1. Introduction
Mitochondria, known for their diverse functions, have emerged as promising targets in anticancer strategies [
1]. The focus of research has shifted towards targeting mitochondrial metabolism, encompassing aspects such as the electron transport chain function, redox signaling pathways, ROS homeostasis, and apoptotic signaling pathways. Notably, mitochondria, despite being integral to the cell, are considered to have originated as independent organisms, separate from the genetic mechanisms of host cells. This unique origin underscores the potential benefits of targeting mitochondria, as it allows for circumventing tumor genetic surveillance mechanisms that often lead to drug resistance [
2,
3]. Consequently, the delivery of anticancer drugs to mitochondria holds significant clinical relevance, as it could enhance the drug selectivity for cancer cells, overcome drug resistance, and greatly augment the anticancer efficacy [
1].
Recent advancements in mitochondrial-targeting therapies for cancer have focused on exploiting the unique characteristics of cancer cell mitochondria, such as their altered membrane potential. A prime example is the triphenylphosphonium (TPP) cation, which has been widely studied for its mitochondrial selectivity due to the electrochemical gradient inherent to these organelles. Beyond TPP, mitochondria-penetrating peptides (MPPs) have garnered attention, characterized by their ability to translocate across biological membranes and localize within the mitochondrial matrix, demonstrating potential as carriers for antitumor agents [
4,
5]. Furthermore, pyridinium derivatives are being explored for their pro-apoptotic activity through mitochondrial pathways. A notable study reported the accumulation of amyloid (Aβ) peptides in mitochondria through an interaction with the TOMM22 channel, a component of the mitochondrial import machinery, indicating a mechanism for peptide accumulation that may extend to therapeutic applications [
6,
7]. Moreover, peptides like SS-31 and those containing a mitochondrial-targeting sequence (MTS) conjugated to cell-penetrating peptides offer a broad landscape of mitochondrial modulators with therapeutic potential [
8]. These studies underscore the diversity of strategies employed to target and manipulate mitochondrial function, revealing vast potential for the development of novel anticancer therapies.
Mito-FF is a synthetic molecule composed of diphenylalanine (FF), triphenylphosphonium (TPP), and pyrene, designed to selectively target mitochondria [
9]. Diphenylalanine enables self-assembly into highly ordered nanostructures through hydrogen bonding and π-π stacking interactions, while TPP, a lipophilic cation, allows for efficient accumulation in the mitochondrial matrix by traversing the inner mitochondrial membrane [
10,
11]. Pyrene, as a fluorescent probe, facilitates the visualization of Mito-FF’s localization and behavior within cells. The reaction underlying Mito-FF formation is driven by the unique self-assembly properties of diphenylalanine, which aggregates into tubular structures when a critical aggregation concentration (CAC) is reached. Within mitochondria, Mito-FF molecules undergo self-assembly into fibrils that disrupt mitochondrial membranes and functions. This process is thought to increase ROS production and initiate apoptotic pathways, making Mito-FF a promising candidate for overcoming chemotherapy resistance in cancer therapy. The antitumor efficacy of Mito-FF has been demonstrated in gastric and liver cancer animal models [
12,
13]. In this study, we aimed to investigate whether the promising antitumor effects of Mito-FF could also be observed in pancreatic cancer models, focusing on its mitochondrial-targeting and apoptosis-inducing mechanisms.
