
Exploring the Therapeutic Potential of Estrogen-Related Receptor γ Inverse Agonists in Atopic Dermatitis-like Lesions

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
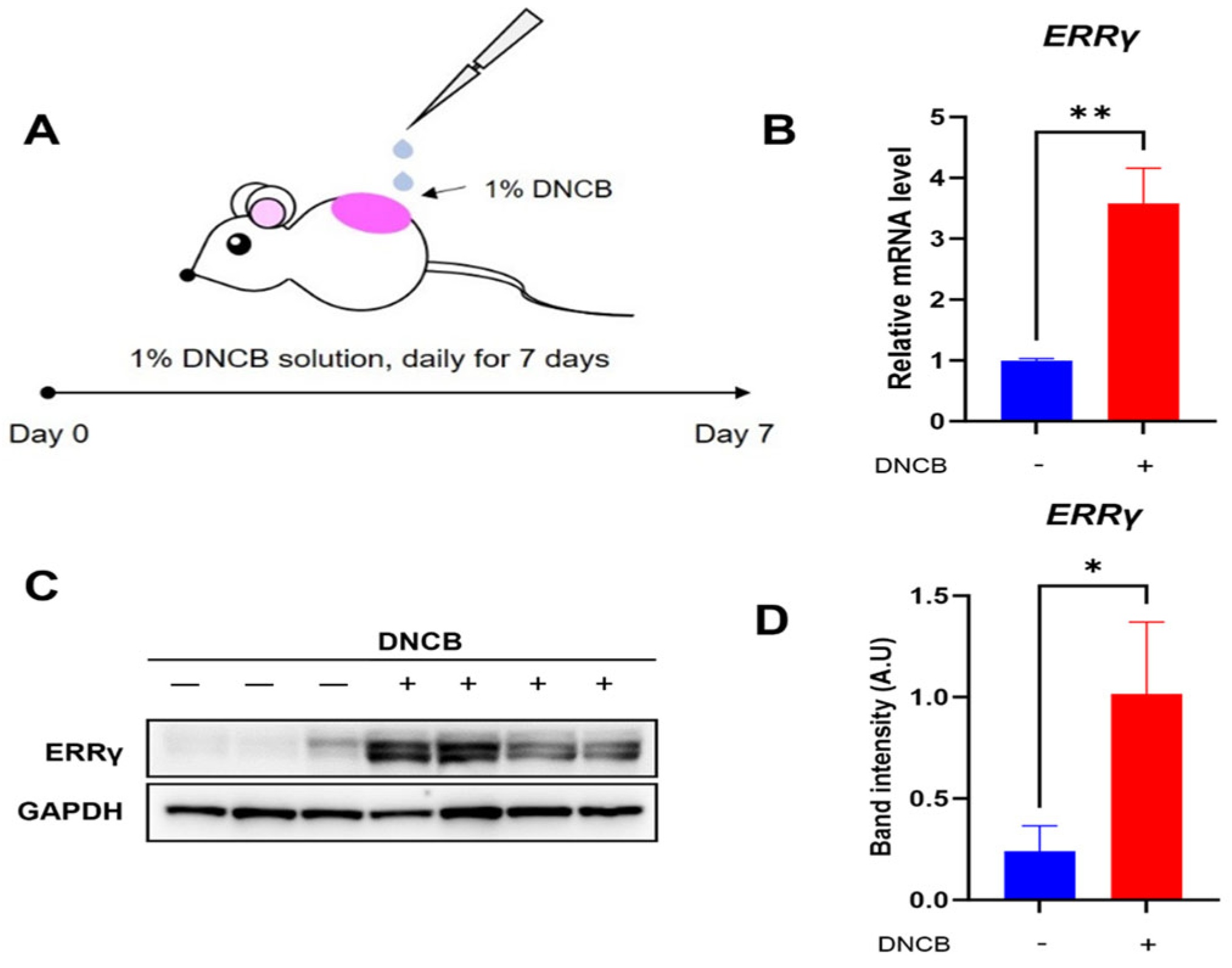
2.1. Increased Endogenous Expression of ERRγ in DNCB-Induced AD Skin Lesion
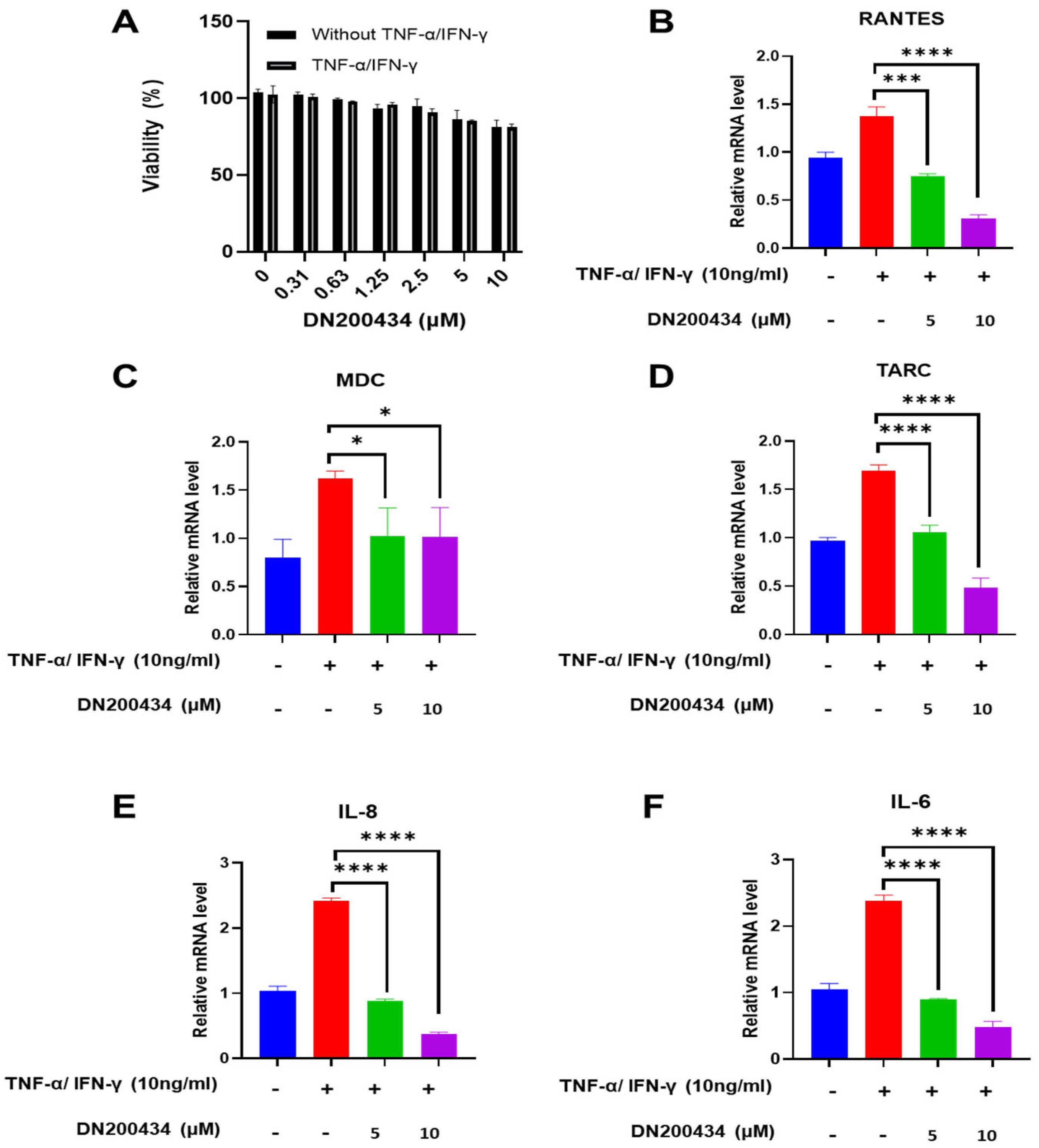
2.2. Reduction in the mRNA Level of Chemokines and Inflammatory Cytokines in TNF-α/IFN-γ-Treated HaCaT Cells by DN200434
2.3. DN200434 Inhibits the Upregulation of Phosphorylated AKT, MAPK, and NF-κB in TNF-α/IFN-γ-Treated HaCaT Cells
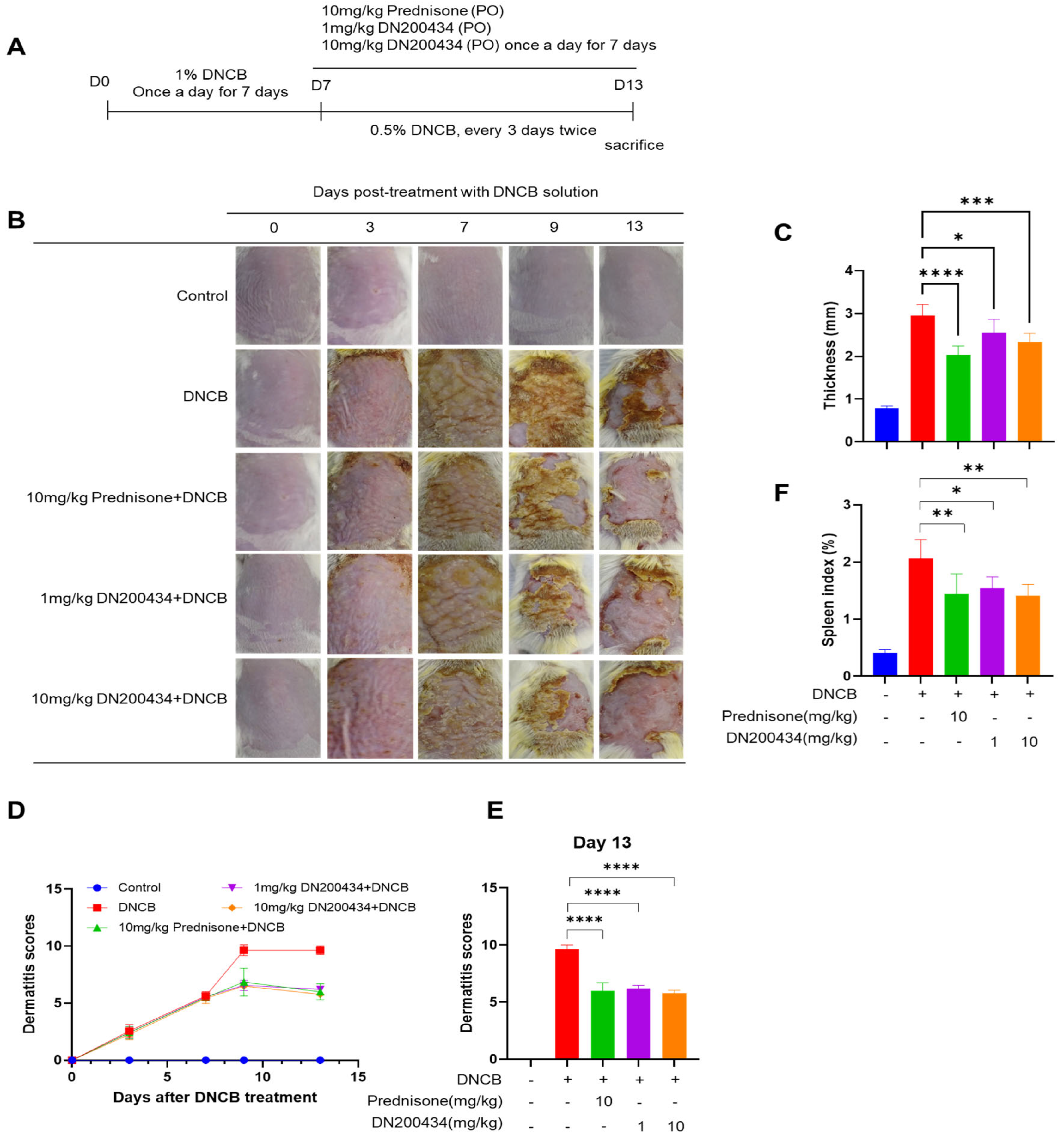
2.4. DN200434 Alleviated DNCB-Induced AD Symptoms
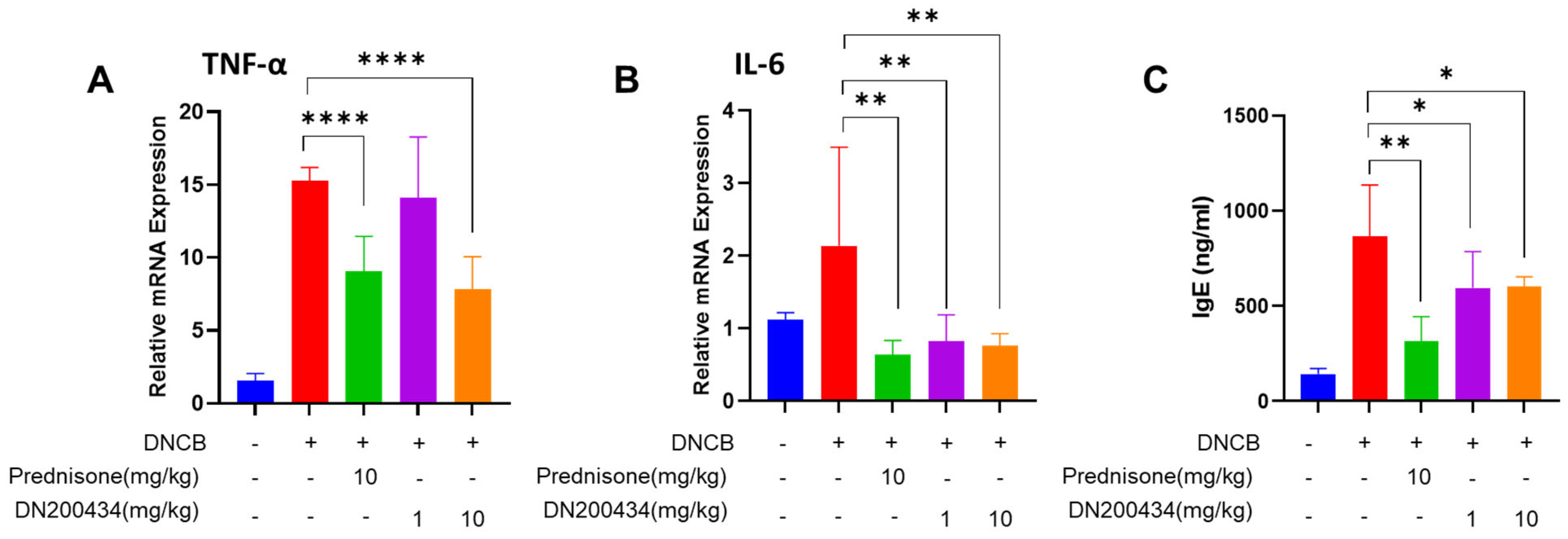
2.5. DN200434 Decrease mRNA Level of Proinflammatory Cytokines in AD Lesions and Levels of Serum IgE in Mice with DNCB-Induced AD
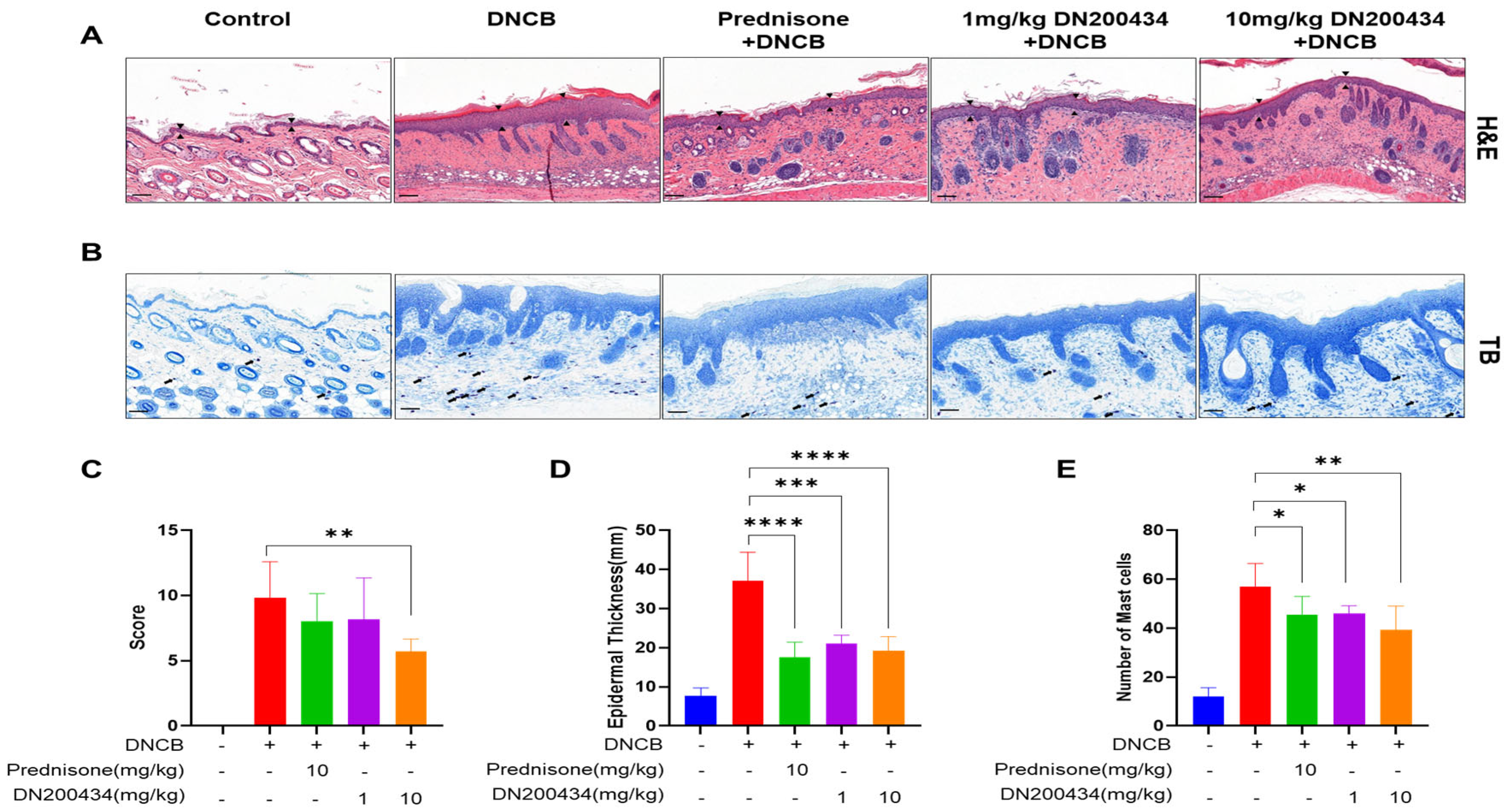
2.6. Reduction of Skin Hyperplasia and Mast Cell Infiltration in AD Lesions by DN200434
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cells
4.3. Cell Viability Assay
4.4. qRT-PCR and IgE Assay
4.5. Western Blot
4.6. In Vivo Study
4.7. Histopathological Evaluation
