
Synthesis and Transformation of Tricyclic KYNA Derivatives




Abstract

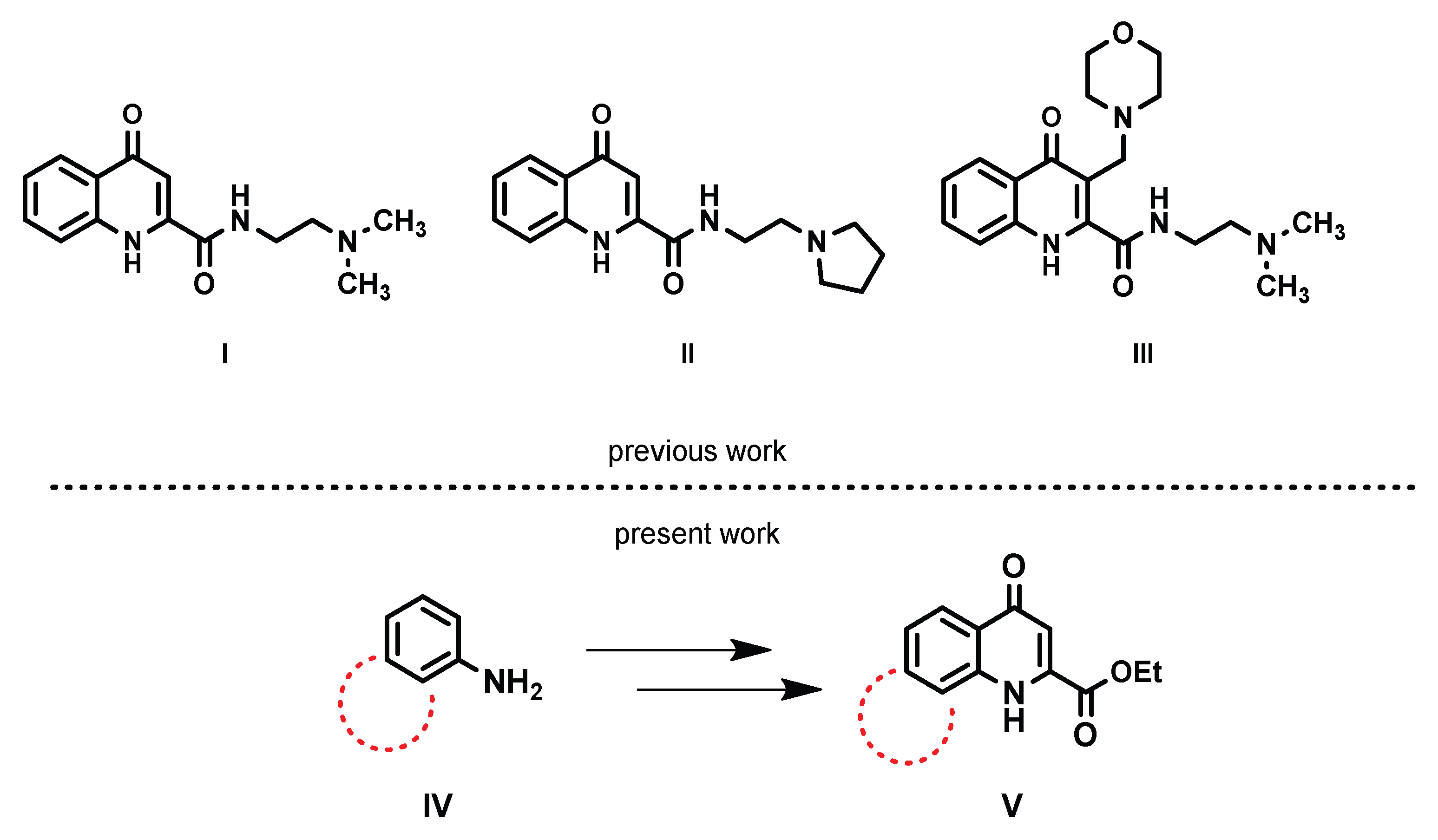
1. Introduction
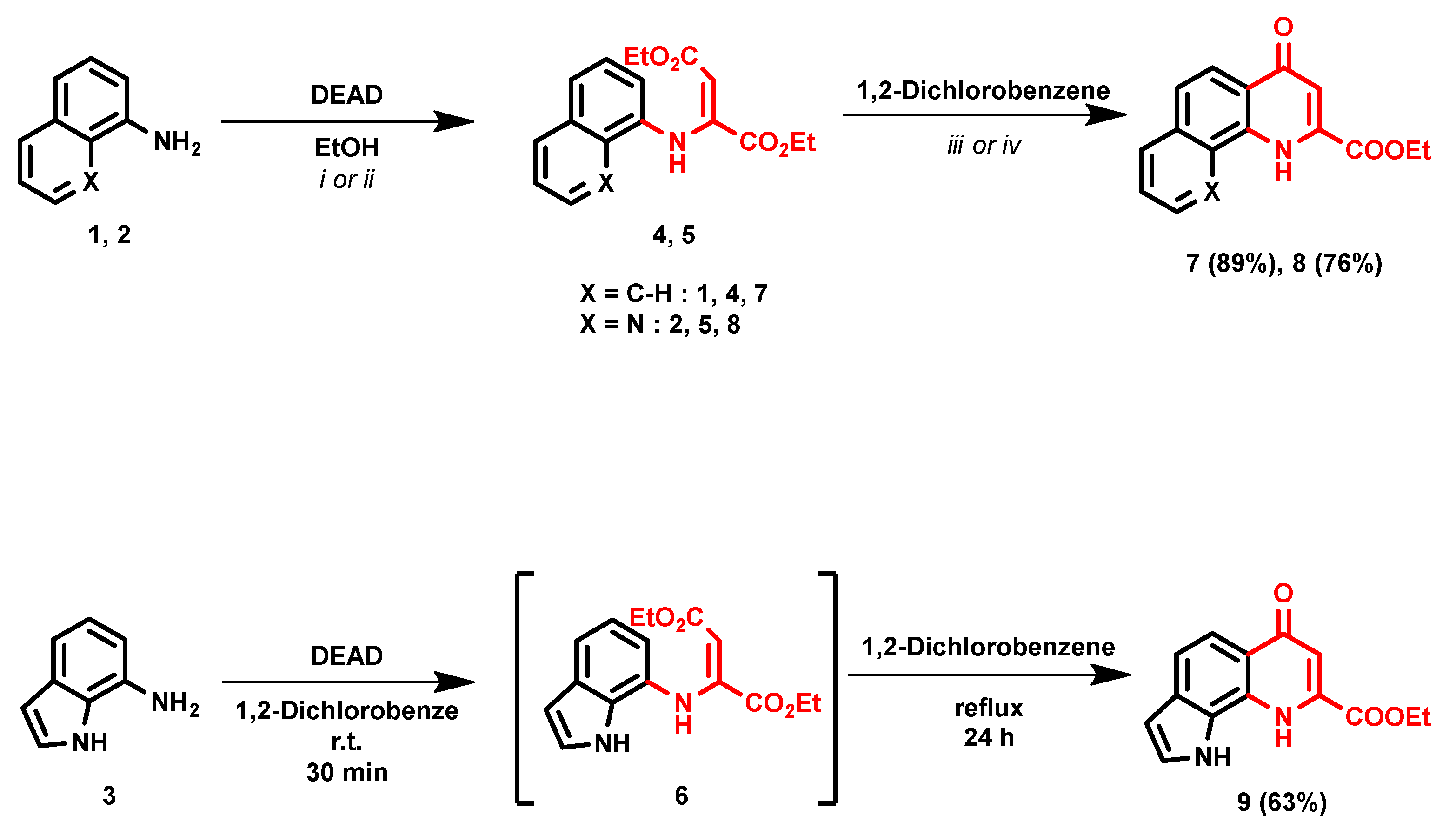
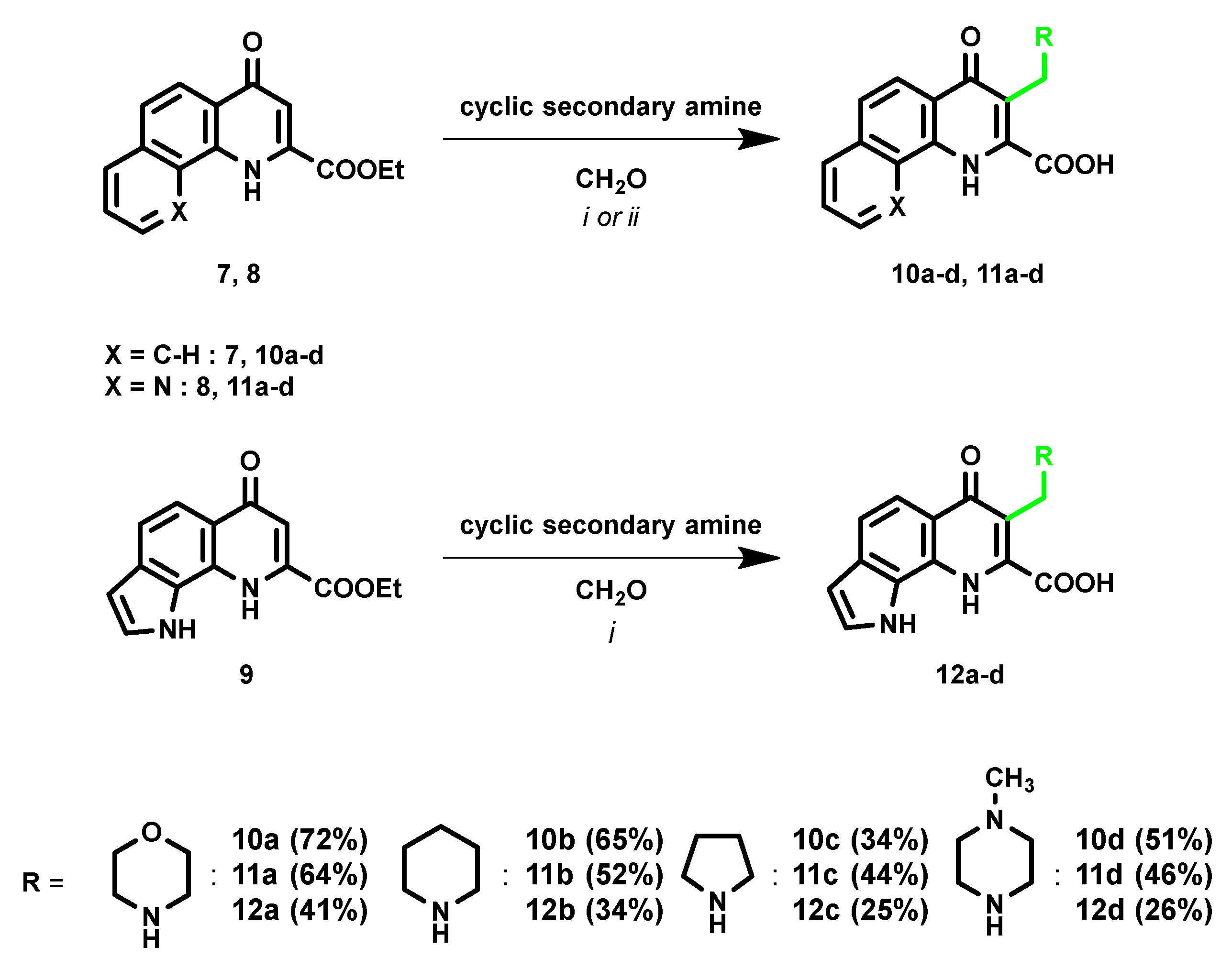
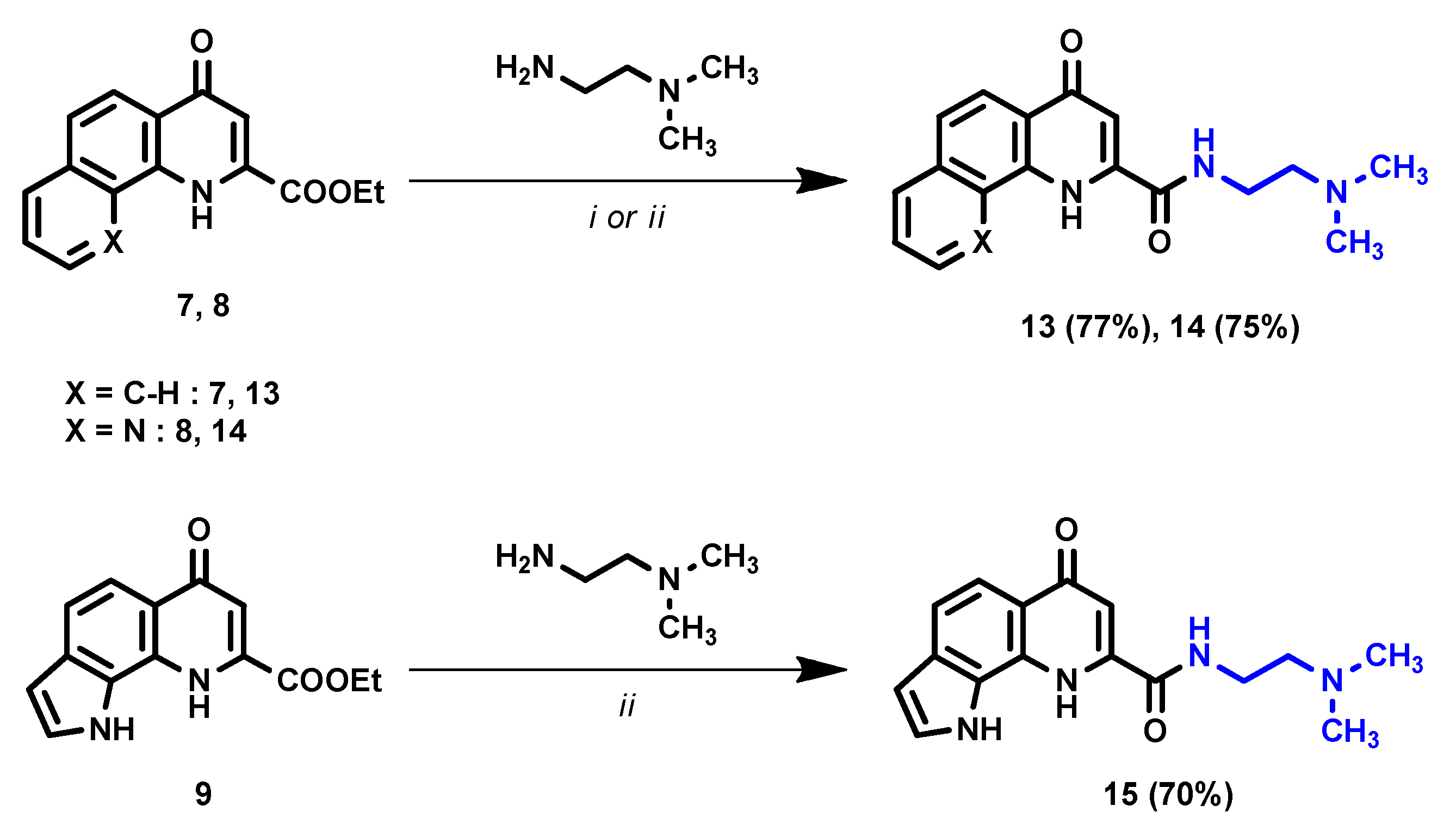
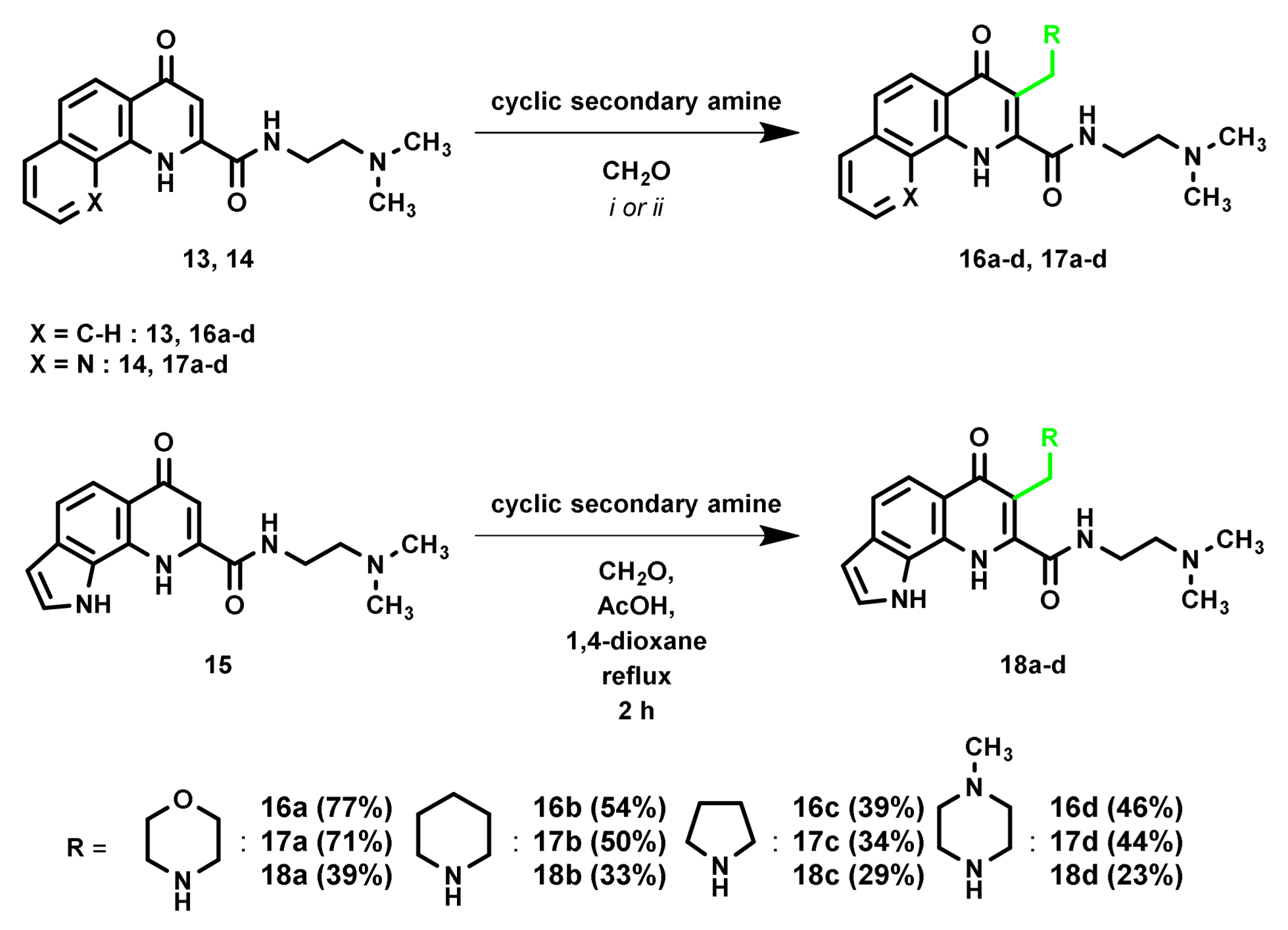
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis
3.1.1. Synthesis of Tricyclic KYNA Derivatives
- Diethyl 2-(naphthalen-1-ylamino)fumarate (4)
- Diethyl 2-(quinolin-8-ylamino)fumarate (5)
- Ethyl 4-oxo-1,4-dihydrobenzo[h]quinoline-2-carboxylate (7)
- Ethyl 4-oxo-1,4-dihydro-1,10-phenanthroline-2-carboxylate (8)
- Ethyl 6-oxo-6,9-dihydro-1H-pyrrolo[3,2-h]quinoline-8-carboxylate (9)
3.1.2. Synthesis of Mannich-Bases from Ester Derivatives
- 3-(Morpholinomethyl)-4-oxo-1,4-dihydrobenzo[h]quinoline-2-carboxylic acid (10a)
- 4-Oxo-3-(piperidin-1-ylmethyl)-1,4-dihydrobenzo[h]quinoline-2-carboxylic acid (10b)
- 4-Oxo-3-(pyrrolidin-1-ylmethyl)-1,4-dihydrobenzo[h]quinoline-2-carboxylic acid (10c)
- 3-((4-Methylpiperazin-1-yl)methyl)-4-oxo-1,4-dihydrobenzo[h]quinoline-2-carboxylic acid (10d)
- 3-(Morpholinomethyl)-4-oxo-1,4-dihydro-1,10-phenanthroline-2-carboxylic acid (11a)
- 4-Oxo-3-(piperidin-1-ylmethyl)-1,4-dihydro-1,10-phenanthroline-2-carboxylic acid (11b)
- 4-Oxo-3-(pyrrolidin-1-ylmethyl)-1,4-dihydro-1,10-phenanthroline-2-carboxylic acid (11c)