3. Discussion
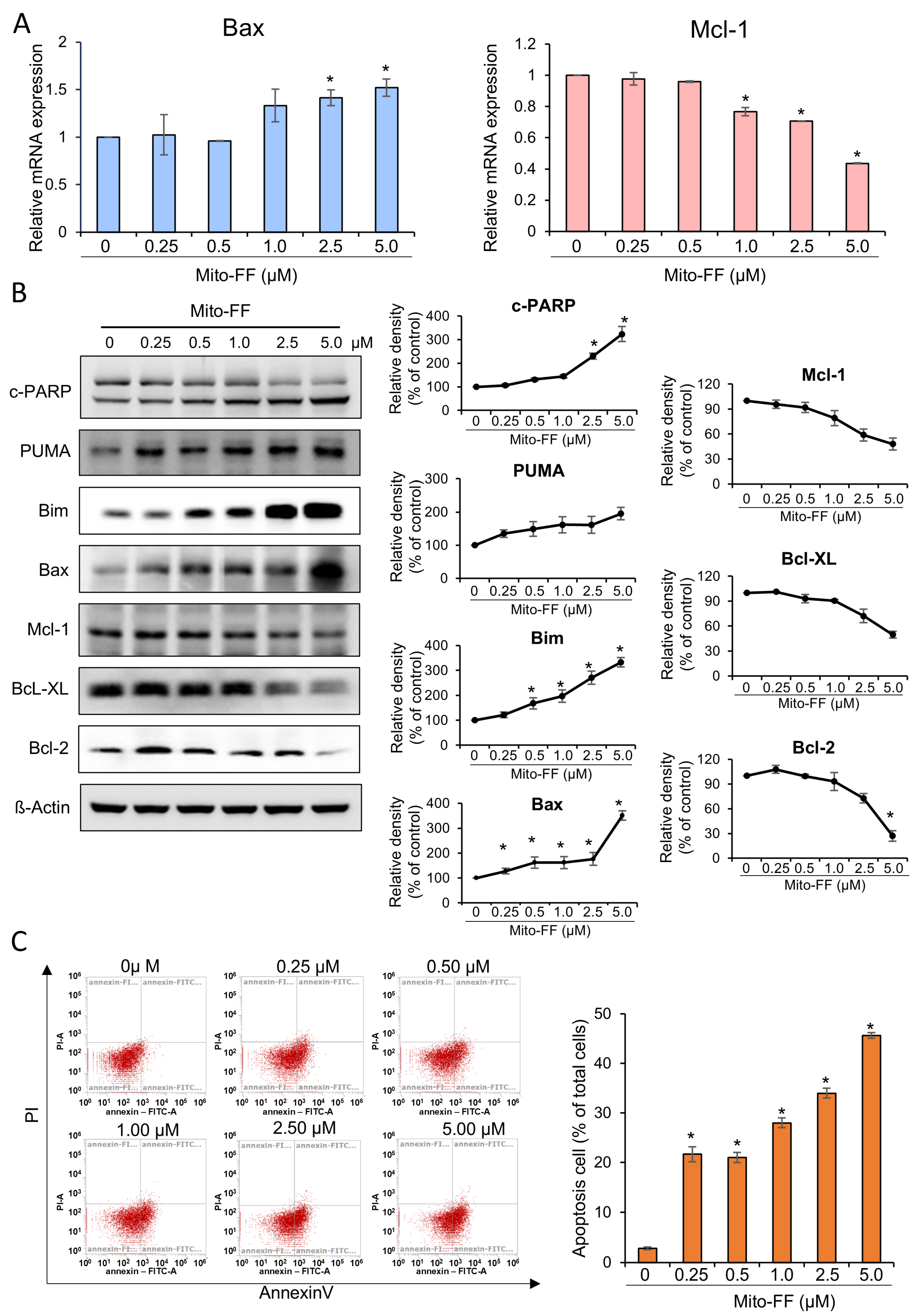
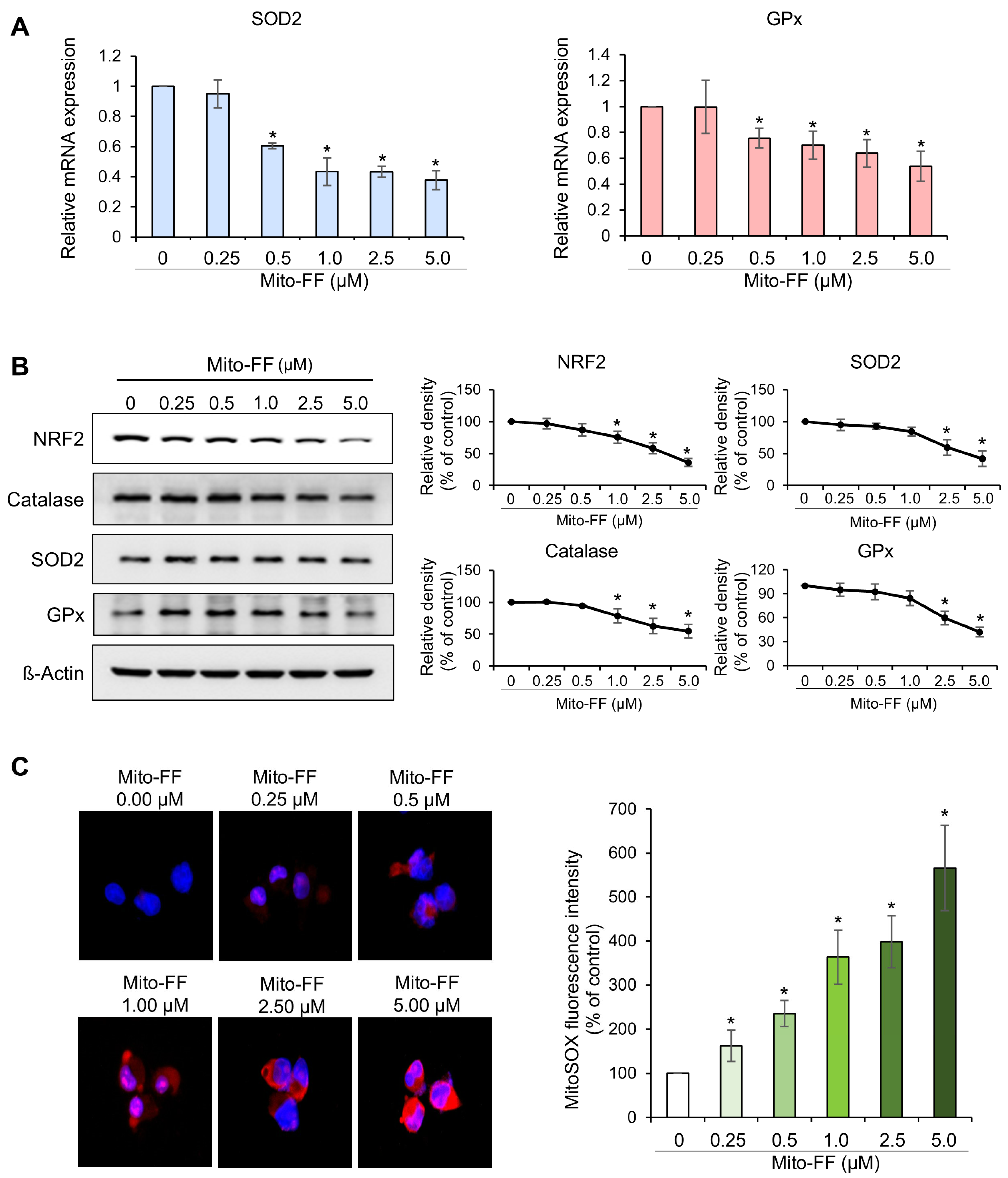
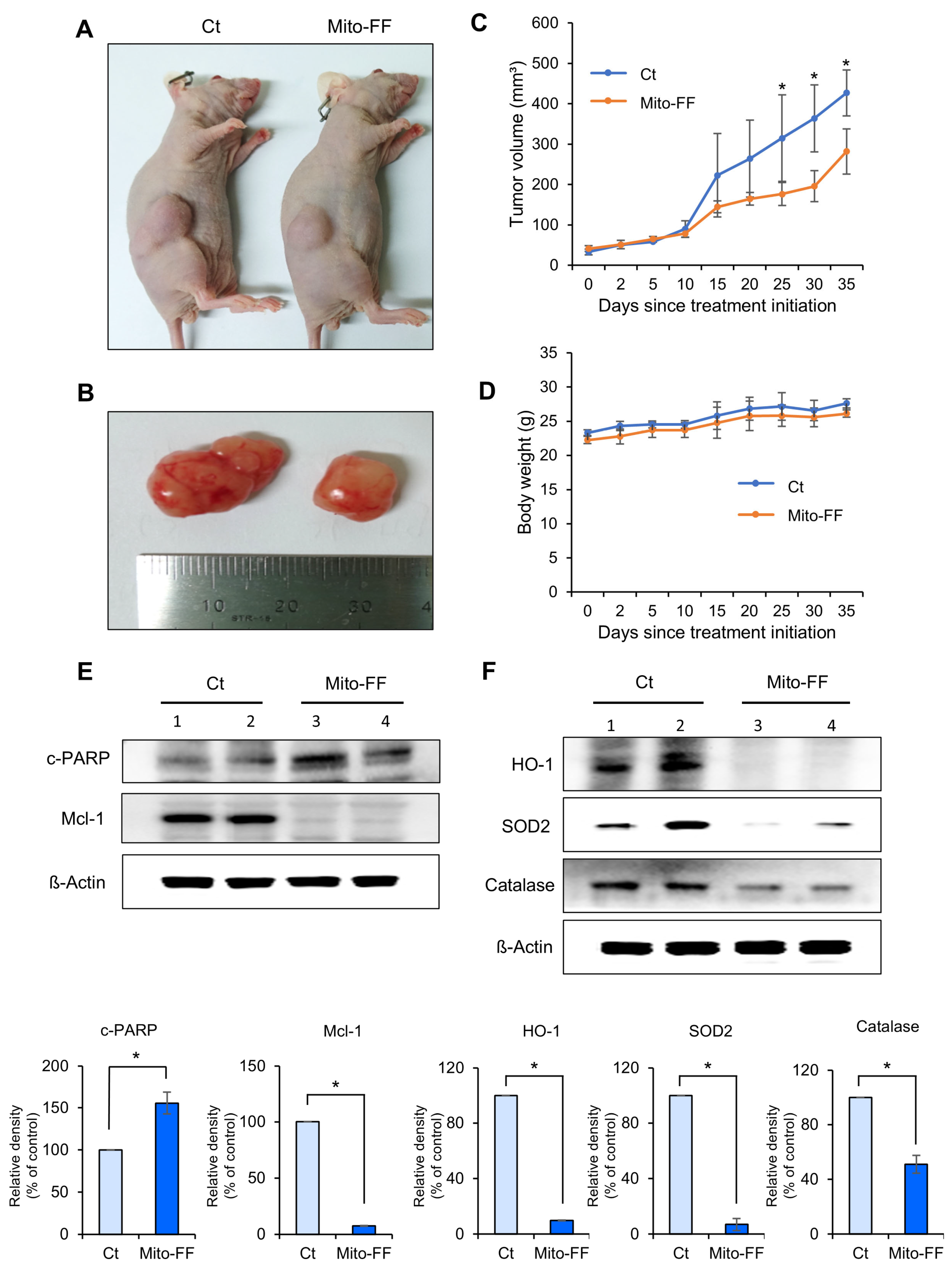
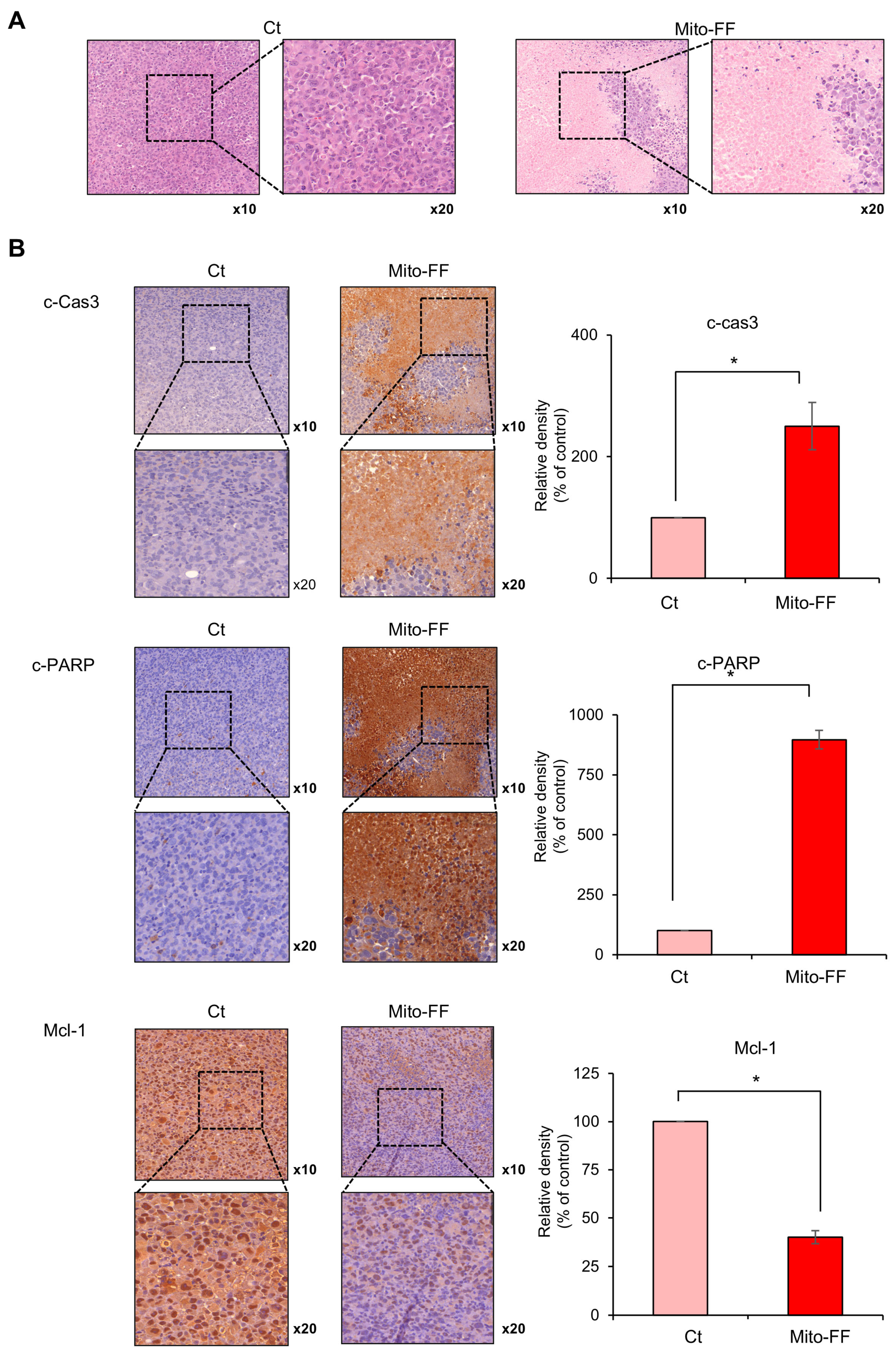
In this study, the therapeutic efficacy of the novel anticancer compound Mito-FF was assessed in both in vitro and in vivo models of pancreatic cancer. Cell viability assays revealed a concentration-dependent reduction in the viability of MIA-PACA2 pancreatic cancer cells in response to Mito-FF treatment (p < 0.05). Following in vitro Mito-FF treatment, several molecular analyses, including Real-time PCR, Western blot analysis, and MitoSOX staining, indicated an increase in apoptosis, a reduction in the expression of antioxidant enzymes, and an elevation in mitochondrial ROS levels. In a mouse xenograft model of pancreatic cancer, the intravenous administration of Mito-FF resulted in a reduced tumor volume. Furthermore, it enhanced the expression of pro-apoptotic markers while reducing the expression of anti-apoptotic markers in excised tumor tissues, as confirmed through various techniques including