
Can Proteomics Play a Significant Role in the Identification of Biomarkers for Alpha1-Antitrypsin Deficiency?

 , ,
, ,  , ,
, ,  and
and
Abstract

1. Introduction
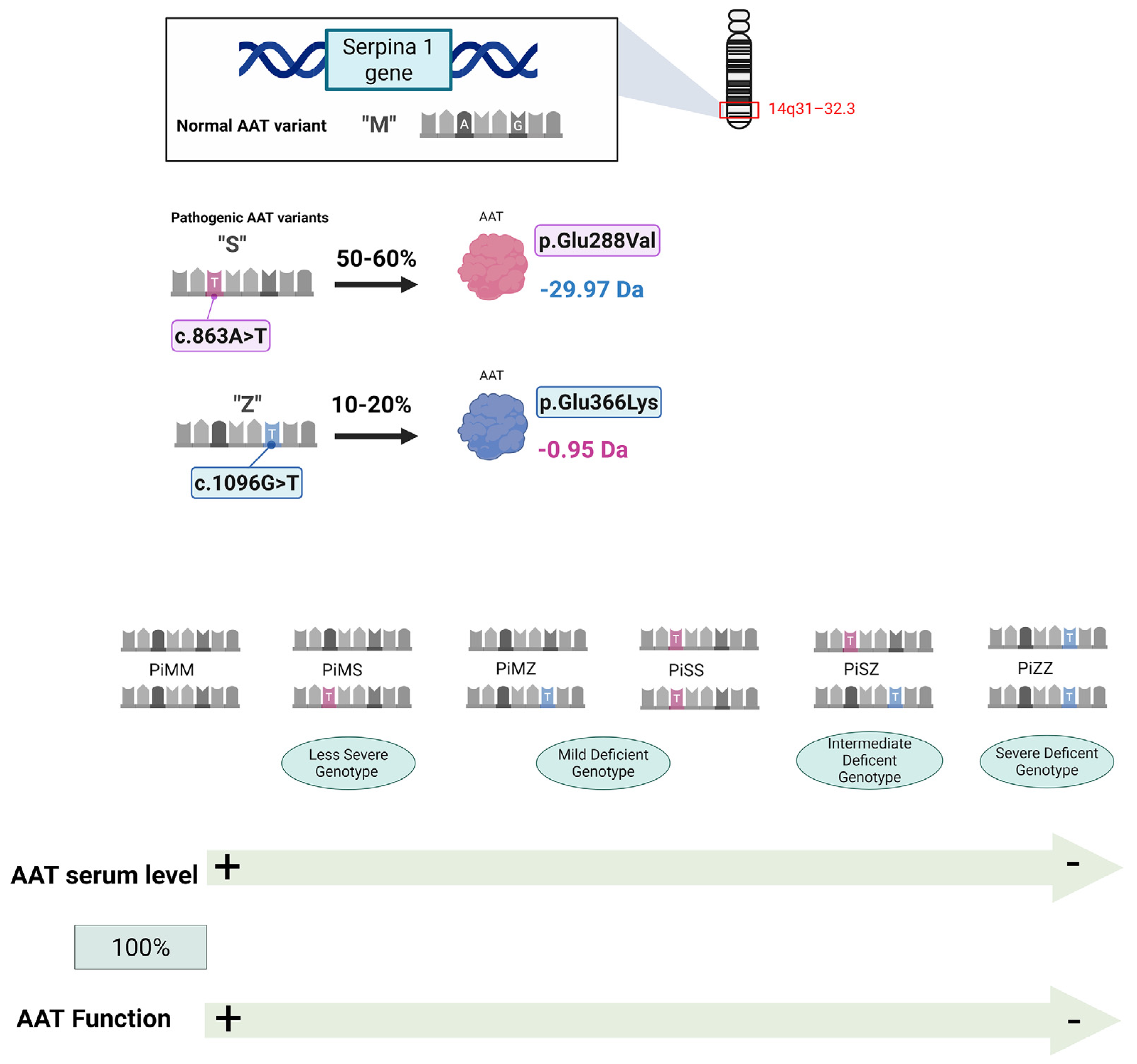
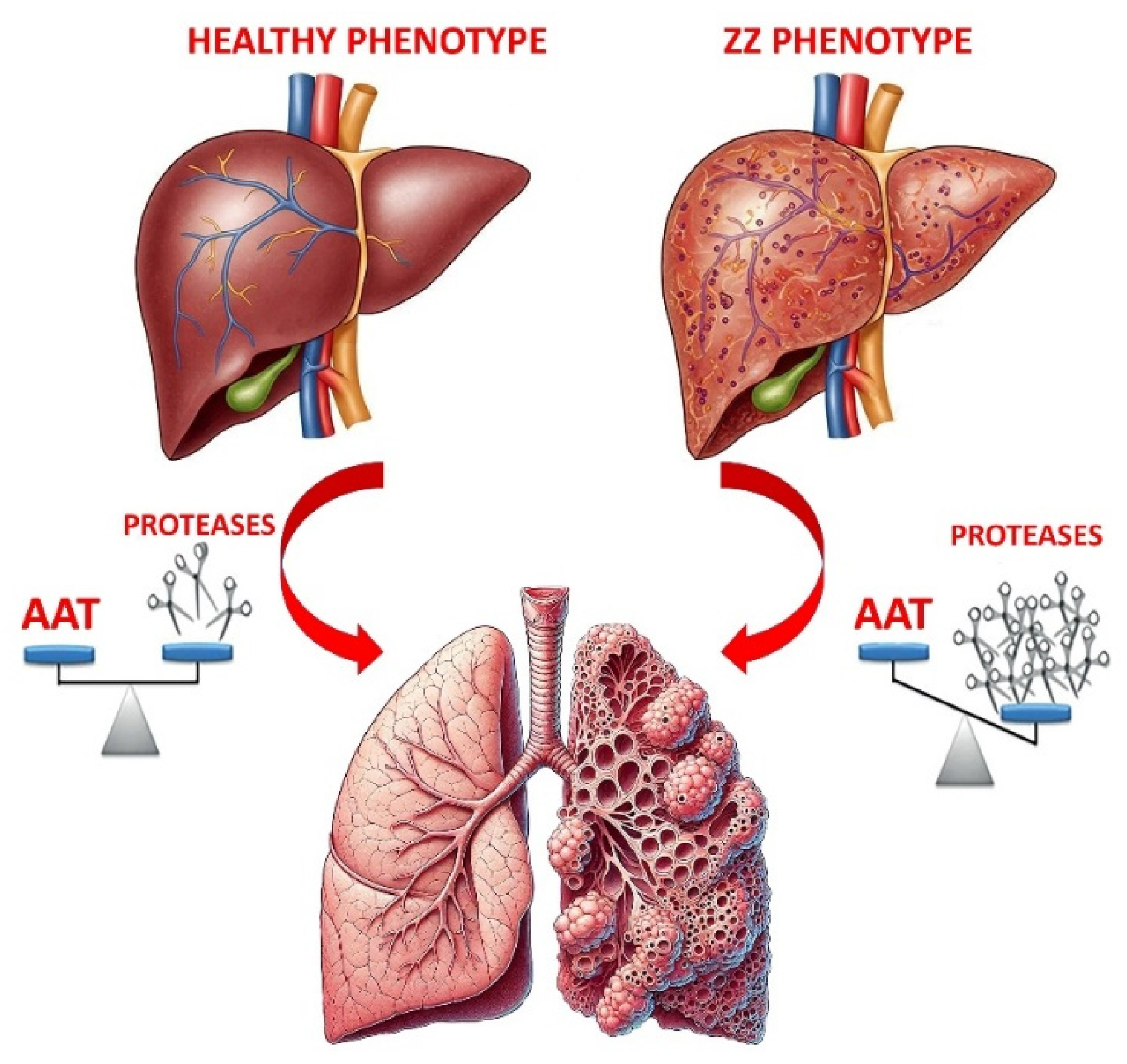
2. Introducing Alpha1-Antitrypsin Deficiency (AATD) and Potential Treatments
3. Proteomics and Biomarker Discovery
4. Proteomic Analysis of AATD
4.1. Application to Lung and Liver Diseases
4.2. Limitations of Current Proteomic Studies
4.3. Unmet Clinical and Research Needs in AATD
5. Is Proteomics Still in the Early Stages of Research on AATD?
5.1. Integrative Multi-Omics Approaches for Predictive Modeling of AATD
5.2. Clinical Translation of Proteomic Biomarkers: Validation and Implementation
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAT | α1-antitrypsin |
| AATD | α1-antitrypsin deficiency |
| COPD | Chronic obstructive pulmonary disease |
| IEF | Isoelectric focusing |
| LC-MS/MS | Liquid chromatography tandem mass spectrometry |
| DIGE | Two-dimensional difference gel electrophoresis |
| IPF | Idiopathic pulmonary fibrosis |
| NS | Non-smokers |
| HS | Healthy smokers |
| ELISA | Enzyme-linked immunosorbent assay |
References
- Viglio, S.; Iadarola, P.; D’Amato, M.; Stolk, J. Methods of Purification and Application Procedures of Alpha1 Antitrypsin: A Long-Lasting History. Molecules 2020, 25, 4014. [Google Scholar] [CrossRef] [PubMed]
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Köhnlein, T.; Welte, T. The Discovery of A1-Antitrypsin and Its Role in Health and Disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef]
- Gettins, P.G.W. Serpin Structure, Mechanism, and Function. Chem. Rev. 2002, 102, 4751–4804. [Google Scholar] [CrossRef]
