
The Role of Inflammation in Neurodegenerative Diseases: Parkinson’s Disease, Alzheimer’s Disease, and Multiple Sclerosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
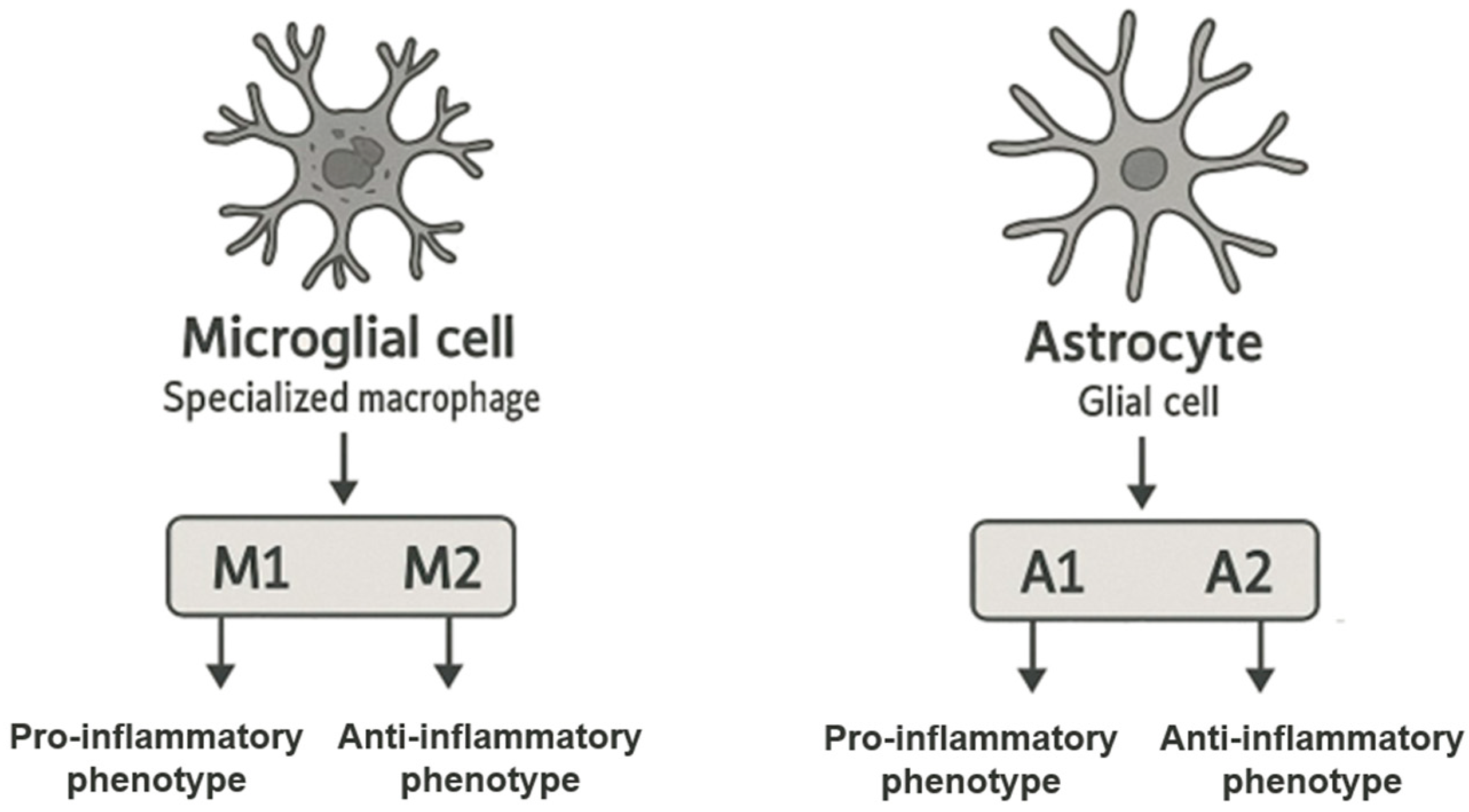
2. The Role of Microglia and Astrocytes in the Inflammatory Process
3. Parkinson’s Disease
3.1. Background Information
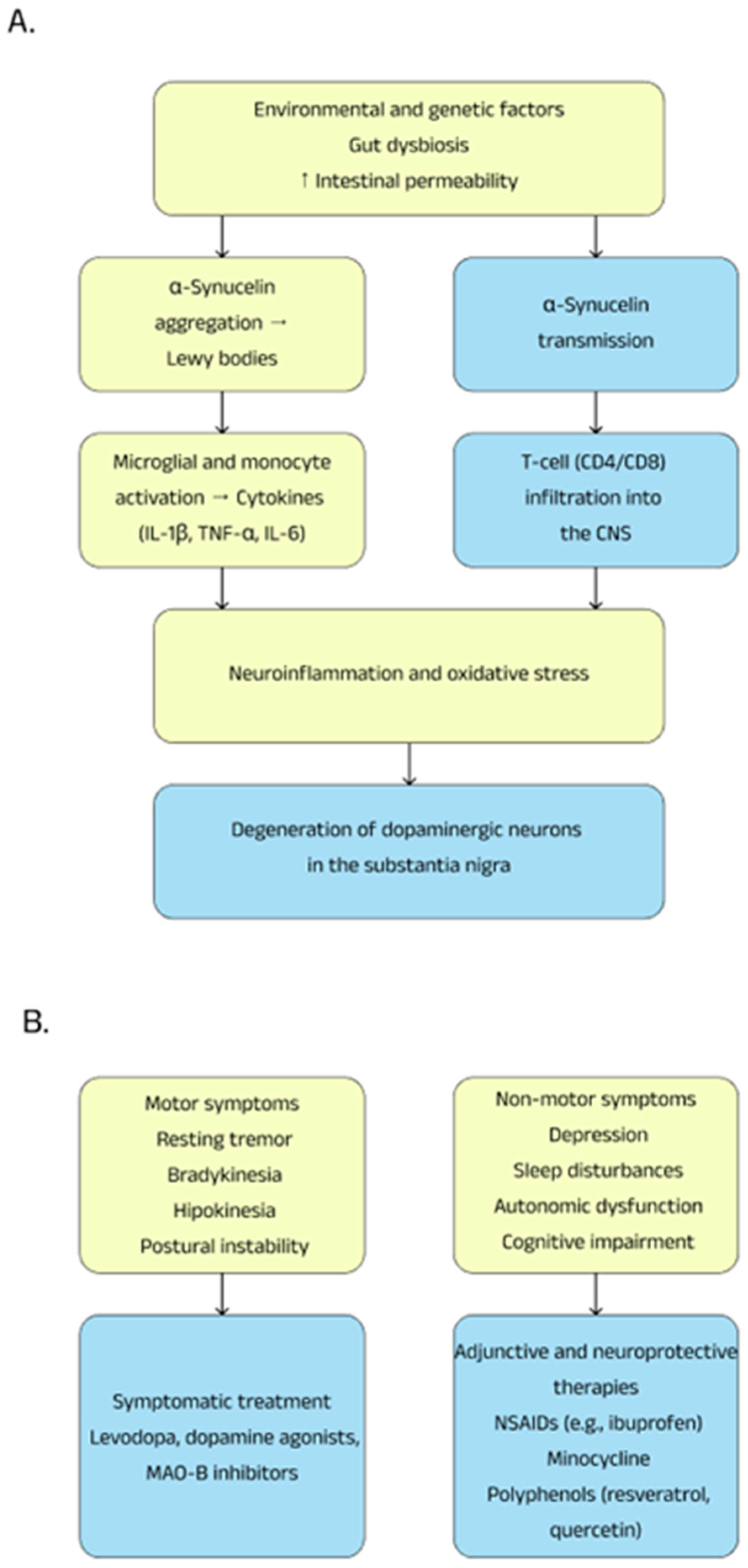
3.2. Pathogenesis
- A reduction in the number of short-chain fatty acid (SCFA)-producing bacteria and an increase in the number of mucin-degrading bacteria such as Akkermansia may lead to increased intestinal permeability, thereby allowing toxins such as lipopolysaccharides—a known pro-inflammatory factor—to access the nervous system and promote α-synuclein aggregation;
- Deficiency of SCFA-producing bacteria can increase inflammation in the central nervous system by activating microglia [65].
3.3. Treatment
4. Alzheimer’s Disease
4.1. Background Information
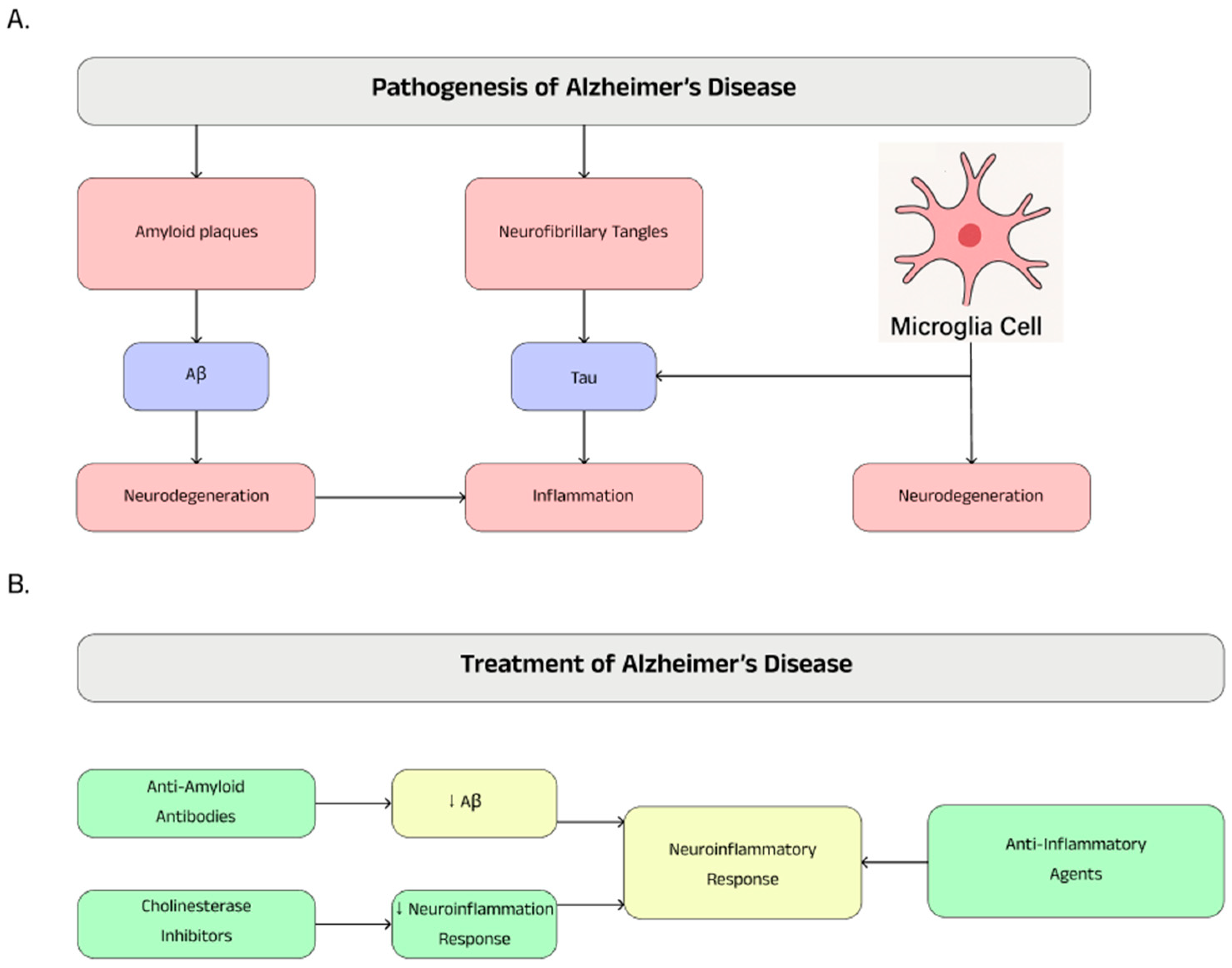
4.2. Pathogenesis
4.3. Treatment
5. Inflammation in Multiple Sclerosis
5.1. Background Information
5.2. Pathogenesis
5.3. Treatment
6. Methods
7. Conclusions
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | Amyloid β |
| AD | Alzheimer’s disease |
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| COX | Cyclooxygenase |
| DMTs | Disease-modifying therapies |
| FDA | Food and Drugs Administration |
| IFN | Interferon |
| IL | Interleukin |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| MS | Multiple sclerosis |
| NO | Nitric oxide |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| PD | Parkinson’s disease |
| SCFA | Short-chain fatty acid |
| TNF-α | Tumor necrosis factor-α |
| TREM2 | Triggering receptor expressed on myeloid cell 2 |
| WHO | World Health Organization |
References
- Culig, L.; Chu, X.; Bohr, V.A. Neurogenesis in aging and age-related neurodegenerative diseases. Ageing Res Rev. 2022, 78, 101636. [Google Scholar] [CrossRef] [PubMed]
- Bondy, S.C. Anthropogenic pollutants may increase the incidence of neurodegenerative disease in an aging population. Toxicology 2016, 341–343, 41–46. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Neri, S.; Mastroianni, G.; Gardella, E.; Aguglia, U.; Rubboli, G. Epilepsy in neurodegenerative diseases. Epileptic Disord. 2022, 24, 249–273. [Google Scholar] [CrossRef]
- Antonioni, A.; Raho, E.M.; Lopriore, P.; Pace, A.P.; Latino, R.R.; Assogna, M.; Mancuso, M.; Gragnaniello, D.; Granieri, E.; Pugliatti, M.; et al. Frontotemporal dementia, where do we stand? A narrative review. Int. J. Mol. Sci. 2023, 24, 11732. [Google Scholar] [CrossRef]
- Sivandzade, F.; Cucullo, L. Regenerative stem cell therapy for neurodegenerative diseases: An overview. Int. J. Mol. Sci. 2021, 22, 2153. [Google Scholar] [CrossRef]
- Gorji, A. Neuroinflammation: The pathogenic mechanism of neurological disorders. Int. J. Mol. Sci. 2022, 23, 5744. [Google Scholar] [CrossRef]
- Suleiman Khoury, Z.; Sohail, F.; Wang, J.; Mendoza, M.; Raake, M.; Tahoor Silat, M.; Reddy Bathinapatta, M.; Sadeghzadegan, A.; Meghana, P.; Paul, J. Neuroinflammation: A critical factor in neurodegenerative disorders. Cureus 2024, 16, 62310. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Niculescu, A.-G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An overview of oxidative stress, neuroinflammation, and neurodegenerative diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative stress in neurodegenerative diseases: From molecular mechanisms to clinical applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Dash, U.C.; Bhol, N.K.; Swain, S.K.; Samal, R.R.; Nayak, P.K.; Raina, V.; Panda, S.K.; Kerry, R.G.; Duttaroy, A.K.; Jena, A.B. Oxidative stress and inflammation in the pathogenesis of neurological disorders: Mechanisms and implications. Acta Pharm. Sin. B 2025, 15, 15–34. [Google Scholar] [CrossRef]
