
Obesity-Related Glomerulosclerosis—How Adiposity Damages the Kidneys

 , ,
, ,
Abstract
1. Introduction
1.1. Obesity-Related Glomerulosclerosis (ORG)—Kidney as a Victim of Adipose Tissue
1.2. Hyperfiltration—Phenomenon of ORG
1.3. The Potential Role of Adipose Tissue Surrounding the Kidneys in Development of ORG
2. Treatment of ORG
2.1. Weight Loss
2.2. RAAS Blockade
2.3. Sodium-Glucose Cotransporter 2 Inhibitors (SGLT2i)
2.4. GLP-1 Receptor Agonists
2.5. Cagrilintide
2.6. Gut Microbiota Manipulation
2.7. Melatonin
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ogden, C.L.; Carroll, M.D.; Kit, B.K.; Flegal, K.M. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014, 311, 806–814. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 Obesity Collaborators; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [PubMed]
- Lobstein, T.; Brinsden, H. Atlas of Childhood Obesity; World Obesity Federation: London, UK, 2019. [Google Scholar]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.; Eriksson, J.G.; Broekman, B.F. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef]
- Kambham, N.; Markowitz, G.S.; Valeri, A.M.; Lin, J.; D’Agati, V.D. Obesity-related glomerulopathy: An emerging epidemic. Kidney Int. 2001, 59, 1498–1509. [Google Scholar] [CrossRef]
- Chander, P.N.; Gealekman, O.; Brodsky, S.V.; Elitok, S.; Tojo, A.; Crabtree, M.; Gross, S.S.; Goligorsky, M.S. Nephropathy in Zucker diabetic fat rat is associated with oxidative and nitrosative stress: Prevention by chronic therapy with a peroxynitrite scavenger ebselen. J. Am. Soc. Nephrol. 2004, 15, 2391–2403. [Google Scholar] [CrossRef]
- Serra, A.; Romero, R.; Lopez, D.; Navarro, M.; Esteve, A.; Perez, N.; Alastrue, A.; Ariza, A. Renal injury in the extremely obese patients with normal renal function. Kidney Int. 2008, 73, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.C.; Hwang, S.J.; Larson, M.G.; Lichtman, J.H.; Parikh, N.I.; Vasan, R.S.; Levy, D.; Fox, C.S. Overweight, obesity, and the development of stage 3 CKD: The Framingham Heart Study. Am. J. Kidney Dis. 2008, 52, 39–48. [Google Scholar] [CrossRef]
- Pinto-Sietsma, S.J.; Navis, G.; Janssen, W.M.; de Zeeuw, D.; Gans, R.O.; de Jong, P.E.; PREVEND Study Group. A central body fat distribution is related to renal function impairment, even in lean subjects. Am. J. Kidney Dis. 2003, 41, 733–741. [Google Scholar] [CrossRef]
- Cohen, A.H. Massive obesity and the kidney. A morphologic and statistical study. Am. J. Pathol. 1975, 81, 117–130. [Google Scholar]
- Chen, H.M.; Liu, Z.H.; Zeng, C.H.; Li, S.J.; Wang, Q.W.; Li, L.S. Podocyte lesions in patients with obesity-related glomerulopathy. Am. J. Kidney Dis. 2006, 48, 772–779. [Google Scholar] [CrossRef]
- Praga, M.; Hernández, E.; Morales, E.; Campos, A.P.; Valero, M.A.; Martínez, M.A.; León, M. Clinical features and long-term outcome of obesity-associated focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2001, 16, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Darouich, S.; Goucha, R.; Jaafoura, M.H.; Zekri, S.; Ben Maiz, H.; Kheder, A. Clinicopathological characteristics of obesity-associated focal segmental glomerulosclerosis. Ultrastruct. Pathol. 2011, 35, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Chagnac, A.; Weinstein, T.; Korzets, A.; Ramadan, E.; Hirsch, J.; Gafter, U. Glomerular hemodynamics in severe obesity. Am. J. Physiol. Renal Physiol. 2000, 278, F817–F822. [Google Scholar] [CrossRef]
- Goumenos, D.S.; Kawar, B.; El Nahas, M.; Conti, S.; Wagner, B.; Spyropoulos, C.; Vlachojannis, J.G.; Benigni, A.; Kalfarentzos, F. Early histological changes in the kidney of people with morbid obesity. Nephrol. Dial. Transplant. 2009, 24, 3732–3738. [Google Scholar] [CrossRef]
- Bobulescu, I.A.; Lotan, Y.; Zhang, J.; Rosenthal, T.R.; Rogers, J.T.; Adams-Huet, B.; Sakhaee, K.; Moe, O.W. Triglycerides in the human kidney cortex: Relationship with body size. PLoS ONE 2014, 9, e101285. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nat. Rev. Drug Discov. 2010, 9, 107–115. [Google Scholar] [CrossRef]