- Ferrarotti, I.; Wencker, M.; Chorostowska-Wynimko, J. Rare Variants in Alpha 1 Antitrypsin Deficiency: A Systematic Literature Review. Orphanet J. Rare Dis. 2024, 19, 82. [Google Scholar] [CrossRef]
- Foil, K.E. Variants of SERPINA1 and the Increasing Complexity of Testing for Alpha-1 Antitrypsin Deficiency. Ther. Adv. Chronic Dis. 2021, 12. [Google Scholar] [CrossRef]
- de Serres, F.J.; Blanco, I. Prevalence of A1-Antitrypsin Deficiency Alleles PI*S and PI*Z Worldwide and Effective Screening for Each of the Five Phenotypic Classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: A Comprehensive Review. Ther. Adv. Respir. Dis. 2012, 6, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Teckman, J.H.; Buchanan, P.; Blomenkamp, K.S.; Heyer-Chauhan, N.; Burling, K.; Lomas, D.A. Biomarkers Associated With Future Severe Liver Disease in Children With Alpha-1-Antitrypsin Deficiency. Gastro Hep Adv. 2024, 3, 842–850. [Google Scholar] [CrossRef]
- Miravitlles, M.; Turner, A.M.; Sucena, M.; Mornex, J.-F.; Greulich, T.; Wencker, M.; McElvaney, N.G. Assessment and Monitoring of Lung Disease in Patients with Severe Alpha 1 Antitrypsin Deficiency: A European Delphi Consensus of the EARCO Group. Respir. Res. 2024, 25, 318. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Crisford, H.; Scott, A.; Sapey, E.; Stockley, R.A. A Novel in Vitro Cell Model of the Proteinase/Antiproteinase Balance Observed in Alpha-1 Antitrypsin Deficiency. Front. Pharmacol. 2024, 15, 1421598. [Google Scholar] [CrossRef]
- Cagnone, M.; Piloni, D.; Ferrarotti, I.; Di Venere, M.; Viglio, S.; Magni, S.; Bardoni, A.; Salvini, R.; Fumagalli, M.; Iadarola, P.; et al. A Pilot Study to Investigate the Balance between Proteases and A1-Antitrypsin in Bronchoalveolar Lavage Fluid of Lung Transplant Recipients. High Throughput 2019, 8, 5. [Google Scholar] [CrossRef]