- Hamzeh, O.; Rabiei, F.; Shakeri, M.; Parsian, H.; Saadat, P.; Rostami-Mansoor, S. Mitochondrial dysfunction and inflammasome activation in neurodegenerative diseases: Mechanisms and therapeutic implications. Mitochondrion 2023, 68, 72–83. [Google Scholar] [CrossRef]
- Henrich, M.T.; Oertel, W.H.; Surmeier, D.J.; Geibl, F.F. Mitochondrial dysfunction in Parkinson’s disease—A key disease hallmark with therapeutic potential. Mol. Neurodegener. 2023, 18, 83. [Google Scholar] [CrossRef]
- Sonninen, T.-M.; Goldsteins, G.; Laham-Karam, N.; Koistinaho, J.; Lehtonen, Š. Proteostasis disturbances and inflammation in neurodegenerative diseases. Cells 2020, 9, 183. [Google Scholar] [CrossRef]
- Kouli, A.; Camacho, M.; Allinson, K.; Williams-Gray, C.H. Neuroinflammation and protein pathology in Parkinson’s disease dementia. Acta Neuropathol. Commun. 2020, 8, 135. [Google Scholar] [CrossRef]
- Song, X.; Ma, F.; Herrup, K. DNA damage-induced neuroinflammation in neurodegenerative disease. Alzheimer’s Dement. 2021, 17, e055175. [Google Scholar] [CrossRef]
- Song, X.; AW, T.M.; Ma, F.; Leung, D.; Herrup, K. DNA damage induced neuroinflammation in neurodegenerative disease. Alzheimer’s Dement. 2020, 16, e047445. [Google Scholar] [CrossRef]
- Kalinowska-Lyszczarz, A.; Losy, J. The role of neurotrophins in multiple sclerosis—Pathological and clinical implications. Int. J. Mol. Sci. 2012, 13, 13713–13725. [Google Scholar] [CrossRef]
- Severini, C. Neurotrophic factors in health and disease. Cells 2023, 12, 47. [Google Scholar] [CrossRef]
- De Chiara, G.; Marcocci, M.E.; Sgarbanti, R.; Civitelli, L.; Ripoli, C.; Piacentini, R.; Garaci, E.; Grassi, C.; Palamara, A.T. Infectious agents and neurodegeneration. Mol. Neurobiol. 2012, 46, 614–638. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Khachaturian, Z.S. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer’s Dement. 2017, 13, 178–182.e17. [Google Scholar] [CrossRef]
- Spiteri, A.G.; Wishart, C.L.; Pamphlett, R.; Locatelli, G.; King, N.J.C. Microglia and monocytes in inflammatory CNS disease: Integrating phenotype and function. Acta Neuropathol. 2022, 143, 179–224. [Google Scholar] [CrossRef]
- Huang, Y.H.; Lin, C.H. Neuroinflammation. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK326720/ (accessed on 13 May 2025).
- Zheng, R.Z.; Lee, K.Y.; Qi, Z.X.; Wang, Z.; Xu, Z.Y.; Wu, X.H.; Mao, Y. Neuroinflammation following traumatic brain injury: Take it seriously or not. Front. Immunol. 2022, 13, 855701. [Google Scholar] [CrossRef]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-faced microglia: Beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci. 2013, 7, 6. [Google Scholar] [CrossRef]
- Wang, C.; Zong, S.; Cui, X.; Wang, X.; Wu, S.; Wang, L.; Liu, Y.; Lu, Z. The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Front Immunol. 2023, 14, 1117172. [Google Scholar] [CrossRef]
- Al-Ghraiybah, N.F.; Wang, J.; Alkhalifa, A.E.; Roberts, A.B.; Raj, R.; Yang, E.; Kaddoumi, A. Glial cell-mediated neuroinflammation in Alzheimer’s disease. Int. J. Mol. Sci. 2022, 23, 10572. [Google Scholar] [CrossRef]
