
Breaking Barriers: The Role of the Bone Marrow Microenvironment in Multiple Myeloma Progression

 , ,
, ,  ,
,  , , , and
, , , and
Abstract
1. Bone Marrow Barrier
2. Abnormal Bone Marrow Barrier
3. Multiple Myeloma
4. Multiple Myeloma and Bone Marrow Barrier
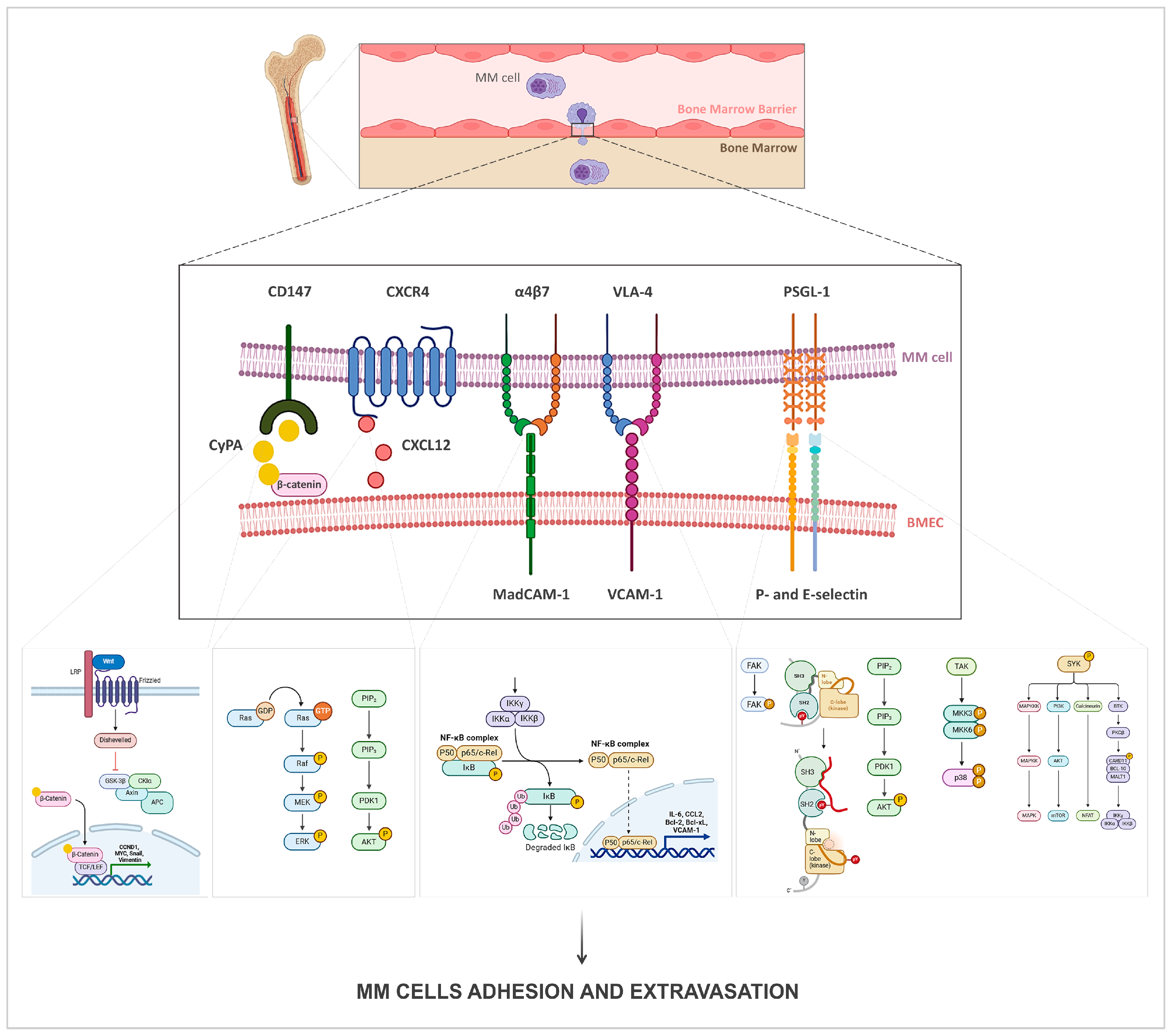
4.1. Homing Pathways
4.2. Proliferation Pathways
5. Diagnosis and Treatments of Multiple Myeloma
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BMB | Bone Marrow Barrier |
| BMECs | Bone Marrow Endothelial Cells |
| BMSCs | Bone Marrow Stromal Cells |
| MM | Multiple Myeloma |
| AML | Acute Myeloid Leukemia |
| IL | Interleukin |
| TNF | Tumor Necrosis Factor |
| ICAM | Intercellular Cell Adhesion Molecule |
| VCAM-1 | Vascular Cell Adhesion Molecule-1 |
| VLA-4 | Very Late Antigen-4 |
| BM | Bone Marrow |
| VEGF | Vascular Endothelial Growth Factor |
| Ang | Angiopoietin |
| NO | Nitric Oxide |
| iNOS | inducible Nitric Oxide Synthase |
| eNOS | endothelial Nitric Oxide Synthase |
| MMPs | Matrix Metalloproteinases |
| EC | Extracellular Matrix |
| EMM | Extramedullary Multiple Myeloma |
| MGUS | Monoclonal Gammopathy of Undetermined Significance |
| SMM | Smoldering Multiple Myeloma |
| CyPA | Cyclophilin A |
| siRNA | Small Interfering RNA |
| AURKA | Aurora Kinase A |
| EMT | Epithelial–Mesenchymal Transition |
| Syk | Spleen Tyrosine Kinase |
| MadCAM-1 | Mucosal Vascular Addressin Cell Adhesion Molecule-1 |
| CAM-DR | Cell-Adhesion-Mediated Drug Resistance |
| SDF-1α | Stromal-Cell-Derived Factor 1α |
| PPARβ/δ | Peroxisome Proliferator-Activated Receptor β/δ |
| PGI2 | Prostacyclin I2 |
| EGFR | Epidermal Growth Factor Receptor |
| HB-EGF | Heparin-Binding EGF-like Growth Factor |
| IGF-1 | Insulin-like Growth Factor-1 |
| Epo | Erythropoietin |
| MRD | Minimal Residual Disease |
| NGS | Next-Generation Sequencing |
| MFC | Multiparameter Flow Cytometry |
| EVs | Extracellular Vesicles |
| miRNA | microRNA |
| CAR-T | Chimeric Antigen Receptor-T |
References
- Lucas, D. Structural Organization of the Bone Marrow and Its Role in Hematopoiesis. Curr. Opin. Hematol. 2021, 28, 36–42. [Google Scholar] [CrossRef]
- Tavassoli, M. The Marrow-Blood Barrier. Br. J. Haematol. 1979, 41, 297–302. [Google Scholar] [CrossRef]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct Bone Marrow Blood Vessels Differentially Regulate Hematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Campbell, F.R. Ultrastructure of Cytoskeletal Element of Endothelial and Stromal Cells of Rat Marrow. Anat. Rec. 1990, 227, 152–158. [Google Scholar] [CrossRef]
- Weiss, L. Transmural Cellular Passage in Vascular Sinuses of Rat Bone Marrow. Blood 1970, 36, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Osmond, D.g. Basement Membrane of Mouse Bone Marrow Sinusoids Shows Distinctive Structure and Proteoglycan Composition: A High Resolution Ultrastructural Study. Anat. Rec. 2001, 264, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hendriks, M.; Chatzis, A.; Ramasamy, S.K.; Kusumbe, A.P. Bone Vasculature and Bone Marrow Vascular Niches in Health and Disease. J. Bone Miner. Res. 2020, 35, 2103–2120. [Google Scholar] [CrossRef] [PubMed]
- Heideveld, E.; van den Akker, E. Digesting the Role of Bone Marrow Macrophages on Hematopoiesis. Immunobiology 2017, 222, 814–822. [Google Scholar] [CrossRef]
- Yusop, N.; Battersby, P.; Alraies, A.; Sloan, A.J.; Moseley, R.; Waddington, R.J. Isolation and Characterisation of Mesenchymal Stem Cells from Rat Bone Marrow and the Endosteal Niche: A Comparative Study. Stem Cells Int. 2018, 2018, 6869128. [Google Scholar] [CrossRef]
- De Bruyn, P.P.H.; Michelson, S.; Thomas, T.B. The Migration of Blood Cells of the Bone Marrow through the Sinusoidal Wall. J. Morphol. 1971, 133, 417–437. [Google Scholar] [CrossRef]
- Möhle, R.; Rafii, S.; Moore, M.A. The Role of Endothelium in the Regulation of Hematopoietic Stem Cell Migration. Stem Cells 1998, 16 (Suppl. 1), 159–165. [Google Scholar] [CrossRef]
- Prisby, R.D. Bone Marrow Microvasculature. Compr. Physiol. 2020, 10, 1009–1046. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, Q.; Bian, W.; Wang, J.; Guan, H.; Niu, J. Imaging Techniques and Clinical Application of the Marrow–Blood Barrier in Hematological Malignancies. Diagnostics 2024, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Rafii, S.; Mohle, R.; Shapiro, F.; Frey, B.M.; Moore, M.A. Regulation of Hematopoiesis by Microvascular Endothelium. Leuk. Lymphoma 1997, 27, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Stucki, A.; Rivier, A.S.; Gikic, M.; Monai, N.; Schapira, M.; Spertini, O. Endothelial Cell Activation by Myeloblasts: Molecular Mechanisms of Leukostasis and Leukemic Cell Dissemination. Blood 2001, 97, 2121–2129. [Google Scholar] [CrossRef]