- 3-((4-Methylpiperazin-1-yl)methyl)-4-oxo-1,4-dihydro-1,10-phenanthroline-2-carboxylic acid (11d)
- 7-(Morpholinomethyl)-6-oxo-6,9-dihydro-1H-pyrrolo[3,2-h]quinoline-8-carboxylic acid (12a)
- 6-oxo-7-(piperidin-1-ylmethyl)-6,9-dihydro-1H-pyrrolo[3,2-h]quinoline-8-carboxylic acid (12b)
- 6-oxo-7-(pyrrolidin-1-ylmethyl)-6,9-dihydro-1H-pyrrolo[3,2-h]quinoline-8-carboxylic acid (12c)
- 7-((4-methylpiperazin-1-yl)methyl)-6-oxo-6,9-dihydro-1H-pyrrolo[3,2-h]quinoline-8-carboxylic acid (12d)
3.1.3. Synthesis of KYNA Amide Derivatives
- N-(2-(dimethylamino)ethyl)-4-oxo-1,4-dihydrobenzo[h]quinoline-2-carboxamide (13)
- N-(2-(dimethylamino)ethyl)-4-oxo-1,4-dihydro-1,10-phenanthroline-2-carboxamide (14)
- N-(2-(dimethylamino)ethyl)-6-oxo-6,9-dihydro-1H-pyrrolo[3,2-h]quinoline-8-carboxamide (15)
3.1.4. Synthesis of Mannich-Bases from Amide Derivatives
- N-(2-(dimethylamino)ethyl)-3-(morpholinomethyl)-4-oxo-1,4-dihydrobenzo[h]quinoline-2-carboxamide (16a)
- N-(2-(dimethylamino)ethyl)-4-oxo-3-(piperidin-1-ylmethyl)-1,4-dihydrobenzo[h]quinoline-2-carboxamide (16b)
- N-(2-(dimethylamino)ethyl)-4-oxo-3-(pyrrolidin-1-ylmethyl)-1,4-dihydrobenzo[h]quinoline-2-carboxamide (16c)
- N-(2-(dimethylamino)ethyl)-3-((4-methylpiperazin-1-yl)methyl)-4-oxo-1,4-dihydrobenzo[h]quinoline-2-carboxamide (16d)
- N-(2-(dimethylamino)ethyl)-3-(morpholinomethyl)-4-oxo-1,4-dihydro-1,10-phenanthroline-2-carboxamide (17a)