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boguniewicz, M.; Leung, D.Y. Atopic dermatitis: A disease of altered skin barrier and immune dysregulation. Immunol. Rev. 2011, 242, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Brar, K.K.; Nicol, N.H.; Boguniewicz, M. Strategies for successful management of severe atopic dermatitis. J. Allergy Clin. Immunol. 2019, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.M.; Giguere, V. The NR3B subgroup: An ovERRview. Nucl. Recept. Signal. 2007, 5, e009. [Google Scholar] [CrossRef] [PubMed]
- Ariazi, E.A.; Jordan, V.C. Estrogen-related receptors as emerging targets in cancer and metabolic disorders. Curr. Top. Med. Chem. 2006, 6, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Horard, B.; Vanacker, J. Estrogen receptor-related receptors: Orphan receptors desperately seeking a ligand. J. Mol. Endocrinol. 2003, 31, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-K.; Kim, J.R.; Koh, M.; Kim, Y.D.; Lee, J.-M.; Chanda, D.; Park, S.B.; Min, J.-J.; Lee, C.-H.; Park, T.-S. Estrogen-related receptor γ (ERRγ) is a novel transcriptional regulator of phosphatidic acid phosphatase, LIPIN1, and inhibits hepatic insulin signaling. J. Biol. Chem. 2011, 286, 38035–38042. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, N.; Shigekawa, T.; Ikeda, K.; Horie-Inoue, K.; Fujimura, T.; Tsuda, H.; Osaki, A.; Saeki, T.; Inoue, S. Estrogen-related receptor γ modulates cell proliferation and estrogen signaling in breast cancer. J. Steroid Biochem. Mol. Biol. 2011, 123, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, S.M.; Wang, X.; Calvo, J.A.; Lindsley, L.; Zhang, Y.; Deyneko, G.; Beaulieu, V.; Gao, J.; Turner, G.; Markovits, J. Estrogen-related receptor γ is a key regulator of muscle mitochondrial activity and oxidative capacity. J. Biol. Chem. 2010, 285, 22619–22629. [Google Scholar] [CrossRef] [PubMed]
- Nolan, L.S.; Maier, H.; Hermans-Borgmeyer, I.; Girotto, G.; Ecob, R.; Pirastu, N.; Cadge, B.A.; HŘbner, C.; Gasparini, P.; Strachan, D.P. Estrogen-related receptor gamma and hearing function: Evidence of a role in humans and mice. Neurobiol. Aging. 2013, 34, 2077.e1-9. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.-O.; Park, S.; Kwak, J.-S.; Won, Y.; Choi, W.-S.; Rhee, J.; Chun, C.-H.; Ryu, J.-H.; Kim, D.-K.; Choi, H.-S. Estrogen-related receptor γ causes osteoarthritis by upregulating extracellular matrix-degrading enzymes. Nat. Commun. 2017, 8, 2133. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Khadka, D.B.; Han, J.; Na, S.