- Sanadgol, N.; Mostafaie, A.; Bahrami, G.; Mansouri, K.; Ghanbari, F.; Bidmeshkipour, A. Elaidic Acid Sustains LPS and TNF-Alpha Induced ICAM-1 and VCAM-I Expression on Human Bone Marrow Endothelial Cells (HBMEC). Clin. Biochem. 2010, 43, 968–972. [Google Scholar] [CrossRef]
- Fereydouni, Z.; Amirinezhad Fard, E.; Mansouri, K.; Mohammadi Motlagh, H.-R.; Mostafaie, A. Saponins from Tribulus terrestris L. Extract Down-Regulate the Expression of ICAM-1, VCAM-1 and E-Selectin in Human Endothelial Cell Lines. Int. J. Mol. Cell. Med. 2020, 9, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Sanadgol, N.; Mostafaie, A.; Mansouri, K.; Bahrami, G. Effect of Palmitic Acid and Linoleic Acid on Expression of ICAM-1 and VCAM-1 in Human Bone Marrow Endothelial Cells (HBMECs). Arch. Med. Sci. 2012, 8, 192–198. [Google Scholar] [CrossRef]
- Mosteo, L.; Storer, J.; Batta, K.; Searle, E.J.; Duarte, D.; Wiseman, D.H. The Dynamic Interface Between the Bone Marrow Vascular Niche and Hematopoietic Stem Cells in Myeloid Malignancy. Front. Cell Dev. Biol. 2021, 9, 635189. [Google Scholar] [CrossRef]
- Barbier, V.; Erbani, J.; Fiveash, C.; Davies, J.M.; Tay, J.; Tallack, M.R.; Lowe, J.; Magnani, J.L.; Pattabiraman, D.R.; Perkins, A.C.; et al. Endothelial E-Selectin Inhibition Improves Acute Myeloid Leukaemia Therapy by Disrupting Vascular Niche-Mediated Chemoresistance. Nat. Commun. 2020, 11, 2042. [Google Scholar] [CrossRef]
- Winkler, I.G.; Barbier, V.; Tay, J.; Levesque, J.-P.; Magnani, J.L.; Fiveash, C.E.; Erbani, J.D. Blocking Vascular Niche E-Selectin Dampens AML Stem Cell Regeneration/Survival Potential In Vivo By Inhibiting MAPK/ERK and PI3K/AKT Signalling Pathways. Blood 2019, 134, 2657. [Google Scholar] [CrossRef]
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal Leukemia-Stroma VCAM-1/VLA-4-Dependent Activation of NF-κB Mediates Chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef]
- Hall, B.M.; Fortney, J.E.; Gibson, L.F. Alteration of Nuclear Factor-kappaB (NF-kappaB) Expression in Bone Marrow Stromal Cells Treated with Etoposide. Biochem. Pharmacol. 2001, 61, 1243–1252. [Google Scholar] [CrossRef]
- Matsunaga, T.; Takemoto, N.; Sato, T.; Takimoto, R.; Tanaka, I.; Fujimi, A.; Akiyama, T.; Kuroda, H.; Kawano, Y.; Kobune, M.; et al. Interaction between Leukemic-Cell VLA-4 and Stromal Fibronectin Is a Decisive Factor for Minimal Residual Disease of Acute Myelogenous Leukemia. Nat. Med. 2003, 9, 1158–1165. [Google Scholar] [CrossRef]
- Layani-Bazar, A.; Skornick, I.; Berrebi, A.; Pauker, M.H.; Noy, E.; Silberman, A.; Albeck, M.; Longo, D.L.; Kalechman, Y.; Sredni, B. Redox Modulation of Adjacent Thiols in VLA-4 by AS101 Converts Myeloid Leukemia Cells from a Drug-Resistant to Drug-Sensitive State. Cancer Res. 2014, 74, 3092–3103. [Google Scholar] [CrossRef]
- Ayala, F.; Dewar, R.; Kieran, M.; Kalluri, R. Contribution of Bone Microenvironment to Leukemogenesis and Leukemia Progression. Leukemia 2009, 23, 2233–2241. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Maxwell, C.A.; Taylor, B.J.; Hendzel, M.J.; Chesi, M.; Bergsagel, P.L.; Larratt, L.M.; Mant, M.J.; Reiman, T.; Belch, A.R.; et al. Overexpression of Transcripts Originating from the MMSET Locus Characterizes All t(4;14)(P16;Q32)-Positive Multiple Myeloma Patients. Blood 2005, 105, 4060–4069. [Google Scholar] [CrossRef]
- Santos, S.C.R.; Dias, S. Internal and External Autocrine VEGF/KDR Loops Regulate Survival of Subsets of Acute Leukemia through Distinct Signaling Pathways. Blood 2004, 103, 3883–3889. [Google Scholar] [CrossRef] [PubMed]
- Weidenaar, A.C.; ter Elst, A.; Koopmans-Klein, G.; Rosati, S.; den Dunnen, W.F.A.; Meeuwsen-de Boer, T.; Kamps, W.A.; Vellenga, E.; de Bont, E.S.J.M. High Acute Myeloid Leukemia Derived VEGFA Levels Are Associated with a Specific Vascular Morphology in the Leukemic Bone Marrow. Cell. Oncol. 2011, 34, 289–296. [Google Scholar] [CrossRef]
