.; Shimpukade, B.; Ulven, T. Treatment with TUG891, a free fatty acid receptor 4 agonist, restores adipose tissue metabolic dysfunction following chronic sleep fragmentation in mice. Int. J. Obes. 2016, 40, 1143–1149. [Google Scholar] [CrossRef]
- Schilperoort, M.; van Dam, A.D.; Hoeke, G.; Shabalina, I.G.; Okolo, A.; Hanyaloglu, A.C.; Dib, L.H.; Mol, I.M.; Caengprasath, N.; Chan, Y.W.; et al. The GPR120 agonist TUG-891 promotes metabolic health by stimulating mitochondrial respiration in brown fat. EMBO Mol. Med. 2018, 10, e8047. [Google Scholar] [CrossRef]
- Murtaza, B.; Hichami, A.; Khan, A.S.; Shimpukade, B.; Ulven, T.; Ozdener, M.H.; Khan, N.A. Novel GPR120 agonist TUG891 modulates fat taste perception and preference and activates tongue-brain-gut axis in mice. J. Lipid Res. 2020, 61, 133–142. [Google Scholar] [CrossRef]
- Zhang, X.; Cai, C.; Sui, Z.; Macielag, M.; Wang, Y.; Yan, W.; Suckow, A.; Hua, H.; Bell, A.; Haug, P.; et al. Discovery of an Isothiazole-Based Phenylpropanoic Acid GPR120 Agonist as a Development Candidate for Type 2 Diabetes. ACS Med. Chem. Lett. 2017, 8, 947–952. [Google Scholar] [CrossRef]
- Ma, J.; Novack, A.; Nashashibi, I.; Pham, P.; Rabbat, C.J.; Song, J.; Shi, D.F.; Zhao, Z.; Choi, Y.J.; Chen, X. Aryl GPR120 Receptor Agonists and Uses Thereof. International Patent WO 2010/048207, 29 April 2010. [Google Scholar]
- Wang, C.; Liu, Y.; Pan, Y.; Jin, H. Effect of GSK-137647A, the first non-carboxylic FFA4 agonist, on the osteogenic and adipogenic differentiation of bone mesenchymal stem cells in db/db mice. J. Pharm. Pharmacol. 2020, 72, 461–469. [Google Scholar] [CrossRef]
- Salaga, M.; Bartoszek, A.; Binienda, A.; Krajewska, J.B.; Fabisiak, A.; Mosińska, P.; Dziedziczak, K.; Niewinna, K.; Talar, M.; Tarasiuk, A.; et al. Activation of Free Fatty Acid Receptor 4 Affects Intestinal Inflammation and Improves Colon Permeability in Mice. Nutrients 2021, 13, 2716. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.U.; Ohmori, K.; Hashimoto, T.; Kamitori, K.; Yamaguchi, F.; Noma, T.; Igarashi, J.; Tsuboi, K.; Tokuda, M.; Nishiyama, A.; et al. GPR120 in adipocytes has differential roles in the production of pro-inflammatory adipocytokines. Biochem. Biophys. Res. Commun. 2017, 486, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Bender, E.; Schamberger, J.; Eitner, F. Pharmacology of Free Fatty Acid Receptors and Their Allosteric Modulators. Int. J. Mol. Sci. 2021, 22, 1763. [Google Scholar] [CrossRef]
- Im, H.; Park, J.H.; Im, S.; Han, J.; Kim, K.; Lee, Y.H. Regulatory roles of G-protein coupled receptors in adipose tissue metabolism and their therapeutic potential. Arch. Pharm. Res. 2021, 44, 133–145. [Google Scholar] [CrossRef]
- Zhang, S.; Roth, B.L. Sensing unsaturated fatty acids: Insights from GPR120 signaling. Cell Res. 2023, 33, 657–658. [Google Scholar] [CrossRef] [PubMed]
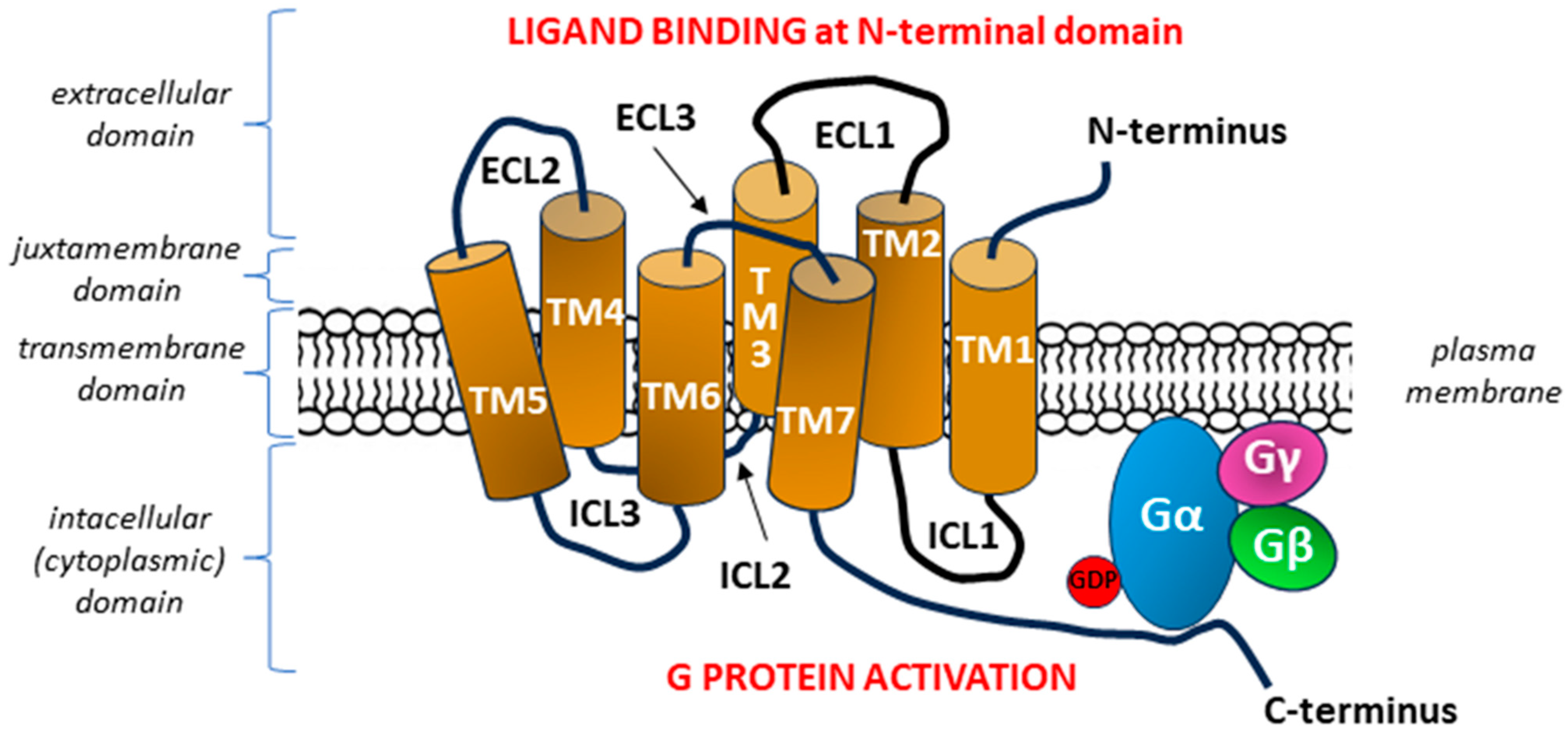
 —ANTI-INFLAMMATORY EFFECTS via beta-arrestin 2 (ARRβ2) signaling in immune pro-inflammatory cells. After receptor stimulation, the adapter protein ARRβ2 is recruited, which, after forming a complex with GPR120, leads to the desensitization of GPR120 as well as other receptors [e.g., Toll-like receptor 4 (TLR4), interleukin 1 receptor (IL1R), and tumor necrosis factor receptor (TNFR)] to pro-inflammatory stimuli. The GPR120- ARRβ2 complex internalizes and interacts with TGF-β-activated kinase 1 (MAP3K7)-binding protein 1 (TAB1) and NLRP3 [nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR)-containing protein (NLR) family member 3] inflammasome, which leads to the inhibition of the subsequent inflammatory cascade. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-кB) essential modulator (NEMO) ubiquitination, which is crucial for the activation of the canonical NF-κB signaling pathway, does not occur. A decrease in the concentration of pro-inflammatory cytokines indirectly modulates adipose tissue cell functions by reducing insulin resistance and, consequently, stimulating glucose uptake [82,94,95,96,97,98].
