
Diclofenac Immune-Mediated Hepatitis: Identification of Innate and Adaptive Immune Responses at Clinically Relevant Doses


Abstract
1. Introduction
2. Results
2.1. Immunogenomics
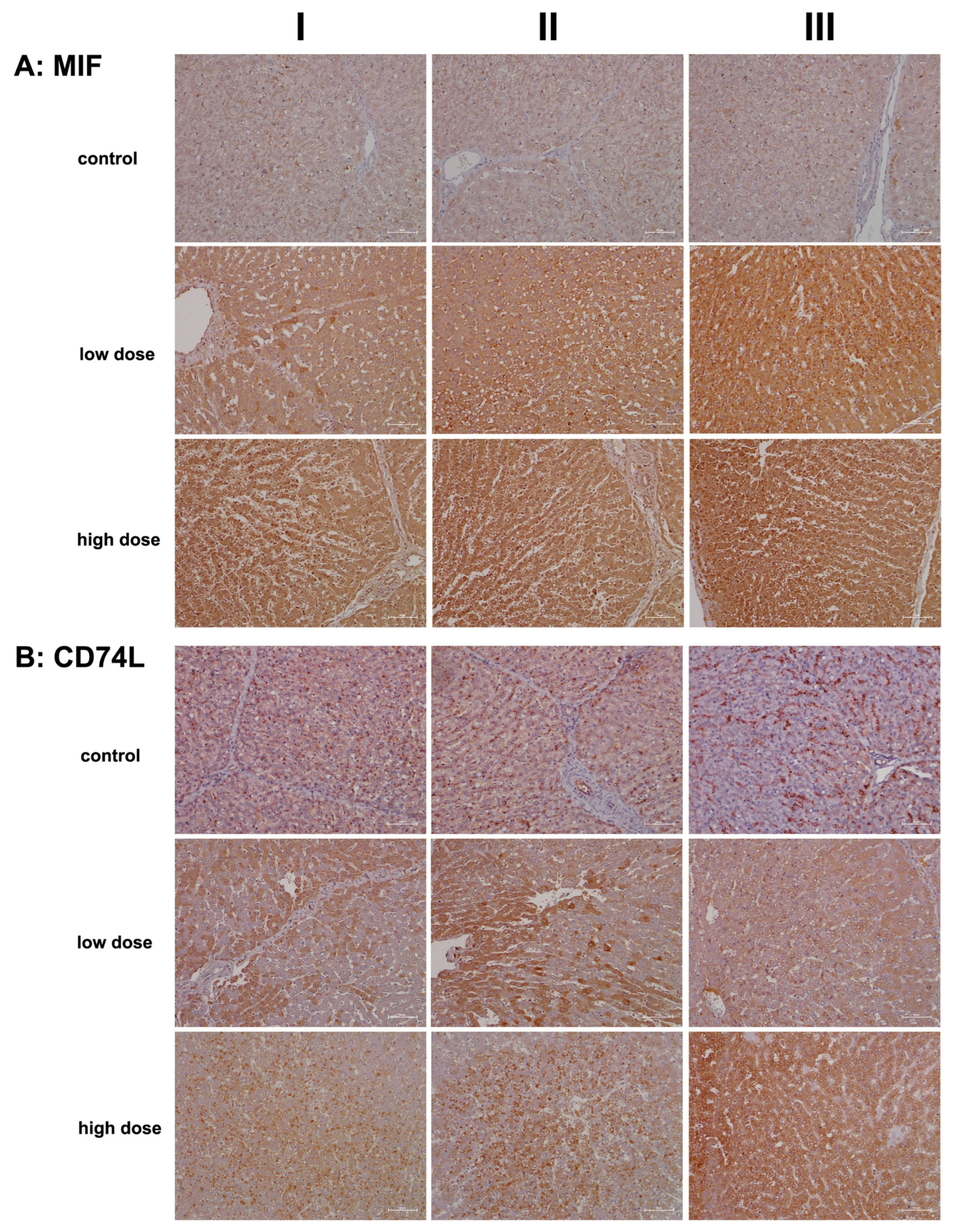
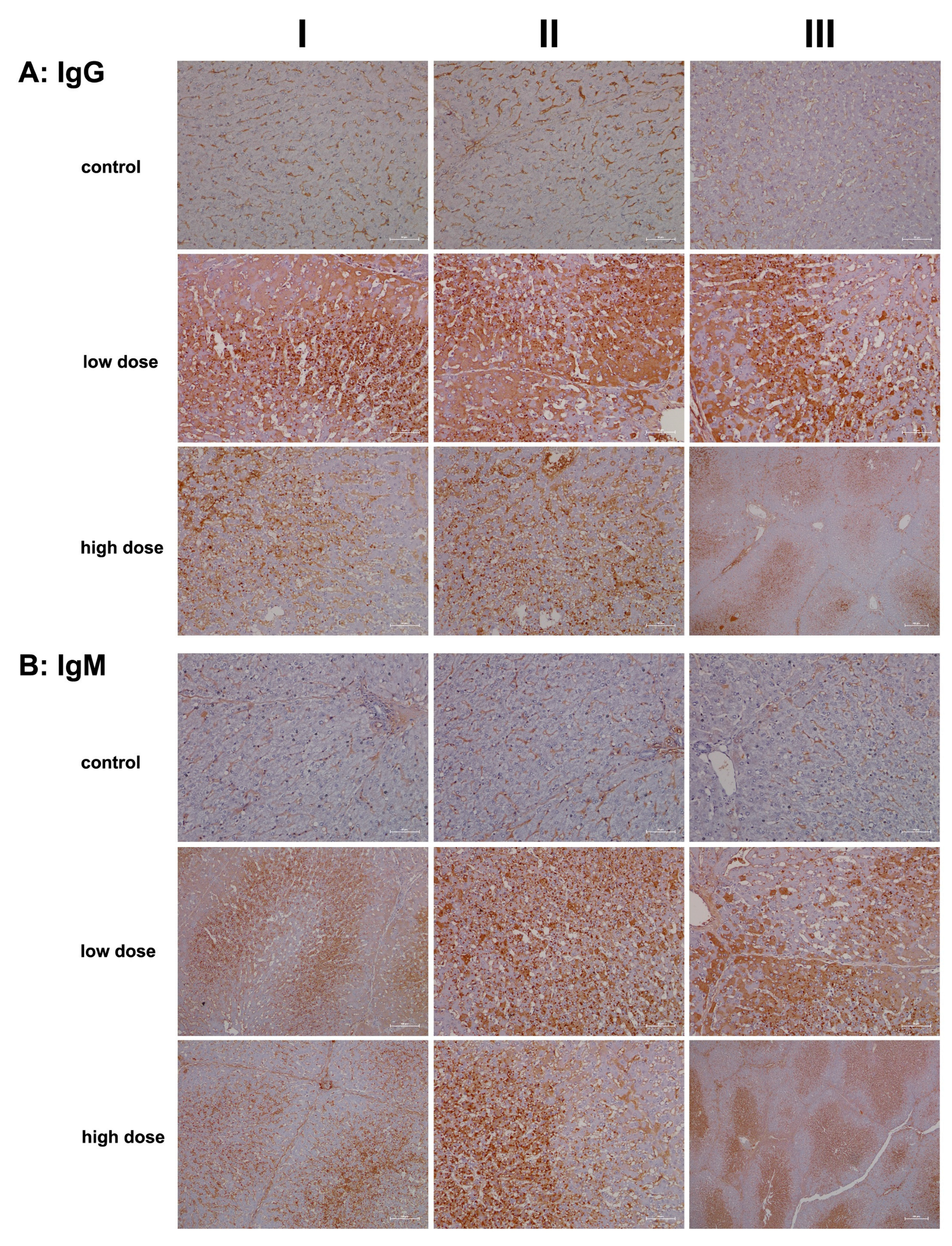
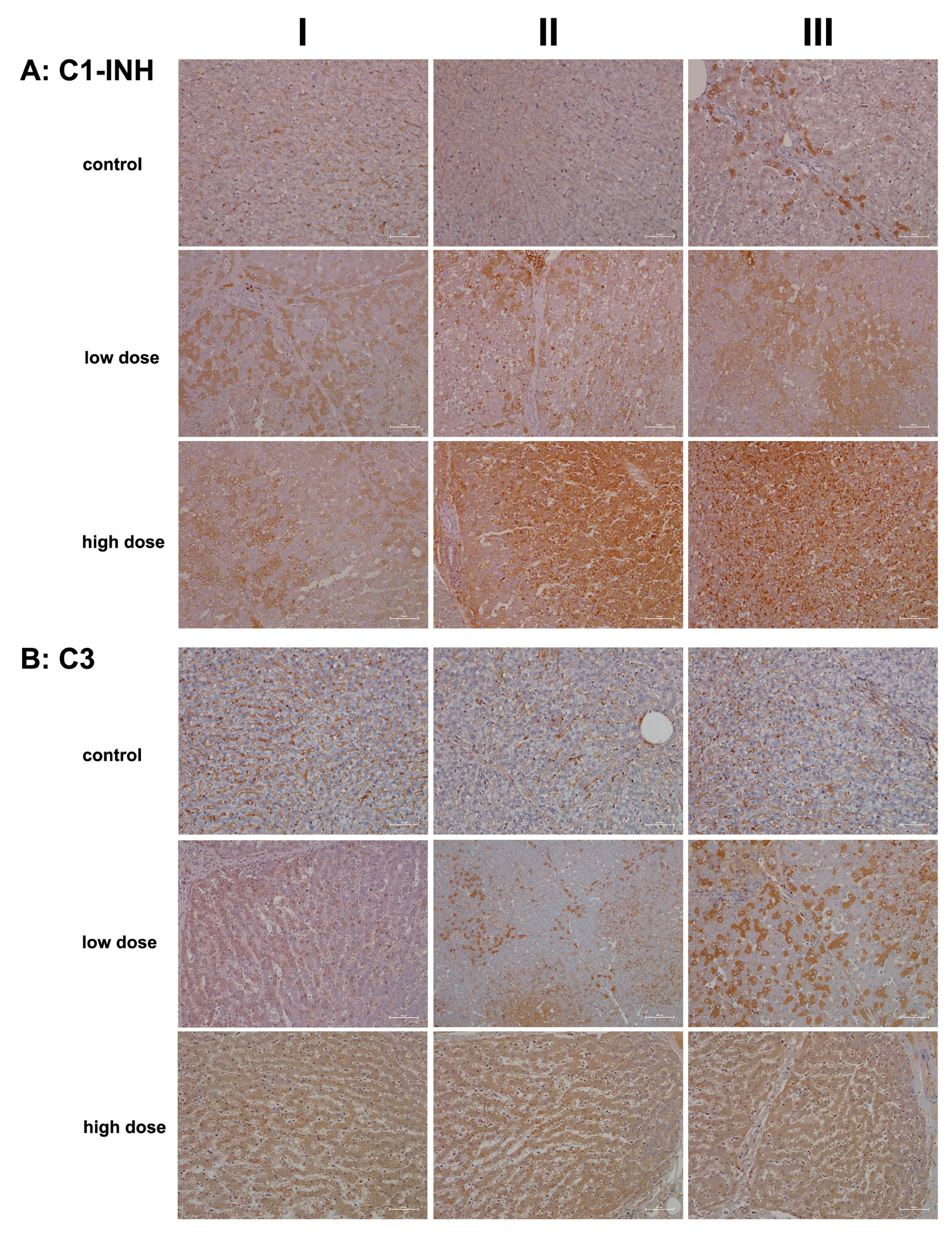
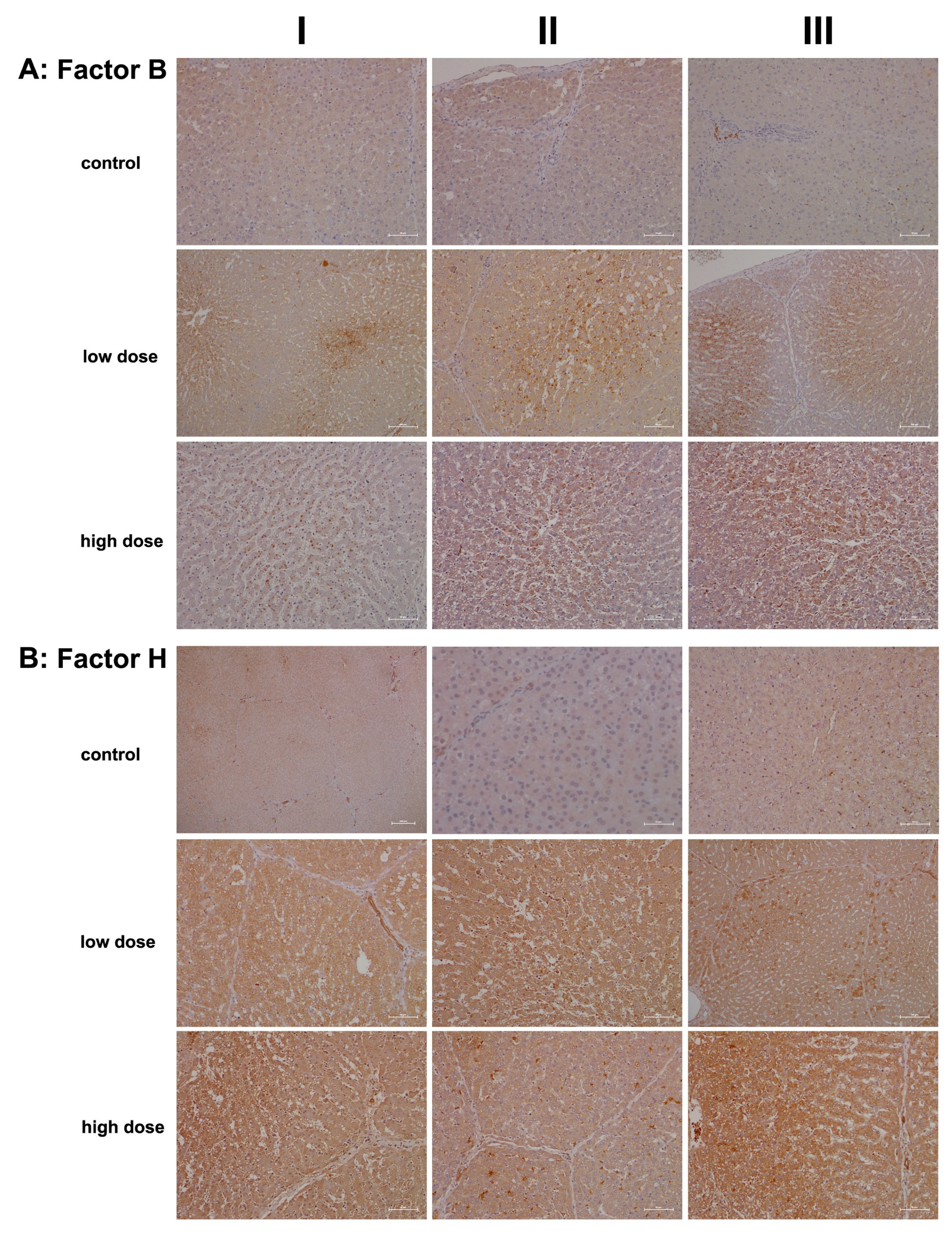
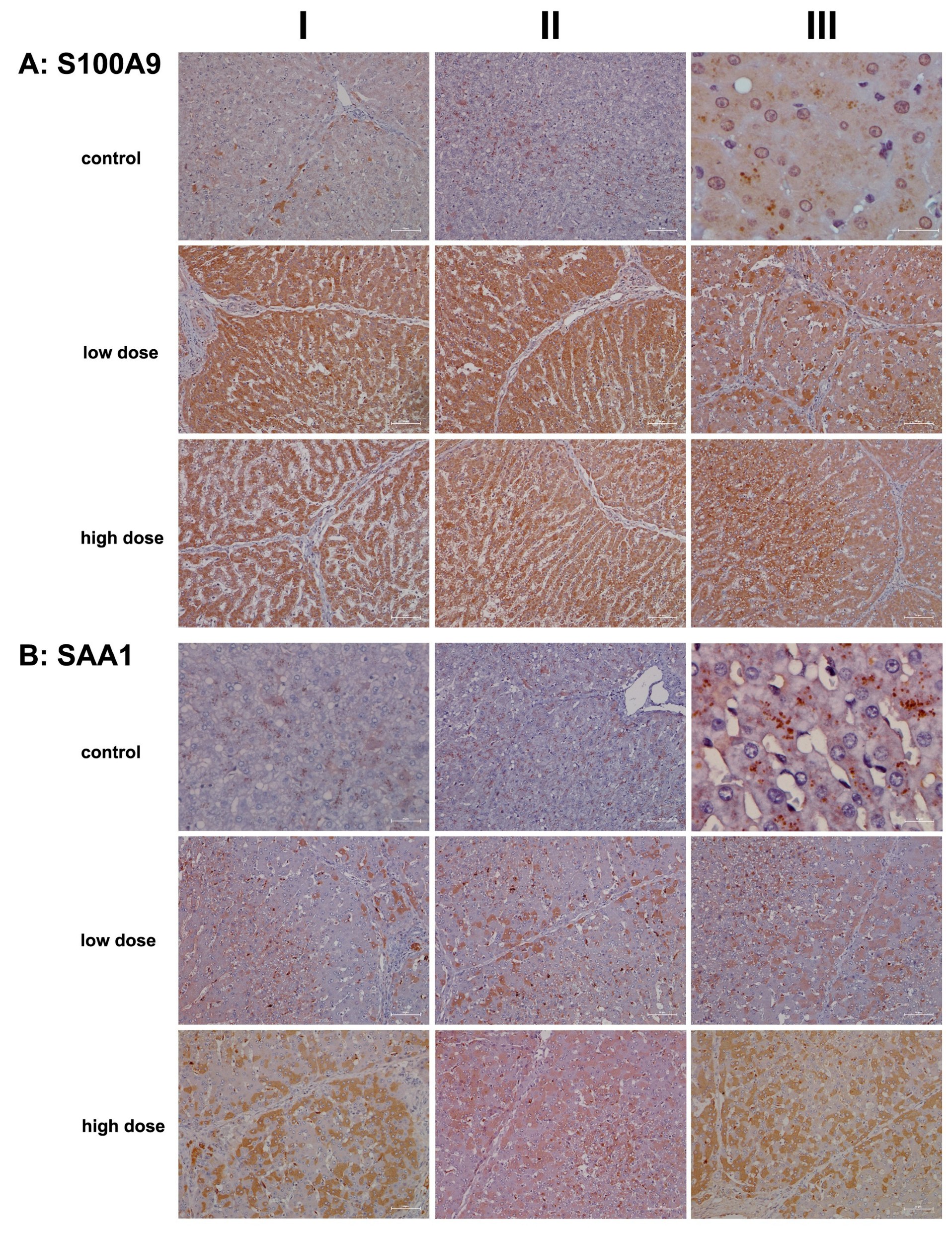
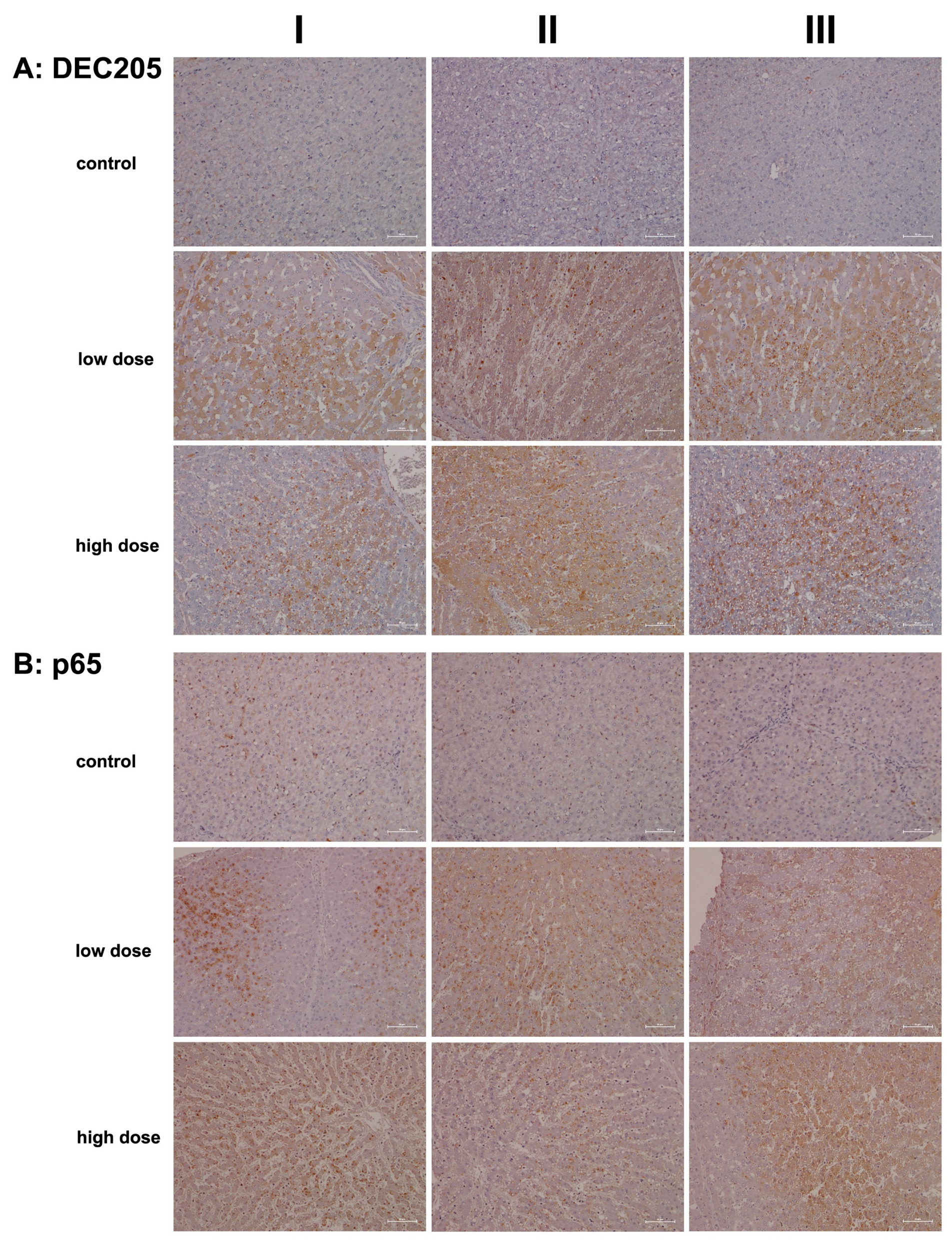
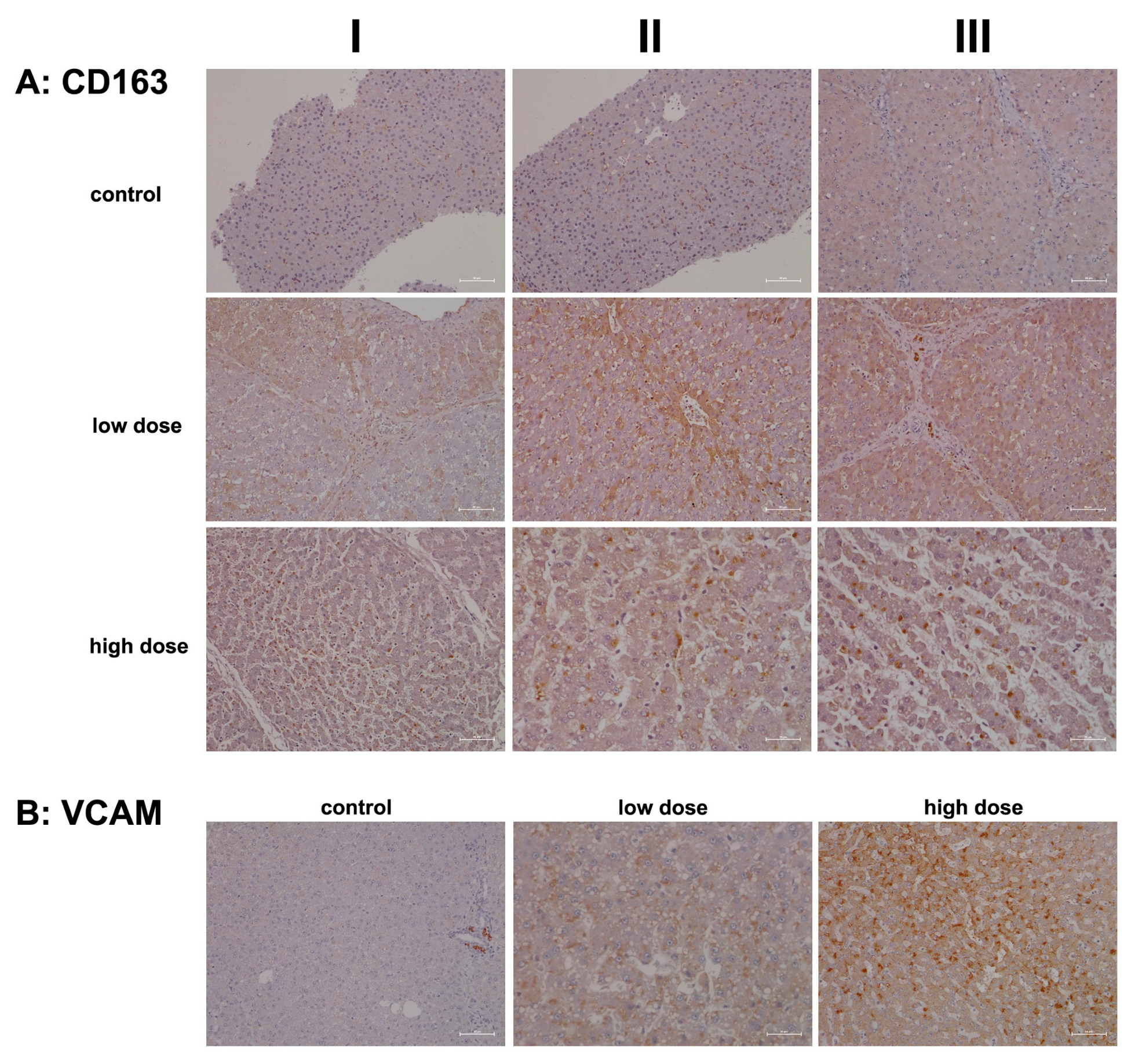
2.2. Immunopathology
3. Discussion
Study Limitations
4. Materials and Methods
4.1. Animals
4.2. Drug Treatment
4.3. RNA Extraction
4.4. Microarray Experiments and Data Analysis
4.5. Immunohistochemistry
| Antibody | Vendor | Cat No. | Lot Number | Dilution | Antigen Retrieval |
| C1INH | Santa Cruz | sc-46298 | B2706 | 1:200 | ph6 |
| C3 | abcam | ab112829 | GR119618-2 | 1:1000 | ph6 |
| CD163 | abcam | ab87099 | GR3197855-1 | 1:100 | ph6 |
| CD74L | Santa Cruz | sc-5441 | I2807 | 1:500 | ph6 |
| DEC-205 | Santa Cruz | sc-14602 | L1813 | 1:250 | ph6 |
| Factor B | Santa Cruz | sc-67141 | G1808 | 1:25 | ph9 |
| Factor H | abcam | ab170036 | GR179267-14 | 1:300 | ph6 |
| IgG | Dako | A0423 | 20020059 | 1:10,000 | Pronase |
| IgM | Dako | A0425 | 86531 | 1:1000 | Pronase |
| KLF6 | Santa Cruz | sc-7158 | D2613 | 1:50 | ph6 |
| Lysozyme | abcam | ab74666 | GR3185036-1 | RTU | ph6 |
| MIF | Abbiotec | 251415 | 13030401 | 1:500 | ph9 |
| NF-kB p65 | abcam | ab86299 | GR3204852-3 | 1:1500 | ph9 |
| S100A9 | abcam | ab63818 | GR126635-1 | 1:300 | ph6 |
| SAA1 | abcam | ab171030 | GR147621-10 | 1:100 | ph6 |
| VCAM-1 | Santa Cruz | sc-1504 | H1215 | 1:50 | ph6 |
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADR | adverse drug reactions |
| AIH | autoimmune hepatitis |
| CLR | C-type lectin receptors. |
| CXCL | C-X-C motif chemokine ligand |
| DAMP | death-associated molecular pattern |
| DEGs | differentially expressed genes |
| DILI | drug-induced liver injury |
| GR | glucocorticoid receptor |
| HSC | hepatic stellate cells |
| IgG | immunoglobulin G |
| IgE | immunoglobulin E |
| IgM | immunoglobulin M |
| MCP-2 | monocyte chemotactic protein-2 |
| MIF | macrophage migration inhibitory factor |
| NSAID | nonsteroidal anti-inflammatory diclofenac |
| PBC | primary biliary cholangitis |
| PRRs | pattern recognition receptors |
| Rgcc | regulator of cell cycle |
| SAA1 | serum amyloid A1 |
| sMR | soluble mannose receptor |
| TFBS | transcription factor binding sites |
| VCAM1 | vascular cell adhesion molecule 1 |
References
- Gan, T.J. Diclofenac: An Update on its Mechanism of Action and Safety Profile. Curr. Med. Res. Opin. 2010, 26, 1715–1731. [Google Scholar] [CrossRef] [PubMed]
- Coxib and traditional NSAID Trialists’ (CNT) Collaboration; Bhala, N.; Emberson, J.; Merhi, A.; Abramson, S.; Arber, N.; Baron, J.A.; Bombardier, C.; Cannon, C.; Farkouh, M.E.; et al. Vascular and Upper Gastrointestinal Effects of Non-Steroidal Anti-Inflammatory Drugs: Meta-Analyses of Individual Participant Data from Randomised Trials. Lancet 2013, 382, 769–779. [Google Scholar]
- Boelsterli, U.A. Diclofenac-Induced Liver Injury: A Paradigm of Idiosyncratic Drug Toxicity. Toxicol. Appl. Pharmacol. 2003, 192, 307–322. [Google Scholar] [CrossRef]
- Lee, E.H.; Oh, J.H.; Selvaraj, S.; Park, S.M.; Choi, M.S.; Spanel, R.; Yoon, S.; Borlak, J. Immunogenomics Reveal Molecular Circuits of Diclofenac Induced Liver Injury in Mice. Oncotarget 2016, 7, 14983–15017. [Google Scholar] [CrossRef]
- Selvaraj, S.; Oh, J.; Spanel, R.; Langer, F.; Han, H.; Lee, E.; Yoon, S.; Borlak, J. The Pathogenesis of Diclofenac Induced Immunoallergic Hepatitis in a Canine Model of Liver Injury. Oncotarget 2017, 8, 107763–107824. [Google Scholar] [CrossRef]
- Yano, A.; Higuchi, S.; Tsuneyama, K.; Fukami, T.; Nakajima, M.; Yokoi, T. Involvement of Immune-Related Factors in Diclofenac-Induced Acute Liver Injury in Mice. Toxicology 2012, 293, 107–114. [Google Scholar] [CrossRef]
- Deng, X.; Liguori, M.J.; Sparkenbaugh, E.M.; Waring, J.F.; Blomme, E.A.G.; Ganey, P.E.; Roth, R.A. Gene Expression Profiles in Livers from Diclofenac-Treated Rats Reveal Intestinal Bacteria-Dependent and -Independent Pathways Associated with Liver Injury. J. Pharmacol. Exp. Ther. 2008, 327, 634–644. [Google Scholar] [CrossRef]
- Hickey, E.J.; Raje, R.R.; Reid, V.E.; Gross, S.M.; Ray, S.D. Diclofenac Induced in Vivo Nephrotoxicity may Involve Oxidative Stress-Mediated Massive Genomic DNA Fragmentation and Apoptotic Cell Death. Free Radic. Biol. Med. 2001, 31, 139–152. [Google Scholar] [CrossRef]
- Laine, L.; Goldkind, L.; Curtis, S.P.; Connors, L.G.; Yanqiong, Z.; Cannon, C.P. How Common is Diclofenac-Associated Liver Injury? Analysis of 17,289 Arthritis Patients in a Long-Term Prospective Clinical Trial. Am. J. Gastroenterol. 2009, 104, 356–362. [Google Scholar] [CrossRef]