- Kampen, K.R.; ter Elst, A.; de Bont, E.S.J.M. Vascular Endothelial Growth Factor Signaling in Acute Myeloid Leukemia. Cell. Mol. Life Sci. 2012, 70, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Bellamy, W.T.; Richter, L.; Sirjani, D.; Roxas, C.; Glinsmann-Gibson, B.; Frutiger, Y.; Grogan, T.M.; List, A.F. Vascular Endothelial Cell Growth Factor Is an Autocrine Promoter of Abnormal Localized Immature Myeloid Precursors and Leukemia Progenitor Formation in Myelodysplastic Syndromes. Blood 2001, 97, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Chand, R.; Chandra, H.; Chandra, S.; Verma, S.K. Role of Microvessel Density and Vascular Endothelial Growth Factor in Angiogenesis of Hematological Malignancies. Bone Marrow Res. 2016, 2016, 5043483. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, M.; Miwa, H.; Shikami, M.; Hiramatsu, A.; Ikai, T.; Tajima, E.; Yamamoto, H.; Miura, K.; Satoh, A.; Itoh, M.; et al. Autocrine Pathway of Angiopoietins-Tie2 System in AML Cells: Association with Phosphatidyl-Inositol 3 Kinase. Hematol. J. 2004, 5, 353–360. [Google Scholar] [CrossRef]
- Reikvam, H.; Hatfield, K.J.; Lassalle, P.; Kittang, A.O.; Ersvaer, E.; Bruserud, Ø. Targeting the Angiopoietin (Ang)/Tie-2 Pathway in the Crosstalk between Acute Myeloid Leukaemia and Endothelial Cells: Studies of Tie-2 Blocking Antibodies, Exogenous Ang-2 and Inhibition of Constitutive Agonistic Ang-1 Release. Expert. Opin. Investig. Drugs 2010, 19, 169–183. [Google Scholar] [CrossRef]
- Passaro, D.; Di Tullio, A.; Abarrategi, A.; Rouault-Pierre, K.; Foster, K.; Ariza-McNaughton, L.; Montaner, B.; Chakravarty, P.; Bhaw, L.; Diana, G.; et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 324–341.e6. [Google Scholar] [CrossRef]
- Koistinen, P.; Siitonen, T.; Mäntymaa, P.; Säily, M.; Kinnula, V.; Savolainen, E.R.; Soini, Y. Regulation of the Acute Myeloid Leukemia Cell Line OCI/AML-2 by Endothelial Nitric Oxide Synthase under the Control of a Vascular Endothelial Growth Factor Signaling System. Leukemia 2001, 15, 1433–1441. [Google Scholar] [CrossRef]
- Brandão, M.M.; Soares, E.; Salles, T.S.; Saad, S.T. Expression of Inducible Nitric Oxide Synthase Is Increased in Acute Myeloid Leukaemia. Acta Haematol. 2001, 106, 95–99. [Google Scholar] [CrossRef]
- Verma, D.; Zanetti, C.; Godavarthy, P.S.; Kumar, R.; Minciacchi, V.R.; Pfeiffer, J.; Metzler, M.; Lefort, S.; Maguer-Satta, V.; Nicolini, F.E.; et al. Bone Marrow Niche-Derived Extracellular Matrix-Degrading Enzymes Influence the Progression of B-Cell Acute Lymphoblastic Leukemia. Leukemia 2020, 34, 1540–1552. [Google Scholar] [CrossRef]
- Chen, H.; Shen, Y.; Gong, F.; Jiang, Y.; Zhang, R. HIF-α Promotes Chronic Myelogenous Leukemia Cell Proliferation by Upregulating P21 Expression. Cell Biochem. Biophys. 2015, 72, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Singh, R.; Kushwaha, R.; Verma, S.P.; Tripathi, A.K.; Mahdi, A.A. The Molecular Role of HIF1α Is Elucidated in Chronic Myeloid Leukemia. Front. Oncol. 2022, 12, 912942. [Google Scholar] [CrossRef]
- Abdul-Aziz, A.M.; Shafat, M.S.; Sun, Y.; Marlein, C.R.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Zhou, Z.; Collins, A.; Bowles, K.M.; et al. HIF1α Drives Chemokine Factor Pro-Tumoral Signaling Pathways in Acute Myeloid Leukemia. Oncogene 2018, 37, 2676–2686. [Google Scholar] [CrossRef]
- Rohde, D.; Vandoorne, K.; Lee, I.-H.; Grune, J.; Zhang, S.; McAlpine, C.S.; Schloss, M.J.; Nayar, R.; Courties, G.; Frodermann, V.; et al. Bone Marrow Endothelial Dysfunction Promotes Myeloid Cell Expansion in Cardiovascular Disease. Nat. Cardiovasc. Res. 2022, 1, 28–44. [Google Scholar] [CrossRef]
- Alaarg, A.; Pérez-Medina, C.; Metselaar, J.M.; Nahrendorf, M.; Fayad, Z.A.; Storm, G.; Mulder, W.J.M. Applying Nanomedicine in Maladaptive Inflammation and Angiogenesis. Adv. Drug Deliv. Rev. 2017, 119, 143–158. [Google Scholar] [CrossRef]
- Castro, P.R.; Barbosa, A.S.; Pereira, J.M.; Ranfley, H.; Felipetto, M.; Gonçalves, C.A.X.; Paiva, I.R.; Berg, B.B.; Barcelos, L.S. Cellular and Molecular Heterogeneity Associated with Vessel Formation Processes. Biomed. Res. Int. 2018, 2018, 6740408. [Google Scholar] [CrossRef]
- Kuehl, W.M.; Bergsagel, P.L. Multiple Myeloma: Evolving Genetic Events and Host Interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Tamura, A.; Nakazawa, N.; Ueda, Y.; Abe, T.; Matsuda, F.; Kashima, K.; Taniwaki, M. The Ig Heavy Chain Gene Is Frequently Involved in Chromosomal Translocations in Multiple Myeloma and Plasma Cell Leukemia as Detected by In Situ Hybridization. Blood 1997, 90, 526–534. [Google Scholar] [CrossRef]
