
Exploring Carboxamide Derivatives as Promising Anticancer Agents: Design, In Vitro Evaluation, and Mechanistic Insights

 , , , , , , , ,
, , , , , , , ,  ,
,  , , and
, , and
Abstract
1. Introduction
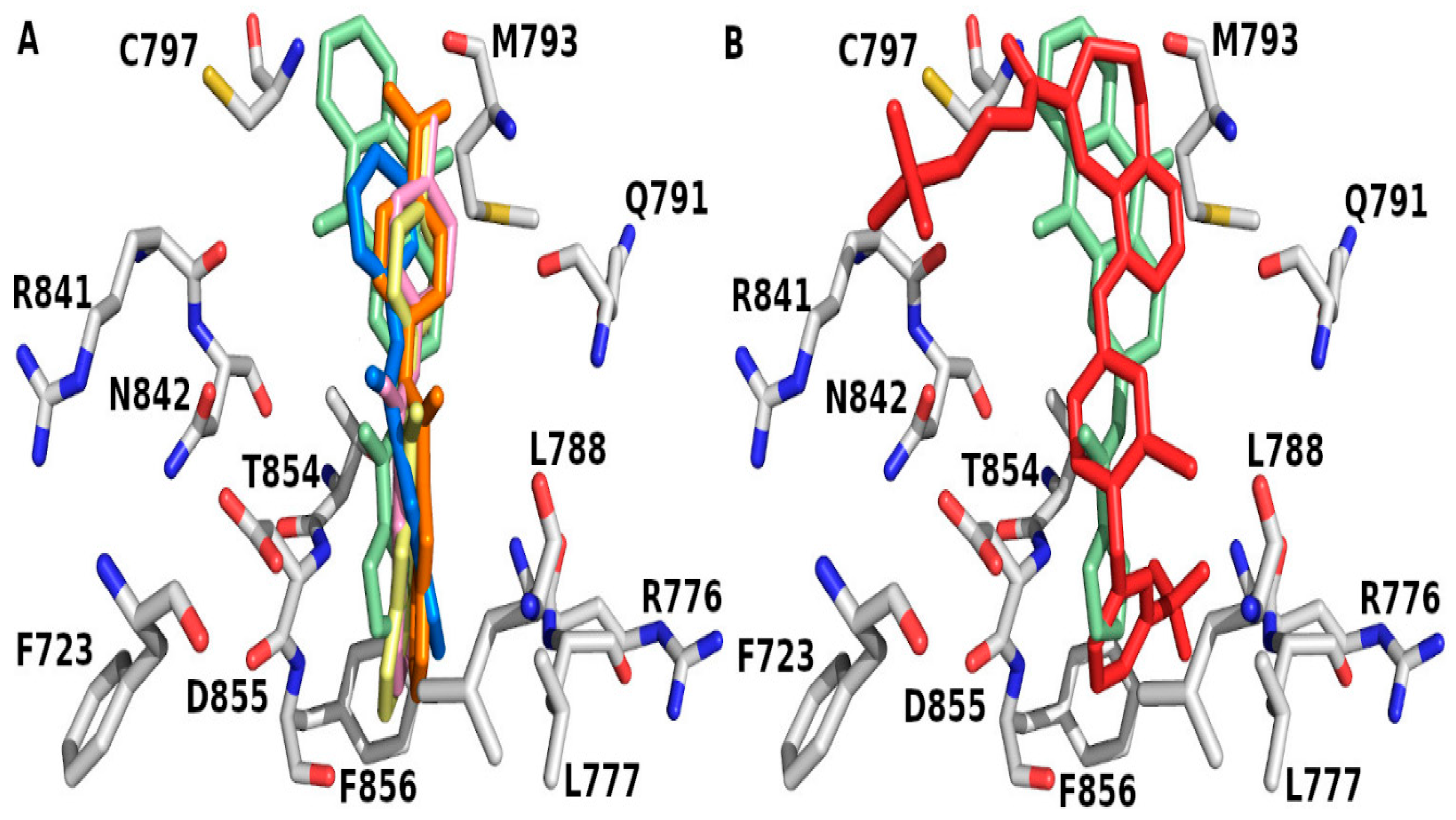
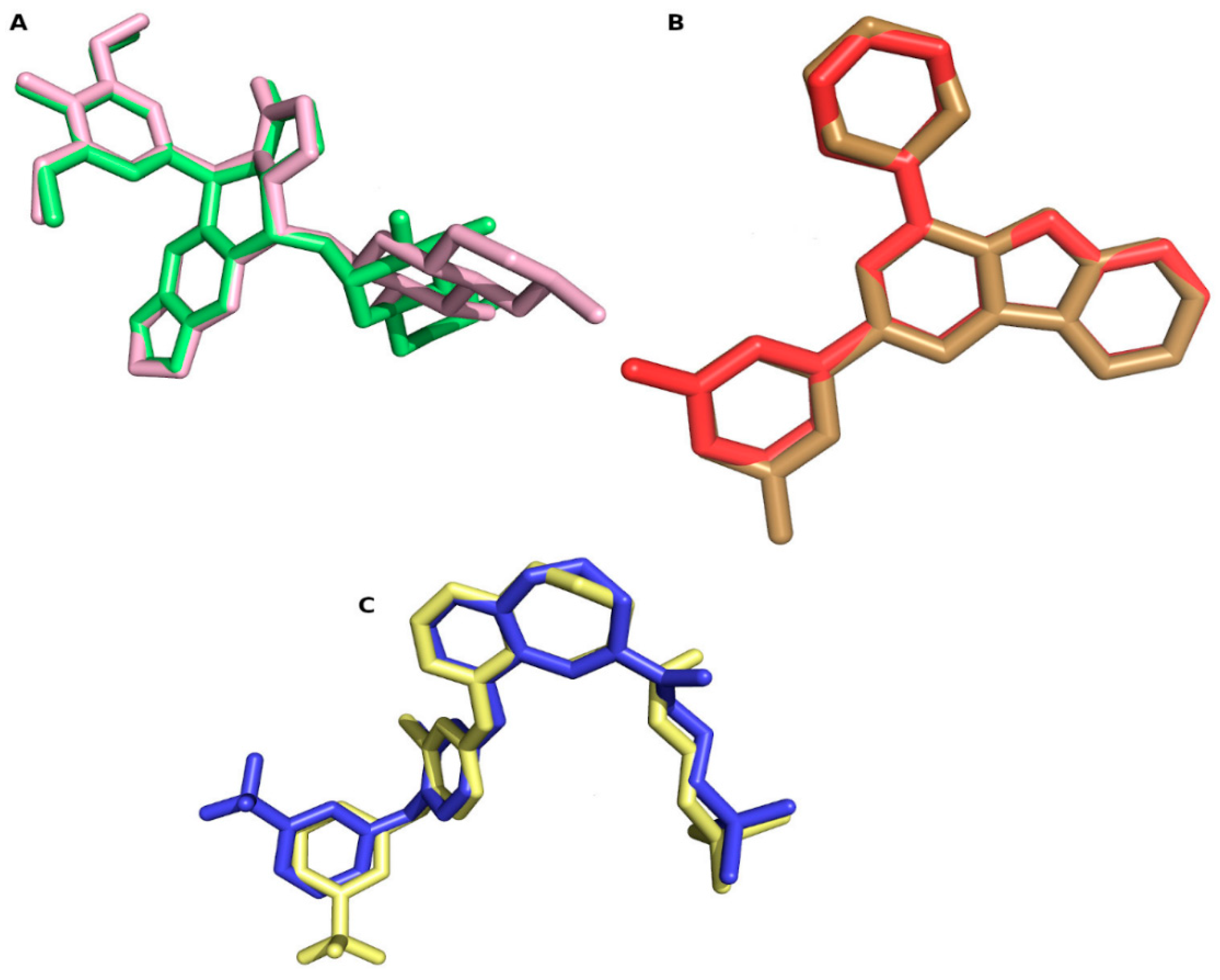
2. Results and Discussion
3. Materials and Methods
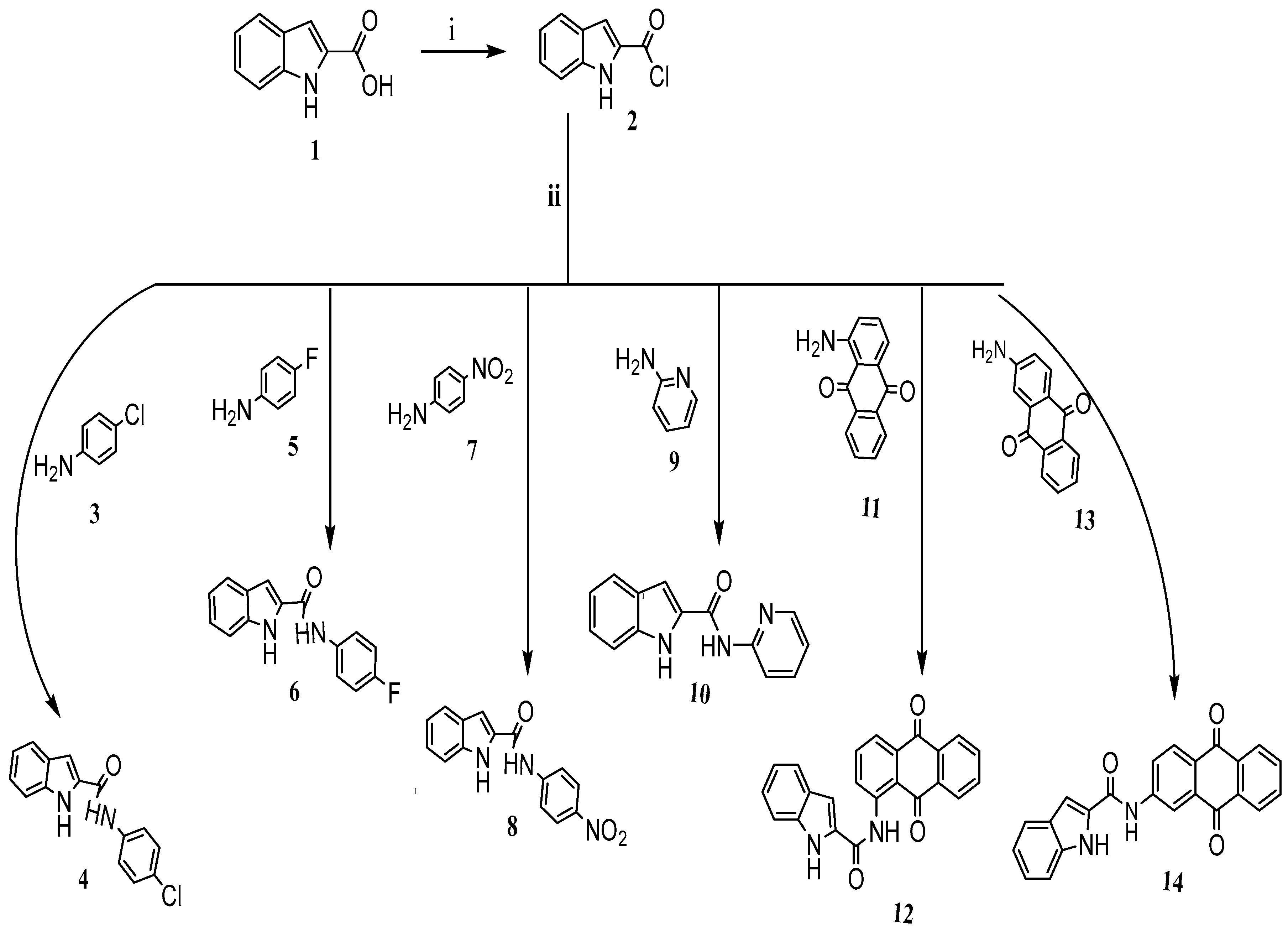
3.1. Synthesis of Target Compounds and Materials
3.2. Spectroscopic and Characterization Techniques
3.3. Synthesis of Target Compounds and Data Analysis
- Compound (2), Indole-2-carbonyl chloride
- Compound (4), N-(4-Chlorophenyl)-1H-indole-2-carboxamide
- ▪
- Rf value: 0.60 (cyclohexane/ethyl acetate, 6:4).
- ▪
- Melting point: 259 °C.
- ▪
- 1H -NMR (300 MHz, DMSO-d6) showed the following peaks, δ ppm: 11.79 (br s, 1H, NH-indole), 10.35 (s, 1H, NHCO), 7.86 (d, J = 9.0 Hz, 2H, Ar-H), 7.69 (d, J = 8.1 Hz, 1H, Ar-H), 7.49 (s, 1H, Ar-H), 7.47–7.39 (m, 3H, Ar-H), 7.23 (dd, J = 8.1, 7.2 Hz, 1H, Ar-H), 7.08 (dd, J = 7.2, 7.8 Hz, 1H, Ar-H).
- ▪
- 13C -NMR (DMSO-d6, δ ppm): 160.26 (CONH), 138.43, 137.36, 131.66, 129.09 (C-3′, 5′), 127.57, 127.44, 124.37 (CH-Ar), 122.26 (CH-Ar), 122.05 (C-2′, 6′), 120.44 (CH-Ar), 112.88 (CH-Ar), 104.60 (CH-Ar).
- ▪
- IR (thin film, cm−1): 3406 (NH-indole), 3318 (NHCO), 1651 (CO), 1593, 1539, 1493, 1400, 1315, 1238, 1192, 1099, 1015, 937, 826, 748.
- ▪
- Elemental analysis (C15H11N2OCl):
- ▪
- Theoretical values (calculated): C, 66.55%; H, 4.10%; N, 10.35%.
- ▪
- Experimental values (found): C, 66.40%; H, 3.92%; N, 10.12%.
- Compound (6), N-(4-Florophenyl)-1H-indole-2-carboxamide