- Berfield, A.K.; Andress, D.L.; Abrass, C.K. IGF-1-induced lipid accumulation impairs mesangial cell migration and contractile function. Kidney Int. 2002, 62, 1229–1237. [Google Scholar] [CrossRef]
- van Zonneveld, A.J.; Rabelink, T.J. Mesangial cells defy LDL receptor paradigm. Kidney Int. 2001, 60, 2037–2048. [Google Scholar] [CrossRef]
- Coimbra, T.M.; Janssen, U.; Gröne, H.J.; Ostendorf, T.; Kunter, U.; Schmidt, H.; Brabant, G.; Floege, J. Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int. 2000, 57, 167–182. [Google Scholar] [CrossRef]
- Jalovaara, K.; Santaniemi, M.; Timonen, M.; Jokelainen, J.; Kesäniemi, Y.A.; Ukkola, O.; Keinänen-Kiukaanniemi, S.; Rajala, U. Low serum adiponectin level as a predictor of impaired glucose regulation and type 2 diabetes mellitus in a middle-aged Finnish population. Metabolism 2008, 57, 1130–1144. [Google Scholar] [CrossRef]
- Johnson, D.W.; Armstrong, K.; Campbell, S.B.; Mudge, D.W.; Hawley, C.M.; Coombes, J.S.; Prins, J.B.; Isbel, N.M. Metabolic syndrome in severe chronic kidney disease: Prevalence, predictors, prognostic significance and effects of risk factor modification. Nephrology 2007, 12, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Iwatani, H.; Kihara, S.; Nakagawa, Y.; Komura, N.; Fujita, K.; Maeda, N.; Nishida, M.; Katsube, F.; Shimomura, I.; et al. Exacerbation of albuminuria and renal fibrosis in subtotal renal ablation model of adiponectin-knockout mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1910–1917. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Yamauchi, T.; Kubota, N.; Hara, K.; Ueki, K.; Tobe, K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest. 2006, 116, 1784–1792. [Google Scholar] [CrossRef]
- Meyvis, K.; Verrijken, A.; Wouters, K.; Van Gaal, L. Plasma adiponectin level is inversely correlated with albuminuria in overweight and obese nondiabetic individuals. Metabolism 2013, 62, 1570–1576. [Google Scholar] [CrossRef]
- Choi, H.M.; Doss, H.M.; Kim, K.S. Multifaceted Physiological Roles of Adiponectin in Inflammation and Diseases. Int. J. Mol. Sci. 2020, 21, 1219. [Google Scholar] [CrossRef]
- Rovin, B.H.; Song, H. Chemokine induction by the adipocyte-derived cytokine adiponectin. Clin. Immunol. 2006, 120, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Song, S.H.; Oh, T.R.; Choi, H.S.; Kim, C.S.; Ma, S.K.; Oh, K.H.; Ahn, C.; Kim, S.W.; Bae, E.H. High serum adiponectin as a biomarker of renal dysfunction: Results from the KNOW-CKD study. Sci. Rep. 2020, 10, 5598. [Google Scholar] [CrossRef]
- Sharma, K.; Ramachandrarao, S.; Qiu, G.; Usui, H.K.; Zhu, Y.; Dunn, S.R.; Ouedraogo, R.; Hough, K.; McCue, P.; Chan, L.; et al. Adiponectin regulates albuminuria and podocyte function in mice. J. Clin. Invest. 2008, 118, 1645–1656. [Google Scholar] [CrossRef]
- Rutkowski, J.M.; Wang, Z.V.; Park, A.S.; Zhang, J.; Zhang, D.; Hu, M.C.; Moe, O.W.; Susztak, K.; Scherer, P.E. Adiponectin promotes functional recovery after podocyte ablation. J. Am. Soc. Nephrol. 2013, 24, 268–282. [Google Scholar] [CrossRef]
- Su, Y.X.; Deng, H.C.; Zhang, M.X.; Long, J.; Peng, Z.G. Adiponectin inhibits PDGF-induced mesangial cell proliferation: Regulation of mammalian target of rapamycin-mediated survival pathway by adenosine 5-monophosphate-activated protein kinase. Horm. Metab. Res. 2012, 44, 21–27. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, Y.; Heiman, M.; Dimarchi, R. Leptin: Structure, function and biology. Vitam. Horm. 2005, 71, 345–372. [Google Scholar] [PubMed]
- Teixeira, T.M.; da Costa, D.C.; Resende, A.C.; Soulage, C.O.; Bezerra, F.F.; Daleprane, J.B. Activation of Nrf2-Antioxidant Signaling by 1,25-Dihydroxycholecalciferol Prevents Leptin-Induced Oxidative Stress and Inflammation in Human Endothelial Cells. J. Nutr. 2017, 147, 506–513. [Google Scholar] [CrossRef]