- N-(2-(dimethylamino)ethyl)-4-oxo-3-(piperidin-1-ylmethyl)-1,4-dihydro-1,10-phenanthroline-2-carboxamide (17b)
- N-(2-(dimethylamino)ethyl)-4-oxo-3-(pyrrolidin-1-ylmethyl)-1,4-dihydro-1,10-phenanthroline-2-carboxamide (17c)
- N-(2-(dimethylamino)ethyl)-3-((4-methylpiperazin-1-yl)methyl)-4-oxo-1,4-dihydro-1,10-phenanthroline-2-carboxamide (17d)
- N-(2-(dimethylamino)ethyl)-7-(morpholinomethyl)-6-oxo-6,9-dihydro-1H-pyrrolo[3,2-h]quinoline-8-carboxamide (18a)
- N-(2-(dimethylamino)ethyl)-6-oxo-7-(piperidin-1-ylmethyl)-6,9-dihydro-1H-pyrrolo[3,2-h]quinoline-8-carboxamide (18b)
- N-(2-(dimethylamino)ethyl)-6-oxo-7-(pyrrolidin-1-ylmethyl)-6,9-dihydro-1H-pyrrolo[3,2-h]quinoline-8-carboxamide (18c)
- N-(2-(dimethylamino)ethyl)-7-((4-methylpiperazin-1-yl)methyl)-6-oxo-6,9-dihydro-1H-pyrrolo [3,2-h]quinoline-8-carboxamide (18d)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rózsa, É.; Robotka, H.; Vécsei, L.; Toldi, J. The Janus-Face Kynurenic Acid. J. Neural Transm. 2008, 115, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W. Kynurenic Acid Antagonists and Kynurenine Pathway Inhibitors. Expert. Opin. Investig. Drugs 2001, 10, 633–645. [Google Scholar] [CrossRef]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic Acid as a Ligand for Orphan G Protein-Coupled Receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [PubMed]