- Fenton, J.a.L.; Pratt, G.; Rawstron, A.C.; Morgan, G.J. Isotype Class Switching and the Pathogenesis of Multiple Myeloma. Hematol. Oncol. 2002, 20, 75–85. [Google Scholar] [CrossRef]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic Complexity of Multiple Myeloma and Its Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Blood, E.A.; Oken, M.M.; Kyle, R.A.; Dewald, G.W.; Bailey, R.J.; Van Wier, S.A.; Henderson, K.J.; Hoyer, J.D.; Harrington, D.; et al. Myeloma and the t(11;14)(Q13;Q32); Evidence for a Biologically Defined Unique Subset of Patients. Blood 2002, 99, 3735–3741. [Google Scholar] [CrossRef]
- Weinhold, N.; Johnson, D.C.; Chubb, D.; Chen, B.; Försti, A.; Hosking, F.J.; Broderick, P.; Ma, Y.P.; Dobbins, S.E.; Hose, D.; et al. The CCND1 c.870G>A Polymorphism Is a Risk Factor for t(11;14)(Q13;Q32) Multiple Myeloma. Nat. Genet. 2013, 45, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Reiman, T.; Belch, A.R.; Pilarski, L.M. Ten Years and Counting: So What Do We Know about t(4;14)(P16;Q32) Multiple Myeloma. Leuk. Lymphoma 2006, 47, 2289–2300. [Google Scholar] [CrossRef]
- Morito, N.; Yoh, K.; Maeda, A.; Nakano, T.; Fujita, A.; Kusakabe, M.; Hamada, M.; Kudo, T.; Yamagata, K.; Takahashi, S. A Novel Transgenic Mouse Model of the Human Multiple Myeloma Chromosomal Translocation t(14;16)(Q32;Q23). Cancer Res. 2011, 71, 339–348. [Google Scholar] [CrossRef]
- Hurt, E.M.; Wiestner, A.; Rosenwald, A.; Shaffer, A.L.; Campo, E.; Grogan, T.; Bergsagel, P.L.; Kuehl, W.M.; Staudt, L.M. Overexpression of C-Maf Is a Frequent Oncogenic Event in Multiple Myeloma That Promotes Proliferation and Pathological Interactions with Bone Marrow Stroma. Cancer Cell 2004, 5, 191–199. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Gerson, F.; Magrangeas, F.; Minvielle, S.; Harousseau, J.-L.; Bataille, R. Rearrangements of the C-Myc Oncogene Are Present in 15% of Primary Human Multiple Myeloma Tumors. Blood 2001, 98, 3082–3086. [Google Scholar] [CrossRef]
- Szabo, A.G.; Gang, A.O.; Pedersen, M.Ø.; Poulsen, T.S.; Klausen, T.W.; Nørgaard, P. Overexpression of C-Myc Is Associated with Adverse Clinical Features and Worse Overall Survival in Multiple Myeloma. Leuk. Lymphoma 2016, 57, 2526–2534. [Google Scholar] [CrossRef]
- Burroughs Garcìa, J.; Eufemiese, R.A.; Storti, P.; Sammarelli, G.; Craviotto, L.; Todaro, G.; Toscani, D.; Marchica, V.; Giuliani, N. Role of 1q21 in Multiple Myeloma: From Pathogenesis to Possible Therapeutic Targets. Cells 2021, 10, 1360. [Google Scholar] [CrossRef]
- Jovanović, K.K.; Escure, G.; Demonchy, J.; Willaume, A.; Van de Wyngaert, Z.; Farhat, M.; Chauvet, P.; Facon, T.; Quesnel, B.; Manier, S. Deregulation and Targeting of TP53 Pathway in Multiple Myeloma. Front. Oncol. 2019, 8, 665. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.; Durie, B.; Rajkumar, S.; Landgren, O.; Blade, J.; Merlini, G.; Kröger, N.; Einsele, H.; Vesole, D.; Dimopoulos, M.; et al. Monoclonal Gammopathy of Undetermined Significance (MGUS) and Smoldering (Asymptomatic) Multiple Myeloma: IMWG Consensus Perspectives Risk Factors for Progression and Guidelines for Monitoring and Management. Leukemia 2010, 24, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.B.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal Gammopathy of Undetermined Significance (MGUS) Consistently Precedes Multiple Myeloma: A Prospective Study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Landgren, O.; Mateos, M.-V. Smoldering Multiple Myeloma. Blood 2015, 125, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group Updated Criteria for the Diagnosis of Multiple Myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Tibullo, D.; Longo, A.; Vicario, N.; Romano, A.; Barbato, A.; Di Rosa, M.; Barbagallo, I.; Anfuso, C.D.; Lupo, G.; Gulino, R.; et al. Ixazomib Improves Bone Remodeling and Counteracts Sonic Hedgehog Signaling Inhibition Mediated by Myeloma Cells. Cancers 2020, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Bladé, J.; Beksac, M.; Caers, J.; Jurczyszyn, A.; von Lilienfeld-Toal, M.; Moreau, P.; Rasche, L.; Rosiñol, L.; Usmani, S.Z.; Zamagni, E.; et al. Extramedullary Disease in Multiple Myeloma: A Systematic Literature Review. Blood Cancer J. 2022, 12, 45. [Google Scholar] [CrossRef]
- Sammartano, V.; Cerase, A.; Venanzi, V.; Mazzei, M.A.; Vangone, B.E.; Gentili, F.; Chiarotti, I.; Bocchia, M.; Gozzetti, A. Central Nervous System Myeloma and Unusual Extramedullary Localizations: Real Life Practical Guidance. Front. Oncol. 2022, 12, 934240. [Google Scholar] [CrossRef]