- Patrycy, M.; Chodkowski, M.; Krzyzowska, M. Role of microglia in Herpesvirus-related neuroinflammation and neurodegeneration. Pathogens 2022, 11, 809. [Google Scholar] [CrossRef]
- Lukens, J.R.; Eyo, U.B. Microglia and neurodevelopmental disorders. Annu. Rev. Neurosci. 2022, 45, 425–445. [Google Scholar] [CrossRef]
- Wu, Y.; Eisel, U.L.M. Microglia-astrocyte communication in Alzheimer’s disease. J. Alzheimer’s Dis. 2023, 95, 785–803. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.-J.; Zhu, Z.-Q. To re-examine the intersection of microglial activation and neuroinflammation in neurodegenerative diseases from the perspective of pyroptosis. Front. Aging Neurosci. 2023, 15, 1284214. [Google Scholar] [CrossRef]
- Abbott, N.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Zeng, J.; Bao, T.; Yang, K.; Zhu, X.; Wang, S.; Xiang, W.; Ge, A.; Zeng, L.; Ge, J. The mechanism of microglia-mediated immune inflammation in ischemic stroke and the role of natural botanical components in regulating microglia: A review. Front. Immunol. 2023, 13, 1047550. [Google Scholar] [CrossRef]
- Umpierre, A.D.; Wu, L.J. How microglia sense and regulate neuronal activity. Glia 2021, 69, 1637–1653. [Google Scholar] [CrossRef]
- Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS pro-inflammatory response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef]
- Brisch, R.; Wojtylak, S.; Saniotis, A.; Steiner, J.; Gos, T.; Kumaratilake, J.; Henneberg, M.; Wolf, R. The role of microglia in neuropsychiatric disorders and suicide. Eur. Arch. Psychiatry Clin. Neurosci. 2022, 272, 929–945. [Google Scholar] [CrossRef]
- Gradisnik, L.; Velnar, T. Astrocytes in the central nervous system and their functions in health and disease: A review. World J. Clin. Cases 2023, 11, 3385–3394. [Google Scholar] [CrossRef]
- Alhadidi, Q.M.; Bahader, G.A.; Arvola, O.; Kitchen, P.; Shah, Z.A.; Salman, M.M. Astrocytes in functional recovery following central nervous system injuries. J. Physiol. 2024, 602, 3069–3096. [Google Scholar] [CrossRef]
- Gullotta, G.S.; Costantino, G.; Sortino, M.A.; Spampinato, S.F. Microglia and the Blood-Brain Barrier: An external player in acute and chronic neuroinflammatory conditions. Int. J. Mol. Sci. 2023, 24, 9144. [Google Scholar] [CrossRef]
- Lawrence, J.M.; Schardien, K.; Wigdahl, B.; Nonnemacher, M.R. Roles of neuropathology-associated reactive astrocytes: A systematic review. Acta Neuropathol. Commun. 2023, 11, 42. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte crosstalk in CNS inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of microglia and astrocytes in the neurovascular unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, J.; Sun, X.; Shao, L.; Guo, Z.; Li, Y. Morphological and functional alterations of astrocytes responding to traumatic brain injury. J. Integr. Neurosci. 2019, 18, 203–215. [Google Scholar] [CrossRef]
- Takouda, J.; Katada, S.; Nakashima, K. Emerging mechanisms underlying astrogenesis in the developing mammalian brain. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 386–398. [Google Scholar] [CrossRef]
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef]
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Ray Dorsey, E.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and projected future economic burden of Parkinson’s disease in the U.S. npj Parkinson’s Dis. 2020, 6, 15. [Google Scholar] [CrossRef]