- ▪
- Rf value: 0.54 (cyclohexane/ethyl acetate, 7:3).
- ▪
- Melting point: 245 °C (decomposed).
- ▪
- 1H -NMR (300 MHz, DMSO-d6, δ ppm): 11.78 (br s, 1H, NH-indole), 10.80 (s, 1H, NHCO), 7.89–7.79 (m, 2H, Ar-H), 7.67 (d, J = 7.8 Hz, 1H, Ar-H), 7.48 (d, J = 8.0 Hz, 1H, Ar-H), 7.44–7.39 (m, 1H, Ar-H), 7.28–7.15 (m, 3H, Ar-H), 7.11–7.01 (m, 1H, Ar-H).
- ▪
- 13C-NMR (DMSO-d6, δ ppm, rotamers observed): 160.28, 157.09 (d, 1J C-F = 283 Hz, C-4′, CH-Ar), 160.14 (CONH), 157.09, 137.30, 135.82, 131.82, 127.47, 124.25 (CH-Ar), 122.44, 122.19 (d, 2J C-F = 19 Hz, C-3′, 5′, CH-Ar), 122.34 (CH-Ar), 120.38 (CH-Ar), 115.89, 115.59 (d, 3J C-F = 22.13 Hz, C-2′, 6’, CH-Ar), 112.86 (CH-Ar), 104.40 (CH-Ar).
- ▪
- IR (thin film, cm−1): 3422 (NH-indole), 3337 (NHCO), 3271, 1639 (CO), 1543, 1508, 1408, 1339, 1315, 1223, 1188, 1096, 833, 818, 748.
- ▪
- HRMS (ESI, negative mode): m/z (M+-H+) 253.07826 (C15H10N2OF) requires 253.07772.
- ▪
- Elemental analysis (C15H11N2OF):
- ○
- Theoretical values: C, 70.86%; H, 4.36%; N, 11.02%.
- ○
- Experimental values: C, 70.97%; H, 4.15%; N, 10.90%.
- Compound (8), N-(4-Nitrophenyl)-1H-indole-2-carboxamide
- ▪
- Rf value: 0.47 (CHCl3/CH3OH/CH3CO2H, 94:5:1).
- ▪
- Melting point: > 290 °C (decomposed).
- ▪
- 1H-NMR (300 MHz, DMSO-d6, δ ppm): 11.84 (br s, 1H, NH-indole), 10.74 (br s, 1H, NHCO), 8.28 (d, J = 9.2 Hz, 2H, H-3′, 5′, Ar-H), 8.11 (d, J = 9.2 Hz, 2H, H-2′, 6′, Ar-H), 7.71 (d, J = 8.0 Hz, 1H, Ar-H), 7.58–7.45 (m, 2H, Ar-H), 7.76 (dd, J = 7.4, 7.5 Hz, 1H, Ar-H), 7.09 (dd, J = 7.6, 7.3 Hz, 1H, Ar-H).