- Considine, R.V. Human leptin: An adipocyte hormone with weight-regulatory and endocrine functions. Semin. Vasc. Med. 2005, 5, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Han, D.C.; Isono, M.; Chen, S.; Casaretto, A.; Hong, S.W.; Wolf, G.; Ziyadeh, F.N. Leptin stimulates type I collagen production in db/db mesangial cells: Glucose uptake and TGF-beta type II receptor expression. Kidney Int. 2001, 59, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef]
- Declèves, A.E.; Sharma, K. Obesity and kidney disease: Differential effects of obesity on adipose tissue and kidney inflammation and fibrosis. Curr. Opin. Nephrol. Hypertens. 2015, 24, 28–36. [Google Scholar] [CrossRef]
- Coward, R.; Fornoni, A. Insulin signaling: Implications for podocyte biology in diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 104–110. [Google Scholar] [CrossRef]
- Lin, L.; Tan, W.; Pan, X.; Tian, E.; Wu, Z.; Yang, J. Metabolic Syndrome-Related Kidney Injury: A Review and Update. Front. Endocrinol. 2022, 13, 904001. [Google Scholar] [CrossRef]
- Tsuruta, H.; Yasuda-Yamahara, M.; Yoshibayashi, M.; Kuwagata, S.; Yamahara, K.; Tanaka-Sasaki, Y.; Chin-Kanasaki, M.; Matsumoto, S.; Ema, M.; Kume, S. Fructose overconsumption accelerates renal dysfunction with aberrant glomerular endothelial-mesangial cell interactions in db/db mice. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167074. [Google Scholar] [CrossRef]
- Feng, Z.; Zhu, L.; Wu, J. RAGE signalling in obesity and diabetes: Focus on the adipose tissue macrophage. Adipocyte 2020, 9, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Machado-Lima, A.; López-Díez, R.; Iborra, R.T.; Pinto, R.S.; Daffu, G.; Shen, X.; Nakandakare, E.R.; Machado, U.F.; Corrêa-Giannella, M.L.C.; Schmidt, A.M.; et al. RAGE Mediates Cholesterol Efflux Impairment in Macrophages Caused by Human Advanced Glycated Albumin. Int. J. Mol. Sci. 2020, 21, 7265. [Google Scholar] [CrossRef]
- Guan, Y.; Wei, X.; Li, J.; Zhu, Y.; Luo, P.; Luo, M. Obesity-related glomerulopathy: Recent advances in inflammatory mechanisms and related treatments. J. Leukoc. Biol. 2024, 115, 819–839. [Google Scholar] [CrossRef]
- Sharma, R.; Gaze, D.C.; Pellerin, D.; Mehta, R.L.; Gregson, H.; Streather, C.P.; Collinson, P.O.; Brecker, S.J. Ischemia-modified albumin predicts mortality in ESRD. Am. J. Kidney Dis. 2006, 47, 493–502. [Google Scholar] [CrossRef]
- Endlich, N.; Kress, K.R.; Reiser, J.; Uttenweiler, D.; Kriz, W.; Mundel, P.; Endlich, K. Podocytes respond to mechanical stress in vitro. J. Am. Soc. Nephrol. 2001, 12, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Pecly, I.M.; Genelhu, V.; Francischetti, E.A. Renal functional reserve in obesity hypertension. Int. J. Clin. Pract. 2006, 60, 1198–1203. [Google Scholar] [CrossRef]
- Cortinovis, M.; Perico, N.; Ruggenenti, P.; Remuzzi, A.; Remuzzi, G. Glomerular hyperfiltration. Nat. Rev. Nephrol. 2022, 18, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart-Bornstein, M.; Arakelyan, K.; Krug, A.W.; Scherbaum, W.A.; Bornstein, S.R. Fat cells may be the obesity-hypertension link: Human adipogenic factors stimulate aldosterone secretion from adrenocortical cells. Endocr. Res. 2004, 30, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.R.; Burns, K.D. Angiotensin II as a mediator of renal tubular transport. Contrib. Nephrol. 2001, 135, 47–62. [Google Scholar]
- Davy, K.P.; Orr, J.S. Sympathetic nervous system behavior in human obesity. Neurosci. Biobehav. Rev. 2009, 33, 116–124. [Google Scholar] [CrossRef]
- Young, C.N.; Morgan, D.A.; Butler, S.D.; Mark, A.L.; Davisson, R.L. The brain subfornical organ mediates leptin-induced increases in renal sympathetic activity but not its metabolic effects. Hypertension 2013, 61, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Blantz, R.C.; Thomson, S. Glomerular hyperfiltration and the salt paradox in early [corrected] type 1 diabetes mellitus: A tubulo-centric view. J. Am. Soc. Nephrol. 2003, 14, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.E.; Haas, J.A.; Cuche, J.L.; Knox, F.G. Effect of increased peritubule protein concentration on proximal tubule reabsorption in the presence and absence of extracellular volume expansion. J. Clin. Invest. 1975, 55, 612–620. [Google Scholar] [CrossRef]