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, Metabolic Disturbances, Oxidative Stress and the Kynurenine System, with Focus on Neurodegenerative Disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Jellinger, K.; Deecke, L. Kynurenine Metabolism in Alzheimer’s Disease. J. Neural Transm. 1999, 106, 165–181. [Google Scholar] [CrossRef]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine Pathway Abnormalities in Parkinson’s Disease. Neurology 1992, 42, 1702. [Google Scholar] [CrossRef]
- Beal, M.F.; Matson, W.R.; Swartz, K.J.; Gamache, P.H.; Bird, E.D. Kynurenine Pathway Measurements in Huntington’s Disease Striatum: Evidence for Reduced Formation of Kynurenic Acid. J. Neurochem. 1990, 55, 1327–1339. [Google Scholar] [CrossRef]
- Yamamoto, H.; Murakami, H.; Horiguchi, K.; Egawa, B. Studies on Cerebrospinal Fluid Kynurenic Acid Concentrations in Epileptic Children. Brain Dev. 1995, 17, 327–329. [Google Scholar] [CrossRef]
- Schwarcz, R.; Rassoulpour, A.; Wu, H.-Q.; Medoff, D.; Tamminga, C.A.; Roberts, R.C. Increased Cortical Kynurenate Content in Schizophrenia. Biol. Psychiatry 2001, 50, 521–530. [Google Scholar] [CrossRef]
- Gigler, G.; Szénási, G.; Simó, A.; Lévay, G.; Hársing, L.G.; Sas, K.; Vécsei, L.; Toldi, J. Neuroprotective Effect of L-Kynurenine Sulfate Administered before Focal Cerebral Ischemia in Mice and Global Cerebral Ischemia in Gerbils. Eur. J. Pharmacol. 2007, 564, 116–122. [Google Scholar] [CrossRef]
