
In Vitro Correction of Point Mutations in the DYSF Gene Using Prime Editing

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
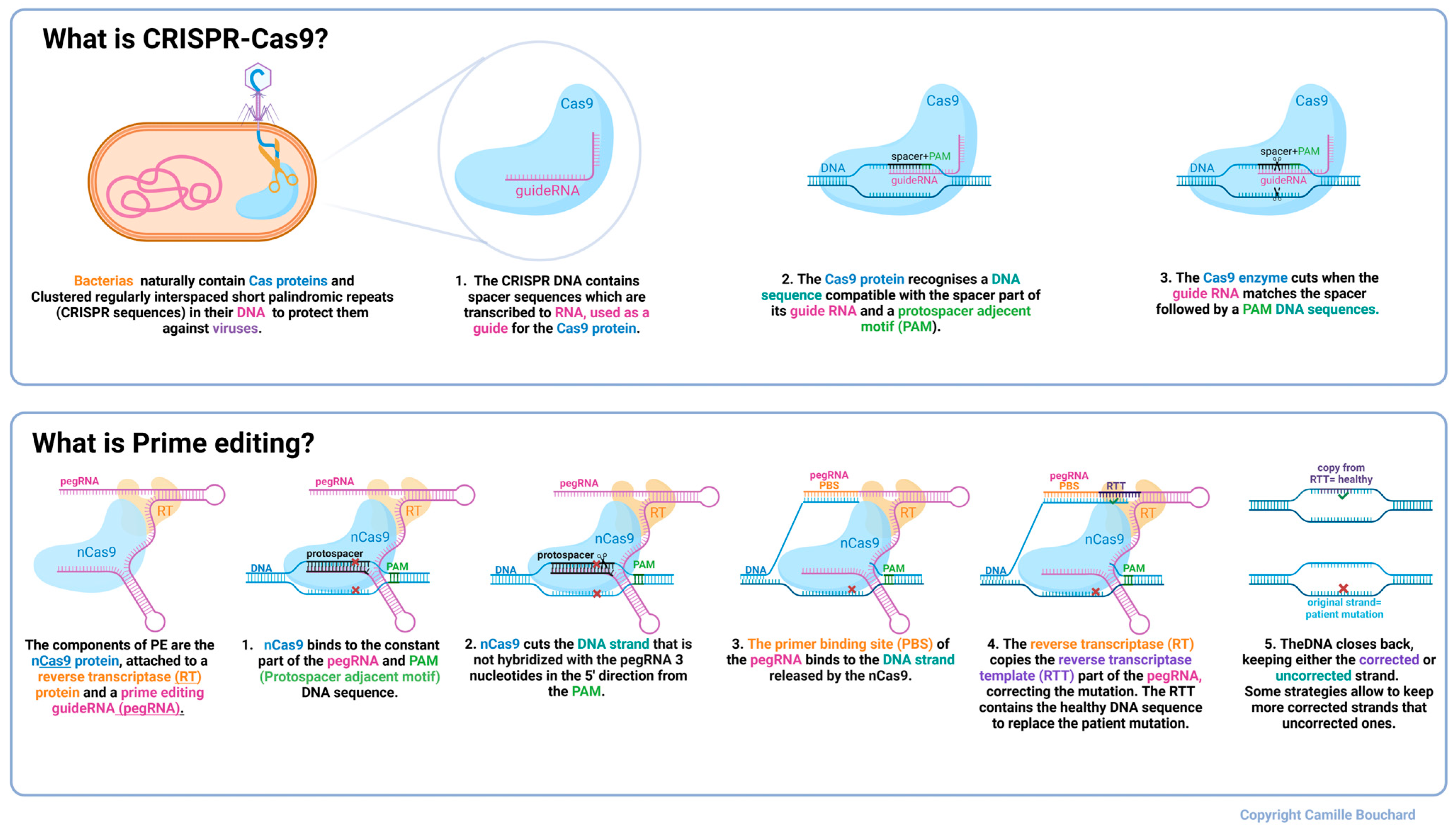
1. Introduction
2. Results
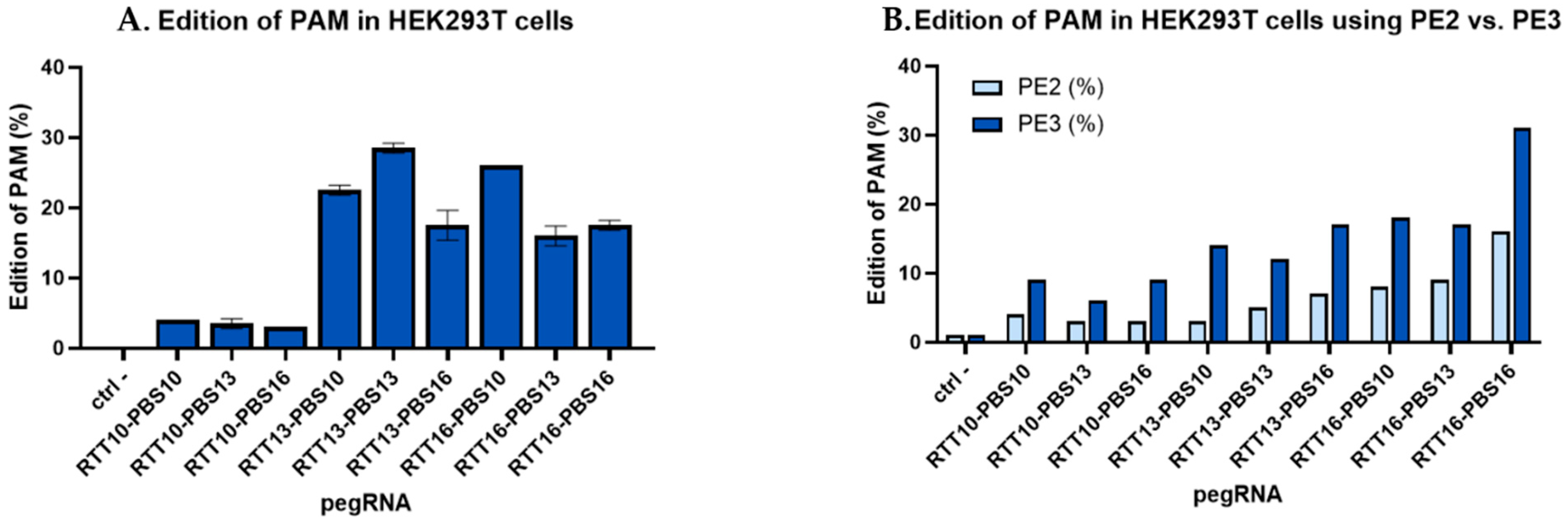
2.1. Insertion of Synonymous Mutations Using Plasmids to Correct Patient Point Mutations in HEK293T Cells
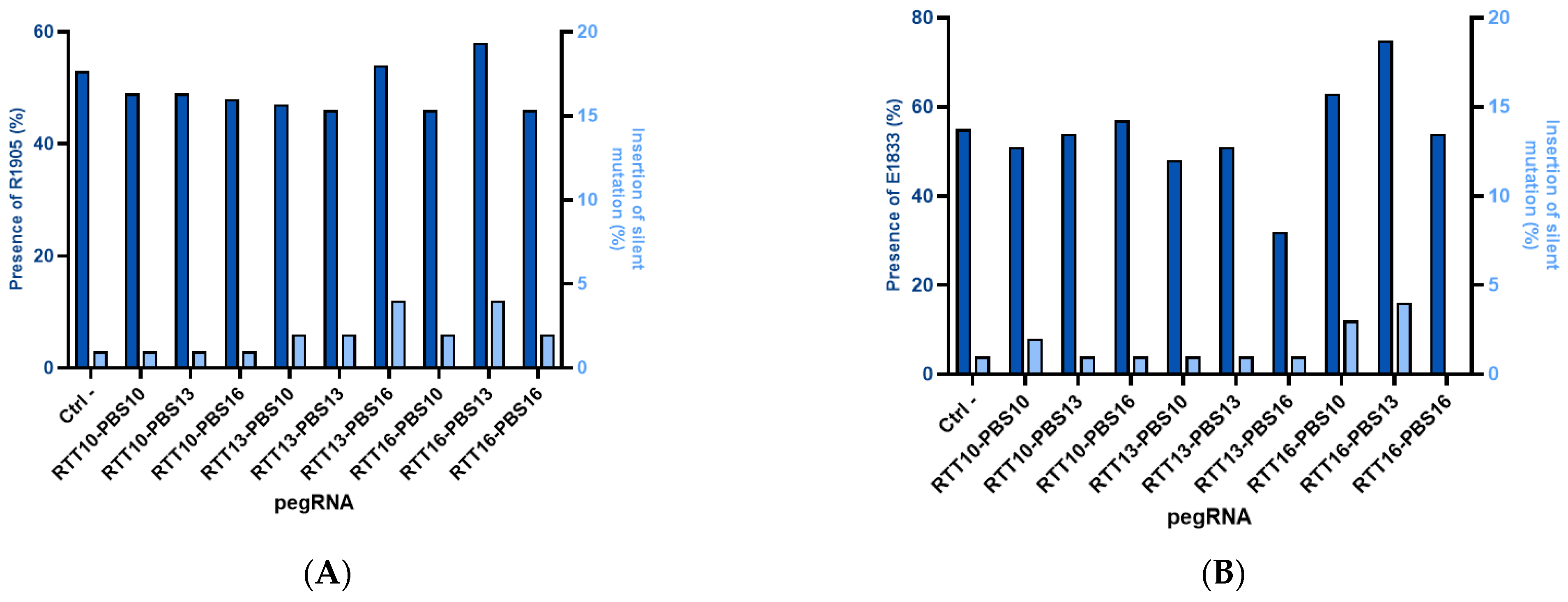
2.2. Correction of Patient Point Mutations in Fibroblast Lines Derived from Patient Skin Biopsies
2.3. Gene Correction in Myoblast-like Cells Derived from Patient Fibroblasts
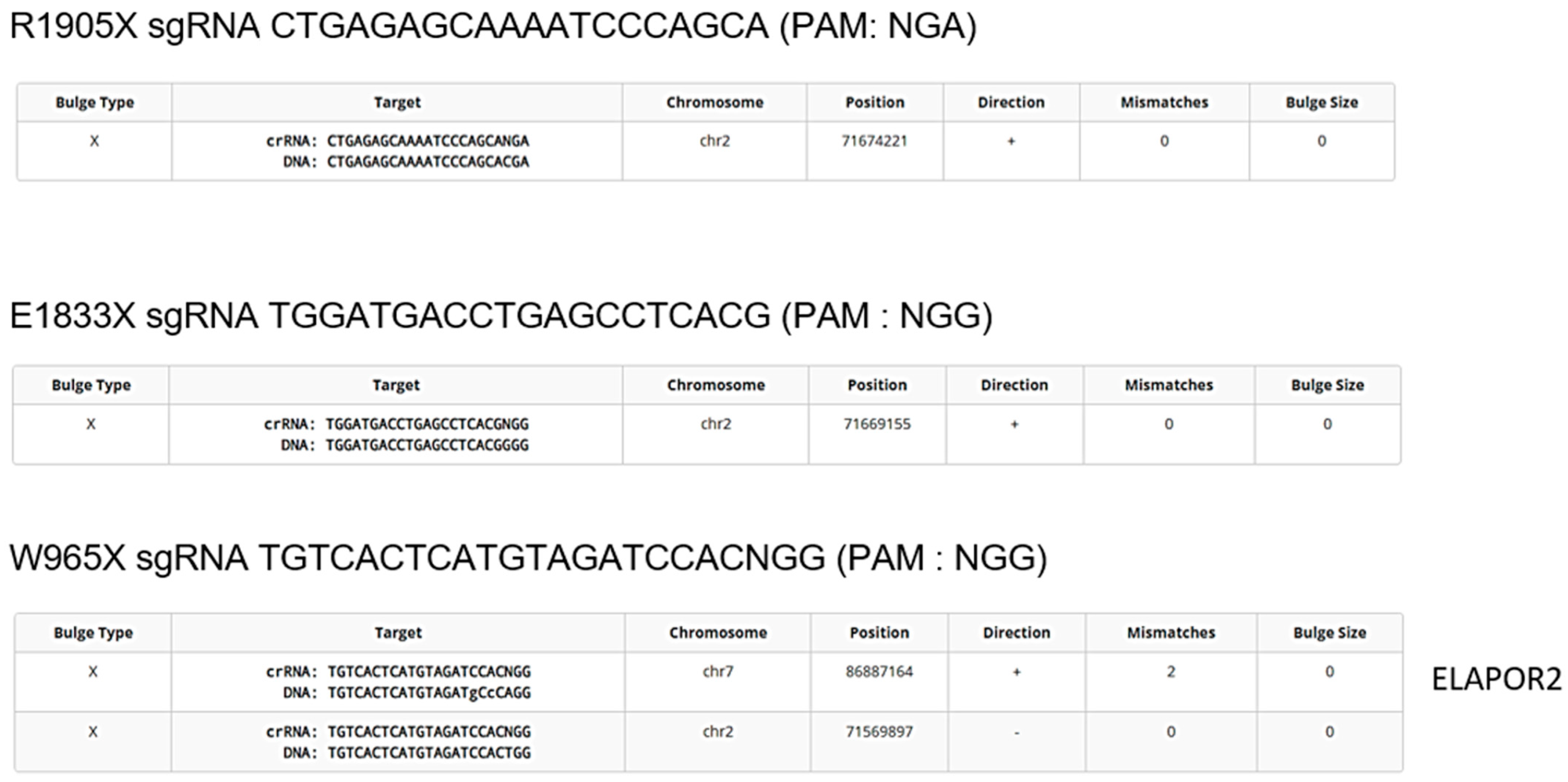
2.4. Avoiding Off-Target Mutation
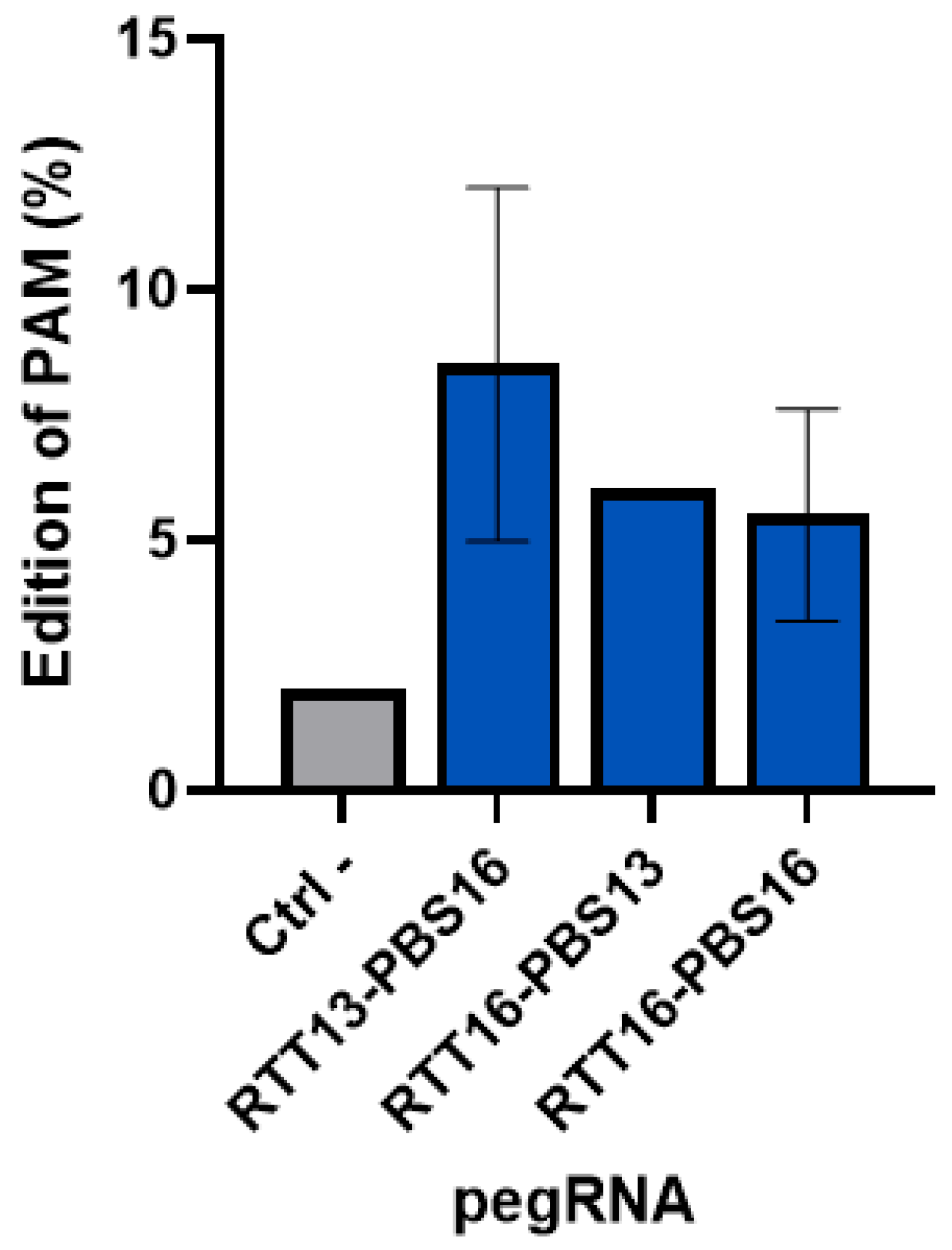
2.5. Tracking Synonymous Mutations Using epegRNA to Insert a Synonymous PAM Mutation in Healthy Myoblasts
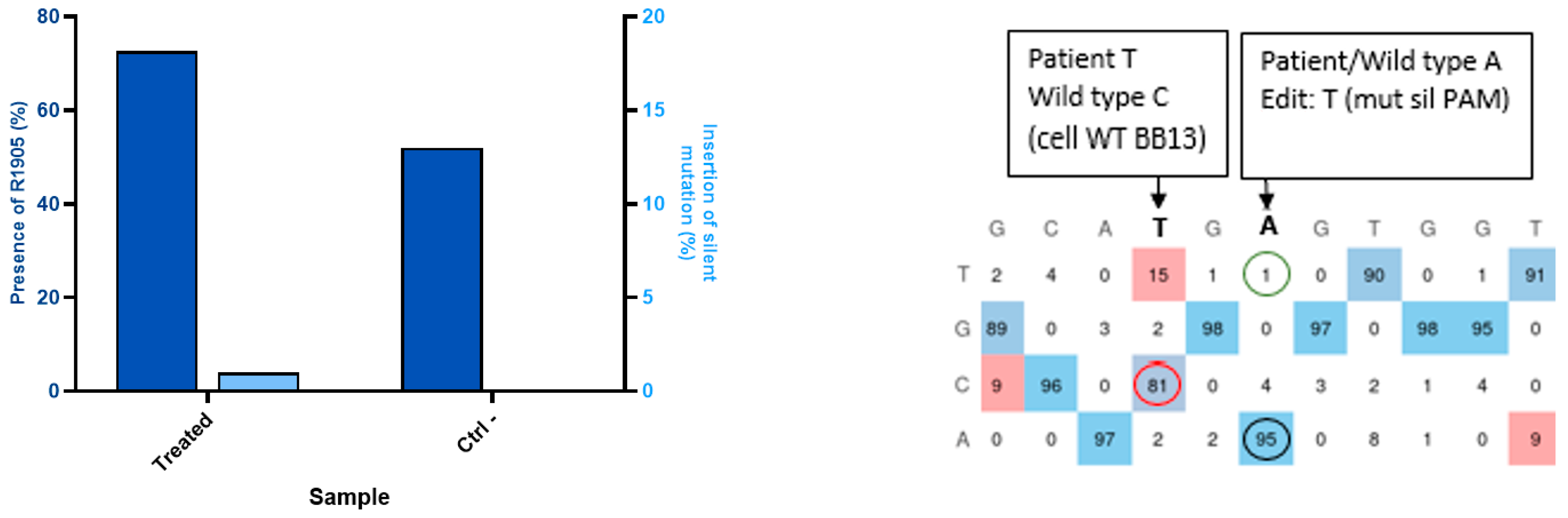
2.6. Creation of a Patient Mutation Cell Line (W965X) from Healthy Myoblasts Modified by Prime Editing
3. Discussion
4. Materials and Methods
4.1. Selecting Patient Mutations
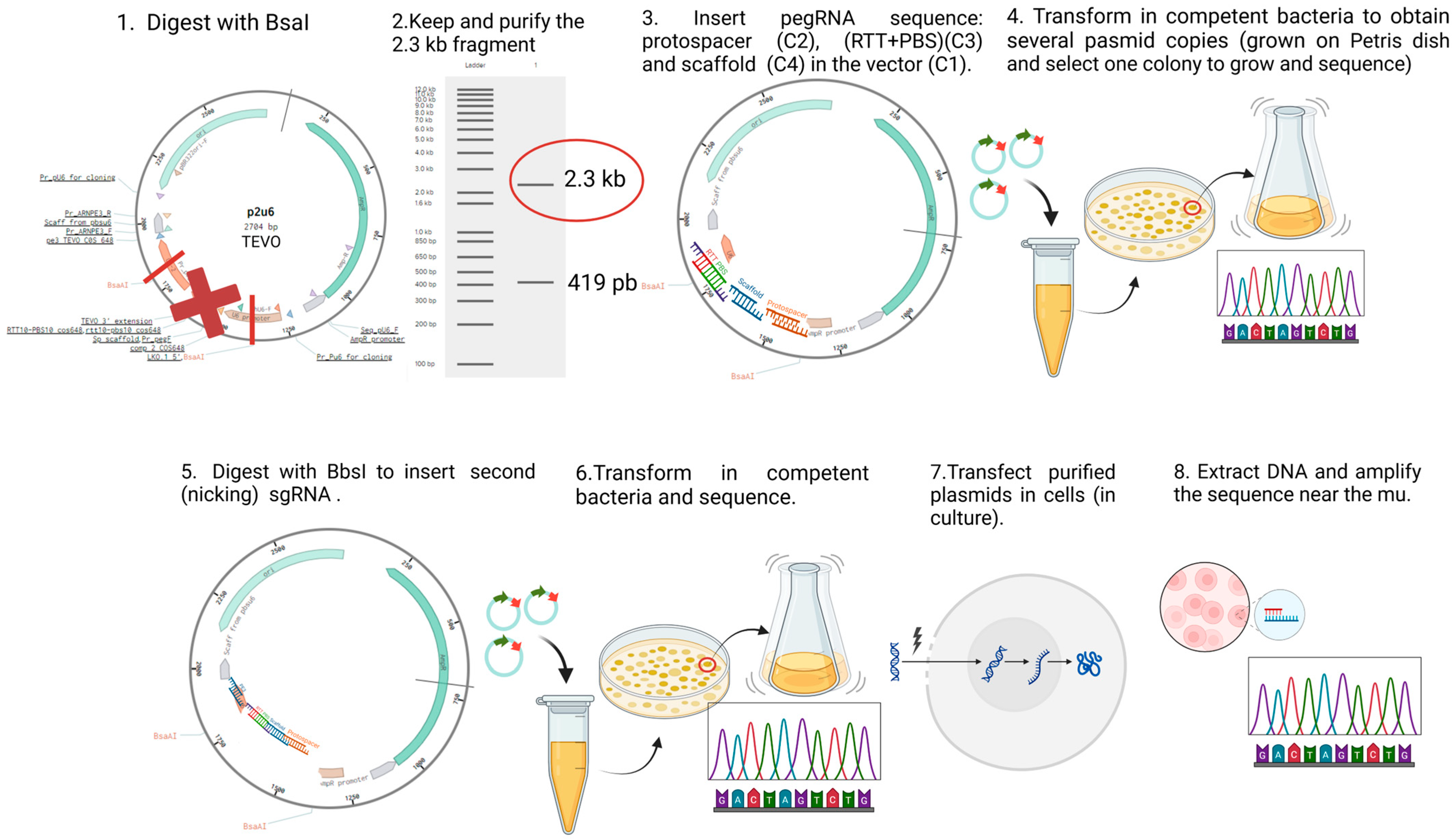
4.2. Plasmid Construction
4.3. In Vitro Prime Editing of HEK293T
4.4. Culture of Patient Fibroblasts
4.5. Plasmid Delivery and Sanger Sequencing
4.6. Generation of Myoblast-like Cell Lines
4.7. Creation of a Patient Mutation Cell Line (W965X) from Healthy Myoblasts Modified by Prime Editing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bouchard, C.; Tremblay, J.P. Limb-girdle muscular dystrophies classification and therapies. J. Clin. Med. 2023, 12, 4769. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Murphy, A.; Udd, B.; Corrado, A.; Aymé, S.; Bönneman, C.; de Visser, M.; Hamosh, A.; Jacobs, L.; Khizanishvili, N. 229th ENMC international workshop: Limb girdle muscular dystrophies-Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef]
- Bouchard, C.; Tremblay, J.P. Portrait of dysferlinopathy: Diagnosis and development of therapy. J. Clin. Med. 2023, 12, 6011. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Urtizberea, J.A.; Hentati, F.; Hamida, M.B. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Muriel, J.; Lukyanenko, V.; Kwiatkowski, T.; Bhattacharya, S.; Garman, D.; Weisleder, N.; Bloch, R.J. The C2 domains of dysferlin: Roles in membrane localization, Ca2+ signalling and sarcolemmal repair. J. Physiol. 2022, 600, 1953–1968. [Google Scholar] [CrossRef]
- Kerr, J.P.; Ziman, A.P.; Mueller, A.L.; Muriel, J.M.; Kleinhans-Welte, E.; Gumerson, J.D.; Vogel, S.S.; Ward, C.W.; Roche, J.A.; Bloch, R.J. Dysferlin stabilizes stress-induced Ca2+ signaling in the transverse tubule membrane. Proc. Natl. Acad. Sci. USA 2013, 110, 20831–20836. [Google Scholar] [CrossRef]
- Zhong, H.; Yu, M.; Lin, P.; Zhao, Z.; Zheng, X.; Xi, J.; Zhu, W.; Zheng, Y.; Zhang, W.; Lv, H. Molecular landscape of DYSF mutations in dysferlinopathy: From a Chinese multicenter analysis to a worldwide perspective. Hum. Mutat. 2021, 42, 1615–1623. [Google Scholar] [CrossRef]