-Y.; Shin, M.; Kim, D.-K.; Oh, B.-C.; Kim, E.Y.; Choi, H.-S.; Cho, W.-J. Structure-based discovery of pyrazolamides as novel ERRγ inverse agonists. Eur. J. Med. Chem. 2023, 250, 115174. [Google Scholar] [CrossRef] [PubMed]
- Chao, E.Y.; Collins, J.L.; Gaillard, S.; Miller, A.B.; Wang, L.; Orband-Miller, L.A.; Nolte, R.T.; McDonnell, D.P.; Willson, T.M.; Zuercher, W.J. Structure-guided synthesis of tamoxifen analogs with improved selectivity for the orphan ERRγ. Bioorg. Med. Chem. Lett. 2006, 16, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Coward, P.; Lee, D.; Hull, M.V.; Lehmann, J.M. 4-Hydroxytamoxifen binds to and deactivates the estrogen-related receptor γ. Proc. Natl. Acad. Sci. USA 2001, 98, 8880–8884. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Jin, J.; Park, H.Y.; Kim, M.-J.; Chin, J.; Lee, S.; Kim, J.; Kim, J.-G.; Choi, Y.-K.; Park, K.-G. DN200434 Inhibits Vascular Smooth Muscle Cell Proliferation and Prevents Neointima Formation in Mice after Carotid Artery Ligation. Endocrinol. Metab. 2022, 37, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Kim, M.-J.; Kim, N.-Y.; Lee, S.; Byun, J.