- Luchowska, E.; Luchowski, P.; Sarnowska, A.; Wielosz, M.; Turski, W.A.; Urbańska, E.M. Endogenous level of kynurenic acid and activities of kynurenine aminotransferases following transient global ischemia in the gerbil hippocampus. Pol. J. Pharmacol. 2003, 55, 443–447. [Google Scholar]
- Zádori, D.; Nyiri, G.; Szőnyi, A.; Szatmári, I.; Fülöp, F.; Toldi, J.; Freund, T.F.; Vécsei, L.; Klivényi, P. Neuroprotective Effects of a Novel Kynurenic Acid Analogue in a Transgenic Mouse Model of Huntington’s Disease. J. Neural Transm. 2011, 118, 865–875. [Google Scholar] [CrossRef]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Toldi, J.; Vécsei, L. Glutamatergic Dysfunctioning in Alzheimer’s Disease and Related Therapeutic Targets. JAD 2014, 42, S177–S187. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Demartini, C.; Zanaboni, A.M.; Redavide, E.; Pampalone, S.; Toldi, J.; Fülöp, F.; Blandini, F.; Nappi, G.; Sandrini, G.; et al. Effects of Kynurenic Acid Analogue 1 (KYNA-A1) in Nitroglycerin-Induced Hyperalgesia: Targets and Anti-Migraine Mechanisms. Cephalalgia 2017, 37, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Fejes-Szabó, A.; Bohár, Z.; Vámos, E.; Nagy-Grócz, G.; Tar, L.; Veres, G.; Zádori, D.; Szentirmai, M.; Tajti, J.; Szatmári, I.; et al. Pre-Treatment with New Kynurenic Acid Amide Dose-Dependently Prevents the Nitroglycerine-Induced Neuronal Activation and Sensitization in Cervical Part of Trigemino-Cervical Complex. J. Neural Transm. 2014, 121, 725–738. [Google Scholar] [CrossRef]