- Diaz-Manera, J.; Fernandez-Torron, R.; LLauger, J.; James, M.K.; Mayhew, A.; Smith, F.E.; Moore, U.R.; Blamire, A.M.; Carlier, P.G.; Rufibach, L. Muscle MRI in patients with dysferlinopathy: Pattern recognition and implications for clinical trials. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1071–1081. [Google Scholar] [CrossRef]
- Woudt, L.; Di Capua, G.A.; Krahn, M.; Castiglioni, C.; Hughes, R.; Campero, M.; Trangulao, A.; González-Hormazábal, P.; Godoy-Herrera, R.; Lévy, N. Toward an objective measure of functional disability in dysferlinopathy. Muscle Nerve 2016, 53, 49–57. [Google Scholar] [CrossRef]
- Patel, N.J.; Van Dyke, K.W.; Espinoza, L.R. Limb-girdle muscular dystrophy 2B and miyoshi presentations of dysferlinopathy. Am. J. Med. Sci. 2017, 353, 484–491. [Google Scholar] [CrossRef]
- Weiler, T.; Bashir, R.; Anderson, L.V.; Davison, K.; Moss, J.A.; Britton, S.; Nylen, E.; Keers, S.; Vafiadaki, E.; Greenberg, C.R. Identical mutation in patients with limb girdle muscular dystrophy type 2B or Miyoshi myopathy suggests a role for modifier gene (s). Hum. Mol. Genet. 1999, 8, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Angelini, C. Progress and challenges in diagnosis of dysferlinopathy. Muscle Nerve 2016, 54, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Moore, U.; Gordish, H.; Diaz-Manera, J.; James, M.K.; Mayhew, A.G.; Guglieri, M.; Fernandez-Torron, R.; Rufibach, L.E.; Feng, J.; Blamire, A.M. Miyoshi myopathy and limb girdle muscular dystrophy R2 are the same disease. Neuromuscul. Disord. 