- Ellis, T.D.; Colón-Semenza, C.; DeAngelis, T.R.; Thomas, C.A.; Hilaire, M.S.; Earhart, G.M.; Dibble, L.E. Evidence for early and regular physical therapy and exercise in Parkinson’s disease. Semin. Neurol. 2021, 41, 189–205. [Google Scholar] [CrossRef]
- Parkinson, J. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef]
- Costa, H.N.; Esteves, A.R.; Empadinhas, N.; Cardoso, S.M. Parkinson’s disease: A multisystem disorder. Neurosci. Bull. 2023, 39, 113–124. [Google Scholar] [CrossRef]
- De Miranda, B.R.; Goldman, S.M.; Miller, G.W.; Greenamyre, J.T.; Dorsey, E.R. Preventing Parkinson’s disease: An environmental agenda. J. Parkinson’s Dis. 2022, 12, 45–68. [Google Scholar] [CrossRef]
- Hariz, M.; Blomstedt, P. Deep brain stimulation for Parkinson’s disease. J. Intern. Med. 2022, 292, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and inflammation- an interesting interplay in Parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Yi, L.X.; Wang, D.Q.; Lim, T.M.; Tan, E.K. Role of dopamine in the pathophysiology of Parkinson’s disease. Transl. Neurodegener. 2023, 12, 44. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell biology and pathophysiology of α-synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Sandal, M.; Valle, F.; Tessari, I.; Mammi, S.; Bergantino, E.; Musiani, F.; Brucale, M.; Bubacco, L.; Samorì, B. Conformational equilibria in monomeric α-synuclein at the single-molecule level. PLoS Biol. 2008, 6, e6. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Yang, D.; Li, X.Q.; Liu, J.; Back, T.C.; Trivett, A.; Karim, B.; Barbut, D.; Zasloff, M.; Oppenheim, J.J. A synuclein, the culprit in Parkinson disease, is required for normal immune function. Cell Rep. 2022, 38, 110090. [Google Scholar] [CrossRef] [PubMed]
- Forloni, G. A Synuclein: Neurodegeneration and inflammation. Int. J. Mol. Sci. 2023, 24, 5914. [Google Scholar] [CrossRef]
- Borghammer, P.; Van Den Berge, N. Brain-first versus gut-first Parkinson’s disease: A hypothesis. J. Parkinson’s Dis. 2019, 9, S281–S295. [Google Scholar] [CrossRef]
- Grozdanov, V.; Bousset, L.; Hoffmeister, M.; Bliederhaeuser, C.; Meier, C.; Madiona, K.; Pieri, L.; Kiechle, M.; McLean, P.J.; Kassubek, J.; et al. Increased immune activation by pathologic α-synuclein in Parkinson’s disease. Ann. Neurol. 2019, 86, 593–606. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, 55375. [Google Scholar] [CrossRef] [PubMed]
- Niskanen, J.; Peltonen, S.; Ohtonen, S.; Fazaludeen, M.F.; Luk, K.C.; Giudice, L.; Koistinaho, J.; Malm, T.; Goldsteins, G.; Albert, K.; et al. Uptake of α-synuclein preformed fibrils is suppressed by inflammation and induces an aberrant phenotype in human microglia. Glia 2025, 73, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.P.; Schonhoff, A.M.; Jurkuvenaite, A.; Gallups, N.J.; Standaert, D.G.; Harms, A.S. CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson’s disease. Brain 2021, 144, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, M.; Ohno, K. Parkinson’s disease and gut microbiota. Ann. Nutr. Metab. 2021, 77, 28–35. [Google Scholar] [CrossRef]
- Hall, D.A.; Voigt, R.M.; Cantu-Jungles, T.M.; Hamaker, B.; Engen, P.A.; Shaikh, M.; Raeisi, S.; Green, S.J.; Naqib, A.; Forsyth, C.B.; et al. An open label, non-randomized study assessing a prebiotic fiber intervention in a small cohort of Parkinson’s disease participants. Nat. Commun. 2023, 14, 926. [Google Scholar] [CrossRef]