Real-time PCR, Western blot analysis, and immunohistochemical staining. Collectively, Mito-FF demonstrated significant anticancer effects against pancreatic cancer cells, primarily achieved by promoting apoptosis, diminishing the expression of antioxidant enzymes, and increasing the mitochondrial ROS levels within tumor tissues.
Cancer cells commonly exhibit deviations from “normal” energy metabolism, enabling their survival and proliferation in the challenging microenvironmental conditions frequently encountered within tumors, such as hypoxia and a limited nutrient availability. Mitochondria, serving as the primary source of energy and metabolites within cells, play a pivotal role in this adaptive response. Mitochondrial abnormalities are thus commonly observed in tumor cells, where they undergo metabolic shifts, transitioning from normal oxidative phosphorylation patterns to glycolysis even under aerobic conditions [
14]. These alterations in mitochondrial metabolism lead to various adverse outcomes, including elevated levels of ROS, the excessive production of free radicals, the dysregulation of ion channels, and other metabolic changes [
3,
15]. Consequently, the investigation of strategies aimed at modulating these altered mitochondrial metabolic pathways has emerged as a prominent focus in cancer research. Given the multifaceted roles of mitochondria in energy metabolism, apoptosis regulation, and cell signaling, mitigating the toxic effects on normally metabolizing cells represents an area where mitochondria-targeted agents hold significant therapeutic promise, distinguishing them from other targeted anticancer agents.
Self-assembly represents an equilibrium process involving the transition from individual building units to their aggregated state, necessitating a concentration surpassing the critical aggregation concentration (CAC) [
16,
17]. However, achieving the CAC within living cells presents challenges due to the complex chemical environment, which hinders interactions among synthetic building units. Intracellular self-assembly thus demands higher molecular concentrations than the CAC, limiting its practical applications. To address this limitation, the concept of Organelle-Localized Induced Supramolecular Self-Assembly (OLISA) was utilized in this study as a general strategy to induce self-assembly within cells by elevating the local concentrations of self-assembling molecules without the need for additional external treatments [
9]. This approach relies on small molecules capable of diffusing through the cell membrane reaching specific organelles or subcellular compartments based on their targeting moieties. Within these targeted organelles, the molecules undergo self-assembly due to the increased local concentration, exploiting the fact that the accumulation of molecules inside organelles, such as mitochondria, can be 500–1000 times higher than in the extracellular space [
9]. Unlike Enzyme-Instructed Intracellular Self-Assembly (EISA), which typically requires very high concentrations of molecules (several hundreds of micromoles) and often occurs within the cell or near the cell surface [
18,
19,
20], OLISA operates at lower dosages (several tens of micromoles), providing a distinct advantage.