- Abdallah, A.; Atrash, S.; Shahid, Z.; Jameel, M.; Grazziutti, M.; Apewokin, S.; Kumar, N.S.; Restrepo, A.; Waheed, S.; Van Rhee, F.; et al. Patterns of Central Nervous System Involvement in Relapsed and Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2014, 14, 211–214. [Google Scholar] [CrossRef]
- Bladé, J.; Fernández de Larrea, C.; Rosiñol, L.; Cibeira, M.T.; Jiménez, R.; Powles, R. Soft-Tissue Plasmacytomas in Multiple Myeloma: Incidence, Mechanisms of Extramedullary Spread, and Treatment Approach. J. Clin. Oncol. 2011, 29, 3805–3812. [Google Scholar] [CrossRef] [PubMed]
- Tohami, T.; Drucker, L.; Shapiro, H.; Radnay, J.; Lishner, M. Overexpression of Tetraspanins Affects Multiple Myeloma Cell Survival and Invasive Potential. FASEB J. 2007, 21, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; MacLeod, V.; Bendre, M.; Huang, Y.; Theus, A.M.; Miao, H.-Q.; Kussie, P.; Yaccoby, S.; Epstein, J.; Suva, L.J.; et al. Heparanase Promotes the Spontaneous Metastasis of Myeloma Cells to Bone. Blood 2005, 105, 1303–1309. [Google Scholar] [CrossRef]
- Ghobrial, I.M. Myeloma as a Model for the Process of Metastasis: Implications for Therapy. Blood 2012, 120, 20–30. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Zhang, L.-Y.; Xiang, Y.-H.; Li, D.; Zhang, J. Matrix Metalloproteinases and Tissue Inhibitors in Multiple Myeloma: Promote or Inhibit? Front. Oncol. 2023, 13, 1127407. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Rakshit, S.; Kumar, S. Extramedullary Disease in Multiple Myeloma. Blood Cancer J. 2021, 11, 161. [Google Scholar] [CrossRef]
- Li, Y.; Sun, Z.; Qu, X. Advances in the Treatment of Extramedullary Disease in Multiple Myeloma. Transl. Oncol. 2022, 22, 101465. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, P.P.G.; Figueroa-Espada, C.G.; Riley, R.S.; Gong, N.; Xue, L.; Sewastianik, T.; Dennis, P.S.; Loebel, C.; Chung, A.; Shepherd, S.J.; et al. In Vivo Bone Marrow Microenvironment siRNA Delivery Using Lipid–Polymer Nanoparticles for Multiple Myeloma Therapy. Proc. Natl. Acad. Sci. USA 2023, 120, e2215711120. [Google Scholar] [CrossRef]
- Figueroa-Espada, C.G.; Guimarães, P.P.G.; Riley, R.S.; Xue, L.; Wang, K.; Mitchell, M.J. siRNA Lipid–Polymer Nanoparticles Targeting E-Selectin and Cyclophilin A in Bone Marrow for Combination Multiple Myeloma Therapy. Cell. Mol. Bioeng. 2023, 16, 383–392. [Google Scholar] [CrossRef] [PubMed]
- van Andel, H.; Kocemba, K.A.; Spaargaren, M.; Pals, S.T. Aberrant Wnt Signaling in Multiple Myeloma: Molecular Mechanisms and Targeting Options. Leukemia 2019, 33, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Guo, M.; Gu, C.; Yang, Y. The Role of Wnt/β-Catenin Signaling Pathway in the Pathogenesis and Treatment of Multiple Myeloma (Review). Am. J. Transl. Res. 2021, 13, 9932–9949. [Google Scholar]
- Zhu, D.; Wang, Z.; Zhao, J.-J.; Calimeri, T.; Meng, J.; Hideshima, T.; Fulciniti, M.; Kang, Y.; Ficarro, S.; Tai, Y.-T.; et al. The Cyclophilin A-CD147 Complex Promotes Bone Marrow Colonization of B-Cell Malignancies: Implications for Therapy. Nat. Med. 2015, 21, 572–580. [Google Scholar] [CrossRef]
- Wang, G.; Shen, J.; Sun, J.; Jiang, Z.; Fan, J.; Wang, H.; Yu, S.; Long, Y.; Liu, Y.; Bao, H.; et al. Cyclophilin A Maintains Glioma-Initiating Cell Stemness by Regulating Wnt/β-Catenin Signaling. Clin. Cancer Res. 2017, 23, 6640–6649. [Google Scholar] [CrossRef]
- Fang, F.; Li, Q.; Wu, M.; Nie, C.; Xu, H.; Wang, L. CD147 Promotes Epithelial-Mesenchymal Transition of Prostate Cancer Cells via the Wnt/β-Catenin Pathway. Exp. Ther. Med. 2020, 20, 3154–3160. [Google Scholar] [CrossRef]
- Natoni, A.; Smith, T.A.G.; Keane, N.; McEllistrim, C.; Connolly, C.; Jha, A.; Andrulis, M.; Ellert, E.; Raab, M.S.; Glavey, S.V.; et al. E-Selectin Ligands Recognised by HECA452 Induce Drug Resistance in Myeloma, Which Is Overcome by the E-Selectin Antagonist, GMI-1271. Leukemia 2017, 31, 2642–2651. [Google Scholar] [CrossRef]
- Natoni, A.; Macauley, M.S.; O’Dwyer, M.E. Targeting Selectins and Their Ligands in Cancer. Front. Oncol. 2016, 6, 93. [Google Scholar] [CrossRef]
- Natoni, A.; Cerreto, M.; De Propris, M.S.; Petrucci, M.T.; Fazio, F.; Intoppa, S.; Milani, M.L.; Kirkham-McCarthy, L.; Henderson, R.; Swan, D.; et al. Sialofucosylation Enables Platelet Binding to Myeloma Cells via P-Selectin and Suppresses NK Cell-Mediated Cytotoxicity. Cancers 2023, 15, 2154. [Google Scholar] [CrossRef]