- Nagatsu, T.; Sawada, M. Inflammatory process in Parkinson’s disease: Role for cytokines. Curr. Pharm. Des. 2005, 11, 999–1016. [Google Scholar] [CrossRef]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef]
- Elsworth, J.D. Parkinson’s disease treatment: Past, present, and future. J. Neural Transm. 2020, 127, 785–791. [Google Scholar] [CrossRef]
- Ren, L.; Yi, J.; Yang, J.; Li, P.; Cheng, X.; Mao, P. Nonsteroidal anti-inflammatory drugs use and risk of Parkinson disease: A dose-response meta-analysis. Medicine 2018, 97, 12172. [Google Scholar] [CrossRef]
- Singh, A.; Tripathi, P.; Singh, S. Neuroinflammatory responses in Parkinson’s disease: Relevance of ibuprofen in therapeutics. Inflammopharmacology 2021, 29, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Le, W.D. Minocycline: Neuroprotective mechanisms in Parkinson’s disease. Curr. Pharm. Des. 2004, 10, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Cankaya, S.; Cankaya, B.; Kilic, U.; Kilic, E.; Yulug, B. The therapeutic role of minocycline in Parkinson’s disease. Drugs Context 2019, 8, 212553. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Le, W.D.; Jankovic, J. Minocycline and other tetracycline derivatives: A neuroprotective strategy in Parkinson’s disease and Huntington’s disease. Clin. Neuropharmacol. 2003, 26, 18–23. [Google Scholar] [CrossRef]
- Giuliano, C.; Cerri, S.; Blandini, F. Potential therapeutic effects of polyphenols in Parkinson’s disease: In vivo and in vitro pre-clinical studies. Neural Regen. Res. 2021, 16, 234–241. [Google Scholar] [CrossRef]
- Zhang, F.; Shi, J.S.; Zhou, H.; Wilson, B.; Hong, J.S.; Gao, H.M. Resveratrol protects dopamine neurons against lipopolysaccharide-induced neurotoxicity through its anti-inflammatory actions. Mol. Pharmacol. 2010, 78, 466–477. [Google Scholar] [CrossRef]
- Passeri, E.; Elkhoury, K.; Morsink, M.; Broersen, K.; Linde, M.; Tamayol, A.; Malaplate, C.; Yen, F.T.; Arab-Tehrany, E. Alzheimer’s disease: Treatment strategies and their limitations. Int. J. Mol. Sci. 2022, 23, 13954. [Google Scholar] [CrossRef]
- Alzheimer’s Disease International. World Alzheimer Report 2024 Global Changes in Attitudes to Dementia September. 2024. Available online: https://www.alzint.org/u/World-Alzheimer-Report-2024.pdf (accessed on 26 February 2025).
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s disease: Past, present, and future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef]
- Ahmed, T.F.; Ahmed, A.; Imtiaz, F. History in perspective: How Alzheimer’s disease came to be where it is? Brain Res. 2021, 1758, 147342. [Google Scholar] [CrossRef]
- Alzheimer, A. Uber eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychiatr. 1907, 64, 146–148. [Google Scholar]
- Korolev, I.O. Alzheimer’s disease: A clinical and basic science review. Med. Stud. Res. J. 2014, 4, 24–33. [Google Scholar]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent advancements in pathogenesis, diagnostics and treatment of Alzheimer’s disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Andrews, J.S.; Beach, T.G.; Buracchio, T.; Dunn, B.; Graf, A.; Hansson, O.; Ho, C.; Jagust, W.; McDade, E.; et al. Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s association workgroup. Alzheimer’s Dement. 2024, 20, 5143–5169. [Google Scholar] [CrossRef]