- Lőrinczi, B.; Csámpai, A.; Fülöp, F.; Szatmári, I. Synthesis of New C-3 Substituted Kynurenic Acid Derivatives. Molecules 2020, 25, 937. [Google Scholar] [CrossRef]
- Lőrinczi, B.; Csámpai, A.; Fülöp, F.; Szatmári, I. Synthetic- and DFT Modelling Studies on Regioselective Modified Mannich Reactions of Hydroxy-KYNA Derivatives. RSC Adv. 2021, 11, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Molnár, K.; Lőrinczi, B.; Fazakas, C.; Szatmári, I.; Fülöp, F.; Kmetykó, N.; Berkecz, R.; Ilisz, I.; Krizbai, I.A.; Wilhelm, I.; et al. SZR-104, a Novel Kynurenic Acid Analogue with High Permeability through the Blood–Brain Barrier. Pharmaceutics 2021, 13, 61. [Google Scholar] [CrossRef]
- Fehér, E.; Szatmári, I.; Dudás, T.; Zalatnai, A.; Farkas, T.; Lőrinczi, B.; Fülöp, F.; Vécsei, L.; Toldi, J. Structural Evaluation and Electrophysiological Effects of Some Kynurenic Acid Analogs. Molecules 2019, 24, 3502. [Google Scholar] [CrossRef]
- Mullins, S.T.; Sammes, P.G.; West, R.M.; Yahioglu, G. Preparation of Some New Intercalating Europium(III) Sensitizers. J. Chem. Soc. Perkin Trans. 1 1996, 75. [Google Scholar] [CrossRef]
- Ziessel, R.; Weibel, N.; Charbonnière, L. Stepwise Construction of Polysubstituted Phenanthroline-Based Glutamate Pockets for Lanthanide Complexation. Synthesis 2006, 2006, 3127–3133. [Google Scholar] [CrossRef]