—ANTI-INFLAMMATORY EFFECTS via beta-arrestin 2 (ARRβ2) signaling in immune pro-inflammatory cells. After receptor stimulation, the adapter protein ARRβ2 is recruited, which, after forming a complex with GPR120, leads to the desensitization of GPR120 as well as other receptors [e.g., Toll-like receptor 4 (TLR4), interleukin 1 receptor (IL1R), and tumor necrosis factor receptor (TNFR)] to pro-inflammatory stimuli. The GPR120- ARRβ2 complex internalizes and interacts with TGF-β-activated kinase 1 (MAP3K7)-binding protein 1 (TAB1) and NLRP3 [nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR)-containing protein (NLR) family member 3] inflammasome, which leads to the inhibition of the subsequent inflammatory cascade. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-кB) essential modulator (NEMO) ubiquitination, which is crucial for the activation of the canonical NF-κB signaling pathway, does not occur. A decrease in the concentration of pro-inflammatory cytokines indirectly modulates adipose tissue cell functions by reducing insulin resistance and, consequently, stimulating glucose uptake [82,94,95,96,97,98].  —METABOLIC EFFECTS via classical Gα q/11 signaling. In this pathway, omega-3-fatty acids (Ω-3 FAs), acting as a ligand and agonist of GPR120, activate the Gαq/11-linked phospholipase C-beta (PLCβ). After PLCβ activation, phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolyzes into diacyl glycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), which serve as second messengers. IP3 binds to receptors, the conformational changes of which, after activation, lead to the mobilization of calcium ions (Ca2+) from intracellular stores. The following cytosolic Ca2+ influx stimulates the release of both cholecystokinin (CCK) and incretin hormones, including glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP, also known as glucose-dependent insulinotropic polypeptide). Incretin hormones support the survival and proliferation of pancreatic β-cells, which translates into increased insulin secretion. Moreover, the increase in Ca2+ concentration activates the phosphoinositide 3-kinase (PI3K) pathway including Akt, which stimulates downstream anabolic signaling, favoring cell growth (adipogenesis), proliferation, (angiogenesis), and survival. Upregulated glucose transporter type 4 (GLUT4) activity promotes increased glucose uptake, whereas augmented vascular endothelial growth factor (VEGF) expression induced by NF-кB enhances angiogenesis. Increased DAG synthesis activates protein kinase C (PKC), which results in the further activation of various intracellular signal transduction systems, including extracellular signal-regulated kinases ERK1 and ERK2 (ERK1/2). ERK1/2 has anti-apoptotic effects promoting cell growth and survival and indirectly reduces ghrelin activity by inhibiting proprotein convertase 1/3 (PC1/3), an enzyme necessary for the cleavage of pro-ghrelin into mature ghrelin in the Golgi apparatus [91,92,93,96,99,100,101,102,103,104].
—METABOLIC EFFECTS via classical Gα q/11 signaling. In this pathway, omega-3-fatty acids (Ω-3 FAs), acting as a ligand and agonist of GPR120, activate the Gαq/11-linked phospholipase C-beta (PLCβ). After PLCβ activation, phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolyzes into diacyl glycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), which serve as second messengers. IP3 binds to receptors, the conformational changes of which, after activation, lead to the mobilization of calcium ions (Ca2+) from intracellular stores. The following cytosolic Ca2+ influx stimulates the release of both cholecystokinin (CCK) and incretin hormones, including glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP, also known as glucose-dependent insulinotropic polypeptide). Incretin hormones support the survival and proliferation of pancreatic β-cells, which translates into increased insulin secretion. Moreover, the increase in Ca2+ concentration activates the phosphoinositide 3-kinase (PI3K) pathway including Akt, which stimulates downstream anabolic signaling, favoring cell growth (adipogenesis), proliferation, (angiogenesis), and survival. Upregulated glucose transporter type 4 (GLUT4) activity promotes increased glucose uptake, whereas augmented vascular endothelial growth factor (VEGF) expression induced by NF-кB enhances angiogenesis. Increased DAG synthesis activates protein kinase C (PKC), which results in the further activation of various intracellular signal transduction systems, including extracellular signal-regulated kinases ERK1 and ERK2 (ERK1/2). ERK1/2 has anti-apoptotic effects promoting cell growth and survival and indirectly reduces ghrelin activity by inhibiting proprotein convertase 1/3 (PC1/3), an enzyme necessary for the cleavage of pro-ghrelin into mature ghrelin in the Golgi apparatus [91,92,93,96,99,100,101,102,103,104].  —METABOLIC EFFECTS via Gα i/o signaling. Gαi/o affects related signaling pathways through the inhibition of adenylate cyclase (AC) activity, influencing intracellular cyclic adenosine 3,5-monophosphate (cAMP) levels in ghrelin-producing cells with a subsequent decrease in ghrelin synthesis. In this mechanism, the inhibitory effect of ghrelin on insulin secretion in pancreatic islet β-cells is reduced [99,105]. a PC1/3 is synthesized and expressed in a variety of tissues such as the neuroendocrine cells in the arcuate and paraventricular nuclei of the hypothalamus, the enteroendocrine cells (gut), and in β cells of the pancreas. b CCK is produced in the intestinal epithelial endocrine I-cells of the small intestine (mainly in the duodenum) and various neurons in the gastrointestinal tract and central nervous system (CNS). c GLP-1 and GIP are produced in the intestinal epithelial endocrine L cells and K cells, respectively. Since 2003, when Fredriksson et al. discovered the GPR120/FFAR4 protein [43,106], interest in this receptor has consistently increased as further reports of the anti-inflammatory and metabolic effects of its agonists have appeared [99,107,108,109,110,111,112].