- Nasb, M.; Tao, W.; Chen, N. Alzheimer’s disease puzzle: Delving into pathogenesis hypotheses. Aging Dis. 2024, 15, 43–73. [Google Scholar] [CrossRef]
- Chen, Z.R.; Huang, J.B.; Yang, S.L.; Hong, F.F. Role of cholinergic signaling in Alzheimer’s disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and tau in the pathogenesis of Alzheimer’s disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Van Hoecke, L.; Vandenbroucke, R.E. The impact of systemic inflammation on Alzheimer’s disease pathology. Front. Immunol. 2022, 12, 796867. [Google Scholar] [CrossRef]
- Mangold, C.A.; Wronowski, B.; Du, M.; Masser, D.R.; Hadad, N.; Bixler, G.V.; Brucklacher, R.M.; Ford, M.M.; Sonntag, W.E.; Freeman, W.M. Sexually divergent induction of microglial-associated neuroinflammation with hippocampal aging. J. Neuroinflamm. 2017, 14, 141. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, M. Inflammation in Alzheimer’s disease. Curr. Alzheimer Res. 2020, 17, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Tam, W.W.; Zhang, M.W.; Ho, C.S.; Husain, S.F.; McIntyre, R.S.; Ho, R.C. IL-1β, IL-6, TNF- α and CRP in elderly patients with depression or Alzheimer’s disease: Systematic review and meta-analysis. Sci. Rep. 2018, 8, 12050. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, X.; So, K.F.; Jiang, W.; Chiu, K. Targeting microglia in Alzheimer’s disease: Pathogenesis and potential therapeutic strategies. Biomolecules 2024, 14, 833. [Google Scholar] [CrossRef]
- Patel, N.S.; Paris, D.; Mathura, V.; Quadros, A.N.; Crawford, F.C.; Mullan, M.J. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflamm. 2005, 2, 9. [Google Scholar] [CrossRef]
- Niso-Santano, M.; Fuentes, J.M.; Galluzzi, L. Immunological aspects of central neurodegeneration. Cell Discov. 2024, 10, 41. [Google Scholar] [CrossRef]
- Doens, D.; Fernández, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef]
- Nizami, S.; Hall-Roberts, H.; Warrier, S.; Cowley, S.A.; Di Daniel, E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharmacol. 2019, 176, 3515–3532. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, L.; Fan, Y.; Ji, W. The pathogenesis in Alzheimer’s disease: TREM2 as a potential target. J. Integr. Neurosci. 2023, 22, 150. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Gutierrez, R.A.; Bhat, M.A. Microglia, Trem2, and neurodegeneration. Neuroscientist 2024, 31, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Sun, H.; Cai, Q.; Tai, H.C. The enigma of tau protein aggregation: Mechanistic insights and future challenges. Int. J. Mol. Sci. 2024, 25, 4969. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhi, W.; Wang, L. Role of tau protein in neurodegenerative diseases and development of its targeted drugs: A literature review. Molecules 2024, 29, 2812. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Lane-Donovan, C.; Boxer, A.L. Disentangling tau: One protein, many therapeutic approaches. Neurotherapeutics 2024, 21, e00321. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Kitazawa, M.; Oddo, S.; Yamasaki, T.R.; Green, K.N.; LaFerla, F.M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 8843–8853. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Weston, L.L.; Jiang, S.; Chisholm, D.; Jantzie, L.L.; Bhaskar, K. Interleukin-10 deficiency exacerbates inflammation-induced tau pathology. J. Neuroinflamm. 2021, 18, 161. [Google Scholar] [CrossRef]
- Wojtunik-Kulesza, K.; Rudkowska, M.; Orzeł-Sajdłowska, A. Aducanumab-hope or disappointment for Alzheimer’s disease. Int. J. Mol. Sci. 2023, 24, 4367. [Google Scholar] [CrossRef]