- Neidle, S.; Parkinson, G.N. Quadruplex DNA Crystal Structures and Drug Design. Biochimie 2008, 90, 1184–1196. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. Prevalence of Quadruplexes in the Human Genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [PubMed]
- Fonseca Guerra, C.; Zijlstra, H.; Paragi, G.; Bickelhaupt, F.M. Telomere Structure and Stability: Covalency in Hydrogen Bonds, Not Resonance Assistance, Causes Cooperativity in Guanine Quartets. Chem. A Eur. J. 2011, 17, 12612–12622. [Google Scholar] [CrossRef] [PubMed]
- Butovskaya, E.; Heddi, B.; Bakalar, B.; Richter, S.N.; Phan, A.T. Major G-Quadruplex Form of HIV-1 LTR Reveals a (3 + 1) Folding Topology Containing a Stem-Loop. J. Am. Chem. Soc. 2018, 140, 13654–13662. [Google Scholar] [CrossRef]
- Terrell, J.R.; Le, T.T.; Paul, A.; Brinton, M.A.; Wilson, W.D.; Poon, G.M.K.; Germann, M.W.; Siemer, J.L. Structure of an RNA G-Quadruplex from the West Nile Virus Genome. Nat. Commun. 2024, 15, 5428. [Google Scholar] [CrossRef]
- Santos, T.; Salgado, G.F.; Cabrita, E.J.; Cruz, C. G-Quadruplexes and Their Ligands: Biophysical Methods to Unravel G-Quadruplex/Ligand Interactions. Pharmaceuticals 2021, 14, 769. [Google Scholar] [CrossRef]
- Jaumot, J.; Gargallo, R. Experimental Methods for Studying the Interactions between G-Quadruplex Structures and Ligands. CPD 2012, 18, 1900–1916. [Google Scholar] [CrossRef]