—ANTI-INFLAMMATORY EFFECTS via beta-arrestin 2 (ARRβ2) signaling in immune pro-inflammatory cells. After receptor stimulation, the adapter protein ARRβ2 is recruited, which, after forming a complex with GPR120, leads to the desensitization of GPR120 as well as other receptors [e.g., Toll-like receptor 4 (TLR4), interleukin 1 receptor (IL1R), and tumor necrosis factor receptor (TNFR)] to pro-inflammatory stimuli. The GPR120- ARRβ2 complex internalizes and interacts with TGF-β-activated kinase 1 (MAP3K7)-binding protein 1 (TAB1) and NLRP3 [nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR)-containing protein (NLR) family member 3] inflammasome, which leads to the inhibition of the subsequent inflammatory cascade. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-кB) essential modulator (NEMO) ubiquitination, which is crucial for the activation of the canonical NF-κB signaling pathway, does not occur. A decrease in the concentration of pro-inflammatory cytokines indirectly modulates adipose tissue cell functions by reducing insulin resistance and, consequently, stimulating glucose uptake [82,94,95,96,97,98]. —METABOLIC EFFECTS via classical Gα q/11 signaling. In this pathway, omega-3-fatty acids (Ω-3 FAs), acting as a ligand and agonist of GPR120, activate the Gαq/11-linked phospholipase C-beta (PLCβ). After PLCβ activation, phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolyzes into diacyl glycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), which serve as second messengers. IP3 binds to receptors, the conformational changes of which, after activation, lead to the mobilization of calcium ions (Ca2+) from intracellular stores. The following cytosolic Ca2+ influx stimulates the release of both cholecystokinin (CCK) and incretin hormones, including glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP, also known as glucose-dependent insulinotropic polypeptide). Incretin hormones support the survival and proliferation of pancreatic β-cells, which translates into increased insulin secretion. Moreover, the increase in Ca2+ concentration activates the phosphoinositide 3-kinase (PI3K) pathway including Akt, which stimulates downstream anabolic signaling, favoring cell growth (adipogenesis), proliferation, (angiogenesis), and survival. Upregulated glucose transporter type 4 (GLUT4) activity promotes increased glucose uptake, whereas augmented vascular endothelial growth factor (VEGF) expression induced by NF-кB enhances angiogenesis. Increased DAG synthesis activates protein kinase C (PKC), which results in the further activation of various intracellular signal transduction systems, including extracellular signal-regulated kinases ERK1 and ERK2 (ERK1/2). ERK1/2 has anti-apoptotic effects promoting cell growth and survival and indirectly reduces ghrelin activity by inhibiting proprotein convertase 1/3 (PC1/3), an enzyme necessary for the cleavage of pro-ghrelin into mature ghrelin in the Golgi apparatus [91,92,93,96,99,100,101,102,103,104]. —METABOLIC EFFECTS via Gα i/o signaling. Gαi/o affects related signaling pathways through the inhibition of adenylate cyclase (AC) activity, influencing intracellular cyclic adenosine 3,5-monophosphate (cAMP) levels in ghrelin-producing cells with a subsequent decrease in ghrelin synthesis. In this mechanism, the inhibitory effect of ghrelin on insulin secretion in pancreatic islet β-cells is reduced [99,105]. a PC1/3 is synthesized and expressed in a variety of tissues such as the neuroendocrine cells in the arcuate and paraventricular nuclei of the hypothalamus, the enteroendocrine cells (gut), and in β cells of the pancreas. b CCK is produced in the intestinal epithelial endocrine I-cells of the small intestine (mainly in the duodenum) and various neurons in the gastrointestinal tract and central nervous system (CNS). c GLP-1 and GIP are produced in the intestinal epithelial endocrine L cells and K cells, respectively. Since 2003, when Fredriksson et al. discovered the GPR120/FFAR4 protein [43,106], interest in this receptor has consistently increased as further reports of the anti-inflammatory and metabolic effects of its agonists have appeared [99,107,108,109,110,111,112].
—METABOLIC EFFECTS via Gα i/o signaling. Gαi/o affects related signaling pathways through the inhibition of adenylate cyclase (AC) activity, influencing intracellular cyclic adenosine 3,5-monophosphate (cAMP) levels in ghrelin-producing cells with a subsequent decrease in ghrelin synthesis. In this mechanism, the inhibitory effect of ghrelin on insulin secretion in pancreatic islet β-cells is reduced [99,105]. a PC1/3 is synthesized and expressed in a variety of tissues such as the neuroendocrine cells in the arcuate and paraventricular nuclei of the hypothalamus, the enteroendocrine cells (gut), and in β cells of the pancreas. b CCK is produced in the intestinal epithelial endocrine I-cells of the small intestine (mainly in the duodenum) and various neurons in the gastrointestinal tract and central nervous system (CNS). c GLP-1 and GIP are produced in the intestinal epithelial endocrine L cells and K cells, respectively. Since 2003, when Fredriksson et al. discovered the GPR120/FFAR4 protein [43,106], interest in this receptor has consistently increased as further reports of the anti-inflammatory and metabolic effects of its agonists have appeared [99,107,108,109,110,111,112].