- Bjornsson, E.S.; Bergmann, O.M.; Bjornsson, H.K.; Kvaran, R.B.; Olafsson, S. Incidence, Presentation, and Outcomes in Patients with Drug-Induced Liver Injury in the General Population of Iceland. Gastroenterology 2013, 144, 1419–1420. [Google Scholar] [CrossRef]
- Rubic-Schneider, T.; Christen, B.; Brees, D.; Kammuller, M. Minipigs in Translational Immunosafety Sciences: A Perspective. Toxicol. Pathol. 2016, 44, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Oh, J.; Yoon, S.; Borlak, J. Diclofenac Disrupts the Circadian Clock and through Complex Cross-Talks Aggravates Immune-Mediated Liver Injury—A Repeated Dose Study in Minipigs for 28 Days. Int. J. Mol. Sci. 2023, 24, 1445. [Google Scholar] [CrossRef]
- Iveson, T.J.; Ryley, N.G.; Kelly, P.M.; Trowell, J.M.; McGee, J.O.; Chapman, R.W. Diclofenac Associated Hepatitis. J. Hepatol. 1990, 10, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Sriuttha, P.; Sirichanchuen, B.; Permsuwan, U. Hepatotoxicity of Nonsteroidal Anti-Inflammatory Drugs: A Systematic Review of Randomized Controlled Trials. Int. J. Hepatol. 2018, 2018, 5253623. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Lu, W. Defensins: A Double-Edged Sword in Host Immunity. Front. Immunol. 2020, 11, 764. [Google Scholar] [CrossRef]
- Klag, T.; Thomas, M.; Ehmann, D.; Courth, L.; Mailander-Sanchez, D.; Weiss, T.S.; Dayoub, R.; Abshagen, K.; Vollmar, B.; Thasler, W.E.; et al. Beta-Defensin 1 is Prominent in the Liver and Induced during Cholestasis by Bilirubin and Bile Acids Via Farnesoid X Receptor and Constitutive Androstane Receptor. Front. Immunol. 2018, 9, 1735. [Google Scholar] [CrossRef]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Ragland, S.A.; Criss, A.K. From Bacterial Killing to Immune Modulation: Recent Insights into the Functions of Lysozyme. PLoS Pathog. 2017, 13, e1006512. [Google Scholar] [CrossRef]
- Manifold, I.H.; Bishop, F.M.; Cloke, P.; Triger, D.R.; Underwood, J.C. Lysozyme in Chronic Liver Disease: A Biochemical and Histological Study. J. Clin. Pathol. 1982, 35, 815–819. [Google Scholar] [CrossRef]
- Cha, H.H.; Cram, E.J.; Wang, E.C.; Huang, A.J.; Kasler, H.G.; Firestone, G.L. Glucocorticoids Stimulate p21 Gene Expression by Targeting Multiple Transcriptional Elements within a Steroid Responsive Region of the p21waf1/Cip1 Promoter in Rat Hepatoma Cells. J. Biol. Chem. 1998, 273, 1998–2007. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, M.; Gao, B. Hepatocytes: A Key Cell Type for Innate Immunity. Cell. Mol. Immunol. 2016, 13, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Calandra, T.; Roger, T. Macrophage Migration Inhibitory Factor: A Regulator of Innate Immunity. Nat. Rev. Immunol. 2003, 3, 791–800. [Google Scholar] [CrossRef]
- Wen, Y.; Cai, W.; Yang, J.; Fu, X.; Putha, L.; Xia, Q.; Windsor, J.A.; Phillips, A.R.; Tyndall, J.D.A.; Du, D.; et al. Targeting Macrophage Migration Inhibitory Factor in Acute Pancreatitis and Pancreatic Cancer. Front. Pharmacol. 2021, 12, 638950. [Google Scholar] [CrossRef]
- Marin, V.; Poulsen, K.; Odena, G.; McMullen, M.R.; Altamirano, J.; Sancho-Bru, P.; Tiribelli, C.; Caballeria, J.; Rosso, N.; Bataller, R.; et al. Hepatocyte-Derived Macrophage Migration Inhibitory Factor Mediates Alcohol-Induced Liver Injury in Mice and Patients. J. Hepatol. 2017, 67, 1018–1025. [Google Scholar] [CrossRef]
- Heinrichs, D.; Berres, M.; Coeuru, M.; Knauel, M.; Nellen, A.; Fischer, P.; Philippeit, C.; Bucala, R.; Trautwein, C.; Wasmuth, H.E.; et al. Protective Role of Macrophage Migration Inhibitory Factor in Nonalcoholic Steatohepatitis. FASEB J. 2014, 28, 5136–5147. [Google Scholar] [CrossRef]
- Schroder, B. The Multifaceted Roles of the Invariant Chain CD74—More than just a Chaperone. Biochim. Biophys. Acta 2016, 1863, 1269–1281. [Google Scholar] [CrossRef]
- Su, H.; Na, N.; Zhang, X.; Zhao, Y. The Biological Function and Significance of CD74 in Immune Diseases. Inflamm. Res. 2017, 66, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Huang, T.; Su, M.; Luo, J.; Korteweg, C.; Li, J.; Chen, Z.; Qiu, Y.; Liu, X.; Yan, M.; et al. Expression and Distribution of Immunoglobulin G in the Normal Liver, Hepatocarcinoma and Postpartial Hepatectomy Liver. Lab. Investig. 2014, 94, 1283–1295. [Google Scholar] [CrossRef]
- Shao, W.; Hu, F.; Ma, J.; Zhang, C.; Liao, Q.; Zhu, Z.; Liu, E.; Qiu, X. Epithelial Cells are a Source of Natural IgM that Contribute to Innate Immune Responses. Int. J. Biochem. Cell Biol. 2016, 73, 19–29. [Google Scholar] [CrossRef]
- Renston, R.H.; Jones, A.L.; Christiansen, W.D.; Hradek, G.T.; Underdown, B.J. Evidence for a Vesicular Transport Mechanism in Hepatocytes for Biliary Secretion of Immunoglobulin A. Science 1980, 208, 1276–1278. [Google Scholar] [CrossRef]