2021, 31, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Yokota, T. The dysferlinopathies conundrum: Clinical spectra, disease mechanism and genetic approaches for treatments. Biomolecules 2024, 14, 256. [Google Scholar] [CrossRef]
- Straub, V.; Bushby, K. Therapeutic possibilities in the autosomal recessive limb-girdle muscular dystrophies. Neurotherapeutics 2008, 5, 619–626. [Google Scholar] [CrossRef]
- Leriche-Guérin, K.; Anderson, L.; Wrogemann, K.; Roy, B.; Goulet, M.; Tremblay, J. Dysferlin expression after normal myoblast transplantation in SCID and in SJL mice. Neuromuscul. Disord. 2002, 12, 167–173. [Google Scholar] [CrossRef]
- Walter, M.C.; Reilich, P.; Thiele, S.; Schessl, J.; Schreiber, H.; Reiners, K.; Kress, W.; Müller-Reible, C.; Vorgerd, M.; Urban, P. Treatment of dysferlinopathy with deflazacort: A double-blind, placebo-controlled clinical trial. Orphanet J. Rare Dis. 2013, 8, 26. [Google Scholar] [CrossRef]
- Sondergaard, P.C.; Griffin, D.A.; Pozsgai, E.R.; Johnson, R.W.; Grose, W.E.; Heller, K.N.; Shontz, K.M.; Montgomery, C.L.; Liu, J.; Clark, K.R. AAV. Dysferlin overlap vectors restore function in dysferlinopathy animal models. Ann. Clin. Transl. Neurol. 2015, 2, 256–270. [Google Scholar] [CrossRef]
- Turan, S.; Farruggio, A.P.; Srifa, W.; Day, J.W.; Calos, M.P. Precise correction of disease mutations in induced pluripotent stem cells derived from patients with limb girdle muscular dystrophy. Mol. Ther. 2016, 24, 685–696. [Google Scholar] [CrossRef]