- ▪
- 13C-NMR (DMSO-d6, δ ppm): 160.67 (CONH), 145.84, 142.79, 137.68, 131.15, 127.38, 125.31 (CH-Ar), 124.80 (CH-Ar), 122.45 (CH-Ar), 120.92 (CH-Ar), 120.02 (CH-Ar), 112.97 (CH-Ar), 105.69 (CH-Ar).
- ▪
- Elemental analysis (C15H11N3O3):
- ○
- Theoretical values: C, 64.05%; H, 3.94%; N, 14.94%.
- ○
- Experimental values: C, 63.97%; H, 3.89%; N, 14.78%.
- Compound (10), N-(Pyridin-2-yl)-1H-indole-2-carboxamide
- ▪
- Rf: 0.53 (CHCl3/CH3OH/CH3CO2H, 94:5:1).
- ▪
- Melting point: 260 °C (decomposed).
- ▪
- 1H-NMR (300 MHz, DMSO-d6): rotamers, δ = 13.75 (br s, 1H, NH-indole), 11.8 + 10.91 (2 br s, 1H, NHCO), 8.61–7.78 (m, 3H, Ar-H), 7.75–7.35 (m, 3H, Ar-H), 7.30–6.80 (m, 3H, Ar-H) ppm.
- ▪
- 13C-NMR (DMSO-d6): δ = 160.11 (CONH), 144.10, 137.60, 137.24, 133.32, 127.56, 124.76, 113.56, 112.45, 111.90, 108.37, 104.99.
- ▪
- IR (thin film): ν = 3449 (NH-indole), 3422 (NHCO), 3144, 2924, 1701 (CO), 1558, 1450, 1366, 1346, 1200, 1080, 748 cm−1.
- ▪
- HRMS (ESI, negative mode): m/z (M+-H+) 236.08294 (C14H11N3O) requires 236.08239.
- ▪
- Elemental analysis (C14H11N3O):
- ○
- Theoretical values (calculated): C, 70.87%; H, 4.67%; and N, 17.71%.
- ○
- Experimental values (found): C, 70.62%; H, 4.88%; and N, 17.55%.
- Compound (12), N-(9,10-Dihydro-9,10-dioxoanthracen-1-yl)-1H-indole-2-carboxamide
- ▪
- Rf: 0.59 (cyclohexane/ethyl acetate, 6:4).
- ▪
- Melting point: > 350 °C (decomposed).
- ▪
- 1H-NMR (300 MHz, DMSO-d6) rotamers: δ = 13.23 (br s, 1H, NH-indole), 12.08 (s, 1H, NHCO), 9.15 + 9.13 (2s, 1H, Ar-H), 8.50–7.76 (m, 5H, Ar-H), 7.62–6.89 (m, 5H, Ar-H) ppm.
- ▪
- 13C-NMR (DMSO-d6): δ = 182.60 (CO-ketone), 176.31 (CO-ketone), 163.21 (CONH), 134.37 (CH-Ar), 133.90 (CH-Ar), 132.90, 131.68, 129.37, 128.06 (CH-Ar), 127.69, 127.13, 126.48 (CH-Ar), 125.25, 124.47, 123.85 (CH-Ar), 122.37 (CH-Ar), 121.83, 120.90 (CH-Ar), 118.16 (CH-Ar), 116.20 (CH-Ar), 115.24 (CH-Ar), 112.94 (CH-Ar), 104.18 (CH-Ar) ppm.
- ▪
- IR (thin film): ν = 3314 (NH-indole), 3213 (NHCO), 3125, 1667 (br CO), 1636 (CONH), 1578, 1531, 1412, 1312, 1269, 1238, 1173, 1015, 806, 741, 705 cm⁻¹.
- ▪
- HRMS (ESI, negative mode): m/z (M+-H+) 365.09317 (C23H13N2O3) requires 365.09262.
- ▪
- Elemental analysis (C23H₁4N2O3):
- ○
- Theoretical values (calculated): C, 75.40%; H, 3.85%; and N, 7.65%.
- ○
- Experimental values (found): C, 75.19%; H, 4.01%; and N, 7.96%.
- Compound (14), N-(9,10-Dihydro-9,10-dioxoanthracen-2-yl)-1H-indole-2-carboxamide
- ▪
- Rf: 0.41 (cyclohexane/ethyl acetate, 6:4).
- ▪
- Melting point: > 350 °C (decomposed).
- ▪
- 1H-NMR (300 MHz, DMSO-d6): δ = 11.88 (br s, 1H, NH-indole), 10.79 (s, 1H, NHCO), 8.68 (d, J = 3.0 Hz, 1H, Ar-H), 8.39 (d, J = 9.0 Hz, 1H, Ar-H), 8.31–8.11 (m, 3H, Ar-H), 8.0–7.83 (m, 2H, Ar-H), 7.75 (d, J = 6.0 Hz, 1H, Ar-H), 7.56 (s, 1H, Ar-H), 7.46 (d, J = 6.0 Hz, 1H, Ar-H), 7.37–7.03 (m, 2H, Ar-H) ppm.
- ▪
- 13C-NMR (DMSO-d6): δ = 183.0 (CO-ketone), 181.86 (CO-ketone), 160.67 (CONH), 145.19, 137.65, 135.03 (CH-Ar), 134.68 (CH-Ar), 134.58, 133.67, 131.20, 128.89 (CH-Ar), 128.50, 127.43, 127.21 (CH-Ar), 127.13 (CH-Ar), 124.94 (CH-Ar), 124.77 (CH-Ar), 122.48 (CH-Ar), 120.58 (CH-Ar), 117.21 (CH-Ar), 112.96 (CH-Ar), 105.59 (CH-Ar) ppm.
- ▪
- IR (thin film): ν = 3402 (NH-indole), 3321 (NHCO), 3117, 3067, 1667 (br CO), 1651 (CONH), 1589, 1535, 1416, 1331, 1292, 1238, 954, 840, 755, 710 cm−1.
- ▪
- HRMS (ESI, negative mode): m/z (M+-H+) 365.09317 (C23H13N2O3) requires 365.09262.
- ▪
- Elemental analysis (C23H14N2O3):
- ○
- Theoretical values (calculated): C, 75.40%; H, 3.85%; and N, 7.65%.
- ○
- Experimental values (found): C, 75.72%; H, 3.74%; and N, 7.87%.
- Compound (16), N-(9,10-Dihydro-9,10-dioxoanthracen-1-yl)-2-furamide
- ▪
- Rf: 0.67 (CHCl3/CH3OH, 9:1).
- ▪
- Melting point: 280 °C (decomposed).
- ▪
- 1H-NMR (300 MHz, DMSO-d6) rotamers: δ = 13.18 (br s, 1H, NHCO), 9.15 (br s, 1H, Ar-H, H3-furan), 8.35–8.10 (m, 2H, Ar-H), 7.94–7.72 (m, 4H, Ar-H), 7.46–7.12 (m, 2H, Ar-H), 6.95–6.61 (m, 1H, Ar-H) ppm.
- ▪
- 13C-NMR (DMSO-d6) rotamers: δ = 192.82 (CO-ketone), 186.44 (CO-ketone), 161.22 (CONH), 148.09, 147.51, 147.22, 136.33, 135.25, 127.62, 126.96, 125.71, 122.59, 118.83, 118.18, 116.91, 113.52, 112.77, 112.56 ppm.
- ▪
- Elemental analysis (C19H11NO4):
- ○
- Theoretical values (calculated): C, 71.92%; H, 3.49%; and N, 4.41%.
- ○
- Experimental values (found): C, 71.55%; H, 3.63%; and N, 4.66%.