—ANTI-INFLAMMATORY EFFECTS via beta-arrestin 2 (ARRβ2) signaling in immune pro-inflammatory cells. After receptor stimulation, the adapter protein ARRβ2 is recruited, which, after forming a complex with GPR120, leads to the desensitization of GPR120 as well as other receptors [e.g., Toll-like receptor 4 (TLR4), interleukin 1 receptor (IL1R), and tumor necrosis factor receptor (TNFR)] to pro-inflammatory stimuli. The GPR120- ARRβ2 complex internalizes and interacts with TGF-β-activated kinase 1 (MAP3K7)-binding protein 1 (TAB1) and NLRP3 [nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR)-containing protein (NLR) family member 3] inflammasome, which leads to the inhibition of the subsequent inflammatory cascade. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-кB) essential modulator (NEMO) ubiquitination, which is crucial for the activation of the canonical NF-κB signaling pathway, does not occur. A decrease in the concentration of pro-inflammatory cytokines indirectly modulates adipose tissue cell functions by reducing insulin resistance and, consequently, stimulating glucose uptake [82,94,95,96,97,98]. —METABOLIC EFFECTS via classical Gα q/11 signaling. In this pathway, omega-3-fatty acids (Ω-3 FAs), acting as a ligand and agonist of GPR120, activate the Gαq/11-linked phospholipase C-beta (PLCβ). After PLCβ activation, phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolyzes into diacyl glycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), which serve as second messengers. IP3 binds to receptors, the conformational changes of which, after activation, lead to the mobilization of calcium ions (Ca2+) from intracellular stores. The following cytosolic Ca2+ influx stimulates the release of both cholecystokinin (CCK) and incretin hormones, including glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP, also known as glucose-dependent insulinotropic polypeptide). Incretin hormones support the survival and proliferation of pancreatic β-cells, which translates into increased insulin secretion. Moreover, the increase in Ca2+ concentration activates the phosphoinositide 3-kinase (PI3K) pathway including Akt, which stimulates downstream anabolic signaling, favoring cell growth (adipogenesis), proliferation, (angiogenesis), and survival. Upregulated glucose transporter type 4 (GLUT4) activity promotes increased glucose uptake, whereas augmented vascular endothelial growth factor (VEGF) expression