- Kriz, W.; Lemley, K.V. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J. Am. Soc. Nephrol. 2015, 26, 258–269. [Google Scholar] [CrossRef]
- Huang, C.; Bruggeman, L.A.; Hydo, L.M.; Miller, R.T. Shear stress induces cell apoptosis via a c-Src-phospholipase D-mTOR signaling pathway in cultured podocytes. Exp. Cell Res. 2012, 318, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, T.; Dai, H.; Heruth, D.P.; Alon, U.S.; Garola, R.E.; Zhou, J.; Duncan, R.S.; El-Meanawy, A.; McCarthy, E.T.; Sharma, R.; et al. Mechanotransduction signaling in podocytes from fluid flow shear stress. Am. J. Physiol. Renal Physiol. 2018, 314, F22–F34. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, T.; Celsi, G.E.; Sharma, M.; Dai, H.; McCarthy, E.T.; Ruiz, M.; Cudmore, P.A.; Alon, U.S.; Sharma, R.; Savin, V.A. Fluid flow shear stress over podocytes is increased in the solitary kidney. Nephrol. Dial. Transplant. 2014, 29, 65–72. [Google Scholar] [CrossRef]
- Manno, C.; Campobasso, N.; Nardecchia, A.; Triggiani, V.; Zupo, R.; Gesualdo, L.; Silvestris, F.; De Pergola, G. Relationship of para- and perirenal fat and epicardial fat with metabolic parameters in overweight and obese subjects. Eat. Weight. Disord. 2019, 24, 67–72. [Google Scholar] [CrossRef]
- GBD 2021 Stroke Risk Factor Collaborators. Global, regional, and national burden of stroke and its risk factors, 1990–2021, a systematic analysis for the Global Burden of Disease Study 2021. Lancet Neurol. 2024, 23, 973–1003. [Google Scholar] [CrossRef]
- Liu, B.X.; Sun, W.; Kong, X.Q. Perirenal Fat: A Unique Fat Pad and Potential Target for Cardiovascular Disease. Angiology 2019, 70, 584–593. [Google Scholar] [CrossRef]
- Huang, N.; Mao, E.W.; Hou, N.N.; Liu, Y.P.; Han, F.; Sun, X.D. Novel insight into perirenal adipose tissue: A neglected adipose depot linking cardiovascular and chronic kidney disease. World J. Diabetes 2020, 11, 115–125. [Google Scholar] [CrossRef]
- Choi, J.W.; Lee, C.M.; Kang, B.K.; Kim, M. Perirenal fat thickness is an independent predictor for metabolic syndrome in steatotic liver disease. Sci. Rep. 2024, 14, 26548. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yu, Y.; Han, L. High FFA levels related to microalbuminuria and uncoupling of VEGF-NO axis in obese rats. Int. Urol. Nephrol. 2013, 45, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Luo, M.; Chen, N.; Deng, X.; He, J.; Zhang, L.; Luo, P.; Wu, J. Inhibition of PAI-1 attenuates perirenal fat inflammation and the associated nephropathy in high-fat diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E260–E267. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Biffi, A.; Seravalle, G.; Trevano, F.Q.; Dell’Oro, R.; Corrao, G.; Mancia, G. Sympathetic Neural Overdrive in the Obese and Overweight State. Hypertension 2019, 74, 349–358. [Google Scholar] [CrossRef]
- Cao, Q.; Zhang, J.; Yu, Q.; Wang, J.; Dai, M.; Zhang, Y.; Luo, Q.; Bao, M. Carotid baroreceptor stimulation in obese rats affects white and brown adipose tissues differently in metabolic protection. J. Lipid Res. 2019, 60, 1212–1224. [Google Scholar] [CrossRef]
- Chen, X.; Qin, Y.; Hu, J.; Shen, Y.; Mao, Y.; Xie, L.; Li, J.; Wang, J.; Yang, S.; Li, Q.; et al. Perirenal fat and chronic kidney disease in type 2 diabetes: The mediation role of afferent arteriolar resistance. Diabetes Metab. 2024, 50, 101583. [Google Scholar] [CrossRef]
- Hu, H.; Liang, W.; Zhang, Z.; Liu, Z.; Chu, F.; Bao, Y.; Ran, J.; Ding, G. The Utility of Perirenal Fat in Determining the Risk of Onset and Progression of Diabetic Kidney Disease. Int. J. Endocrinol. 2022, 2022, 2550744. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, Z.; Liu, Z.; Chu, F.; Ran, J.; Liang, W. Thickened Perirenal Fat Predicts Poor Renal Outcome in Patients with Immunoglobulin A Nephropathy: A Population-Based Retrospective Cohort Study. Kidney Dis. 2023, 10, 51–60. [Google Scholar] [CrossRef]