- Kurkinen, M. Lecanemab (Leqembi) is not the right drug for patients with Alzheimer’s disease. Adv. Clin. Exp. Med. 2023, 32, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Buccellato, F.R.; D’Anca, M.; Tartaglia, G.M.; Del Fabbro, M.; Scarpini, E.; Galimberti, D. Treatment of Alzheimer’s disease: Beyond symptomatic therapies. Int. J. Mol. Sci. 2023, 24, 13900. [Google Scholar] [CrossRef]
- Ajenikoko, M.K.; Ajagbe, A.O.; Onigbinde, O.A.; Okesina, A.A.; Tijani, A.A. Review of Alzheimer’s disease drugs and their relationship with neuron-glia interaction. IBRO Neurosci. Rep. 2022, 14, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Kojima, A.; Ishikawa, C.; Arai, K. Anti-inflammatory action of donepezil ameliorates tau pathology, synaptic loss, and neurodegeneration in a tauopathy mouse model. J. Alzheimer’s Dis. 2010, 22, 295–306. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.J.; Park, S.K.; Park, J.H.; Jeong, H.R.; Lee, S.; Lee, H.; Seol, E.; Hoe, H.S. Donepezil regulates LPS and Aβ-stimulated neuroinflammation through MAPK/NLRP3 inflammasome/STAT3 signaling. Int. J. Mol. Sci. 2021, 22, 10637. [Google Scholar] [CrossRef]
- Li, A.; Zhang, J.; Chen, K.; Wang, J.; Xu, A.; Wang, Z. Donepezil attenuates inflammation and apoptosis in ulcerative colitis via regulating LRP1/AMPK/NF-κB signaling. Pathol. Int. 2023, 73, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, E.; Agrawal, R.; Nath, C.; Shukla, R. Effect of anti-dementia drugs on LPS induced neuroinflammation in mice. Life Sci. 2007, 80, 1977–1983. [Google Scholar] [CrossRef]
- Yu, C.; Liu, X.; Ma, B.; Xu, J.; Chen, Y.; Dai, C.; Peng, H.; Zha, D. Novel anti-neuroinflammatory pyranone-carbamate derivatives as selective butyrylcholinesterase inhibitors for treating Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2024, 39, 2313682. [Google Scholar] [CrossRef]
- Jasiecki, J.; Wasąg, B. Butyrylcholinesterase protein ends in the pathogenesis of Alzheimer’s disease-could BCHE genotyping be helpful in Alzheimer’s therapy? Biomolecules 2019, 9, 592. [Google Scholar] [CrossRef]
- Stopschinski, B.E.; Weideman, R.A.; McMahan, D.; Jacob, D.A.; Little, B.B.; Chiang, H.S.; Saez Calveras, N.; Stuve, O. Microglia as a cellular target of diclofenac therapy in Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2023, 16, 17562864231156674. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.; Wang, D.; Zhang, J.; Zhang, F. NSAID exposure and risk of Alzheimer’s disease: An updated meta-analysis from cohort studies. Front. Aging Neurosci. 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology 2021, 29, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Multiple sclerosis pathology. Cold Spring Harb. Perspect. Med. 2018, 8, 028936. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Cree, B.A.C. Treatment of multiple sclerosis: A review. Am. J. Med. 2020, 133, 1380–1390.e2. [Google Scholar] [CrossRef]
- Haki, M.; Al-Biati, H.A.; Al-Tameemi, Z.S.; Ali, I.S.; Al-Hussaniy, H.A. Review of multiple sclerosis: Epidemiology, etiology, pathophysiology, and treatment. Medicine 2024, 103, 37297. [Google Scholar] [CrossRef]
- Liu, R.; Du, S.; Zhao, L.; Jain, S.; Sahay, K.; Rizvanov, A.; Lezhnyova, V.; Khaibullin, T.; Martynova, E.; Khaiboullina, S.; et al. Autoreactive lymphocytes in multiple sclerosis: Pathogenesis and treatment target. Front. Immunol. 2022, 13, 996469. [Google Scholar] [CrossRef]