induced by NF-кB enhances angiogenesis. Increased DAG synthesis activates protein kinase C (PKC), which results in the further activation of various intracellular signal transduction systems, including extracellular signal-regulated kinases ERK1 and ERK2 (ERK1/2). ERK1/2 has anti-apoptotic effects promoting cell growth and survival and indirectly reduces ghrelin activity by inhibiting proprotein convertase 1/3 (PC1/3), an enzyme necessary for the cleavage of pro-ghrelin into mature ghrelin in the Golgi apparatus [91,92,93,96,99,100,101,102,103,104]. —METABOLIC EFFECTS via Gα i/o signaling. Gαi/o affects related signaling pathways through the inhibition of adenylate cyclase (AC) activity, influencing intracellular cyclic adenosine 3,5-monophosphate (cAMP) levels in ghrelin-producing cells with a subsequent decrease in ghrelin synthesis. In this mechanism, the inhibitory effect of ghrelin on insulin secretion in pancreatic islet β-cells is reduced [99,105]. a PC1/3 is synthesized and expressed in a variety of tissues such as the neuroendocrine cells in the arcuate and paraventricular nuclei of the hypothalamus, the enteroendocrine cells (gut), and in β cells of the pancreas. b CCK is produced in the intestinal epithelial endocrine I-cells of the small intestine (mainly in the duodenum) and various neurons in the gastrointestinal tract and central nervous system (CNS). c GLP-1 and GIP are produced in the intestinal epithelial endocrine L cells and K cells, respectively. Since 2003, when Fredriksson et al. discovered the GPR120/FFAR4 protein [43,106], interest in this receptor has consistently increased as further reports of the anti-inflammatory and metabolic effects of its agonists have appeared [99,107,108,109,110,111,112].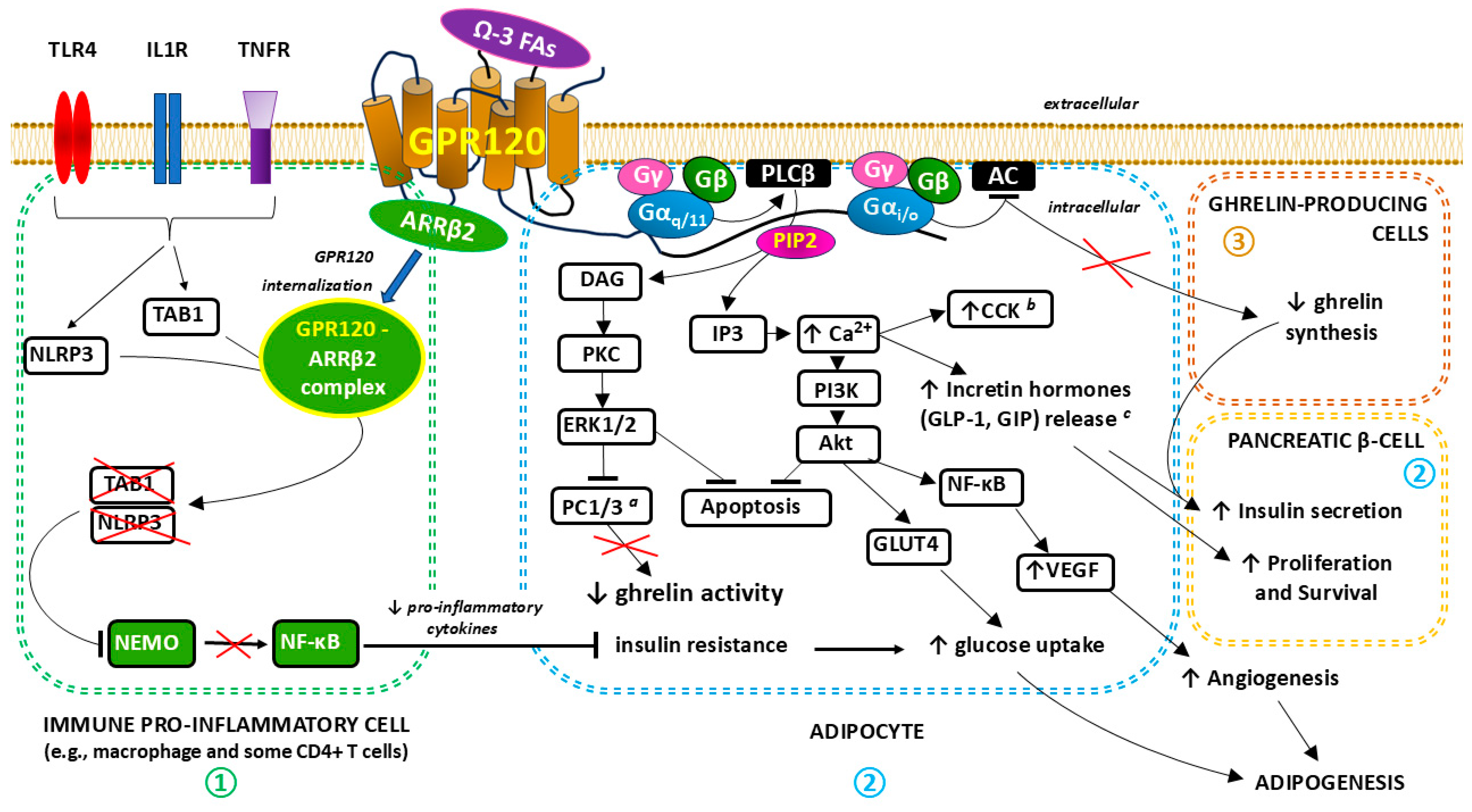
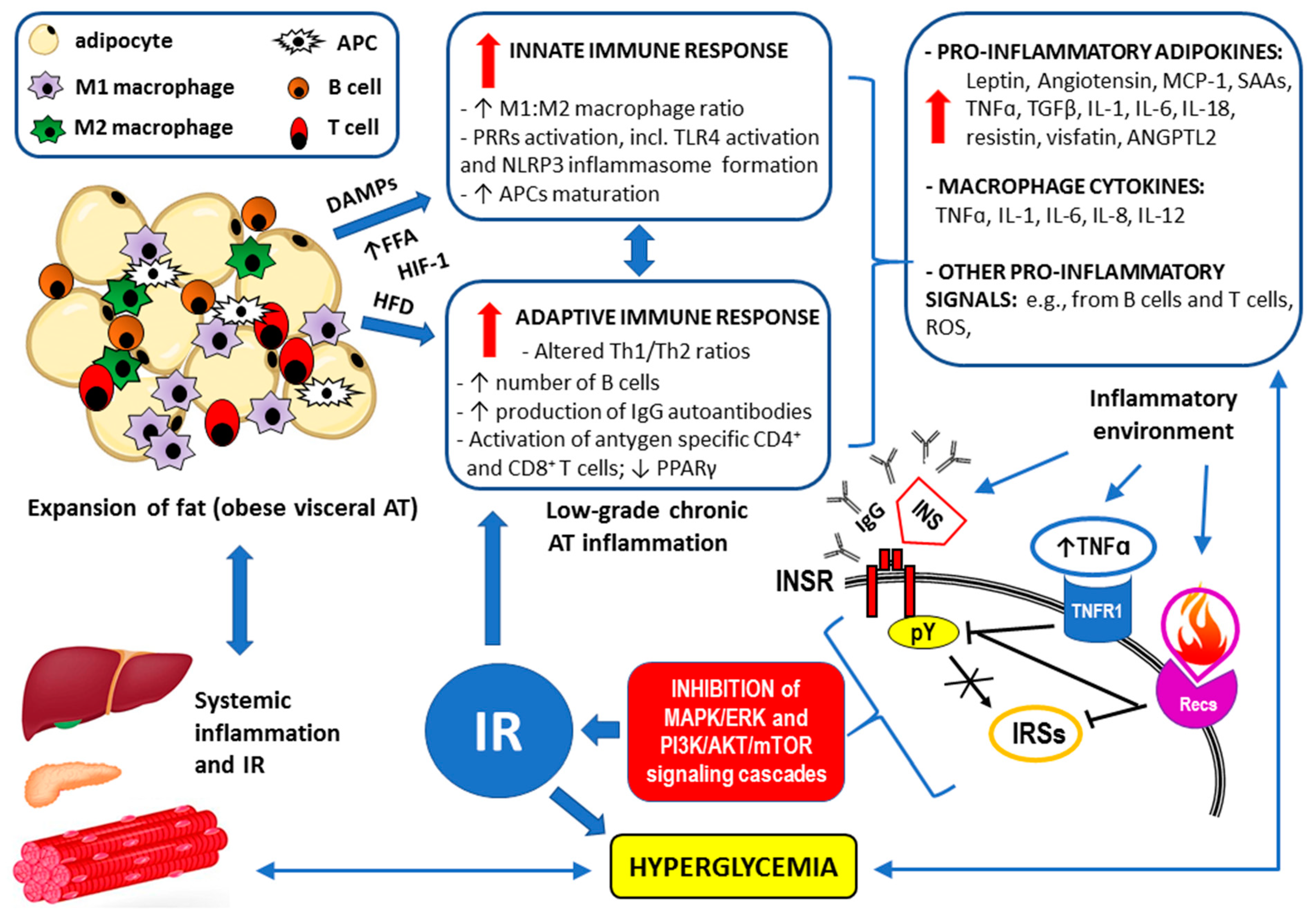

{kind=link}
{kind=link}
{kind=link}
| GPR120/FFAR4 Non-LCFA Agonist from Natural Sources | The Structure of the Molecule and Chemical Nomenclature | Source | Agonist’s Potency (EC50) * | Selectivity | Documented Metabolic Effects in Obesity-Related Disorders |
|---|---|---|---|---|---|