- Navaneethan, S.D.; Yehnert, H.; Moustarah, F.; Schreiber, M.J.; Schauer, P.R.; Beddhu, S. Weight loss interventions in chronic kidney disease: A systematic review and meta-analysis. Clin. J. Am. Soc. Nephrol. 2009, 4, 1565–1574. [Google Scholar] [CrossRef]
- Shulman, A.; Peltonen, M.; Sjöström, C.D.; Andersson-Assarsson, J.C.; Taube, M.; Sjöholm, K.; le Roux, C.W.; Carlsson, L.M.S.; Svensson, P.A. Incidence of end-stage renal disease following bariatric surgery in the Swedish Obese Subjects Study. Int. J. Obes. 2018, 42, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Conley, M.M.; McFarlane, C.M.; Johnson, D.W.; Kelly, J.T.; Campbell, K.L.; MacLaughlin, H.L. Interventions for weight loss in people with chronic kidney disease who are overweight or obese. Cochrane Database Syst. Rev. 2021, 3, CD013119. [Google Scholar] [CrossRef]
- Chang, S.H.; Stoll, C.R.; Song, J.; Varela, J.E.; Eagon, C.J.; Colditz, G.A. The effectiveness and risks of bariatric surgery: An updated systematic review and meta-analysis, 2003–2012. JAMA Surg. 2014, 149, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Taylor, R. Restoring normoglycaemia by use of a very low calorie diet in long- and short-duration Type 2 diabetes. Diabet. Med. 2015, 32, 1149–1155. [Google Scholar] [CrossRef]
- Gregg, E.W.; Chen, H.; Wagenknecht, L.E.; Clark, J.M.; Delahanty, L.M.; Bantle, J.; Pownall, H.J.; Johnson, K.C.; Safford, M.M.; Kitabchi, A.E.; et al. Look AHEAD Research Group. Association of an intensive lifestyle intervention with remission of type 2 diabetes. JAMA 2012, 308, 2489–2496. [Google Scholar] [CrossRef]
- Mallamaci, F.; Ruggenenti, P.; Perna, A.; Leonardis, D.; Tripepi, R.; Tripepi, G.; Remuzzi, G.; Zoccali, C. REIN Study Group. ACE inhibition is renoprotective among obese patients with proteinuria. J. Am. Soc. Nephrol. 2011, 22, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Tofte, N.; Lindhardt, M.; Adamova, K.; Bakker, S.J.L.; Beige, J.; Beulens, J.W.J.; Birkenfeld, A.L.; Currie, G.; Delles, C.; Dimos, I.; et al. Early detection of diabetic kidney disease by urinary proteomics and subsequent intervention with spironolactone to delay progression (PRIORITY): A prospective observational study and embedded randomised placebo-controlled trial. Lancet Diabetes Endocrinol. 2020, 8, 301–312. [Google Scholar] [CrossRef]
- Briones, A.M.; Nguyen Dinh Cat, A.; Callera, G.E.; Yogi, A.; Burger, D.; He, Y.; Corrêa, J.W.; Gagnon, A.M.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: Implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension 2012, 59, 1069–1078. [Google Scholar] [CrossRef]
- Rüster, C.; Wolf, G. The role of the renin-angiotensin-aldosterone system in obesity-related renal diseases. Semin. Nephrol. 2013, 33, 44–53. [Google Scholar] [CrossRef]
- Morales, E.; Gutiérrez, E.; Caro, J.; Sevillano, A.; Rojas-Rivera, J.; Praga, M. Beneficial long-term effect of aldosterone antagonist added to a traditional blockade of the renin-angiotensin-aldosterone system among patients with obesity and proteinuria. Nefrologia 2015, 35, 554–561. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. FIDELIO-DKD Investigators. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 3, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 13, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- The EMPA-KIDNEY Collaborative Group; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 12, 117–127. [Google Scholar]
- DAPA-CKD Trial Committees and Investigators; Wheeler, D.C.; Stefánsson, B.V.; Jongs, N.; Chertow, G.M.; Greene, T.; Hou, F.F.; McMurray, J.J.V.; Correa-Rotter, R.; Rossing, P.; et al. Effects of dapagliflozin on major adverse kidney and cardiovascular events in patients with diabetic and non-diabetic chronic kidney disease: A prespecified analysis from the DAPA-CKD trial. Lancet Diabetes Endocrinol. 2021, 9, 22–31. [Google Scholar] [CrossRef]