- Ford, H. Clinical presentation and diagnosis of multiple sclerosis. Clin. Med. 2020, 20, 380–383. [Google Scholar] [CrossRef]
- Charabati, M.; Wheeler, M.A.; Weiner, H.L.; Quintana, F.J. Multiple sclerosis: Neuroimmune crosstalk and therapeutic targeting. Cell 2023, 186, 1309–1327. [Google Scholar] [CrossRef]
- de Sèze, J.; Maillart, E.; Gueguen, A.; Laplaud, D.A.; Michel, L.; Thouvenot, E.; Zephir, H.; Zimmer, L.; Biotti, D.; Liblau, R. Anti-CD20 therapies in multiple sclerosis: From pathology to the clinic. Front. Immunol. 2023, 14, 1004795. [Google Scholar] [CrossRef]
- Theophanous, S.; Sargiannidou, I.; Kleopa, K.A. Glial cells as key regulators in neuroinflammatory mechanisms associated with multiple sclerosis. Int. J. Mol. Sci. 2024, 25, 9588. [Google Scholar] [CrossRef]
- Lai, S.; Wu, X.; Liu, Y.; Liu, B.; Wu, H.; Ma, K. Interaction between Th17 and central nervous system in multiple sclerosis. Brain Behav. Immun. Health 2024, 43, 100928. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Oukka, M.; Kuchroo, V.K. T(H)-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 2007, 8, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Pozzilli, C.; Pugliatti, M.; Vermersch, P.; Grigoriadis, N.; Alkhawajah, M.; Airas, L.; Oreja-Guevara, C. Diagnosis and treatment of progressive multiple sclerosis: A position paper. Eur. J. Neurol. 2023, 30, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, M.; Sharanova, S.; Sviridova, A.; Rogovskii, V.; Murugina, N.; Nikolaeva, A.; Dagil, Y.; Murugin, V.; Ospelnikova, T.; Boyko, A.; et al. The influence of glatiramer acetate on Th17-immune response in multiple sclerosis. PLoS ONE 2020, 15, e0240305. [Google Scholar] [CrossRef]
- Tacke, S.; Braune, S.; Rovituso, D.M.; Ziemssen, T.; Lehmann, P.V.; Dikow, H.; Bergmann, A.; Kuerten, S. B-Cell activity predicts response to glatiramer acetate and interferon in relapsing-remitting multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e980. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.; Hartung, H.P.; Mycko, M.P.; Selmaj, I.; Cross, A.H. MS treatment de-escalation: Review and commentary. J. Neurol. 2024, 271, 6426–6438. [Google Scholar] [CrossRef]
- Lamb, Y.N. Ocrelizumab: A review in multiple sclerosis. Drugs 2022, 82, 323–334. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fołta, J.; Rzepka, Z.; Wrześniok, D. The Role of Inflammation in Neurodegenerative Diseases: Parkinson’s Disease, Alzheimer’s Disease, and Multiple Sclerosis. Int. J. Mol. Sci. 2025, 26, 5177. https://doi.org/10.3390/ijms26115177
Fołta J, Rzepka Z, Wrześniok D. The Role of Inflammation in Neurodegenerative Diseases: Parkinson’s Disease, Alzheimer’s Disease, and Multiple Sclerosis. International Journal of Molecular Sciences. 2025; 26(11):5177. https://doi.org/10.3390/ijms26115177
Chicago/Turabian StyleFołta, Justyna, Zuzanna Rzepka, and Dorota Wrześniok. 2025. "The Role of Inflammation in Neurodegenerative Diseases: Parkinson’s Disease, Alzheimer’s Disease, and Multiple Sclerosis" International Journal of Molecular Sciences 26, no. 11: 5177. https://doi.org/10.3390/ijms26115177
APA StyleFołta, J., Rzepka, Z., & Wrześniok, D. (2025). The Role of Inflammation in Neurodegenerative Diseases: Parkinson’s Disease, Alzheimer’s Disease, and Multiple Sclerosis. International Journal of Molecular Sciences, 26(11), 5177. https://doi.org/10.3390/ijms26115177