3.4. Cell Proliferation Study
3.4.1. Cells and Cell Culture Conditions
3.4.2. MTT Assay
3.4.3. Statistical Analysis
3.5. Molecular Modeling and Cheminformatics Methods
3.5.1. Protein Preparation
3.5.2. Ligand Structure Preparation
3.5.3. Induced-Fit Docking (IFD)
3.5.4. Molecular Descriptors
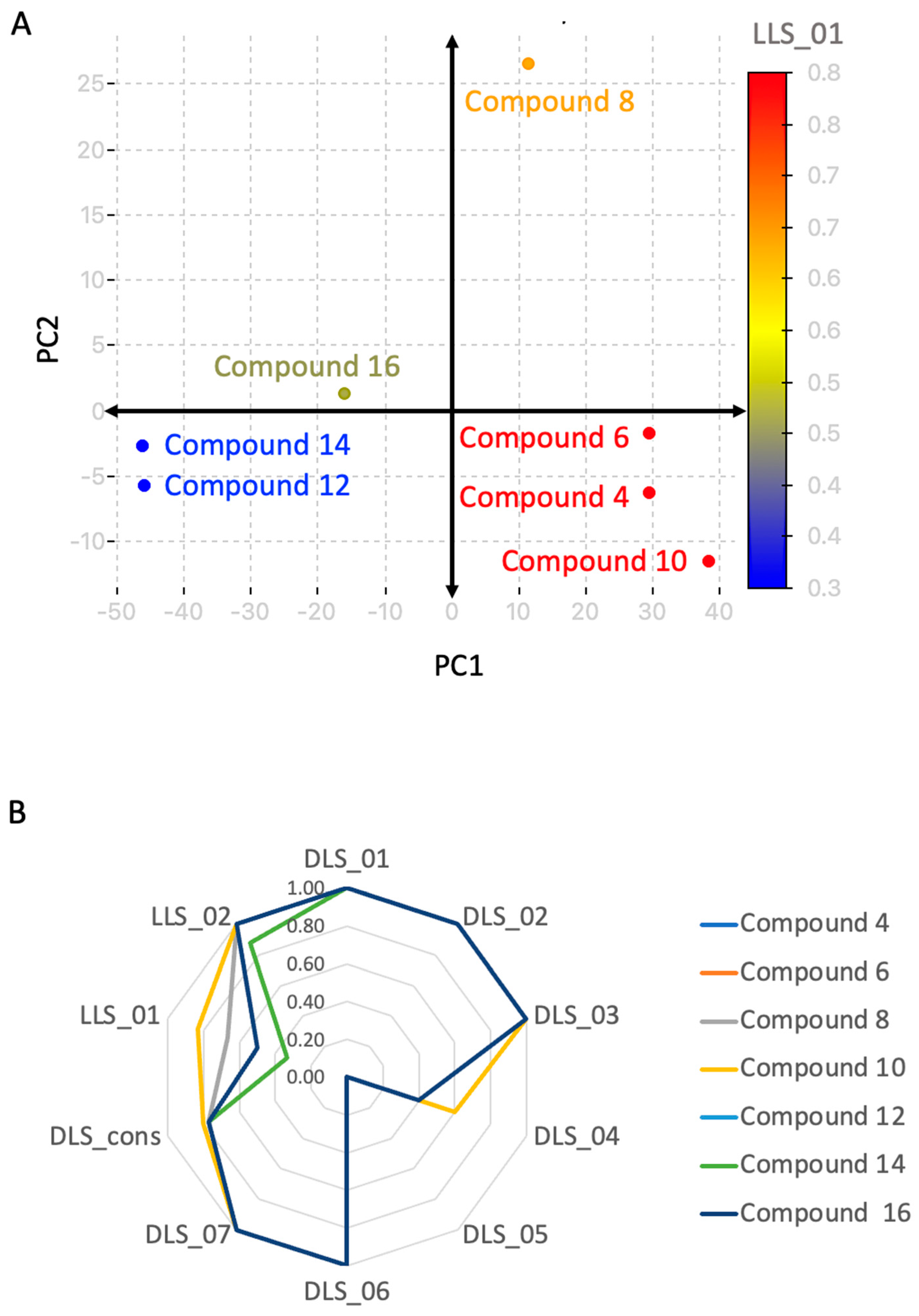
3.5.5. Principal Component Analysis (PCA)
4. Conclusions
Limitation and Future Investigations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| HCT-116 | Human colorectal carcinoma cell line |
| IC50 | 50% Cell proliferation inhibition |
| K-562 | Human chronic myeloid leukemia cell line |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| MCF-7 | Human breast adenocarcinoma |
| OD | Optical density |
| PCA | Principal component analysis |
| SI | Selectivity index |
| RES | Resolution |
| PI3K | Phosphoinositide 3-kinase |
| EGFR | Epidermal growth factor receptor |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2020, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Tarawneh, N.; Hamadneh, L.; Alshaer, W.; Al Bawab, A.Q.; Bustanji, Y.; Abdalla, S. Downregulation of aquaporins and PI3K/AKT and upregulation of PTEN expression induced by the flavone scutellarein in human colon cancer cell lines. Heliyon 2024, 10, e39402. [Google Scholar] [CrossRef] [PubMed]
- Cancer Facts & Figures. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2021/cancer-facts-and-figures-2021.pdf (accessed on 14 September 2023).
- Alsamarat, R.; Sunoqrot, S. A Glucose Oxidase-Curcumin Composite Nanoreactor for Multimodal Synergistic Cancer Therapy. ACS Appl. Bio Mater. 2024, 7, 4611–4621. [Google Scholar] [CrossRef]
- Mestareehi, A.; Zhang, X.; Seyoum, B.; Msallaty, Z.; Mallisho, A.; Burghardt, K.J.; Kowluru, A.; Yi, Z. Metformin Increases Protein Phosphatase 2A Activity in Primary Human Skeletal Muscle Cells Derived from Lean Healthy Participants. J. Diabetes Res. 2021, 2021, 9979234. [Google Scholar] [CrossRef]
- Khan, S.U.; Fatima, K.; Aisha, S.; Malik, F. Unveiling the mechanisms and challenges of cancer drug resistance. J. Cell Commun. Signal. 2024, 22, 109. [Google Scholar] [CrossRef]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef]
- Mestareehi, A.; Abu-Farsakh, N. Impact of Protein Phosphatase Expressions on the Prognosis of Hepatocellular Carcinoma Patients. ACS Omega 2024, 9, 10299–10331. [Google Scholar] [CrossRef]
- Shibuya, M. VEGF-VEGFR Signals in Health and Disease. Biomol. Ther. 2014, 22, 1–9. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Wang, Y.; Lin, C.; Zhang, D.; Chen, J.; Ouyang, L.; Wu, F.; Zhang, J.; Chen, L. Recent progress on vascular endothelial growth factor receptor inhibitors with dual targeting capabilities for tumor therapy. J. Hematol. Oncol. 2022, 15, 89. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.C.; Rini, B.I. Treatment of renal cell carcinoma: Current status and future directions. CA Cancer J. Clin. 2017, 67, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Mestareehi, A. Global Gene Expression Profiling and Bioinformatics Analysis Reveal Downregulated Biomarkers as Potential Indicators for Hepatocellular Carcinoma. ACS Omega 2024, 9, 26075–26096. [Google Scholar] [CrossRef]
- Patel, S.A.; Nilsson, M.B.; Le, X.; Cascone, T.; Jain, R.K.; Heymach, J.V. Molecular Mechanisms and Future Implications of VEGF/VEGFR in Cancer Therapy. Clin. Cancer Res. 2023, 29, 30–39. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Hande, K.R. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef]
- Mestareehi, A.H. Optimized and Validated Stability-Indicating RP-HPLC Method for Comprehensive Profiling of Process-Related Impurities and Stress-Induced Degradation Products in Rivaroxaban (XARELTO)(®). Int. J. Mol. Sci. 2025, 26, 4744. [Google Scholar] [CrossRef]
- Salerno, S.; Da Settimo, F.; Taliani, S.; Simorini, F.; La Motta, C.; Fornaciari, G.; Marini, A.M. Recent advances in the development of dual topoisomerase I and II inhibitors as anticancer drugs. Curr. Med. Chem. 2010, 17, 4270–4290. [Google Scholar] [CrossRef]
- Chen, A.Y.; Liu, L.F. DNA topoisomerases: Essential enzymes and lethal targets. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 191–218. [Google Scholar] [CrossRef]
- Mestareehi, A. Development and validation of a stability-indicating reversed phase high-performance liquid chromatography method for quantifying rivaroxaban (XARELTO). J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2025, 1257, 124573. [Google Scholar] [CrossRef] [PubMed]
- Gaba, M.; Mohan, C. Development of drugs based on imidazole and benzimidazole bioactive heterocycles: Recent advances and future directions. Med. Chem. Res. 2016, 25, 173–210. [Google Scholar] [CrossRef]
- Calcaterra, N.E.; Barrow, J.C. Classics in chemical neuroscience: Diazepam (valium). ACS Chem. Neurosci. 2014, 5, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Dhuguru, J.; Skouta, R. Role of Indole Scaffolds as Pharmacophores in the Development of Anti-Lung Cancer Agents. Molecules 2020, 25, 1615. [Google Scholar] [CrossRef]
- Asif, M.; Srivastava, R.; Fatima, A.; Shakeel, M.; Hassan, F.; Nasibullah, M. A Perspective of the Amide Group Containing FDA Approved Anticancer Drugs from 2021–2022 (A Review). Russ. J. Bioorganic Chem. 2023, 49, 1165–1176. [Google Scholar] [CrossRef]
- Comşa, Ş.; Cîmpean, A.M.; Raica, M. The Story of MCF-7 Breast Cancer Cell Line: 40 years of Experience in Research. Anticancer. Res. 2015, 35, 3147–3154. Available online: https://ar.iiarjournals.org/content/35/6/3147 (accessed on 15 June 2024).