- Goldman, I.S.; Jones, A.L.; Hradek, G.T.; Huling, S. Hepatocyte Handling of Immunoglobulin A in the Rat: The Role of Microtubules. Gastroenterology 1983, 85, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Gerussi, A.; Natalini, A.; Antonangeli, F.; Mancuso, C.; Agostinetto, E.; Barisani, D.; Di Rosa, F.; Andrade, R.; Invernizzi, P. Immune-Mediated Drug-Induced Liver Injury: Immunogenetics and Experimental Models. Int. J. Mol. Sci. 2021, 22, 4557. [Google Scholar] [CrossRef] [PubMed]
- Sebode, M.; Weiler-Normann, C.; Liwinski, T.; Schramm, C. Autoantibodies in Autoimmune Liver Disease-Clinical and Diagnostic Relevance. Front. Immunol. 2018, 9, 609. [Google Scholar] [CrossRef]
- Caliezi, C.; Wuillemin, W.A.; Zeerleder, S.; Redondo, M.; Eisele, B.; Hack, C.E. C1-Esterase Inhibitor: An Anti-Inflammatory Agent and its Potential use in the Treatment of Diseases Other than Hereditary Angioedema. Pharmacol. Rev. 2000, 52, 91–112. [Google Scholar] [CrossRef]
- Dorresteijn, M.J.; Visser, T.; Cox, L.A.E.; Bouw, M.P.; Pillay, J.; Koenderman, A.H.L.; Strengers, P.F.W.; Leenen, L.P.H.; van der Hoeven, J.G.; Koenderman, L.; et al. C1-Esterase Inhibitor Attenuates the Inflammatory Response during Human Endotoxemia. Crit. Care Med. 2010, 38, 2139–2145. [Google Scholar] [CrossRef]
- Panagiotou, A.; Trendelenburg, M.; Osthoff, M. The Lectin Pathway of Complement in Myocardial Ischemia/Reperfusion Injury-Review of its Significance and the Potential Impact of Therapeutic Interference by C1 Esterase Inhibitor. Front. Immunol. 2018, 9, 1151. [Google Scholar] [CrossRef]
- Ricklin, D.; Reis, E.S.; Mastellos, D.C.; Gros, P.; Lambris, J.D. Complement Component C3—The “Swiss Army Knife” of Innate Immunity and Host Defense. Immunol. Rev. 2016, 274, 33–58. [Google Scholar] [CrossRef]
- Thorgersen, E.B.; Barratt-Due, A.; Haugaa, H.; Harboe, M.; Pischke, S.E.; Nilsson, P.H.; Mollnes, T.E. The Role of Complement in Liver Injury, Regeneration, and Transplantation. Hepatology 2019, 70, 725–736. [Google Scholar] [CrossRef]
- Lubbers, R.; van Essen, M.F.; van Kooten, C.; Trouw, L.A. Production of Complement Components by Cells of the Immune System. Clin. Exp. Immunol. 2017, 188, 183–194. [Google Scholar] [CrossRef]
- Li, Y.; Sha, Y.; Wang, H.; He, L.; Li, L.; Wen, S.; Sheng, L.; Hu, W.; Zhou, H. Intracellular C3 Prevents Hepatic Steatosis by Promoting Autophagy and very-Low-Density Lipoprotein Secretion. FASEB J. 2021, 35, e22037. [Google Scholar] [CrossRef]
- Clark, A.; Weymann, A.; Hartman, E.; Turmelle, Y.; Carroll, M.; Thurman, J.M.; Holers, V.M.; Hourcade, D.E.; Rudnick, D.A. Evidence for Non-Traditional Activation of Complement Factor C3 during Murine Liver Regeneration. Mol. Immunol. 2008, 45, 3125–3132. [Google Scholar] [CrossRef] [PubMed]
- Strey, C.W.; Markiewski, M.; Mastellos, D.; Tudoran, R.; Spruce, L.A.; Greenbaum, L.E.; Lambris, J.D. The Proinflammatory Mediators C3a and C5a are Essential for Liver Regeneration. J. Exp. Med. 2003, 198, 913–923. [Google Scholar] [CrossRef]
- Peters, M.G.; Ambrus, J.L.J.; Fauci, A.S.; Brown, E.J. The Bb Fragment of Complement Factor B Acts as a B Cell Growth Factor. J. Exp. Med. 1988, 168, 1225–1235. [Google Scholar] [CrossRef]
- Ferreira, V.P.; Pangburn, M.K.; Cortes, C. Complement Control Protein Factor H: The Good, the Bad, and the Inadequate. Mol. Immunol. 2010, 47, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Barbu, A.; Hamad, O.A.; Lind, L.; Ekdahl, K.N.; Nilsson, B. The Role of Complement Factor C3 in Lipid Metabolism. Mol. Immunol. 2015, 67, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Fan, X.; Yao, J.; Tomlinson, S.; Yuan, G.; He, S. The Role of Complement in Nonalcoholic Fatty Liver Disease. Front. Immunol. 2022, 13, 1017467. [Google Scholar] [CrossRef]
- Shimazaki, R.; Takano, S.; Satoh, M.; Takada, M.; Miyahara, Y.; Sasaki, K.; Yoshitomi, H.; Kagawa, S.; Furukawa, K.; Takayashiki, T.; et al. Complement Factor B Regulates Cellular Senescence and is Associated with Poor Prognosis in Pancreatic Cancer. Cell. Oncol. 2021, 44, 937–950. [Google Scholar] [CrossRef]
- Marzano, C.; Cazals-Hatem, D.; Rautou, P.; Valla, D. The Significance of Nonobstructive Sinusoidal Dilatation of the Liver: Impaired Portal Perfusion Or Inflammatory Reaction Syndrome. Hepatology 2015, 62, 956–963. [Google Scholar] [CrossRef]
- West, E.E.; Kemper, C. Complosome—The Intracellular Complement System. Nat. Rev. Nephrol. 2023, 19, 426–439. [Google Scholar] [CrossRef]
- Kobsel, N.; Ramadori, G. Human Hepatoma Cells Synthesize and Secrete Lysozyme: Modulation by Cytokines. Hepatology 1994, 19, 1182–1188. [Google Scholar]