| Grifolin derivatives: - Grifolic acid and - Grifolic acid methyl ether | 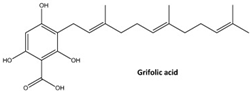 2,4-dihydroxy-6-methyl-3-[(2E,6E)-3,7,11-trimethyldodeca-2,6,10-trienyl] benzoic acid 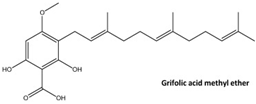 2-hydroxy-4-methoxy-6-methyl-3-[(2E,6E)-3,7,11-trimethyldodeca-2,6,10-trienyl] benzoic acid | First extracted from the mushroom Albatrellus confluens, its content was also confirmed in Albatrellus dispansus, Albatrellus ovinus, Peperomia galioides, a species of plant in the family Piperaceae, and other organisms with data available (i.e., Rhododendron dauricum), including tomato (Solanum lycopersicum L.) [246,253,254] | >30 µM [242,243,255] >30 µM [242,253,255] | Selective partial agonist Selective partial agonist |
|
| KDT501 (isohumulone) |  potassium;(1R,5S)-2-hydroxy-3-(3-methylbutanoyl)-5-(3-methylbutyl)-1-(4-methylpentanoyl)-4-oxocyclopent-2-en-1-olate | Substituted 1,3-cyclopentadione chemically derived from hops (Humulus lupulus L.) extracts [249] | 30.3 µM (mice) [249] | Non-selective, also partial PPARγ agonist [249] |
|
| Teadenol A |  (4aR,10aR)-7,9-dihydroxy-4-methylidene-10,10a-dihydro-4aH-pyrano[3,2-b]chromene-2-carboxylic acid | Polyphenol isolated from both Japanese and Chinese postfermented teas (Camellia sinensis L.) [252] | N.A.D. # | Selective agonist [252] |
|
| Synthetic GPR120/FFAR4 Agonist | The Structure of the Molecule and Chemical Nomenclature | Agonist’s Potency (EC50) * | Selectivity | Documented Metabolic Effects in Obesity-Related Disorders |
|---|---|---|---|---|
| GW9508 | 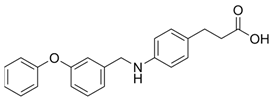 4-{[(3-phenoxyphenyl)methyl]amino} benzenepropanoic acid | 2.2 µM [91] | Non-selective: ~100-fold selectivity for GPR40 over GPR120 |
|
| NCG21 |  4-{4-[2-(phenyl-pyridin-2-yl-amino)-ethoxy]-phenyl}-butyric acid | N.A.D.# [225,233,243,271] | PPAR-γ agonist derivative; high (albeit incomplete) selectivity for GPR120 over GPR40 |
|
| TUG-891 | 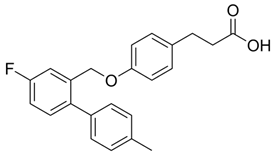 ortho-biphenyl ligand 4-{[4-fluoro-4′-methyl(1,1′-biphenyl)-2-yl]methoxy}-benzenepropanoic acid | 73.74 μM, (bovine neutrophils) [291] | GPR40 non-selective agonist GW9508 derivative; a potent and selective agonist of the GPR120 with 1000-fold selectivity over human GPR40 |
|
| Compound 29 (phenylpropanoic acid) |  3-Phenylpropanoic acid | 42 nM [295] | Highly selective, medium potency |
|
| Compound 18 (chromane propionic acid derivative) | 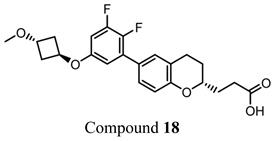 | 35 nM (mice) [275] | Highly selective and potent |
|
| Compound A (Merck) | 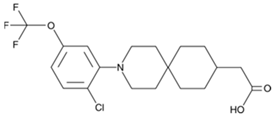 3-[2-chloro-5-(trifluoromethoxy)phenyl]-3-azaspiro[5.5]undecane-9-acetic acid | ~0.35 µM (mice) [277] |
Highly selective, reversible superagonist
|
|