- O’Dwyer, M.; Kirkham-McCarthy, L.; Cerreto, M.; Foà, R.; Natoni, A. PSGL-1 Decorated with Sialyl Lewisa/x Promotes High Affinity Binding of Myeloma Cells to P-Selectin but Is Dispensable for E-Selectin Engagement. Sci. Rep. 2024, 14, 1756. [Google Scholar] [CrossRef]
- Yue, Z.; Wang, A.; Zhu, Z.; Tao, L.; Li, Y.; Zhou, L.; Chen, W.; Lu, Y. Holothurian Glycosaminoglycan Inhibits Metastasis via Inhibition of P-Selectin in B16F10 Melanoma Cells. Mol. Cell. Biochem. 2015, 410, 143–154. [Google Scholar] [CrossRef]
- Nolo, R.; Herbrich, S.; Rao, A.; Zweidler-McKay, P.; Kannan, S.; Gopalakrishnan, V. Targeting P-Selectin Blocks Neuroblastoma Growth. Oncotarget 2017, 8, 86657–86670. [Google Scholar] [CrossRef] [PubMed]
- Azab, A.K.; Quang, P.; Azab, F.; Pitsillides, C.; Thompson, B.; Chonghaile, T.; Patton, J.T.; Maiso, P.; Monrose, V.; Sacco, A.; et al. P-Selectin Glycoprotein Ligand Regulates the Interaction of Multiple Myeloma Cells with the Bone Marrow Microenvironment. Blood 2012, 119, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Abram, C.L.; Hundt, M.; Altman, A.; Lowell, C.A.; Ley, K. PSGL-1 Engagement by E-Selectin Signals through Src Kinase Fgr and ITAM Adapters DAP12 and FcRγ to Induce Slow Leukocyte Rolling. J. Exp. Med. 2008, 205, 2339–2347. [Google Scholar] [CrossRef]
- Chase, S.D.; Magnani, J.L.; Simon, S.I. E-Selectin Ligands as Mechanosensitive Receptors on Neutrophils in Health and Disease. Ann. Biomed. Eng. 2012, 40, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Lowell, C.A.; Ley, K. Syk Signaling Is Necessary for E-Selectin-Induced LFA-1-ICAM-1 Association and Rolling but Not Arrest. Immunity 2007, 26, 773–783. [Google Scholar] [CrossRef]
- Natoni, A.; Farrell, M.L.; Harris, S.; Falank, C.; Kirkham-McCarthy, L.; Macauley, M.S.; Reagan, M.R.; O’Dwyer, M. Sialyltransferase Inhibition Leads to Inhibition of Tumor Cell Interactions with E-Selectin, VCAM1, and MADCAM1, and Improves Survival in a Human Multiple Myeloma Mouse Model. Haematologica 2020, 105, 457–467. [Google Scholar] [CrossRef]
- Rodríguez-García, Y.; Martínez-Moreno, M.; Alonso, L.; Sánchez-Vencells, A.; Arranz, A.; Dagà-Millán, R.; Sevilla-Movilla, S.; Valeri, A.; Martínez-López, J.; Teixidó, J. Regulation of miRNA Expression by A4β1 Integrin-dependent Multiple Myeloma Cell Adhesion. eJHaem 2023, 4, 631–638. [Google Scholar] [CrossRef]
- Martínez-Moreno, M.; Leiva, M.; Aguilera-Montilla, N.; Sevilla-Movilla, S.; Isern de Val, S.; Arellano-Sánchez, N.; Gutiérrez, N.C.; Maldonado, R.; Martínez-López, J.; Buño, I.; et al. In Vivo Adhesion of Malignant B Cells to Bone Marrow Microvasculature Is Regulated by A4β1 Cytoplasmic-Binding Proteins. Leukemia 2016, 30, 861–872. [Google Scholar] [CrossRef]
- Hao, P.; Zhang, C.; Wang, R.; Yan, P.; Peng, R. Expression and Pathogenesis of VCAM-1 and VLA-4 Cytokines in Multiple Myeloma. Saudi J. Biol. Sci. 2020, 27, 1674–1678. [Google Scholar] [CrossRef]
- Hideshima, T.; Anderson, K.C. Signaling Pathway Mediating Myeloma Cell Growth and Survival. Cancers 2021, 13, 216. [Google Scholar] [CrossRef] [PubMed]
- Cippitelli, M.; Stabile, H.; Kosta, A.; Petillo, S.; Lucantonio, L.; Gismondi, A.; Santoni, A.; Fionda, C. Role of NF-κB Signaling in the Interplay between Multiple Myeloma and Mesenchymal Stromal Cells. Int. J. Mol. Sci. 2023, 24, 1823. [Google Scholar] [CrossRef]
- Ullah, T.R. The Role of CXCR4 in Multiple Myeloma: Cells’ Journey from Bone Marrow to beyond. J. Bone Oncol. 2019, 17, 100253. [Google Scholar] [CrossRef] [PubMed]
- García-Ortiz, A.; Rodríguez-García, Y.; Encinas, J.; Maroto-Martín, E.; Castellano, E.; Teixidó, J.; Martínez-López, J. The Role of Tumor Microenvironment in Multiple Myeloma Development and Progression. Cancers 2021, 13, 217. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Mishima, Y.; Sacco, A.; Moschetta, M.; Tai, Y.-T.; Shi, J.; Zhang, Y.; Reagan, M.R.; Huynh, D.; Kawano, Y.; et al. CXCR4 Regulates Extra-Medullary Myeloma through Epithelial-Mesenchymal Transition-like Transcriptional Activation. Cell Rep. 2015, 12, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Alsayed, Y.; Ngo, H.; Runnels, J.; Leleu, X.; Singha, U.K.; Pitsillides, C.M.; Spencer, J.A.; Kimlinger, T.; Ghobrial, J.M.; Jia, X.; et al. Mechanisms of Regulation of CXCR4/SDF-1 (CXCL12)–Dependent Migration and Homing in Multiple Myeloma. Blood 2007, 109, 2708–2717. [Google Scholar] [CrossRef]