- C. Nielsen, M.; Ulven, T. Macrocyclic G-Quadruplex Ligands. CMC 2010, 17, 3438–3448. [Google Scholar] [CrossRef]
- Paritala, H.; Firestine, S.M. Benzo(h)Quinoline Derivatives as G-Quadruplex Binding Agents. Bioorg. Med. Chem. Lett. 2009, 19, 1584–1587. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Temperature (°C) | Conversion to 8 (%) 1 | Conversion to 4-OH-1,10-phenantroline (%) 1 |
|---|---|---|---|
| 1 | 180 | 17 | - |
| 2 | 190 | 76 | - |
| 3 | 200 | 99 | - |
| 4 | 210 | 99 | - |
| 5 | 220 | 99 | - |
| 6 | 230 | 84 | 15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sárik, J.R.; Szatmári, I.; Lőrinczi, B. Synthesis and Transformation of Tricyclic KYNA Derivatives. Int. J. Mol. Sci. 2025, 26, 6248. https://doi.org/10.3390/ijms26136248
Sárik JR, Szatmári I, Lőrinczi B. Synthesis and Transformation of Tricyclic KYNA Derivatives. International Journal of Molecular Sciences. 2025; 26(13):6248. https://doi.org/10.3390/ijms26136248
Chicago/Turabian StyleSárik, Julián Robin, István Szatmári, and Bálint Lőrinczi. 2025. "Synthesis and Transformation of Tricyclic KYNA Derivatives" International Journal of Molecular Sciences 26, no. 13: 6248. https://doi.org/10.3390/ijms26136248
APA StyleSárik, J. R., Szatmári, I., & Lőrinczi, B. (2025). Synthesis and Transformation of Tricyclic KYNA Derivatives. International Journal of Molecular Sciences, 26(13), 6248. https://doi.org/10.3390/ijms26136248