| Metabolex 36 |  3-(3,5-difluoro-4-((3-methyl-1-phenyl-1H-pyrazol-5-yl)methoxy)phenyl)-2-methylpropanoic acid | 570 nM [211] | Selective and potent |
|
| AZ13581837 |  2-(3-Ethynyl-5-(3-pyridyloxy)phenyl)-3H-1,2-benzothiazole 1,1-dioxide | 120 nM [279] | Selective and potent |
|
| GPR120 agonist III | 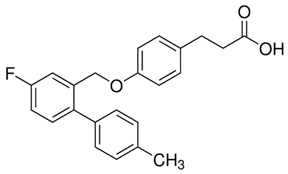 3-(4-((4-Fluoro-4′-methyl-(1,1′-biphenyl)-2-yl)methoxy)-phenyl)propanoic acid | 44 nM [268] | Fully selective for GPR120 and potent |
|
| GSK137647A | 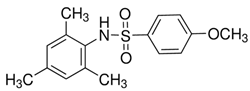 4-Methoxy-N-(2,4,6-trimethylphenyl)-benzenesulfonamide, N-Mesityl-4-methoxybenzenesulfonamide | 501 nM (mice) [297] | Selective and potent |
|
| TUG-1197 (compound 34) | 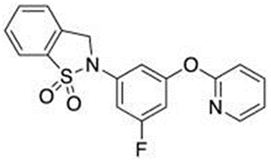 2-(3-fluoro-5-pyridin-2-yloxyphenyl)-3H-1,2-benzothiazole 1,1-dioxide | 24 nM [300] | Fully selective for GPR120 and very potent |
| Origin of the GPR120/FFAR4 Agonist | Example Compounds | General Limitations in Therapeutic Use in Obesity-Related Metabolic Disorders | Future Strategies |
|---|---|---|---|
| NATURAL SOURCES (non-LCFA agonists) | Grifolin derivatives (grifolic acid and grifolic acid methyl ether), KDT501 (isohumulone), Teadenol A |
|
|
| SYNTHETIC | GW9508, NCG21, TUG-891, Compound 29 (phenylpropanoic acid), Compound 18 (chromane propionic acid derivative), Compound A (Merck), Metabolex 36, AZ13581837, GPR120 agonist III, GSK137647A, TUG-1197 (compound 34) |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szukiewicz, D. Potential Therapeutic Exploitation of G Protein-Coupled Receptor 120 (GPR120/FFAR4) Signaling in Obesity-Related Metabolic Disorders. Int. J. Mol. Sci. 2025, 26, 2501. https://doi.org/10.3390/ijms26062501
Szukiewicz D. Potential Therapeutic Exploitation of G Protein-Coupled Receptor 120 (GPR120/FFAR4) Signaling in Obesity-Related Metabolic Disorders. International Journal of Molecular Sciences. 2025; 26(6):2501. https://doi.org/10.3390/ijms26062501
Chicago/Turabian StyleSzukiewicz, Dariusz. 2025. "Potential Therapeutic Exploitation of G Protein-Coupled Receptor 120 (GPR120/FFAR4) Signaling in Obesity-Related Metabolic Disorders" International Journal of Molecular Sciences 26, no. 6: 2501. https://doi.org/10.3390/ijms26062501
APA StyleSzukiewicz, D. (2025). Potential Therapeutic Exploitation of G Protein-Coupled Receptor 120 (GPR120/FFAR4) Signaling in Obesity-Related Metabolic Disorders. International Journal of Molecular Sciences, 26(6), 2501. https://doi.org/10.3390/ijms26062501