A significant advantage of OLISA lies in its ability to activate the intended function upon self-assembly within the specific targeted organelle, a feature effectively demonstrated through the use of the mitochondria-accumulating amphiphilic peptide Mito-FF [
12,
13]. Mito-FF comprises diphenylalanine as a β-sheet-forming building block, TPP as a mitochondrial-targeting moiety, and pyrene as a fluorescent probe. Mito-FF selectively accumulates in the mitochondria of cancer cells due to their elevated negative membrane potential compared to normal cells [
21,
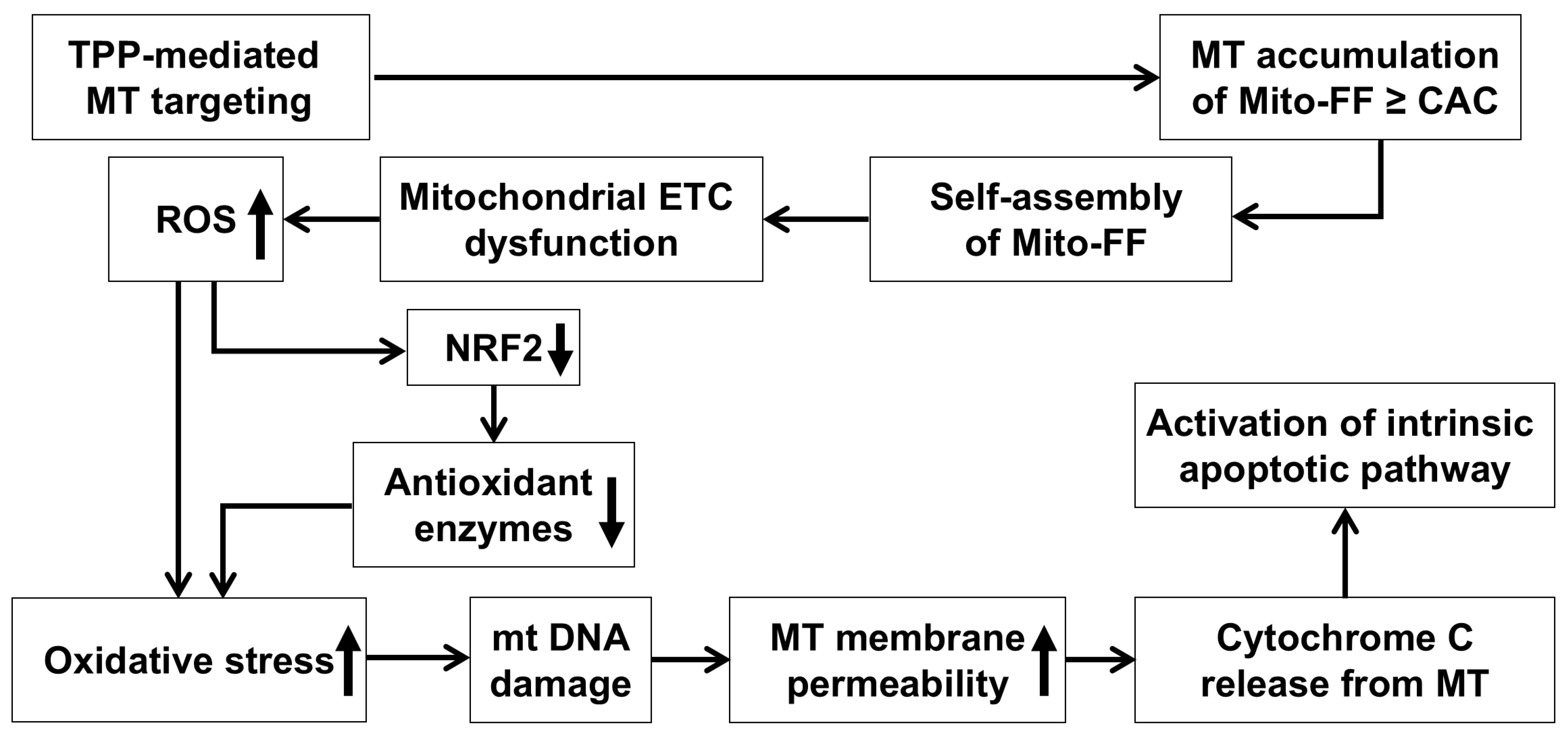
22]. This targeting is facilitated by the TPP cation in Mito-FF, which exploits the mitochondrial membrane potential differences to penetrate and accumulate within cancer cells (
Figure 6). The rapid division and high metabolic demand of cancer cells further amplify this effect, utilizing the Warburg effect, which lowers the membrane potential significantly. Consequently, Mito-FF is less likely to accumulate in normal cells, where the mitochondrial membrane potential is less conducive to the accumulation of TPP-conjugated molecules. The increased concentration of Mito-FF inside the mitochondria drives its self-assembly into a fibrous structure. Notably, this fibril formation is absent in normal cells. The rigid Mito-FF fibrils disrupt the mitochondrial membrane and activate the intrinsic apoptotic pathway in cancer cells.