- Hassan, A.S.; Moustafa, G.O.; Awad, H.M.; Nossier, E.S.; Mady, M.F. Design, Synthesis, Anticancer Evaluation, Enzymatic Assays, and a Molecular Modeling Study of Novel Pyrazole-Indole Hybrids. ACS Omega 2021, 6, 12361–12374. [Google Scholar] [CrossRef]
- Kumar, A.; Anand, T.; Bhattacharyya, J.; Sharma, A.; Jaganathan, B.G. K562 chronic myeloid leukemia cells modify osteogenic differentiation and gene expression of bone marrow stromal cells. J. Cell Commun. Signal. 2018, 12, 441–450. [Google Scholar] [CrossRef]
- Rubin, E.; Shan, K.S.; Dalal, S.; Vu, D.U.D.; Milillo-Naraine, A.M.; Guaqueta, D.; Ergle, A. Molecular Targeting of the Human Epidermal Growth Factor Receptor-2 (HER2) Genes across Various Cancers. Int. J. Mol. Sci. 2024, 25, 1064. [Google Scholar] [CrossRef]
- Miziak, P.; Baran, M.; Błaszczak, E.; Przybyszewska-Podstawka, A.; Kałafut, J.; Smok-Kalwat, J.; Dmoszyńska-Graniczka, M.; Kiełbus, M.; Stepulak, A. Estrogen Receptor Signaling in Breast Cancer. Cancers 2023, 15, 4689. [Google Scholar] [CrossRef]
- Li, L.; Liu, S.; Wang, B.; Liu, F.; Xu, S.; Li, P.; Chen, Y. An Updated Review on Developing Small Molecule Kinase Inhibitors Using Computer-Aided Drug Design Approaches. Int. J. Mol. Sci. 2023, 24, 13953. [Google Scholar] [CrossRef] [PubMed]
- Mestareehi, A.; Li, H.; Zhang, X.; Venkata, S.P.M.; Jaiswal, R.; Yu, F.S.; Yi, Z.; Wang, J.M. Quantitative Proteomics Reveals Transforming Growth Factor β Receptor Targeted by Resveratrol and Hesperetin Coformulation in Endothelial Cells. ACS Omega 2023, 8, 16206–16217. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-F.; Huang, N.-L.; Lin, J.-H.; Wu, C.-C.; Wang, Y.-R.; Yu, Y.-J.; Gilson, M.K.; Chan, N.-L. Structural insights into the gating of DNA passage by the topoisomerase II DNA-gate. Nat. Commun. 2018, 9, 3085. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, X.; Chen, Y.; Lu, S.; Peng, Y.; Wang, X.; Guo, C.; Zhou, A.; Zhang, J.; Luo, Y.; et al. Crystal structures of PI3Kalpha complexed with PI103 and Its derivatives: New directions for inhibitors design. ACS Med. Chem. Lett. 2013, 5, 138–142. [Google Scholar] [CrossRef]
- Kawakita, Y.; Seto, M.; Ohashi, T.; Tamura, T.; Yusa, T.; Miki, H.; Iwata, H.; Kamiguchi, H.; Tanaka, T.; Sogabe, S.; et al. Design and synthesis of novel pyrimido [4,5-b]azepine derivatives as HER2/EGFR dual inhibitors. Bioorganic Med. Chem. 2013, 21, 2250–2261. [Google Scholar] [CrossRef]
- Schrödinger. Protein Preparation Wizard, Maestro, Macromodel, QPLD-Dock, and Pymol; Schrödinger, LLC: Portland, OR, USA, 2022. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Jadhav, A.K.; Karuppayil, S.M. Molecular docking studies on thirteen fluoroquinolines with human topoisomerase II a and b. Silico Pharmacol. 2016, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.K.; Singh, B.; Mujwar, S.; Bisen, P.S. Molecular Docking Based Analysis to Elucidate the DNA Topoisomerase IIβ as the Potential Target for the Ganoderic Acid; A Natural Therapeutic Agent in Cancer Therapy. Curr. Comput.-Aided Drug Des. 2020, 16, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Hu, J.; Luyten, W.; Sun, D.; Jiang, T. Identification of novel topoisomerase II alpha inhibitors by virtual screening, molecular docking, and bioassay. Chem. Biol. Drug Des. 2022, 99, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Arthur, D.E. Molecular docking studies of some topoisomerase II inhibitors: Implications in designing of novel anticancer drugs. Radiol. Infect. Dis. 2019, 6, 68–79. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Samarat, H.H.; Al-Shalabi, E.; Bardaweel, S.K.; Hajjo, R.; Sweidan, K.; Khalaf, R.A.; Al-Zuheiri, A.M.; Abushaikha, G. Design, Synthesis, and Biological Examination of N-Phenyl-6-fluoro-4-hydroxy-2-quinolone-3-carboxamides as Anticancer Agents. ChemistrySelect 2022, 7, e202200662. [Google Scholar] [CrossRef]