- Peled, A.; Klein, S.; Beider, K.; Burger, J.A.; Abraham, M. Role of CXCL12 and CXCR4 in the Pathogenesis of Hematological Malignancies. Cytokine 2018, 109, 11–16. [Google Scholar] [CrossRef]
- Du, S.; Wagner, N.; Wagner, K.-D. The Emerging Role of PPAR Beta/Delta in Tumor Angiogenesis. PPAR Res. 2020, 2020, 3608315. [Google Scholar] [CrossRef] [PubMed]
- Piqueras, L.; Reynolds, A.R.; Hodivala-Dilke, K.M.; Alfranca, A.; Redondo, J.M.; Hatae, T.; Tanabe, T.; Warner, T.D.; Bishop-Bailey, D. Activation of PPARbeta/Delta Induces Endothelial Cell Proliferation and Angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lei, F.; Lin, Y.; Han, Y.; Yang, L.; Tan, H. Peroxisome Proliferator-activated Receptors as Therapeutic Target for Cancer. J. Cell. Mol. Med. 2023, 28, e17931. [Google Scholar] [CrossRef]
- Leone, P.; Solimando, A.G.; Prete, M.; Malerba, E.; Susca, N.; Derakhshani, A.; Ditonno, P.; Terragna, C.; Cavo, M.; Silvestris, N.; et al. Unraveling the Role of Peroxisome Proliferator-Activated Receptor β/Δ (PPAR β/Δ) in Angiogenesis Associated with Multiple Myeloma. Cells 2023, 12, 1011. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Giannico, D.; Leone, P.; Solimando, A.G.; Maiorano, E.; Caporusso, C.; Duda, L.; Tamma, R.; Mallamaci, R.; Susca, N.; et al. HB-EGF–EGFR Signaling in Bone Marrow Endothelial Cells Mediates Angiogenesis Associated with Multiple Myeloma. Cancers 2020, 12, 173. [Google Scholar] [CrossRef]
- Ramakrishnan, V.; D’Souza, A. Signaling Pathways and Emerging Therapies in Multiple Myeloma. Curr. Hematol. Malig. Rep. 2016, 11, 156–164. [Google Scholar] [CrossRef]
- John, L.; Krauth, M.T.; Podar, K.; Raab, M.-S. Pathway-Directed Therapy in Multiple Myeloma. Cancers 2021, 13, 1668. [Google Scholar] [CrossRef]
- Jasrotia, S.; Gupta, R.; Sharma, A.; Halder, A.; Kumar, L. Cytokine Profile in Multiple Myeloma. Cytokine 2020, 136, 155271. [Google Scholar] [CrossRef]
- Chong, P.S.Y.; Chng, W.-J.; de Mel, S. STAT3: A Promising Therapeutic Target in Multiple Myeloma. Cancers 2019, 11, 731. [Google Scholar] [CrossRef]
- Ogiya, D.; Liu, J.; Ohguchi, H.; Kurata, K.; Samur, M.K.; Tai, Y.-T.; Adamia, S.; Ando, K.; Hideshima, T.; Anderson, K.C. The JAK-STAT Pathway Regulates CD38 on Myeloma Cells in the Bone Marrow Microenvironment: Therapeutic Implications. Blood 2020, 136, 2334–2345. [Google Scholar] [CrossRef]
- Mielnik, M.; Szudy-Szczyrek, A.; Homa-Mlak, I.; Mlak, R.; Podgajna-Mielnik, M.; Gorący, A.; Małecka-Massalska, T.; Hus, M. The Clinical Relevance of Selected Cytokines in Newly Diagnosed Multiple Myeloma Patients. Biomedicines 2023, 11, 3012. [Google Scholar] [CrossRef]
- Melaccio, A.; Reale, A.; Saltarella, I.; Desantis, V.; Lamanuzzi, A.; Cicco, S.; Frassanito, M.A.; Vacca, A.; Ria, R. Pathways of Angiogenic and Inflammatory Cytokines in Multiple Myeloma: Role in Plasma Cell Clonal Expansion and Drug Resistance. J. Clin. Med. 2022, 11, 6491. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Hernandez, L.; Davis, R.E.; Zingone, A.; Lamy, L.; Lam, L.T.; Hurt, E.M.; Shaffer, A.L.; Kuehl, W.M.; Staudt, L.M. A Mechanistic Rationale for MEK Inhibitor Therapy in Myeloma Based on Blockade of MAF Oncogene Expression. Blood 2011, 117, 2396–2404. [Google Scholar] [CrossRef]
- Ramakrishnan, V.; Kumar, S. PI3K/AKT/mTOR Pathway in Multiple Myeloma: From Basic Biology to Clinical Promise. Leuk. Lymphoma 2018, 59, 2524–2534. [Google Scholar] [CrossRef] [PubMed]
- Lamanuzzi, A.; Saltarella, I.; Ferrucci, A.; Ria, R.; Ruggieri, S.; Racanelli, V.; Rao, L.; Annese, T.; Nico, B.; Vacca, A.; et al. Role of Erythropoietin in the Angiogenic Activity of Bone Marrow Endothelial Cells of MGUS and Multiple Myeloma Patients. Oncotarget 2016, 7, 14510–14521. [Google Scholar] [CrossRef]
- Ribatti, D. Angiogenic Effects of Erythropoietin. Int. Rev. Cell Mol. Biol. 2012, 299, 199–234. [Google Scholar] [CrossRef]
- De Luisi, A.; Binetti, L.; Ria, R.; Ruggieri, S.; Berardi, S.; Catacchio, I.; Racanelli, V.; Pavone, V.; Rossini, B.; Vacca, A.; et al. Erythropoietin Is Involved in the Angiogenic Potential of Bone Marrow Macrophages in Multiple Myeloma. Angiogenesis 2013, 16, 963–973. [Google Scholar] [CrossRef]
- Peng, Y.; Li, F.; Zhang, P.; Wang, X.; Shen, Y.; Feng, Y.; Jia, Y.; Zhang, R.; Hu, J.; He, A. IGF-1 Promotes Multiple Myeloma Progression through PI3K/Akt-Mediated Epithelial-Mesenchymal Transition. Life Sci. 2020, 249, 117503. [Google Scholar] [CrossRef] [PubMed]