- Kluter, T.; Fitschen-Oestern, S.; Lippross, S.; Weuster, M.; Mentlein, R.; Steubesand, N.; Neunaber, C.; Hildebrand, F.; Pufe, T.; Tohidnezhad, M.; et al. The Antimicrobial Peptide Lysozyme is Induced After Multiple Trauma. Mediators Inflamm. 2014, 2014, 303106. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Li, Y.; Wang, L.; Guo, J.; Liu, W.; Meng, G.; Zhang, L.; Li, M.; Cong, L.; Sun, M. Recent Insights into the Prognostic and Therapeutic Applications of Lysozymes. Front. Pharmacol. 2021, 12, 767642. [Google Scholar] [CrossRef] [PubMed]
- Date, D.; Das, R.; Narla, G.; Simon, D.I.; Jain, M.K.; Mahabeleshwar, G.H. Kruppel-Like Transcription Factor 6 Regulates Inflammatory Macrophage Polarization. J. Biol. Chem. 2014, 289, 10318–10329. [Google Scholar] [CrossRef]
- Markowitz, J.; Carson, W.E. 3. Review of S100A9 Biology and its Role in Cancer. Biochim. Biophys. Acta 2013, 1835, 100–109. [Google Scholar] [CrossRef]
- Zhou, C.; Gao, Y.; Ding, P.; Wu, T.; Ji, G. The Role of CXCL Family Members in Different Diseases. Cell. Death Discov. 2023, 9, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, L.; Nemeth, J.; Pusterla, T.; Bauer, C.; De Ponti, A.; Manthey, S.; Marhenke, S.; Vogel, A.; Klingmuller, U.; Hess, J.; et al. Hepatocyte-Specific S100a8 and S100a9 Transgene Expression in Mice Causes Cxcl1 Induction and Systemic Neutrophil Enrichment. Cell. Commun. Signal. 2012, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Miller, A.L.; Rebelatto, M.; Brewah, Y.; Rowe, D.C.; Clarke, L.; Czapiga, M.; Rosenthal, K.; Imamichi, T.; Chen, Y.; et al. S100A9 Induced Inflammatory Responses are Mediated by Distinct Damage Associated Molecular Patterns (DAMP) Receptors in Vitro and in Vivo. PLoS ONE 2015, 10, e0115828. [Google Scholar] [CrossRef]
- Xia, P.; Ji, X.; Yan, L.; Lian, S.; Chen, Z.; Luo, Y. Roles of S100A8, S100A9 and S100A12 in Infection, Inflammation and Immunity. Immunology 2024, 171, 365–376. [Google Scholar] [CrossRef]
- Chakraborty, P.; Bjork, P.; Kallberg, E.; Olsson, A.; Riva, M.; Morgelin, M.; Liberg, D.; Ivars, F.; Leanderson, T. Vesicular Location and Transport of S100A8 and S100A9 Proteins in Monocytoid Cells. PLoS ONE 2015, 10, e0145217. [Google Scholar] [CrossRef]
- Zhou, L.; Li, C.; Gao, L.; Wang, A. High-Density Lipoprotein Synthesis and Metabolism (Review). Mol. Med. Rep. 2015, 12, 4015–4021. [Google Scholar] [CrossRef]
- Getachew, A.; Abbas, N.; You, K.; Yang, Z.; Hussain, M.; Huang, X.; Cheng, Z.; Tan, S.; Tao, J.; Yu, X.; et al. SAA1/TLR2 Axis Directs Chemotactic Migration of Hepatic Stellate Cells Responding to Injury. iScience 2021, 24, 102483. [Google Scholar] [CrossRef] [PubMed]
- Sack, G.H.J. Serum Amyloid A—A Review. Mol. Med. 2018, 24, 46. [Google Scholar] [CrossRef]
- Shrimpton, R.E.; Butler, M.; Morel, A.; Eren, E.; Hue, S.S.; Ritter, M.A. CD205 (DEC-205): A Recognition Receptor for Apoptotic and Necrotic Self. Mol. Immunol. 2009, 46, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- van der Zande, H.J.P.; Nitsche, D.; Schlautmann, L.; Guigas, B.; Burgdorf, S. The Mannose Receptor: From Endocytic Receptor and Biomarker to Regulator of (Meta)Inflammation. Front. Immunol. 2021, 12, 765034. [Google Scholar] [CrossRef]
- Kato, M.; McDonald, K.J.; Khan, S.; Ross, I.L.; Vuckovic, S.; Chen, K.; Munster, D.; MacDonald, K.P.A.; Hart, D.N.J. Expression of Human DEC-205 (CD205) Multilectin Receptor on Leukocytes. Int. Immunol. 2006, 18, 857–869. [Google Scholar] [CrossRef]
- Kato, M.; Khan, S.; Gonzalez, N.; O’Neill, B.P.; McDonald, K.J.; Cooper, B.J.; Angel, N.Z.; Hart, D.N.J. Hodgkin’s Lymphoma Cell Lines Express a Fusion Protein Encoded by Intergenically Spliced mRNA for the Multilectin Receptor DEC-205 (CD205) and a Novel C-Type Lectin Receptor DCL-1. J. Biol. Chem. 2003, 278, 34035–34041. [Google Scholar] [CrossRef]
- Kato, M.; Khan, S.; d’Aniello, E.; McDonald, K.J.; Hart, D.N.J. The Novel Endocytic and Phagocytic C-Type Lectin Receptor DCL-1/CD302 on Macrophages is Colocalized with F-Actin, Suggesting a Role in Cell Adhesion and Migration. J. Immunol. 2007, 179, 6052–6063. [Google Scholar] [CrossRef] [PubMed]
- Lo, T.; Silveira, P.A.; Fromm, P.D.; Verma, N.D.; Vu, P.A.; Kupresanin, F.; Adam, R.; Kato, M.; Cogger, V.C.; Clark, G.J.; et al. Characterization of the Expression and Function of the C-Type Lectin Receptor CD302 in Mice and Humans Reveals a Role in Dendritic Cell Migration. J. Immunol. 2016, 197, 885–898. [Google Scholar] [CrossRef]
- Schuette, V.; Embgenbroich, M.; Ulas, T.; Welz, M.; Schulte-Schrepping, J.; Draffehn, A.M.; Quast, T.; Koch, K.; Nehring, M.; Konig, J.; et al. Mannose Receptor Induces T-Cell Tolerance via Inhibition of CD45 and Up-Regulation of CTLA-4. Proc. Natl. Acad. Sci. USA 2016, 113, 10649–10654. [Google Scholar] [CrossRef]
- Etzerodt, A.; Moestrup, S.K. CD163 and Inflammation: Biological, Diagnostic, and Therapeutic Aspects. Antioxid. Redox Signal. 2013, 18, 2352–2363. [Google Scholar] [CrossRef]