The mechanism of action for Mito-FF can be understood through its ability to selectively target mitochondria and disrupt their function, as demonstrated in our study. Mito-FF is believed to accumulate in mitochondria due to its TPP moiety, which takes advantage of the negative mitochondrial membrane potential [
23]. Once localized, Mito-FF likely undergoes self-assembly into nanostructures, disrupting the mitochondrial membrane integrity and depolarizing the mitochondrial membrane [
24]. This disruption is proposed to impair the electron transport chain (ETC), leading to an overproduction of ROS, including superoxide (O
2⁻), hydrogen peroxide (H
2O
2), and hydroxyl radicals (OH•), as evidenced by the significant reduction in NRF2, SOD2, catalase, and GPx in our study, indicating a depletion of antioxidant defenses [
25]. The excessive ROS levels are expected to cause oxidative damage to mitochondrial DNA and proteins, creating a feedback loop that exacerbates mitochondrial dysfunction [
26]. This is likely to increase the mitochondrial membrane permeability, as seen from the release of pro-apoptotic factors such as cytochrome c, which then triggers downstream apoptotic cascades. Cytochrome c is hypothesized to interact with Apaf-1, forming the apoptosome, which activates caspase-9 and subsequently caspase-3 and caspase-7, as supported by our data showing an increased expression of cleaved caspases (c-Cas8, c-Cas9, and c-Cas3) [
27,
28]. Simultaneously, ROS overproduction appears to downregulate anti-apoptotic proteins, such as Bcl-2 and Bcl-XL, while upregulating pro-apoptotic proteins like Bax and Bak, further amplifying the apoptotic signaling pathway [
29,
30]. This dual mechanism—ROS-mediated mitochondrial dysfunction and the activation of intrinsic apoptotic pathways—highlights the therapeutic potential of Mito-FF in overcoming chemotherapy resistance in pancreatic cancer. These findings are consistent with the observed dose-dependent reduction in antioxidant proteins and the increase in apoptotic markers in our experiments.
4. Materials and Methods
4.1. Cell Culture
MIA-PACA2, AsPC-1, Capan-1, and Capan-2 pancreatic cancer cell lines were sourced from the Korea Cell Line Bank (KCLB, Seoul, Republic of Korea). These cells were cultured in a DMEM/High (Hyclone; Logan, UT, USA) medium supplemented with 10% fetal bovine serum (Hyclone) and 1% Penicillin–Streptomycin (GibcoBRL; Carlsbad, CA, USA). Human pancreatic stellate cells (HPSCs, iXCells Biotechnologies; San Diego, CA, USA) were also included in the study to evaluate off-target effects and were maintained under identical culture conditions. The cell cultures were maintained at 37 °C in a humidified environment with 5% CO2 in an incubator. To verify the authenticity of our cell lines and prevent the potential issue of cell line misidentification, we conducted short tandem repeat (STR) profiling on all cell lines before initiating experiments. This step ensured the validity of our results and supported the reproducibility of our study.
4.2. Mito-FF Preparation
Mito-FF was synthesized following standard solid-phase peptide synthesis methods. The peptide backbone, comprising diphenylalanine (FF) and lysine (K), was conjugated with 1-pyrene-carboxylic acid to enhance self-assembly through π-π stacking interactions and hydrophobic effects. The lysine side chain was further conjugated with TPP for efficient mitochondrial targeting, leveraging the mitochondrial membrane potential. The resulting product, Mito-FF, was purified using high-performance liquid chromatography (HPLC) to achieve >99% purity. For experimental use, Mito-FF was dissolved in dimethyl sulfoxide (DMSO) to prepare a 10 mM stock solution. For in vitro assays, the stock solution was diluted with a culture medium to a final DMSO concentration of <0.1%, ensuring there was no solvent-induced cytotoxicity. For in vivo studies, the peptide was further diluted in sterile PBS and administered intravenously to mice at 50 μg/kg body weight twice per week for 35 days. Each solution was freshly prepared before use to maintain the stability and biological activity.
4.3. Cell Viability Assay
The cell viability of MIA-PACA2 pancreatic cancer cells was assessed utilizing the Ez-Cytox Cell Viability Assay Kit (Itsbio, Seoul, Republic of Korea) following the guidelines provided by the manufacturer. In all in vitro experiments, DMSO-treated cells served as the vehicle control to ensure that the effects observed were specifically due to Mito-FF and not to the solvent. This control was critical for validating the specificity and efficacy of the treatment.
4.4. Colony Formation Assay
A colony-forming assay was conducted using 6-well plates. Cells were seeded at a density of 1000 cells per well in 4 mL of a complete culture medium. Two distinct concentrations of Mito-FF were administered to the cells for a duration of 7 days. Subsequently, the formed colonies were stained with a 0.2% methylene blue solution and quantified.