- Mack, E.K.M.; Hartmann, S.; Ross, P.; Wollmer, E.; Mann, C.; Neubauer, A.; Brendel, C.; Hoffmann, J. Monitoring Multiple Myeloma in the Peripheral Blood Based on Cell-Free DNA and Circulating Plasma Cells. Ann. Hematol. 2022, 101, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.T.; Yashar, W.M.; Swords, R. Breaking the Bone Marrow Barrier: Peripheral Blood as a Gateway to Measurable Residual Disease Detection in Acute Myelogenous Leukemia. Am. J. Hematol. 2025, 100, 638–651. [Google Scholar] [CrossRef]
- Allegra, A.; Cancemi, G.; Mirabile, G.; Tonacci, A.; Musolino, C.; Gangemi, S. Circulating Tumour Cells, Cell Free DNA and Tumour-Educated Platelets as Reliable Prognostic and Management Biomarkers for the Liquid Biopsy in Multiple Myeloma. Cancers 2022, 14, 4136. [Google Scholar] [CrossRef]
- Wylie, C.; Rowan, R.; Malinova, D.; Crawford, L. Extracellular Vesicles in Multiple Myeloma: Pathogenesis and Therapeutic Application. FEBS J. 2025. [Google Scholar] [CrossRef]
- Lu, Q.; Yang, D.; Li, H.; Niu, T.; Tong, A. Multiple Myeloma: Signaling Pathways and Targeted Therapy. Mol. Biomed. 2024, 5, 25. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, M.; Zheng, S.; Tong, Y.; Tan, Y. Therapeutic Progress in Relapsed/Refractory Multiple Myeloma. Ann. Hematol. 2024, 103, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Firestone, R.; Lesokhin, A.M.; Usmani, S.Z. An Embarrassment of Riches: Three FDA-Approved Bispecific Antibodies for Relapsed Refractory Multiple Myeloma. Blood Cancer Discov. 2023, 4, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Mouhieddine, T.H.; Costa, B.A.; Richter, J. Advancements in Bispecific Antibodies for Multiple Myeloma: What’s New and What Lies Ahead. Semin. Hematol. 2025, 62, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Neri, P.; Bahlis, N.J. BCMA- or GPRC5D-Targeting Bispecific Antibodies in Multiple Myeloma: Efficacy, Safety, and Resistance Mechanisms. Blood 2024, 143, 1211–1217. [Google Scholar] [CrossRef]
- Devasia, A.J.; Chari, A.; Lancman, G. Bispecific Antibodies in the Treatment of Multiple Myeloma. Blood Cancer J. 2024, 14, 158. [Google Scholar] [CrossRef]

{kind=link}
| Homing | Expressed by | Interaction with | Signaling Pathways | Ref. |
|---|---|---|---|---|
| CyPA | BMECs | CD147 | Wnt/β-catenin | [74,77,78] |
| P-selectin | BMECs/BMSCs | PSGL-1 | FAK, Src and PI3K/AKT | [84,85,86] |
| E-selectin | BMECs | PSGL-1 | Src, p38 MAPK and Syk | [87,88,89] |
| VCAM-1 | BMECs/BMSCs | VLA-4, CD44, CD56 | NF-κB | [90,91,92,93] |
| MadCAM-1 | BMECs | α4β7 | NF-κB | [97] |
| ICAM | BMSCs | LFA-1 | NF-κB | [95,97] |
| CXCL12 | BMECs/BMSCs | CXCR4 | PI3K/Akt and Raf/MEK/ERK | [96,98,99,100] |
| Proliferation | Expressed by | Signaling Pathways | Ref. |
|---|---|---|---|
| BCL9 | BMECs | Wnt/β-catenin | [76] |
| PGI2 | BMECs | PPAR β/δ | [104] |
| HB-EGF | BMECs | HB-EGF-EGFR | [105] |
| VEGF | BMSCs | PI3K/Akt and Raf/MEK/ERK | [112] |
| IL-6 | BMSCs | JAK/STAT, Ras/Raf/MEK/MAPK and PI3K/Akt/mTOR | [108,109,110,111] |
| Epo | BMECs | PI3K/Akt and JAK/STAT | [115,116,117] |
| IGF-1 | BMSCs | PI3K/Akt | [118] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agafonova, A.; Prinzi, C.; Trovato Salinaro, A.; Ledda, C.; Cosentino, A.; Cambria, M.T.; Anfuso, C.D.; Lupo, G. Breaking Barriers: The Role of the Bone Marrow Microenvironment in Multiple Myeloma Progression. Int. J. Mol. Sci. 2025, 26, 7301. https://doi.org/10.3390/ijms26157301
Agafonova A, Prinzi C, Trovato Salinaro A, Ledda C, Cosentino A, Cambria MT, Anfuso CD, Lupo G. Breaking Barriers: The Role of the Bone Marrow Microenvironment in Multiple Myeloma Progression. International Journal of Molecular Sciences. 2025; 26(15):7301. https://doi.org/10.3390/ijms26157301
Chicago/Turabian StyleAgafonova, Aleksandra, Chiara Prinzi, Angela Trovato Salinaro, Caterina Ledda, Alessia Cosentino, Maria Teresa Cambria, Carmelina Daniela Anfuso, and Gabriella Lupo. 2025. "Breaking Barriers: The Role of the Bone Marrow Microenvironment in Multiple Myeloma Progression" International Journal of Molecular Sciences 26, no. 15: 7301. https://doi.org/10.3390/ijms26157301
APA StyleAgafonova, A., Prinzi, C., Trovato Salinaro, A., Ledda, C., Cosentino, A., Cambria, M. T., Anfuso, C. D., & Lupo, G. (2025). Breaking Barriers: The Role of the Bone Marrow Microenvironment in Multiple Myeloma Progression. International Journal of Molecular Sciences, 26(15), 7301. https://doi.org/10.3390/ijms26157301