- Pereira, M.J.; Eriksson, J.W. Emerging Role of SGLT-2 Inhibitors for the Treatment of Obesity. Drugs 2019, 79, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Bolinder, J.; Ljunggren, Ö.; Kullberg, J.; Johansson, L.; Wilding, J.; Langkilde, A.M.; Sugg, J.; Parikh, S. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J. Clin. Endocrinol. Metab. 2012, 97, 1020–1031. [Google Scholar] [CrossRef]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Farooqui, K.J.; Singh, M.K.; Wasir, J.S.; Bansal, B.; Kaur, P.; Jevalikar, G.; Gill, H.K.; et al. Effect of Empagliflozin on Liver Fat in Patients With Type 2 Diabetes and Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial (E-LIFT Trial). Diabetes Care 2018, 41, 1801–1808. [Google Scholar] [CrossRef]
- Yagi, S.; Hirata, Y.; Ise, T.; Kusunose, K.; Yamada, H.; Fukuda, D.; Salim, H.M.; Maimaituxun, G.; Nishio, S.; Takagawa, Y.; et al. Canagliflozin reduces epicardial fat in patients with type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2017, 4, 9. [Google Scholar] [CrossRef]
- Wu, P.; Wen, W.; Li, J.; Xu, J.; Zhao, M.; Chen, H.; Sun, J. Systematic Review and Meta-Analysis of Randomized Controlled Trials on the Effect of SGLT2 Inhibitor on Blood Leptin and Adiponectin Level in Patients with Type 2 Diabetes. Horm. Metab. Res. 2019, 51, 487–494. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Apperloo, E.; Davies, M.; Dicker, D.; Kandler, K.; Rosenstock, J.; Sørrig, R.; Lawson, J.; Zeuthen, N.; Cherney, D. Effects of Semaglutide on Albuminuria and Kidney Function in People With Overweight or Obesity With or Without Type 2 Diabetes: Exploratory Analysis From the STEP 1, 2, and 3 Trials. Diabetes Care 2023, 46, 801–810. [Google Scholar] [CrossRef]
- Wilding, J.P.H.; Batterham, R.L.; Calanna, S.; Davies, M.; Van Gaal, L.F.; Lingvay, I.; McGowan, B.M.; Rosenstock, J.; Tran, M.T.D.; Wadden, T.A.; et al. STEP 1 Study Group. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N. Engl. J. Med. 2021, 18, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- FLOW Trial Committees and Investigators; Perkovic, V.; Tuttle, K.R.; Rossing, P.; Mahaffey, K.W.; Mann, J.F.E.; Bakris, G.; Baeres, F.M.M.; Idorn, T.; Bosch-Traberg, H. Effects of Semaglutide on Chronic Kidney Disease in Patients with Type 2 Diabetes. N. Engl. J. Med. 2024, 11, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Apperloo, E.M.; Cherney, D.Z.I.; Kuhlman, A.B.; Mann, J.F.E.; Rasmussen, S.; Rossing, P.; Tuttle, K.R.; Vrhnjak, B.; Heerspink, H.J.L. Effect of semaglutide on kidney function across different levels of baseline HbA1c, blood pressure, body weight and albuminuria in SUSTAIN 6 and PIONEER 6. Nephrol. Dial. Transplant. 2025, 4, 352–359. [Google Scholar] [CrossRef]
- SELECT Trial Investigators; Lincoff, A.M.; Brown-Frandsen, K.; Colhoun, H.M.; Deanfield, J.; Emerson, S.S.; Esbjerg, S.; Hardt-Lindberg, S.; Hovingh, G.K.; Kahn, S.E.; et al. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N. Engl. J. Med. 2023, 14, 2221–2232. [Google Scholar] [CrossRef] [PubMed]
- Colhoun, H.M.; Lingvay, I.; Brown, P.M.; Deanfield, J.; Brown-Frandsen, K.; Kahn, S.E.; Plutzky, J.; Node, K.; Parkhomenko, A.; Rydén, L.; et al. Long-term kidney outcomes of semaglutide in obesity and cardiovascular disease in the SELECT trial. Nat. Med. 2024, 30, 2058–2066. [Google Scholar] [CrossRef]
- SURMOUNT-1 Investigators; Jastreboff, A.M.; le Roux, C.W.; Stefanski, A.; Aronne, L.J.; Halpern, B.; Wharton, S.; Wilding, J.P.H.; Perreault, L.; Zhang, S.; et al. Tirzepatide for Obesity Treatment and Diabetes Prevention. N. Engl. J. Med. 2025, 6, 958–971. [Google Scholar] [CrossRef]
- SYNERGY-NASH Investigators; Loomba, R.; Hartman, M.L.; Lawitz, E.J.; Vuppalanchi, R.; Boursier, J.; Bugianesi, E.; Yoneda, M.; Behling, C.; Cummings, O.W.; et al. Tirzepatide for Metabolic Dysfunction-Associated Steatohepatitis with Liver Fibrosis. N. Engl. J. Med. 2024, 25, 299–310. [Google Scholar] [CrossRef]