- Sun, N.; Zhao, H. Transcription activator-like effector nucleases (TALENs): A highly efficient and versatile tool for genome editing. Biotechnol. Bioeng. 2013, 110, 1811–1821. [Google Scholar] [CrossRef]
- Durai, S.; Mani, M.; Kandavelou, K.; Wu, J.; Porteus, M.H.; Chandrasegaran, S. Zinc finger nucleases: Custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res. 2005, 33, 5978–5990. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Progress and prospects: Zinc-finger nucleases as gene therapy agents. Gene Ther. 2008, 15, 1463–1468. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base editing: Advances and therapeutic opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef] [PubMed]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The biology of CRISPR-Cas: Backward and forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Murray, J.B.; Harrison, P.T.; Scholefield, J. Prime editing: Therapeutic advances and mechanistic insights. Gene Ther. 2025, 32, 83–92. [Google Scholar] [CrossRef]
- Happi Mbakam, C.; Rousseau, J.; Tremblay, G.; Yameogo, P.; Tremblay, J.P. Prime editing permits the introduction of specific mutations in the gene responsible for duchenne muscular dystrophy. Int. J. Mol. Sci. 2022, 23, 6160. [Google Scholar] [CrossRef]
- Cao, B.-R.; Huang, Y.-M.; Tian, F.-Y.; Li, J.-H.; Xu, C.-L.; Wei, Y.; Liu, J.-K.; Guo, Q.-B.; Xu, H.-Y.; Zhan, L. Prime editing-based gene correction alleviates the hyperexcitable phenotype and seizures of a genetic epilepsy mouse model. Acta Pharmacol. Sin. 2023, 44, 2342–2345. [Google Scholar] [CrossRef]