-K.; Yun, J.W.; Lee, J.; Jin, J.; Kim, J.; Chin, J. DN200434, an orally available inverse agonist of estrogen-related receptor γ, induces ferroptosis in sorafenib-resistant hepatocellular carcinoma. BMB Rep. 2022, 55, 547. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.D.; Song, J.; Kim, J.; Chin, J.; Ji, H.D.; Lee, J.-E.; Lee, S.B.; Yoon, H.; Yu, J.H.; Kim, S.K. A novel orally active inverse agonist of Estrogen-related Receptor Gamma (ERRγ), DN200434, a booster of NIS in anaplastic thyroid cancer. Clin. Cancer Res. 2019, 25, 5069–5081. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Thoudam, T.; Sinam, I.S.; Lim, C.W.; Kim, M.; Wang, J.; Lee, K.M.; Ma, J.; Saxena, R.; Choi, J.; et al. Upregulation of the ERRgamma-VDAC1 axis underlies the molecular pathogenesis of pancreatitis. Proc. Natl. Acad. Sci. USA 2023, 120, e2219644120. [Google Scholar] [CrossRef] [PubMed]
- Schuler IV, C.F.; Billi, A.C.; Maverakis, E.; Tsoi, L.C.; Gudjonsson, J.E. Novel insights into atopic dermatitis. J. Allergy Clin. Immunol. 2023, 151, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Z.; Zhang, H.; Guo, Y.; Yao, Z. Update on the pathogenesis and therapy of atopic dermatitis. Clin. Rev. Allergy Immunol. 2021, 61, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-S.; Jeong, E.