- SURMOUNT-OSA Investigators; Malhotra, A.; Grunstein, R.R.; Fietze, I.; Weaver, T.E.; Redline, S.; Azarbarzin, A.; Sands, S.A.; Schwab, R.J.; Dunn, J.P.; et al. Tirzepatide for the Treatment of Obstructive Sleep Apnea and Obesity. N. Engl. J. Med. 2024, 3, 1193–1205. [Google Scholar] [CrossRef]
- Kamrul-Hasan, A.; Patra, S.; Dutta, D.; Nagendra, L.; Muntahi-Reza, A.; Borozan, S.; Pappachan, J.M. Renal effects and safety of tirzepatide in subjects with and without diabetes: A systematic review and meta-analysis. World J. Diabetes 2025, 15, 101282. [Google Scholar] [CrossRef]
- Apperloo, E.M.; Tuttle, K.R.; Pavo, I.; Haupt, A.; Taylor, R.; Wiese, R.J.; Hemmingway, A.; Cherney, D.Z.I.; Sattar, N.; Heerspink, H.J.L. Tirzepatide Associated With Reduced Albuminuria in Participants With Type 2 Diabetes: Pooled Post Hoc Analysis From the Randomized Active- and Placebo-Controlled SURPASS-1-5 Clinical Trials. Diabetes Care 2025, 48, 430–436. [Google Scholar] [CrossRef]
- Karakasis, P.; Patoulias, D.; Fragakis, N.; Klisic, A.; Rizzo, M. Effect of tirzepatide on albuminuria levels and renal function in patients with type 2 diabetes mellitus: A systematic review and multilevel meta-analysis. Diabetes Obes. Metab. 2024, 26, 1090–1104. [Google Scholar] [CrossRef] [PubMed]
- Frias, J.P.; Deenadayalan, S.; Erichsen, L.; Knop, F.K.; Lingvay, I.; Macura, S.; Mathieu, C.; Pedersen, S.D.; Davies, M. Efficacy and safety of co-administered once-weekly cagrilintide 2·4 mg with once-weekly semaglutide 2·4 mg in type 2 diabetes: A multicentre, randomised, double-blind, active-controlled, phase 2 trial. Lancet 2023, 402, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Popkov, V.A.; Zharikova, A.A.; Demchenko, E.A.; Andrianova, N.V.; Zorov, D.B.; Plotnikov, E.Y. Gut Microbiota as a Source of Uremic Toxins. Int. J. Mol. Sci. 2022, 23, 483. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef]
- Alard, J.; Lehrter, V.; Rhimi, M.; Mangin, I.; Peucelle, V.; Abraham, A.L.; Mariadassou, M.; Maguin, E.; Waligora-Dupriet, A.J.; Pot, B.; et al. Beneficial metabolic effects of selected probiotics on diet-induced obesity and insulin resistance in mice are associated with improvement of dysbiotic gut microbiota. Environ. Microbiol. 2016, 18, 1484–1497. [Google Scholar] [CrossRef]
- Alard, J.; Cudennec, B.; Boutillier, D.; Peucelle, V.; Descat, A.; Decoin, R.; Kuylle, S.; Jablaoui, A.; Rhimi, M.; Wolowczuk, I. Multiple Selection Criteria for Probiotic Strains with High Potential for Obesity Management. Nutrients 2021, 13, 713. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Zhou, D.D.; Gan, R.Y.; Huang, S.Y.; Zhao, C.N.; Shang, A.; Xu, X.Y.; Li, H.B. Effects and Mechanisms of Probiotics, Prebiotics, Synbiotics, and Postbiotics on Metabolic Diseases Targeting Gut Microbiota: A Narrative Review. Nutrients 2021, 13, 3211. [Google Scholar] [CrossRef]
- Ou, T.H.; Tung, Y.T.; Yang, T.H.; Chien, Y.W. Melatonin Improves Fatty Liver Syndrome by Inhibiting the Lipogenesis Pathway in Hamsters with High-Fat Diet-Induced Hyperlipidemia. Nutrients 2019, 11, 748. [Google Scholar] [CrossRef]
- Ramirez, A.V.G.; Filho, D.R.; de Sá, L.B.P.C. Melatonin and its Relationships with Diabetes and Obesity: A Literature Review. Curr. Diabetes Rev. 2021, 17, e072620184137. [Google Scholar] [CrossRef]
- McMullan, C.J.; Curhan, G.C.; Schernhammer, E.S.; Forman, J.P. Association of nocturnal melatonin secretion with insulin resistance in nondiabetic young women. Am. J. Epidemiol. 2013, 15, 231–238. [Google Scholar] [CrossRef]
- Bonomini, F.; Dos Santos, M.; Veronese, F.V.; Rezzani, R. NLRP3 Inflammasome Modulation by Melatonin Supplementation in Chronic Pristane-Induced Lupus Nephritis. Int. J. Mol. Sci. 2019, 20, 3466. [Google Scholar] [CrossRef]
- Wu, C.C.; Lu, K.C.; Lin, G.J.; Hsieh, H.Y.; Chu, P.; Lin, S.H.; Sytwu, H.K. Melatonin enhances endogenous heme oxygenase-1 and represses immune responses to ameliorate experimental murine membranous nephropathy. J. Pineal Res. 2012, 52, 460–469. [Google Scholar] [CrossRef]