- Lin, F.-L.; Wang, P.-Y.; Chuang, Y.-F.; Wang, J.-H.; Wong, V.H.; Bui, B.V.; Liu, G.-S. Gene therapy intervention in neovascular eye disease: A recent update. Mol. Ther. 2020, 28, 2120–2138. [Google Scholar] [CrossRef]
- Abraham, A.A.; Tisdale, J.F. Gene therapy for sickle cell disease: Moving from the bench to the bedside. Blood J. Am. Soc. Hematol. 2021, 138, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.S.; Loeckermann, S.; Fritsch, A.; Hausser, I.; Roth, W.; Magin, T.M.; Mack, C.; Müller, M.L.; Paul, O.; Ruther, P. Mechanisms of fibroblast cell therapy for dystrophic epidermolysis bullosa: High stability of collagen VII favors long-term skin integrity. Mol. Ther. 2009, 17, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Everette, K.A.; Newby, G.A.; Levine, R.M.; Mayberry, K.; Jang, Y.; Mayuranathan, T.; Nimmagadda, N.; Dempsey, E.; Li, Y.; Bhoopalan, S.V. Ex vivo prime editing of patient haematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nat. Biomed. Eng. 2023, 7, 616–628. [Google Scholar] [CrossRef]
- Schene, I.F.; Joore, I.P.; Oka, R.; Mokry, M.; van Vugt, A.H.; van Boxtel, R.; van der Doef, H.P.; van der Laan, L.J.; Verstegen, M.M.; van Hasselt, P.M. Prime editing for functional repair in patient-derived disease models. Nat. Commun. 2020, 11, 5352. [Google Scholar] [CrossRef]