- Parra-Sanchez, H.; Puebla-Clark, L.; Resendiz, M.; Valenzuela, O.; Hernandez, J. Characterization and Expression of DEC205 in the cDC1 and cDC2 Subsets of Porcine Dendritic Cells from Spleen, Tonsil, and Submaxillary and Mesenteric Lymph Nodes. Mol. Immunol. 2018, 96, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.L.; Baeuerle, P.A. The p65 Subunit is Responsible for the Strong Transcription Activating Potential of NF-Kappa B. EMBO J. 1991, 10, 3805–3817. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.C.; Hvidbjerg Gantzel, R.; Claria, J.; Trebicka, J.; Moller, H.J.; Gronbaek, H. Macrophage Activation Markers, CD163 and CD206, in Acute-on-Chronic Liver Failure. Cells 2020, 9, 1175. [Google Scholar] [CrossRef]
- Hiraoka, A.; Horiike, N.; Akbar, S.M.F.; Michitaka, K.; Matsuyama, T.; Onji, M. Soluble CD163 in Patients with Liver Diseases: Very High Levels of Soluble CD163 in Patients with Fulminant Hepatic Failure. J. Gastroenterol. 2005, 40, 52–56. [Google Scholar] [CrossRef]
- Kong, D.; Kim, Y.K.; Kim, M.R.; Jang, J.H.; Lee, S. Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef]
- Hiraoka, A.; Horiike, N.; Akbar, S.M.F.; Michitaka, K.; Matsuyama, T.; Onji, M. Expression of CD163 in the Liver of Patients with Viral Hepatitis. Pathol. Res. Pract. 2005, 201, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Akahori, H.; Harari, E.; Smith, S.L.; Polavarapu, R.; Karmali, V.; Otsuka, F.; Gannon, R.L.; Braumann, R.E.; Dickinson, M.H.; et al. CD163+ Macrophages Promote Angiogenesis and Vascular Permeability Accompanied by Inflammation in Atherosclerosis. J. Clin. Investig. 2018, 128, 1106–1124. [Google Scholar] [CrossRef]
- Higa, Y.; Oshiro, S.; Kino, K.; Tsunoo, H.; Nakajima, H. Catabolism of Globin-Haptoglobin in Liver Cells After Intravenous Administration of Hemoglobin-Haptoglobin to Rats. J. Biol. Chem. 1981, 256, 12322–12328. [Google Scholar] [CrossRef]
- Kino, K.; Tsunoo, H.; Higa, Y.; Takami, M.; Hamaguchi, H.; Nakajima, H. Hemoglobin-Haptoglobin Receptor in Rat Liver Plasma Membrane. J. Biol. Chem. 1980, 255, 9616–9620. [Google Scholar] [CrossRef]
- Chow, E.C.Y.; Liu, L.; Ship, N.; Kluger, R.H.; Pang, K.S. Role of Haptoglobin on the Uptake of Native and Beta-Chain [Trimesoyl-(Lys82)Beta-(Lys82)Beta] Cross-Linked Human Hemoglobins in Isolated Perfused Rat Livers. Drug Metab. Dispos. 2008, 36, 937–945. [Google Scholar] [CrossRef]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The Cell Biology of the Hepatocyte: A Membrane Trafficking Machine. J. Cell Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef] [PubMed]
- Skytthe, M.K.; Graversen, J.H.; Moestrup, S.K. Targeting of CD163(+) Macrophages in Inflammatory and Malignant Diseases. Int. J. Mol. Sci. 2020, 21, 5497. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, E.; Kalaitzakis, E.; Olsson, R. The Impact of Eosinophilia and Hepatic Necrosis on Prognosis in Patients with Drug-Induced Liver Injury. Aliment. Pharmacol. Ther. 2007, 25, 1411–1421. [Google Scholar] [CrossRef]
- Charfi, O.; Aouinti, I.; Zaiem, A.; Lakhoua, G.; El Aidli, S.; Kastalli, S. Diclofenac Induced Drug Rash with Eosinophilia and Systemic Symptoms. Therapie 2018, 73, 551–553. [Google Scholar] [CrossRef]
- Um, S.J.; Lee, S.K.; Kim, Y.H.; Kim, K.H.; Son, C.H.; Roh, M.S.; Lee, M.K. Clinical Features of Drug-Induced Hypersensitivity Syndrome in 38 Patients. J. Investig. Allergol. Clin. Immunol. 2010, 20, 556–562. [Google Scholar]
- Farr, L.; Ghosh, S.; Moonah, S. Role of MIF Cytokine/CD74 Receptor Pathway in Protecting Against Injury and Promoting Repair. Front. Immunol. 2020, 11, 1273. [Google Scholar] [CrossRef]
- Assis, D.N.; Leng, L.; Du, X.; Zhang, C.K.; Grieb, G.; Merk, M.; Garcia, A.B.; McCrann, C.; Chapiro, J.; Meinhardt, A.; et al. The Role of Macrophage Migration Inhibitory Factor in Autoimmune Liver Disease. Hepatology 2014, 59, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Lammert, C.; Zhu, C.; Lian, Y.; Raman, I.; Eckert, G.; Li, Q.; Chalasani, N. Exploratory Study of Autoantibody Profiling in Drug-Induced Liver Injury with an Autoimmune Phenotype. Hepatol. Commun. 2020, 4, 1651–1663. [Google Scholar] [CrossRef]
- De Roza, M.A.; Lamba, M.; Goh, G.B.; Lum, J.H.; Cheah, M.C.; Ngu, J.H.J. Immunoglobulin G in Non-Alcoholic Steatohepatitis Predicts Clinical Outcome: A Prospective Multi-Centre Cohort Study. World J. Gastroenterol. 2021, 27, 7563–7571. [Google Scholar] [CrossRef]
- Embgenbroich, M.; van der Zande, H.J.P.; Hussaarts, L.; Schulte-Schrepping, J.; Pelgrom, L.R.; Garcia-Tardon, N.; Schlautmann, L.; Stoetzel, I.; Handler, K.; Lambooij, J.M.; et al. Soluble Mannose Receptor Induces Proinflammatory Macrophage Activation and Metaflammation. Proc. Natl. Acad. Sci. USA 2021, 118, e2103304118. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S. NF-kappaB Signaling in Inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Schwabe, R.F. NF-kappaB in the Liver--Linking Injury, Fibrosis and Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Nejak-Bowen, K.; Moghe, A.; Cornuet, P.; Preziosi, M.; Nagarajan, S.; Monga, S.P. Role and Regulation of p65/Beta-Catenin Association during Liver Injury and Regeneration: A “Complex” Relationship. Gene Expr. 2017, 17, 219–235. [Google Scholar] [CrossRef]