- Chalikias, G.; Drosos, I.; Tziakas, D.N. Prevention of Contrast-Induced Acute Kidney Injury: An Update. Cardiovasc. Drugs Ther. 2016, 30, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.S.; Briscese, K.; Yuan, M.; Deshpande, K.; Aleksunes, L.M.; Brunetti, L. Renoprotective Effects of Melatonin against Vancomycin-Related Acute Kidney Injury in Hospitalized Patients: A Retrospective Cohort Study. Antimicrob. Agents Chemother. 2021, 65, e0046221. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Jo, J.; Kim, J.Y.; Choe, M.; Leem, J.; Park, J.H. Melatonin Attenuates Cisplatin-Induced Acute Kidney Injury through Dual Suppression of Apoptosis and Necroptosis. Biology 2019, 30, 64. [Google Scholar] [CrossRef] [PubMed]
- Mok, J.X.; Ooi, J.H.; Ng, K.Y.; Koh, R.Y.; Chye, S.M. A new prospective on the role of melatonin in diabetes and its complications. Horm. Mol. Biol. Clin. Investig. 2019, 6, 20190036. [Google Scholar] [CrossRef]
- Promsan, S.; Lungkaphin, A. The roles of melatonin on kidney injury in obese and diabetic conditions. Biofactors 2020, 46, 531–549. [Google Scholar] [CrossRef]
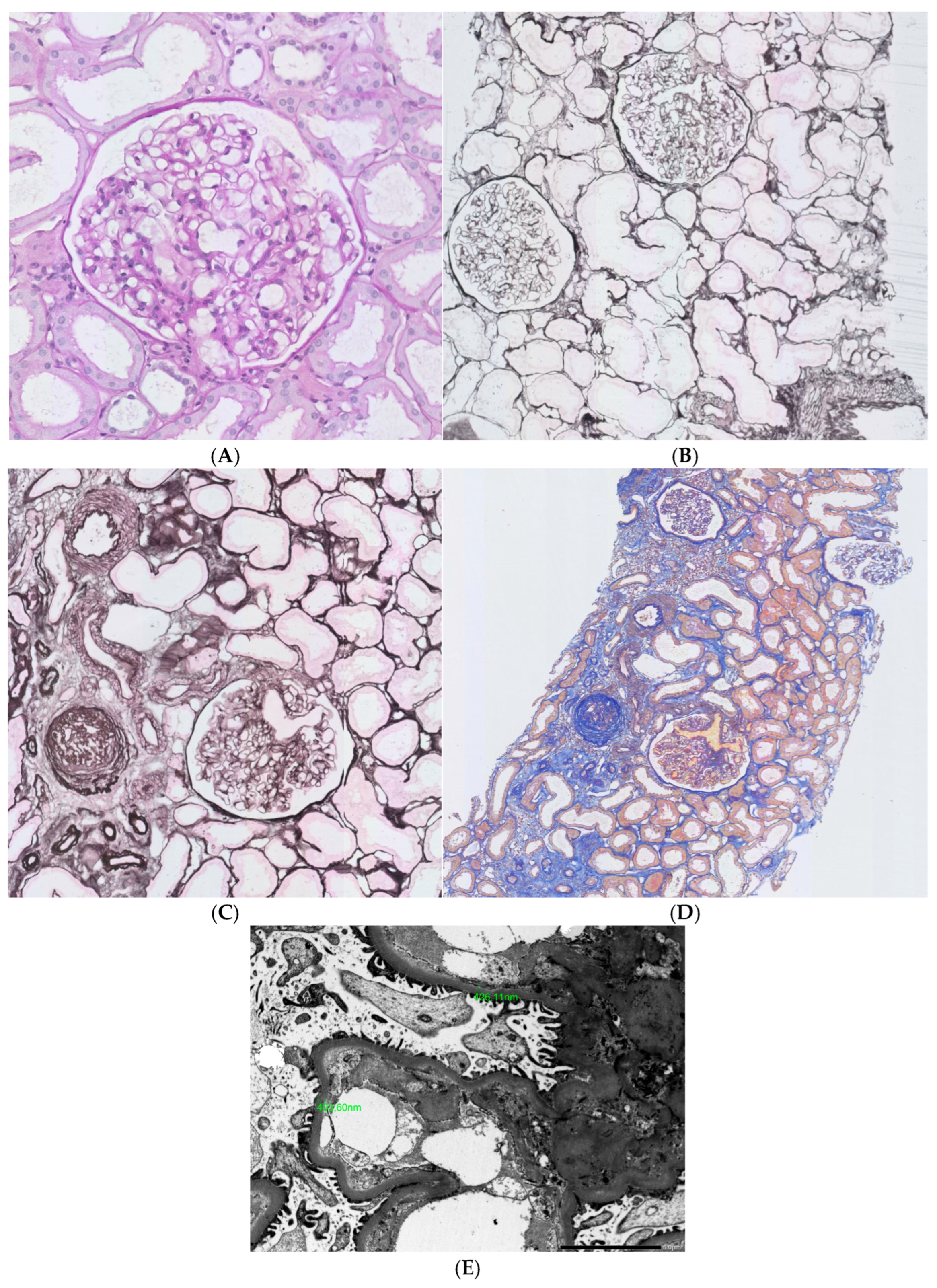

{kind=link}
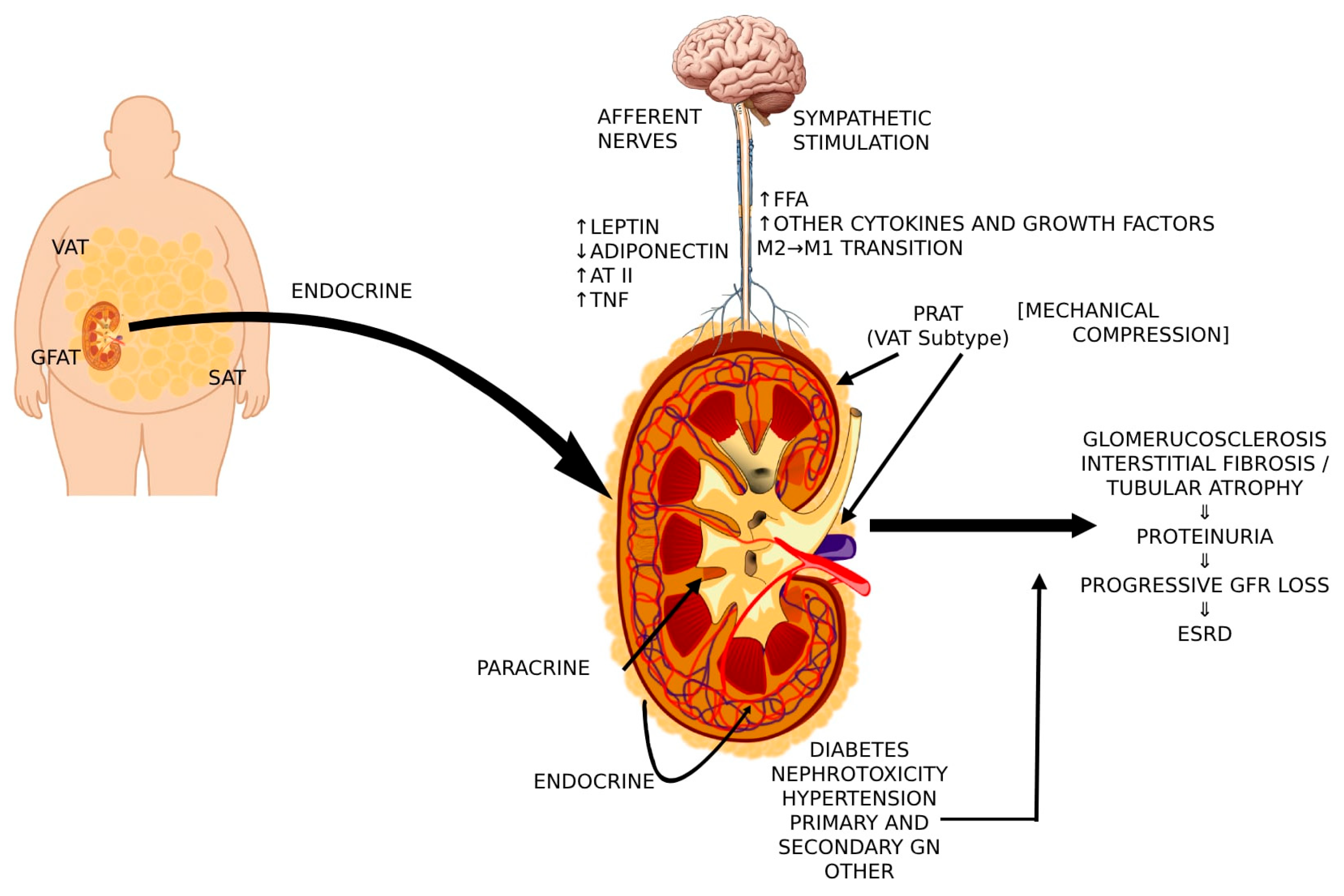
{kind=link}
| Name of the Trial | Study Drug and Dose | N | Concomitant Therapy | Comparator | BMI [kg/m2] | BMI ≥ 35 kg/m2 | Reduction in UACR [%] |
|---|---|---|---|---|---|---|---|
| SURPASS-1 [101,102] | Tirzepatide 5 mg 10 mg 15 mg | 478 | diet and exercise | placebo | 31.9 (6.6) | 131 (27.4%) | −43.4 −43 −31.3 |
| SURPASS-2 [101,102] | Tirzepatide 5 mg 10 mg 15 mg | 1878 | metformin | semaglutide 1 mg | 34.2 (6.9) | 690 (36.7%) | −4.8 2.8 −11.1 |
| SURPASS-3 [101,102] | Tirzepatide 5 mg 10 mg 15 mg | 1437 | metformin with or without SGLT2i | insulin degludec | 33.5 (6.1) | 495 (34.4%) | −12.7 −14.9 −23.3 |
| SURPASS-4 [101,102] | Tirzepatide 5 mg 10 mg 15 mg | 1995 | metformin or sulfonylurea or SGLT2i | insulin glargine | 32.6 (5.5) | 556 (27.9%) | −23.3 −32.3 −35.2 |
| SURPASS-5 [101,102] | Tirzepatide 5 mg 10 mg 15 mg | 475 | insulin glargine with or without metformin | placebo | 33.4 (6.1) | 180 (37.9%) | −10.7 −33.8 −29.4 |
| SURMOUNT-1 [102] | Tirzepatide 5 mg 10 mg 15 mg | 1032 | placebo | 38.8 (7.10) | 674 (65.3%) | 6.2 −9.5 −13.9 | |
| FLOW [93,102] | Semaglutide | 3533 | RAASi with or without SGLT2i | placebo | 32.0 (6.3) | 957 (27%) | −40 |
| SELECT [96] | Semaglutide 2.4 mg | 17,605 | local clinical practice of CV risk factors management | placebo | 33.34 (5.04) | 5106 (29%) | −10.7 |
| STEP 1 [91] | semaglutide 2.4 mg | 1961 | lifestyle intervention | placebo | 37.9 (6.6) | 1201 (61.2%) | No data available |
| STEP 2 [91] | semaglutide 1 mg 2.4 mg | 1210 | lifestyle intervention | placebo | 35.7 (6.3) | 561 (46.3%) | −14.8 −20.6 |
| STEP 3 [91] | semaglutide 2.4 mg | 611 | intensive behavioral therapy | placebo | 38.0 (6.7) | 389 (63.7%) | No data available |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zbrzeźniak-Suszczewicz, J.; Winiarska, A.; Perkowska-Ptasińska, A.; Stompór, T. Obesity-Related Glomerulosclerosis—How Adiposity Damages the Kidneys. Int. J. Mol. Sci. 2025, 26, 6247. https://doi.org/10.3390/ijms26136247
Zbrzeźniak-Suszczewicz J, Winiarska A, Perkowska-Ptasińska A, Stompór T. Obesity-Related Glomerulosclerosis—How Adiposity Damages the Kidneys. International Journal of Molecular Sciences. 2025; 26(13):6247. https://doi.org/10.3390/ijms26136247
Chicago/Turabian StyleZbrzeźniak-Suszczewicz, Justyna, Agata Winiarska, Agnieszka Perkowska-Ptasińska, and Tomasz Stompór. 2025. "Obesity-Related Glomerulosclerosis—How Adiposity Damages the Kidneys" International Journal of Molecular Sciences 26, no. 13: 6247. https://doi.org/10.3390/ijms26136247
APA StyleZbrzeźniak-Suszczewicz, J., Winiarska, A., Perkowska-Ptasińska, A., & Stompór, T. (2025). Obesity-Related Glomerulosclerosis—How Adiposity Damages the Kidneys. International Journal of Molecular Sciences, 26(13), 6247. https://doi.org/10.3390/ijms26136247