- Godbout, K.; Rousseau, J.; Tremblay, J.P. Successful correction by prime editing of a mutation in the RYR1 gene responsible for a myopathy. Cells 2023, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Brüggemann, A.; Eriksson, N.; Michaëlsson, K.; Hailer, N.P. Risk of revision after arthroplasty associated with specific gene loci: A genomewide association study of single-nucleotide polymorphisms in 1,130 twins treated with arthroplasty. JBJS 2022, 104, 610–620. [Google Scholar] [CrossRef]
- Mbakam, C.H.; Rousseau, J.; Lu, Y.; Bigot, A.; Mamchaoui, K.; Mouly, V.; Tremblay, J.P. Prime editing optimized RTT permits the correction of the c. 8713C> T mutation in DMD gene. Mol. Ther. Nucleic Acids 2022, 30, 272–285. [Google Scholar] [CrossRef]
- Rottner, A.K.; Lundin, A.; Li, S.; Firth, M.; Maresca, M.; Sienski, G. Optimized prime editing of the Alzheimer’s disease-associated APOE4 mutation. Stem Cell Rep. 2025, 20, 102372. [Google Scholar] [CrossRef]
- Trojan, J.; Zeuzem, S.; Randolph, A.; Hemmerle, C.; Brieger, A.; Raedle, J.; Plotz, G.; Jiricny, J.; Marra, G. Functional analysis of hMLH1 variants and HNPCC-related mutations using a human expression system. Gastroenterology 2002, 122, 211–219. [Google Scholar] [CrossRef]
- Hu, X.; Wang, C.; Fu, Y.; Liu, Q.; Jiao, X.; Wang, K. Expanding the range of CRISPR/Cas9 genome editing in rice. Mol. Plant 2016, 9, 943–945. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, H.; Zhang, Y.; Wang, Y.; Gan, J.; Ji, Q. Molecular basis for the PAM expansion and fidelity enhancement of an evolved Cas9 nuclease. PLoS Biol. 2019, 17, e3000496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yin, J.; Zhang-Ding, Z.; Xin, C.; Liu, M.; Wang, Y.; Ai, C.; Hu, J. In-depth assessment of the PAM compatibility and editing activities of Cas9 variants. Nucleic Acids Res. 2021, 49, 8785–8795. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouchard, C.; Rousseau, J.; Lamothe, G.; Dubost, M.; Bastrenta, L.; Ramezani, S.; Tremblay, J.P. In Vitro Correction of Point Mutations in the DYSF Gene Using Prime Editing. Int. J. Mol. Sci. 2025, 26, 5647. https://doi.org/10.3390/ijms26125647
Bouchard C, Rousseau J, Lamothe G, Dubost M, Bastrenta L, Ramezani S, Tremblay JP. In Vitro Correction of Point Mutations in the DYSF Gene Using Prime Editing. International Journal of Molecular Sciences. 2025; 26(12):5647. https://doi.org/10.3390/ijms26125647
Chicago/Turabian StyleBouchard, Camille, Joël Rousseau, Gabriel Lamothe, Marie Dubost, Laura Bastrenta, Sina Ramezani, and Jacques P. Tremblay. 2025. "In Vitro Correction of Point Mutations in the DYSF Gene Using Prime Editing" International Journal of Molecular Sciences 26, no. 12: 5647. https://doi.org/10.3390/ijms26125647
APA StyleBouchard, C., Rousseau, J., Lamothe, G., Dubost, M., Bastrenta, L., Ramezani, S., & Tremblay, J. P. (2025). In Vitro Correction of Point Mutations in the DYSF Gene Using Prime Editing. International Journal of Molecular Sciences, 26(12), 5647. https://doi.org/10.3390/ijms26125647