- Stockley, R.A.; Parr, D.G. Antitrypsin Deficiency: Still More to Learn about the Lung after 60 Years. ERJ Open Res. 2024, 10, 00139–02024. [Google Scholar] [CrossRef]
- Mulkareddy, V.; Roman, J. Pulmonary Manifestations of Alpha 1 Antitrypsin Deficiency. Am. J. Med. Sci. 2024, 368, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mornex, J.-F.; Traclet, J.; Guillaud, O.; Dechomet, M.; Lombard, C.; Ruiz, M.; Revel, D.; Reix, P.; Cottin, V. Alpha1-Antitrypsin Deficiency: An Updated Review. Presse Med. 2023, 52, 104170. [Google Scholar] [CrossRef]
- Marin-Hinojosa, C.; Fatela-Cantillo, D.; Lopez-Campos, J.L. Measuring of Alpha-1 Antitrypsin Concentration by Nephelometry or Turbidimetry. Methods Mol. Biol. 2024, 2750, 123–133. [Google Scholar] [CrossRef]
- Scarlata, S.; Ottaviani, S.; Villa, A.; Baglioni, S.; Basile, F.; Annunziata, A.; Santangelo, S.; Francesconi, M.; Arcoleo, F.; Balderacchi, A.M.; et al. Improving the Diagnosis of AATD with Aid of Serum Protein Electrophoresis: A Prospective, Multicentre, Validation Study. Clin. Chem. Lab. Med. 2024, 62, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Strnad, P.; Brantly, M.L.; Bals, R. Alpha-1-Antitrypsin Deficiency. In ERS Monograph, 1st ed.; European Respiratory Society: Sheffield, UK, 2019; ISBN 978-1-84984-108-5. [Google Scholar]
- Gruntman, A.M.; Xue, W.; Flotte, T.R. Alpha-1 Antitrypsin Deficiency. Methods Mol. Biol. 2024, 2750, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dasí, F. Alpha-1 Antitrypsin Deficiency. Med. Clin. 2024, 162, 336–342. [Google Scholar] [CrossRef]