- Geisler, F.; Algul, H.; Paxian, S.; Schmid, R.M. Genetic Inactivation of RelA/p65 Sensitizes Adult Mouse Hepatocytes to TNF-Induced Apoptosis in Vivo and in Vitro. Gastroenterology 2007, 132, 2489–2503. [Google Scholar] [CrossRef]
- Semple, F.; Dorin, J.R. Beta-Defensins: Multifunctional Modulators of Infection, Inflammation and More? J. Innate Immun. 2012, 4, 337–348. [Google Scholar] [CrossRef]
- Bautista, A.P. Chronic Alcohol Intoxication Induces Hepatic Injury through Enhanced Macrophage Inflammatory Protein-2 Production and Intercellular Adhesion Molecule-1 Expression in the Liver. Hepatology 1997, 25, 335–342. [Google Scholar]
- Deng, X.; Stachlewitz, R.F.; Liguori, M.J.; Blomme, E.A.G.; Waring, J.F.; Luyendyk, J.P.; Maddox, J.F.; Ganey, P.E.; Roth, R.A. Modest Inflammation Enhances Diclofenac Hepatotoxicity in Rats: Role of Neutrophils and Bacterial Translocation. J. Pharmacol. Exp. Ther. 2006, 319, 1191–1199. [Google Scholar] [CrossRef]
- Deng, X.; Luyendyk, J.P.; Ganey, P.E.; Roth, R.A. Inflammatory Stress and Idiosyncratic Hepatotoxicity: Hints from Animal Models. Pharmacol. Rev. 2009, 61, 262–282. [Google Scholar] [CrossRef]
- Jaeschke, H.; Ho, Y.S.; Fisher, M.A.; Lawson, J.A.; Farhood, A. Glutathione Peroxidase-Deficient Mice are More Susceptible to Neutrophil-Mediated Hepatic Parenchymal Cell Injury during Endotoxemia: Importance of an Intracellular Oxidant Stress. Hepatology 1999, 29, 443–450. [Google Scholar] [CrossRef]
- Ramaiah, S.K.; Jaeschke, H. Role of Neutrophils in the Pathogenesis of Acute Inflammatory Liver Injury. Toxicol. Pathol. 2007, 35, 757–766. [Google Scholar] [CrossRef]
- Romano, A.; Quaratino, D.; Papa, G.; Di Fonso, M.; Artesani, M.C.; Venuti, A. Delayed Hypersensitivity to Diclofenac: A Report on Two Cases. Ann. Allergy Asthma Immunol. 1998, 81, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Harrer, A.; Lang, R.; Grims, R.; Braitsch, M.; Hawranek, T.; Aberer, W.; Vogel, L.; Schmid, W.; Ferreira, F.; Himly, M. Diclofenac Hypersensitivity: Antibody Responses to the Parent Drug and Relevant Metabolites. PLoS ONE 2010, 5, e13707. [Google Scholar] [CrossRef]
- Salama, A.; Gottsche, B.; Mueller-Eckhardt, C. Autoantibodies and Drug- Or Metabolite-Dependent Antibodies in Patients with Diclofenac-Induced Immune Haemolysis. Br. J. Haematol. 1991, 77, 546–549. [Google Scholar] [CrossRef]
- Publicover, J.; Gaggar, A.; Nishimura, S.; Van Horn, C.M.; Goodsell, A.; Muench, M.O.; Reinhardt, R.L.; van Rooijen, N.; Wakil, A.E.; Peters, M.; et al. Age-Dependent Hepatic Lymphoid Organization Directs Successful Immunity to Hepatitis B. J. Clin. Investig. 2013, 123, 3728–3739. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Kang, D.; Sun, X.; Liu, Y.; Wang, J.; Gao, P. The Effect of C-X-C Motif Chemokine 13 on Hepatocellular Carcinoma Associates with Wnt Signaling. BioMed Res. Int. 2015, 2015, 345413. [Google Scholar] [CrossRef] [PubMed]
- Wehr, A.; Baeck, C.; Heymann, F.; Niemietz, P.M.; Hammerich, L.; Martin, C.; Zimmermann, H.W.; Pack, O.; Gassler, N.; Hittatiya, K.; et al. Chemokine Receptor CXCR6-Dependent Hepatic NK T Cell Accumulation Promotes Inflammation and Liver Fibrosis. J. Immunol. 2013, 190, 5226–5236. [Google Scholar] [CrossRef]
- Wang, H.; Shao, Y.; Zhang, S.; Xie, A.; Ye, Y.; Shi, L.; Jin, L.; Pan, X.; Lin, Z.; Li, X.; et al. CXCL16 Deficiency Attenuates Acetaminophen-Induced Hepatotoxicity through Decreasing Hepatic Oxidative Stress and Inflammation in Mice. Acta Biochim. Biophys. Sin. 2017, 49, 541–549. [Google Scholar] [CrossRef]
- Xu, H.; Gong, Y.; Cheng, J.; Chu, Y.; Xiong, S. CXCL16 Participates in Pathogenesis of Immunological Liver Injury by Regulating T Lymphocyte Infiltration in Liver Tissue. World J. Gastroenterol. 2005, 11, 4979–4985. [Google Scholar] [CrossRef]
- Marra, F.; Tacke, F. Roles for Chemokines in Liver Disease. Gastroenterology 2014, 147, 577–594.e1. [Google Scholar] [CrossRef]
- Si, Y.; Tsou, C.; Croft, K.; Charo, I.F. CCR2 Mediates Hematopoietic Stem and Progenitor Cell Trafficking to Sites of Inflammation in Mice. J. Clin. Investig. 2010, 120, 1192–1203. [Google Scholar] [CrossRef]