- Sweidan, K.; Elfadel, H.; Sabbah, D.A.; Bardaweel, S.K.; Hajjo, R.; Anjum, S.; Sinoj, J.; Nair, V.A.; Abu-Gharbieh, E.; El-Huneidi, W. Novel Derivatives of 4,6-Dihydroxy-2-Quinolone-3-Carboxamides as Potential PI3Kα Inhibitors. ChemistrySelect 2022, 7, e202202263. [Google Scholar] [CrossRef]
- Sweidan, K.; Sabbah, D.A.; Engelmann, J.; Halim, H.A.; Sheikha, G.A. Computational docking studies of novel heterocyclic carboxamides as potential PI3Kα inhibitors. Lett. Drug Des. Discov. 2015, 12, 856–863. [Google Scholar] [CrossRef]
- Sweidan, K.; Zalloum, H.; Sabbah, D.A.; Idris, G.; Abudosh, K.; Mubarak, M.S. Synthesis, characterization, and anticancer evaluation of some new N1-(anthraquinon-2-yl) amidrazone derivatives. Can. J. Chem. 2018, 96, 1123–1128. [Google Scholar] [CrossRef]
- Sweidan, K.; Sabbah, D.A.; Bardaweel, S.; Dush, K.A.; Sheikha, G.A.; Mubarak, M.S. Computer-aided design, synthesis, and biological evaluation of new indole-2-carboxamide derivatives as PI3Kα/EGFR inhibitors. Bioorganic Med. Chem. Lett. 2016, 26, 2685–2690. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Ibrahim, A.H.; Talib, W.H.; Alqaisi, K.M.; Sweidan, K.; Bardaweel, S.K.; Sheikha, G.A.; Zhong, H.A.; Al-Shalabi, E.; Khalaf, R.A. Ligand-based drug design: Synthesis and biological evaluation of substituted benzoin derivatives as potential antitumor agents. Med. Chem. 2019, 15, 417–429. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Hasan, S.E.; Abu Khalaf, R.; Bardaweel, S.K.; Hajjo, R.; Alqaisi, K.M.; Sweidan, K.A.; Al-Zuheiri, A.M. Molecular modeling, synthesis and biological evaluation of N-phenyl-4-hydroxy-6-methyl-2-quinolone-3-carboxamides as anticancer agents. Molecules 2020, 25, 5348. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Haroon, R.A.; Bardaweel, S.K.; Hajjo, R.; Sweidan, K. N-phenyl-6-chloro-4-hydroxy-2-quinolone-3-carboxamides: Molecular Docking, Synthesis, and Biological Investigation as Anticancer Agents. Molecules 2021, 26, 73. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Al-Tarawneh, F.; Talib, W.H.; Sweidan, K.; Bardaweel, S.K.; Al-Shalabi, E.; Zhong, H.A.; Sheikha, G.A.; Khalaf, R.A.; Mubarak, M.S. Benzoin schiff bases: Design, synthesis, and biological evaluation as potential antitumor agents. Med. Chem. 2018, 14, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Vennerstrom, J.L.; Zhong, H.A. Binding selectivity studies of phosphoinositide 3-kinases using free energy calculations. J. Chem. Inf. Model. 2012, 52, 3213–3224. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Vennerstrom, J.L.; Zhong, H. Docking studies on isoform-specific inhibition of phosphoinositide-3-kinases. J. Chem. Inf. Model. 2010, 50, 1887–1898. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Simms, N.A.; Wang, W.; Dong, Y.; Ezell, E.L.; Brattain, M.G.; Vennerstrom, J.L.; Zhong, H.A. N-Phenyl-4-hydroxy-2-quinolone-3-carboxamides as selective inhibitors of mutant H1047R phosphoinositide-3-kinase (PI3Kα). Bioorganic Med. Chem. 2012, 20, 7175–7183. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Simms, N.A.; Brattain, M.G.; Vennerstrom, J.L.; Zhong, H. Biological evaluation and docking studies of recently identified inhibitors of phosphoinositide-3-kinases. Bioorganic Med. Chem. Lett. 2012, 22, 876–880. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Saada, M.; Khalaf, R.A.; Bardaweel, S.; Sweidan, K.; Al-Qirim, T.; Al-Zughier, A.; Halim, H.A.; Sheikha, G.A. Molecular modeling based approach, synthesis, and cytotoxic activity of novel benzoin derivatives targeting phosphoinostide 3-kinase (PI3Kα). Bioorganic Med. Chem. Lett. 2015, 25, 3120–3124. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Hishmah, B.; Sweidan, K.; Bardaweel, S.; AlDamen, M.; Zhong, H.A.; Khalaf, R.A.; Ibrahim, A.H.; Al-Qirim, T.; Sheikha, G.A. Structure-based design: Synthesis, X-ray crystallography, and biological evaluation of N-substituted-4-hydroxy-2-quinolone-3-carboxamides as potential cytotoxic agents. Anticancer Agents Med. Chem. 2018, 18, 263–276. [Google Scholar] [CrossRef]