-S.; Heo, S.-H.; Seo, J.-H.; Jeong, D.-G.; Choi, Y.-K. A novel model for human atopic dermatitis: Application of repeated DNCB patch in BALB/c mice, in comparison with NC/Nga mice. Lab. Anim. Res. 2010, 26, 95–102. [Google Scholar] [CrossRef]
- Yang, H.R.; Lee, H.; Kim, J.-H.; Hong, I.-H.; Hwang, D.H.; Rho, I.R.; Kim, G.S.; Kim, E.; Kang, C. Therapeutic effect of Rumex japonicus Houtt. on DNCB-induced atopic dermatitis-like skin lesions in Balb/c mice and human keratinocyte HaCaT cells. Nutrients 2019, 11, 573. [Google Scholar] [CrossRef] [PubMed]
- Armario-Hita, J.C.; Galan-Gutierrez, M.; Dodero-Anillo, J.M.; Carrascosa, J.M.; Ruiz-Villaverde, R. Updated Review on Treatment of Atopic Dermatitis. J. Investig. Allergol. Clin. Immunol. 2023, 33, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Bantz, S.K.; Zhu, Z.; Zheng, T. The atopic march: Progression from atopic dermatitis to allergic rhinitis and asthma. J. Clin. Cell Immunol. 2014, 5, 202. [Google Scholar] [PubMed]
- Hanifin, J.; Thurston, M.; Omoto, M.; Cherill, R.; Tofte, S.; Graeber, M.; Evaluator Group, T.E. The eczema area and severity index (EASI): Assessment of reliability in atopic dermatitis. Exp. Dermatol. 2001, 10, 11–18. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, J.H.; Lee, S.; Lee, J.-E.; Kim, S.K.; Jeon, J.-H.; Jeon, Y.H. Exploring the Therapeutic Potential of Estrogen-Related Receptor γ Inverse Agonists in Atopic Dermatitis-like Lesions. Int. J. Mol. Sci. 2025, 26, 6959. https://doi.org/10.3390/ijms26146959
Bae JH, Lee S, Lee J-E, Kim SK, Jeon J-H, Jeon YH. Exploring the Therapeutic Potential of Estrogen-Related Receptor γ Inverse Agonists in Atopic Dermatitis-like Lesions. International Journal of Molecular Sciences. 2025; 26(14):6959. https://doi.org/10.3390/ijms26146959
Chicago/Turabian StyleBae, Ju Hyeon, Sijoon Lee, Jae-Eon Lee, Sang Kyoon Kim, Jae-Han Jeon, and Yong Hyun Jeon. 2025. "Exploring the Therapeutic Potential of Estrogen-Related Receptor γ Inverse Agonists in Atopic Dermatitis-like Lesions" International Journal of Molecular Sciences 26, no. 14: 6959. https://doi.org/10.3390/ijms26146959
APA StyleBae, J. H., Lee, S., Lee, J.-E., Kim, S. K., Jeon, J.-H., & Jeon, Y. H. (2025). Exploring the Therapeutic Potential of Estrogen-Related Receptor γ Inverse Agonists in Atopic Dermatitis-like Lesions. International Journal of Molecular Sciences, 26(14), 6959. https://doi.org/10.3390/ijms26146959