- Wasmuth, H.E.; Tacke, F.; Trautwein, C. Chemokines in Liver Inflammation and Fibrosis. Semin. Liver Dis. 2010, 30, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Luster, A.D. Chemokines and their Receptors: Drug Targets in Immunity and Inflammation. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Hellier, S.; Frodsham, A.J.; Hennig, B.J.W.; Klenerman, P.; Knapp, S.; Ramaley, P.; Satsangi, J.; Wright, M.; Zhang, L.; Thomas, H.C.; et al. Association of Genetic Variants of the Chemokine Receptor CCR5 and its Ligands, RANTES and MCP-2, with Outcome of HCV Infection. Hepatology 2003, 38, 1468–1476. [Google Scholar] [PubMed]
- Shouval, D.S.; Ouahed, J.; Biswas, A.; Goettel, J.A.; Horwitz, B.H.; Klein, C.; Muise, A.M.; Snapper, S.B. Interleukin 10 Receptor Signaling: Master Regulator of Intestinal Mucosal Homeostasis in Mice and Humans. Adv. Immunol. 2014, 122, 177–210. [Google Scholar]
- Thurman, J.M.; Holers, V.M. The Central Role of the Alternative Complement Pathway in Human Disease. J. Immunol. 2006, 176, 1305–1310. [Google Scholar] [CrossRef]
- Lin, J.C.; Hwang, S.W.; Luo, H.; Mohamud, Y. Double-Edged Sword: Exploring the Mitochondria-Complement Bidirectional Connection in Cellular Response and Disease. Biology 2024, 13, 431. [Google Scholar] [CrossRef]
- Londhe, K.B.; Borlak, J. A Cross-Platform Comparison of Genome-Wide Expression Changes of Laser Microdissected Lung Tissue of C-Raf Transgenic Mice using 3’IVT and Exon Array. PLoS ONE 2012, 7, e40778. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immune/Inflammatory Response/Leukocyte Migration | ||
|---|---|---|
| Probeset ID | Gene Symbol | Fold Change X ± SD |
| Ssc.27693.1.A1_a_at | ADCY8 | −1.51 ± 0.08 |
| Ssc.6943.1.A1_at | ANGPT1 | −1.51 ± 0.39 |
| Ssc.3703.1.S1_at | APOA2 | −1.57 ± 0.07 |
| Ssc.14503.1.S1_at | APOA4 | 1.56 ± 0.29 |
| Ssc.8659.2.S1_a_at | ATG12 | 1.53 ± 0.15 |
| Ssc.17615.1.S1_at | ATP1B1 | 1.5 ± 0.37 |
| Ssc.12348.2.S1_at | B2M | −1.53 ± 0.34 |
| Ssc.15518.1.A1_at | BCL6 | 2.38 ± 0.48 |
| Ssc.8594.1.A1_at | BLNK | −1.74 ± 0.48 |
| Ssc.9957.1.A1_at | CCL8 | −2.43 ± 1.23 |
| Ssc.5053.1.S1_at | CD163 | 2.04 ± 0.42 |
| Ssc.19817.1.S1_at | CD27 | −1.53 ± 0.15 |
| Ssc.26104.1.S1_at | CD46 | 1.63 ± 0.35 |
| Ssc.3593.1.S1_at | CTSH | −1.72 ± 0.53 |
| Ssc.22002.2.A1_at | CXCL13 | 1.54 ± 0.16 |
| Ssc.19692.1.S1_at | CXCL2 | 2.56 ± 1.05 |
| Ssc.15885.1.S1_at | DDX58 | −1.57 ± 0.07 |
| Ssc.21145.1.S1_at | DEFB1 (PBD2) | 8.32 ± 4.48 |
| Ssc.2714.3.S1_at | FYN | −1.56 ± 0.24 |
| Ssc.6646.1.S1_at | GRB7 | −1.54 ± 0.19 |
| Ssc.376.1.S1_at | HAMP | −1.8 ± 0.25 |
| Ssc.7558.1.A1_at | HERC6 | −1.69 ± 0.23 |
| Ssc.12191.1.A1_at | HSP90AA1 | 2.01 ± 0.3 |
| Ssc.10588.1.A1_at | IFI44L | −2.03 ± 0.41 |
| Ssc.5955.1.A1_at | IL10RB | 2.6 ± 0.28 |
| Ssc.528.1.S1_at | IL5 | −1.52 ± 0.15 |
| Ssc.286.1.S1_s_at | IRG6 (RSAD2) | −3.24 ± 1.23 |
| Ssc.11557.1.A1_at | ISG15 | −1.86 ± 0.11 |
| Ssc.23054.1.S1_at | JAK3 | 1.52 ± 0.19 |
| Ssc.18557.1.S1_at | KNG1 | −1.61 ± 0.15 |
| Ssc.15980.1.S1_at | LBP | 1.87 ± 0.76 |
| Ssc.14340.1.S1_at | LITAF | 1.81 ± 0.22 |
| Ssc.670.1.S1_at | LYZ | 5.89 ± 1.04 |
| Ssc.18928.1.A1_at | MADCAM1 | 1.71 ± 0.38 |
| Ssc.13711.1.S1_at | MAP2K6 | −1.61 ± 0.13 |
| Ssc.24291.1.A1_s_at | MAPK14 | 1.53 ± 0.08 |
| Ssc.3033.1.S1_a_at | MAPKAPK3 | 1.55 ± 0.11 |
| Ssc.18868.1.S1_at | MBL1 (MBL2) | −2.61 ± 1.59 |
| Ssc.6463.2.S1_at | MYD88 | 1.53 ± 0.19 |
| Ssc.1031.1.S1_at | OAS1 | −2.04 ± 0.66 |
| Ssc.10256.1.A1_at | PDE4B | 1.59 ± 0.07 |
| Ssc.16110.1.A1_at | PIAP (BIRC3) | 1.64 ± 0.23 |
| Ssc.11206.1.A1_at | PLCG1 | −1.54 ± 0.07 |
| Ssc.9170.1.A1_at | PRKD1 | −2.52 ± 0.42 |
| Ssc.23963.1.S1_at | RGC32 (RGCC) | −2.87 ± 1.39 |
| Ssc.2381.1.A1_at | S100A9 | 4.13 ± 2.55 * |
| Ssc.18849.1.A1_at | SH2D1A | −1.52 ± 0.09 |
| Ssc.222.1.S1_at | SLA-DRA (HLA-DRA) | −1.54 ± 0.26 |
| Ssc.6583.1.S1_at | SR-PSOX (CXCL16) | −1.51 ± 0.15 |
| Ssc.2594.1.S1_at | SUGT1 | 1.56 ± 0.11 |
| Ssc.16640.1.A1_at | VSIG4 | 1.58 ± 0.17 |
| Ssc.27161.1.A1_at | ZC3HAV1 | −1.57 ± 0.13 |
| Response to Glucocorticoid Stimulus | ||
|---|---|---|
| Probeset ID | Gene Symbol | Fold Change (Average) ± SD |
| Ssc.3703.1.S1_at | APOA2 | −1.57 ± 0.07 |
| Ssc.5737.1.S1_at | CDKN1A (p21) | 2.74 ± 1.11 |
| Ssc.14393.2.S1_x_at | HSD3B1 | −1.91 ± 0.14 |
| Ssc.16231.3.S1_a_at | IGF1 | 1.52 ± 0.19 |
| Ssc.47.1.S1_at | IGFBP2 | −1.85 ± 0.37 |
| Ssc.15986.2.A1_at | INSR | −1.55 ± 0.07 |
| Ssc.7297.1.S1_at | MAOB | −1.86 ± 0.22 |
| Ssc.6988.1.A1_at | PAM | −1.51 ± 0.29 |
| Ssc.9781.1.S1_at | SERPINE1 | 1.58 ± 0.04 |
| Ssc.2464.1.S1_at | STC1 | 1.58 ± 0.35 |
| Ssc.14066.2.S1_at | TAT | −2.15 ± 0.45 |
| Ssc.21161.1.S1_at | UGT1A6 | −2.17 ± 0.44 |
| Cytokine-Mediated Signaling Pathway | ||
| Probeset ID | Gene Symbol | Fold Change (Average) ± SD |
| Ssc.12348.2.S1_at | B2M | −1.53 ± 0.34 |
| Ssc.28997.1.S1_at | CSF2RB | 1.51 ± 0.11 |
| Ssc.29054.1.A1_at | GBP1 | −2.55 ± 0.54 |
| Ssc.9565.1.S1_at | IFNGR1 | 1.55 ± 0.18 |
| Ssc.528.1.S1_at | IL5 | −1.52 ± 0.15 |
| Ssc.12504.1.A1_at | ISG12 (IFI27) | −2.45 ± 0.82 |
| Ssc.11557.1.A1_at | ISG15 | −1.86 ± 0.11 |