- Huang, C.-H.; Mandelker, D.; Schmidt-Kittler, O.; Samuels, Y.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Gabelli, S.B.; Amzel, L.M. The structure of a human p110 alpha/p85 alpha complex elucidates the effects of oncogenic PI3K alpha mutations. Science 2007, 318, 1744–1748. [Google Scholar] [CrossRef]
- Liao, Q.-H.; Gao, Q.-Z.; Wei, J.; Chou, K.-C. Docking and molecular dynamics study on the inhibitory activity of novel inhibitors on epidermal growth factor receptor (EGFR). Med. Chem. 2011, 7, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Celik, İ.; Ayhan-Kılcıgil, G.; Guven, B.; Kara, Z.; Gurkan-Alp, A.S.; Karayel, A.; Onay-Besikci, A. Design, synthesis and docking studies of benzimidazole derivatives as potential EGFR inhibitors. Eur. J. Med. Chem. 2019, 173, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, P.; Sharma, V.; Alam, O.; Manaithiya, A.; Alam, P.; Alam, M.T.; Imran, M. Novel quinazoline-based EGFR kinase inhibitors: A review focussing on SAR and molecular docking studies (2015–2019). Eur. J. Med. Chem. 2020, 204, 112640. [Google Scholar] [CrossRef] [PubMed]
- alvaDesc 2.0. Available online: http://dev.kode-datacenter.net:10050/pf/alvadesc-2-0/ (accessed on 10 October 2023).
- Hopkins, C.R.; O’neil, S.V.; Laufersweiler, M.C.; Wang, Y.; Pokross, M.; Mekel, M.; Evdokimov, A.; Walter, R.; Kontoyianni, M.; Petrey, M.E.; et al. Design and synthesis of novel N-sulfonyl-2-indole carboxamides as potent PPAR-gamma binding agents with potential application to the treatment of osteoporosis. Bioorganic Med. Chem. Lett. 2006, 16, 5659–5663. [Google Scholar] [CrossRef]
- Al-Najdawi, M.; Al-Hiari, Y.; Al-Qirim, T.; Shattat, G.; Al-Zweri, M.; Sheikha, G.A. Synthesis and pharmacological evaluation of novel unsubstituted indole-anthraquinone carboxamide derivatives as potent antihyperlipidemic agents. Z. Naturforsch. C 2014, 69, 21–28. [Google Scholar] [CrossRef]
- Saleh, M.M.; Abuarqoub, D.A.; Hammad, A.M.; Hossan, M.S.; Ahmed, N.; Aslam, N.; Naser, A.Y.; Moody, C.J.; Laughton, C.A.; Bradshaw, T.D. In Vitro Anticancer Properties of Novel Bis-Triazoles. Curr. Issues Mol. Biol. 2023, 45, 175–196. [Google Scholar] [CrossRef]
- Saleh, M.M.; Abuirmeileh, A.N.; Al-Rousan, R.M.; Abudoleh, S.M.; Hassouneh, L.K.; Zihlif, M.A.; Taha, M.O.; Abutayeh, R.F.; Mansour, H.; Abu-Irmaileh, B. Biological Evaluation and Reverse Pharmacophore Mapping of Innovative Bis-Triazoles as Promising Anticancer Agents. Open J. Med. Chem. 2022, 16, e187410452207200. [Google Scholar] [CrossRef]
- Saleh, M.M.; Laughton, C.A.; Bradshaw, T.D.; Moody, C.J. Development of a series of bis-triazoles as G-quadruplex ligands. RSC Adv. 2017, 7, 47297–47308. [Google Scholar] [CrossRef]
- Al-Sammarra’e, A.; Al-Najdawi, M.M.; Saleh, M.M.; Al-Hiari, Y.M.; Al-Bashiti, R. Synthesis and Biological Evaluation of Furyl-Carboxamide Derivatives as Potential Anticancer Agents. J. Turk. Chem. Soc. A Chem. 2022, 9, 909–918. [Google Scholar] [CrossRef]
- Al-Ali, L.; Al-Ani, R.J.; Saleh, M.M.; Hammad, A.M.; Abuarqoub, D.A.; Abu-Irmaileh, B.; Naser, A.Y.; Najdawi, M.M.; Abbas, M.M.; Alkrad, J.A. Biological evaluation of combinations of tyrosine kinase inhibitors with Inecalcitol as novel treatments for human chronic myeloid leukemia. Saudi Pharm. J. 2024, 32, 101931. [Google Scholar] [CrossRef]
- Zalloum, H.; Zalloum, W.; Hameduh, T.; ALSalamat, H.; Zihlif, M. Anti-Proliferative Effect of Potential LSD1/CoREST Inhibitors Based on Molecular Dynamics Model for Treatment of SH-SY5Y Neuroblastoma Cancer Cell Line. Asian Pac. J. Cancer Prev. 2022, 23, 3533–3540. [Google Scholar] [CrossRef] [PubMed]