4.5. Real-Time PCR
Total RNA was extracted from both MIA-Paca2 cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Reverse transcription was carried out using an RT premix kit (TOYOBO, Osaka, Japan) with 1 µg of RNA, following the manufacturer’s instructions. For the SYBR Green real-time quantitative polymerase chain reaction (PCR), the following primers were used: human SOD2, forward 5′-GCATCTGTTGGTGTCCAAGG-3′ and reverse 5′-CTGTTGTTCCTTGCAGTGG-3′; human GPx, forward 5′-TCGAGAAGTGCGAGGTGAAC-3′ and reverse 5′-AGCTTGGGGTCGGTCATAAG-3′; human Mcl-1, forward 5′-GGGCAGGATTGTGACTCTCATT-3′ and reverse 5′-GATGCAGCTTTCTTGGTTTATGG-3′; human Bax, forward 5′-TGGAGCTGCAGAGGATGATTG-3′ and reverse 5′-GAAGTTGCCGTCAGAAAACATG-3′; and human GAPDH, forward 5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse 5′-TGGTGAAGACGCCAGTGGA-3′. PCR reactions were conducted using the Applied Biosystems StepOnePlus Real-time PCR system (Thermo, Carlsbad, CA, USA).
4.6. Western Blot Analysis
MIA-PACA2 pancreatic cancer cells and mouse tissues were lysed using the EzRIPA Lysis kit (ATTO Corporation; Tokyo, Japan) and quantified using the Bradford reagent (Bio-Rad, Hercules, CA, USA). Western analysis was performed with primary antibodies (dilution of 1:1000) from Cell Signaling Technology (Beverly, MA, USA), followed by HRP-conjugated secondary antibodies (dilution 1:2000) from Vector Laboratories (Burlingame, CA, USA). Specific immune complexes were visualized using the Western Blotting Plus Chemiluminescence Reagent (Millipore, Bedford, MA, USA). Primary antibodies against NRF2, catalase, SOD2, GPx, cleaved PARP, MCL-1, PUMA, HO-1, and β-actin, as well as horseradish peroxidase (HRP)-conjugated secondary antibodies, were sourced from Cell Signaling Technology (Beverly, MA, USA).
4.7. Quantification of Apoptosis Using Flow Cytometry
Apoptosis detection involved staining cells with annexin V/PI, utilizing the FITC Annexin V apoptosis detection kit from BD Biosciences (Flanklin Lakes, NJ, USA). Following a 15 min incubation in darkness at 25 °C, a flow cytometric analysis (Thermo) was performed to analyze the cells.
4.8. MitoSOX Red Staining
MIA-PACA2 pancreatic cancer cells were grown on Lab-Tek chamber slides (Thermo Fisher Scientific; Waltham, MA, USA). Following a 24 h treatment with Mito-FF, the cells were subjected to staining with 10 µM MitoSOX reagent (Thermo Fisher Scientific) at 37 °C for 10 min. Subsequently, the mitochondrial ROS levels were visualized using the M5000 fluorescence imaging system (EVOS, Invitrogen, CA, USA).
4.9. Animals and Study Design
Five-week-old male BALB/c nude mice (Orient Bio, Seongnam, Republic of Korea) were utilized to establish a comparative model of subcutaneous tumor growth. MIA-PACA2 pancreatic cancer cells (5 × 106 cells) were subcutaneously inoculated into each mouse. All animal procedures were conducted in accordance with the guidelines of the Institute for Laboratory Animal Research, the Catholic University of Korea (IRB No: CUMC-2020-0113-03). Two weeks following the injection of tumor cells, all mice developed measurable tumors. To evaluate the in vivo efficacy, the mice were randomly divided into groups (n = 5 per group) and subjected to intravenous treatment with a control and Mito-FF (50 µg/mL, equivalent to 11.80 μM, in 100 µL PBS, administered twice a week) for 35 days. The tumor dimensions were measured biweekly using a caliper, and the tumor volume (V) was determined using the formula length × width2 × 0.5236. Upon the completion of the treatment period, all mice were euthanized.
4.10. Immunohistochemical Analysis
For the immunohistochemical analysis, tissue sections that had been formalin-fixed and paraffin-embedded underwent deparaffinization and rehydration through an ethanol series, followed by epitope retrieval using standard protocols. Antibodies specific to cleaved caspase 3, cleaved PARP, and MCL-1 were employed for immunofluorescence staining (all antibodies were sourced from Cell Signaling Technology). Subsequently, the samples were examined under a laser scanning microscope (Eclipse TE300; Nikon, Tokyo, Japan) to assess the expression of these antibodies.
4.11. Statistical Analysis
All data were analyzed utilizing SPSS 11.0 software (SPSS Inc., Chicago, IL, USA) and were presented as the mean ± the standard deviation (SD). Statistical comparisons among groups were performed using the Kruskal–Wallis test. Probability values of p < 0.05 were considered statistically significant.



 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}