| Ssc.23054.1.S1_at | JAK3 | 1.52 ± 0.19 |
| Ssc.5991.1.A1_at | KRT18 | 1.67 ± 0.25 |
| Ssc.263.1.S1_at | LEPR | 1.59 ± 0.27 |
| Ssc.15640.1.S1_at | MT2A | 1.65 ± 0.37 |
| Ssc.6463.2.S1_at | MYD88 | 1.53 ± 0.19 |
| Ssc.1031.1.S1_at | OAS1 | −2.04 ± 0.66 |
| Ssc.11206.1.A1_at | PLCG1 | −1.54 ± 0.07 |
| Ssc.222.1.S1_at | SLA-DRA (HLA-DRA) | −1.54 ± 0.26 |
| Ssc.7207.3.A1_at | SP100 | 1.74 ± 0.57 |
| Ssc.336.1.S1_at | USP18 | −2.25 ± 0.11 |
| Response to Interferon-Gamma | ||
| Probeset ID | Gene Symbol | Fold Change (Average) ± SD |
| Ssc.12348.2.S1_at | B2M | −1.53 ± 0.34 |
| Ssc.6583.1.S1_at | SR-PSOX (CXCL16) | −1.51 ± 0.15 |
| Ssc.7362.1.S1_at | EPRS | 1.61 ± 0.33 |
| Ssc.29054.1.A1_at | GBP1 | −2.55 ± 0.54 |
| Ssc.222.1.S1_at | SLA-DRA (HLA-DRA) | −1.54 ± 0.26 |
| Ssc.9565.1.S1_at | IFNGR1 | 1.55 ± 0.18 |
| Ssc.15640.1.S1_at | MT2A | 1.65 ± 0.37 |
| Ssc.1031.1.S1_at | OAS1 | −2.04 ± 0.66 |
| Ssc.23553.1.S1_at | SEC61A1 | 1.58 ± 0.24 |
| Ssc.7207.3.A1_at | SP100 | 1.74 ± 0.57 |
| Interferon-Gamma-Mediated Signaling Pathway | ||
| Probeset ID | Gene Symbol | Fold Change (Average) ± SD |
| Ssc.12348.2.S1_at | B2M | −1.53 ± 0.34 |
| Ssc.29054.1.A1_at | GBP1 | −2.55 ± 0.54 |
| Ssc.222.1.S1_at | SLA-DRA (HLA-DRA) | −1.54 ± 0.26 |
| Ssc.9565.1.S1_at | IFNGR1 | 1.55 ± 0.18 |
| Ssc.15640.1.S1_at | MT2A | 1.65 ± 0.37 |
| Ssc.1031.1.S1_at | OAS1 | −2.04 ± 0.66 |
| Ssc.7207.3.A1_at | SP100 | 1.74 ± 0.57 |
| Cell Death | ||
|---|---|---|
| Probeset ID | Gene Symbol | Fold Change X ± SD |
| Ssc.6943.1.A1_at | ANGPT1 | −1.51 ± 0.39 |
| Ssc.8980.1.A1_at | ANGPTL4 | −1.54 ± 0.29 |
| Ssc.14212.1.A1_at | ANKRD13C | −1.65 ± 0.17 |
| Ssc.15518.1.A1_at | BCL6 | 2.38 ± 0.48 |
| Ssc.16110.1.A1_at | BIRC3 | 1.64 ± 0.23 |
| Ssc.6833.1.S1_at | BTG1 | −1.52 ± 0.08 |
| Ssc.717.1.S1_at | CCK | −1.52 ± 0.03 |
| Ssc.19817.1.S1_at | CD27 | −1.53 ± 0.15 |
| Ssc.5737.1.S1_at | CDKN1A (p21) | 2.74 ± 1.11 |
| Ssc.6966.3.S1_a_at | CDKN1B (p27) | −2.39 ± 0.96 |
| Ssc.21845.2.S1_at | C-FLIP (CFLAR) | 1.59 ± 0.29 |
| Ssc.3593.1.S1_at | CTSH | −1.72 ± 0.53 |
| Ssc.22002.2.A1_at | CXCL13 | 1.54 ± 0.16 |
| Ssc.11184.1.S1_at | DAD1 | 1.56 ± 0.33 |
| Ssc.22064.1.S1_at | DFFA | −1.55 ± 0.17 |
| Ssc.10498.1.A1_at | EAF2 | 2.56 ± 0.21 |
| Ssc.4303.1.S1_at | EEF1E1 | 1.55 ± 0.21 |
| Ssc.2714.3.S1_at | FYN | −1.56 ± 0.24 |
| Ssc.31027.1.A1_at | G2E3 | 1.52 ± 0.16 |
| Ssc.17033.1.S1_at | HIGD2A | −1.53 ± 0.18 |
| Ssc.2667.1.S1_a_at | HRG | −2.26 ± 0.53 |
| Ssc.1241.1.S1_at | HSPE1 | 1.58 ± 0.15 |
| Ssc.1231.1.A1_at | HSPH1 | 1.77 ± 0.48 |
| Ssc.16231.3.S1_a_at | IGF1 | 1.52 ± 0.19 |
| Ssc.15588.1.S2_at | IGFBP3 | −1.65 ± 0.13 |
| Ssc.12504.1.A1_at | IFI27 | −2.45 ± 0.82 |
| Ssc.23054.1.S1_at | JAK3 | 1.52 ± 0.19 |
| Ssc.27622.1.S1_at | KLF11 | −1.88 ± 0.38 |
| Ssc.18557.1.S1_at | KNG1 | −1.61 ± 0.15 |
| Ssc.5991.1.A1_at | KRT18 | 1.67 ± 0.25 |
| Ssc.9655.1.A1_at | LOC100518125 (YBX3) | 1.59 ± 0.3 |
| Ssc.13711.1.S1_at | MAP2K6 | −1.61 ± 0.13 |
| Ssc.6463.2.S1_at | MYD88 | 1.53 ± 0.19 |
| Ssc.19546.1.S1_at | NME1 | 1.53 ± 0.2 |
| Ssc.16864.1.S1_at | PPARGC1A | −2.11 ± 0.46 |
| Ssc.25206.1.S1_at | PPIF | 1.74 ± 0.33 |
| Ssc.6371.1.A1_at | PRNP | 1.54 ± 0.15 |
| Ssc.12758.1.A1_at | PSMB9 | −1.54 ± 0.28 |
| Ssc.830.1.S1_at | PSME2 | −1.51 ± 0.26 |
| Ssc.23963.1.S1_at | RGCC | −2.87 ± 1.39 |
| Ssc.9781.1.S1_at | SERPINE1 | 1.58 ± 0.04 |
| Ssc.7604.1.A1_at | SKIL | 1.6 ± 0.41 |
| Ssc.3706.1.S2_at | SOD2 | 1.91 ± 0.53 |
| Ssc.5930.1.S1_at | SORT1 | −1.82 ± 0.76 |
| Ssc.7207.3.A1_at | SP100 | 1.74 ± 0.57 |
| Ssc.30934.1.S1_at | TERF1 | −1.74 ± 0.64 |
| Ssc.6634.1.A1_at | THY1 | −1.54 ± 0.38 |
| Ssc.14506.1.S1_at | TOP2A | −1.77 ± 0.33 |
| Ssc.2095.1.S1_at | VEGFB | −1.57 ± 0.09 |
| Ssc.2884.1.S1_at | VIL1 | −1.79 ± 0.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borlak, J.; Spanel, R. Diclofenac Immune-Mediated Hepatitis: Identification of Innate and Adaptive Immune Responses at Clinically Relevant Doses. Int. J. Mol. Sci. 2025, 26, 5899. https://doi.org/10.3390/ijms26125899
Borlak J, Spanel R. Diclofenac Immune-Mediated Hepatitis: Identification of Innate and Adaptive Immune Responses at Clinically Relevant Doses. International Journal of Molecular Sciences. 2025; 26(12):5899. https://doi.org/10.3390/ijms26125899
Chicago/Turabian StyleBorlak, Jürgen, and Reinhard Spanel. 2025. "Diclofenac Immune-Mediated Hepatitis: Identification of Innate and Adaptive Immune Responses at Clinically Relevant Doses" International Journal of Molecular Sciences 26, no. 12: 5899. https://doi.org/10.3390/ijms26125899
APA StyleBorlak, J., & Spanel, R. (2025). Diclofenac Immune-Mediated Hepatitis: Identification of Innate and Adaptive Immune Responses at Clinically Relevant Doses. International Journal of Molecular Sciences, 26(12), 5899. https://doi.org/10.3390/ijms26125899