- Cousins, K.R. Computer review of ChemDraw Ultra 12.0. J. Am. Chem. Soc. 2011, 133, 8388. [Google Scholar] [CrossRef] [PubMed]
- Hajjo, R.; Grulke, C.M.; Golbraikh, A.; Setola, V.; Huang, X.P.; Roth, B.L.; Tropsha, A. Development, validation, and use of quantitative structure-activity relationship models of 5-hydroxytryptamine (2B) receptor ligands to identify novel receptor binders and putative valvulopathic compounds among common drugs. J. Med. Chem. 2010, 53, 7573–7586. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
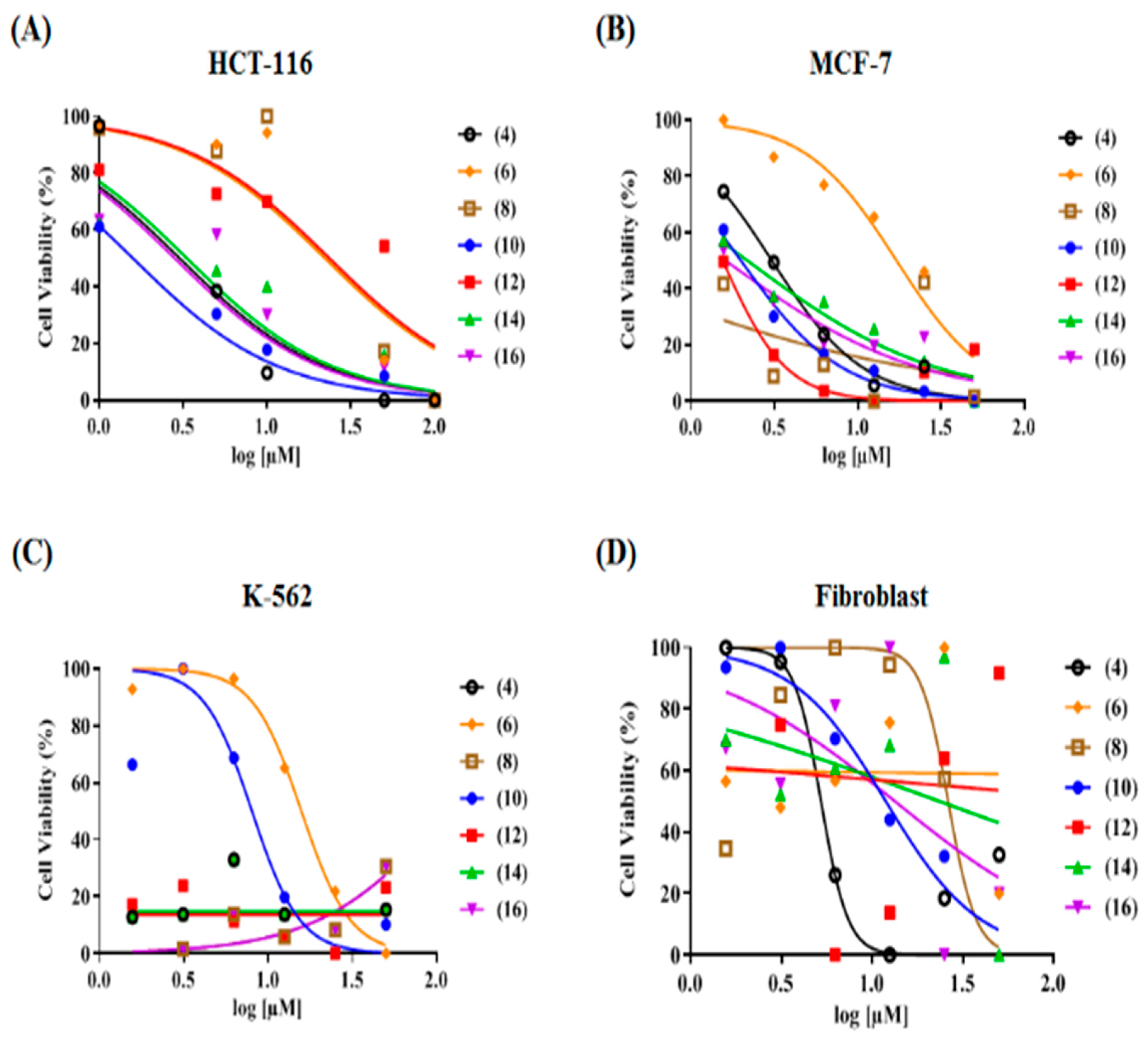
| Compound No. | IC50 (µM) ± S.D. | Selectivity Indices (SIs) | |||
|---|---|---|---|---|---|
| HCT-116 Colon Cancer | MCF-7 Breast Cancer | K-562 Leukemia | Dermal Fibroblast Normal | ||
| 4 | 3.94 ± 0.03 | 10.6 ± 0.53 | 0.61 ± 0.03 | >100 | 25.4, 9.4, 164 |
| 6 | 25.4 ± 0.38 | 80.8 ± 4.04 | 84.6 ± 4.23 | >100 | 3.9, 1.2, 1.2 |
| 8 | 32.0 ± 0.83 | >100 | >100 | >100 | 3.1, 1.0, 1.0 |
| 10 | 1.01 ± 0.04 | 70.3 ± 3.52 | >100 | >100 | 99.4, 1.4, 1.0 |
| 12 | 3.98 ± 0.06 | 9.07 ± 0.45 | 0.33 ± 0.02 | >100 | 25.1, 11.0, 303 |
| 14 | 2.64 ± 0.07 | 7.16 ± 0.36 | 0.61 ± 0.03 | >100 | 37.9, 14.0, 164 |
| 16 | 3.11 ± 0.049 | 33.1 ± 1.66 | >100 | >100 | 32.2, 3.0, 1.0 |
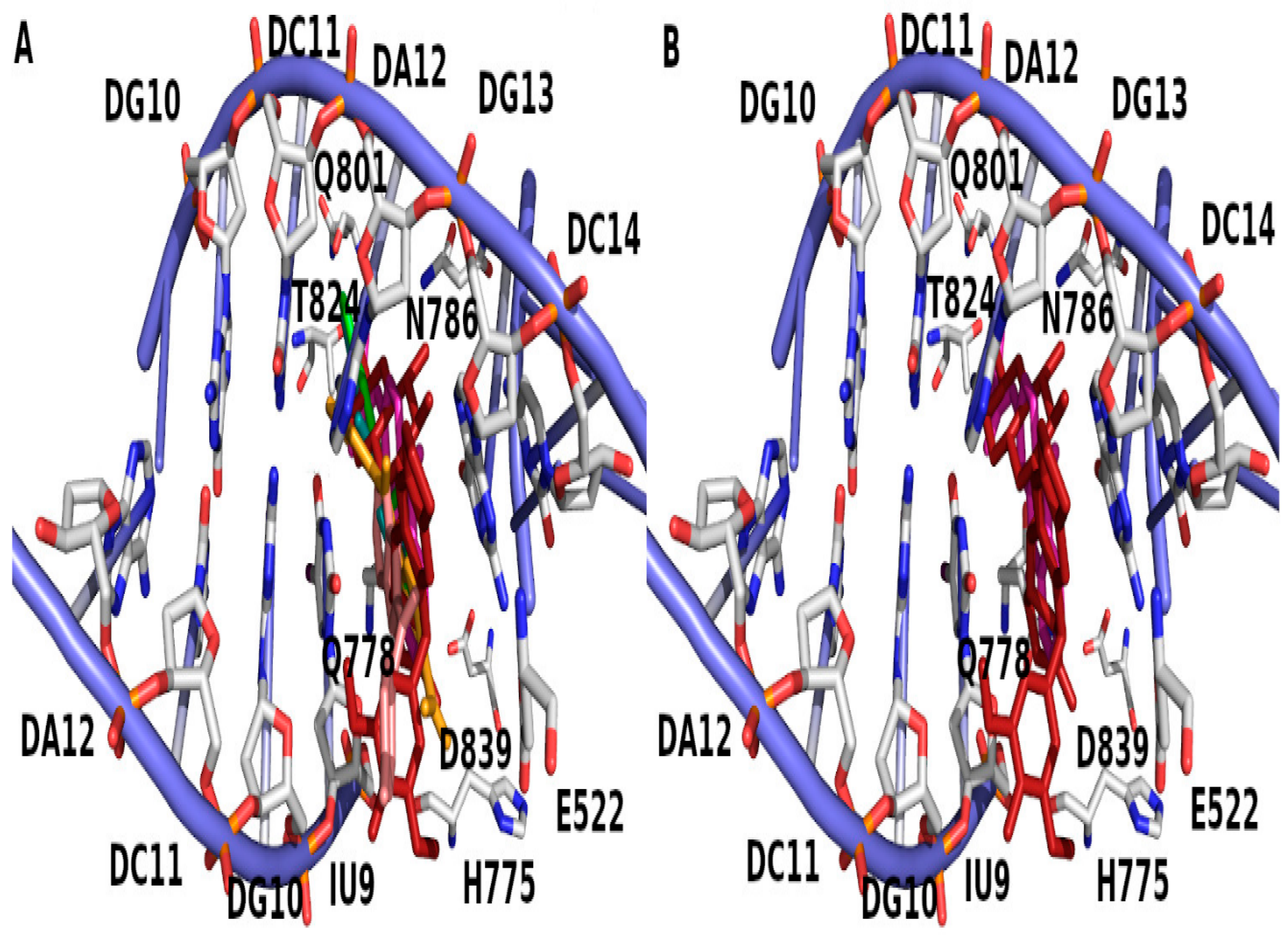
| Compound No. (Moiety) | Docking Score | Binding Residue | Binding Nucleotide |
|---|---|---|---|
| 4 | −7.35 | NA | DG13 |
| 6 | −7.83 | Q778 | NA |
| 8 | −8.88 | NA | DC8, DG13 |
| 10 | −6.79 | Q778 | NA |
| 12 | −10.16 | R503 | NA |
| 14 | −9.35 | NA | DC8, DA12 |
| 16 | −8.95 | H2O-Q778 | DG13 |
| EVP | −13.76 | D479 | DG7, DG13 |
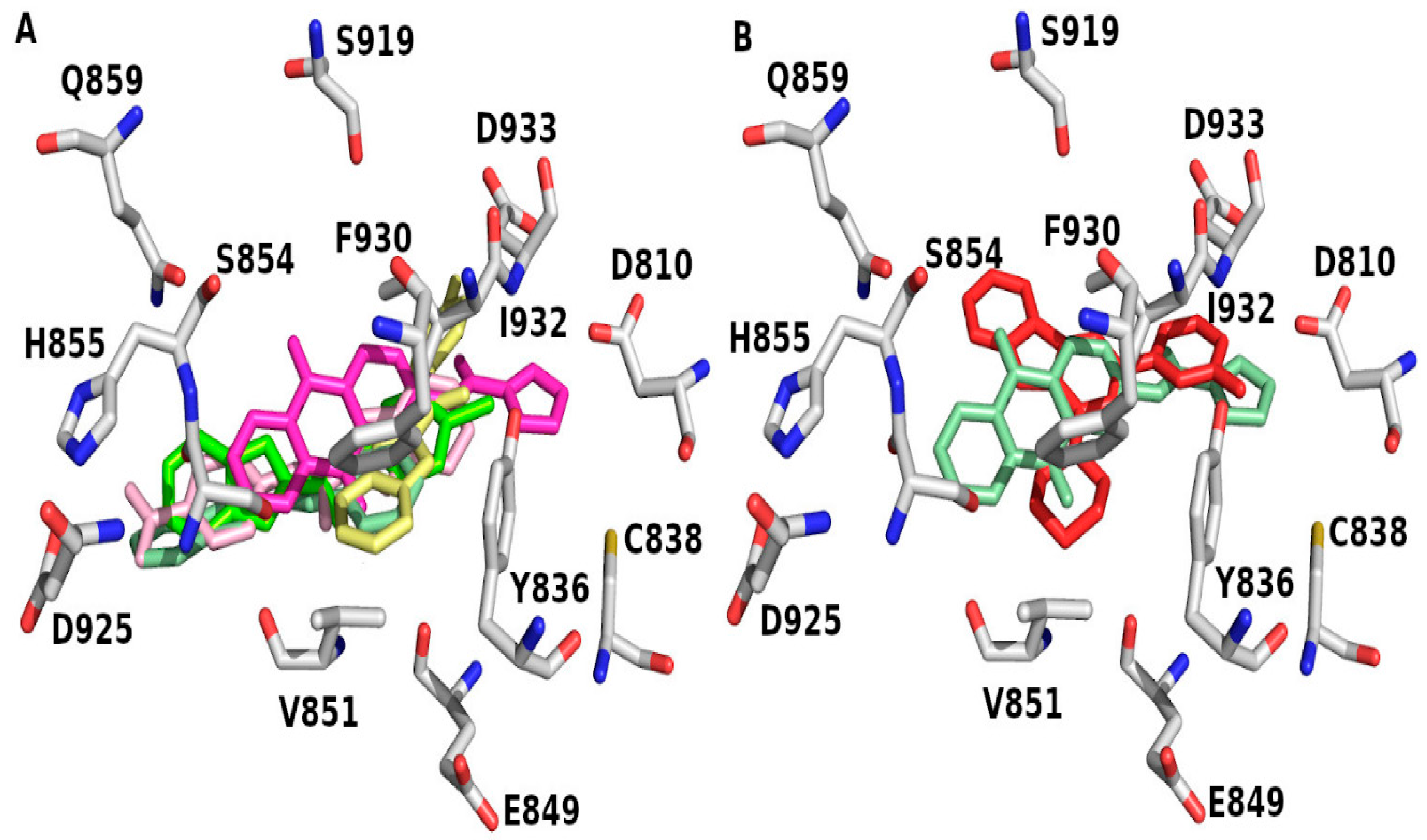
| Compound No. (Moiety) | Docking Score | Binding Residue |
|---|---|---|
| 4 | −6.85 | V851 |
| 6 | −6.71 | Y836 |
| 8 | −7.11 | E849, V851 |
| 10 | −7.29 | V851 |
| 12 | −10.10 | E849, V851, Q859 |
| 14 | −8.85 | W780, E798, V851, S854 |
| 16 | −9.11 | Y836, V851 |
| X6K | −10.16 | S773, Y836, V851 |
| Compound No. (Moiety) | Docking Score | Binding Residue |
|---|---|---|
| 4 | −8.50 | T854 |
| 6 | −8.60 | T854 |
| 8 | −9.04 | M793, T854 |
| 10 | −8.05 | K745, T854 |
| 12 | −9.99 | K745, D855 |
| 14 | −11.74 | M793, T854 |
| 16 | −10.12 | M793, T854 |
| W32 | −10.71 | R841 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Najdawi, M.M.; Saleh, M.M.; Sabbah, D.A.; Hajjo, R.; Zalloum, H.; Abudoleh, S.M.; Abuarqoub, D.A.; Al-Hiari, Y.M.; Mohammad, M.Y.; ALSalamat, H.; et al. Exploring Carboxamide Derivatives as Promising Anticancer Agents: Design, In Vitro Evaluation, and Mechanistic Insights. Int. J. Mol. Sci. 2025, 26, 5903. https://doi.org/10.3390/ijms26125903
Al-Najdawi MM, Saleh MM, Sabbah DA, Hajjo R, Zalloum H, Abudoleh SM, Abuarqoub DA, Al-Hiari YM, Mohammad MY, ALSalamat H, et al. Exploring Carboxamide Derivatives as Promising Anticancer Agents: Design, In Vitro Evaluation, and Mechanistic Insights. International Journal of Molecular Sciences. 2025; 26(12):5903. https://doi.org/10.3390/ijms26125903
Chicago/Turabian StyleAl-Najdawi, Manal M., Maysaa M. Saleh, Dima A. Sabbah, Rima Hajjo, Hiba Zalloum, Suha M. Abudoleh, Duaa A. Abuarqoub, Yusuf M. Al-Hiari, Mohammad Yasin Mohammad, Husam ALSalamat, and et al. 2025. "Exploring Carboxamide Derivatives as Promising Anticancer Agents: Design, In Vitro Evaluation, and Mechanistic Insights" International Journal of Molecular Sciences 26, no. 12: 5903. https://doi.org/10.3390/ijms26125903
APA StyleAl-Najdawi, M. M., Saleh, M. M., Sabbah, D. A., Hajjo, R., Zalloum, H., Abudoleh, S. M., Abuarqoub, D. A., Al-Hiari, Y. M., Mohammad, M. Y., ALSalamat, H., Mansour, H., Aljbour, N. D., & Mestareehi, A. H. (2025). Exploring Carboxamide Derivatives as Promising Anticancer Agents: Design, In Vitro Evaluation, and Mechanistic Insights. International Journal of Molecular Sciences, 26(12), 5903. https://doi.org/10.3390/ijms26125903