- Quinn, M.; Ellis, P.; Pye, A.; Turner, A.M. Obstacles to Early Diagnosis and Treatment of Alpha-1 Antitrypsin Deficiency: Current Perspectives. Ther. Clin. Risk Manag. 2020, 16, 1243–1255. [Google Scholar] [CrossRef]
- Torres-Durán, M.; Lopez-Campos, J.L.; Barrecheguren, M.; Miravitlles, M.; Martinez-Delgado, B.; Castillo, S.; Escribano, A.; Baloira, A.; Navarro-Garcia, M.M.; Pellicer, D.; et al. Alpha-1 Antitrypsin Deficiency: Outstanding Questions and Future Directions. Orphanet J. Rare Dis. 2018, 13, 114. [Google Scholar] [CrossRef]
- Kemp, J.; Ladwig, P.M.; Snyder, M.R. Alpha-1-Antitrypsin (A1AT) Proteotyping by LC-MS/MS. Methods Mol. Biol. 2024, 2750, 95–106. [Google Scholar] [CrossRef]
- Feitosa, P.H. Diagnosis and Augmentation Therapy for Alpha-1 Antitrypsin Deficiency: Current Knowledge and Future Potential. Drugs Context 2023, 12, 2023-3-1. [Google Scholar] [CrossRef]
- Brantly, M. Treatment for Alpha-1 Antitrypsin Deficiency: Does Augmentation Therapy Work? Am. J. Respir. Crit. Care Med. 2023, 208, 948–949. [Google Scholar] [CrossRef]
- Mischak, H.; Ioannidis, J.P.A.; Argiles, A.; Attwood, T.K.; Bongcam-Rudloff, E.; Broenstrup, M.; Charonis, A.; Chrousos, G.P.; Delles, C.; Dominiczak, A.; et al. Implementation of Proteomic Biomarkers: Making It Work. Eur. J. Clin. Investig. 2012, 42, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Birhanu, A.G. Mass Spectrometry-Based Proteomics as an Emerging Tool in Clinical Laboratories. Clin. Proteom. 2023, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, J.S. Protein Identification Using MS/MS Data. J. Proteom. 2011, 74, 1842–1851. [Google Scholar] [CrossRef]
- Serban, K.A.; Pratte, K.A.; Strange, C.; Sandhaus, R.A.; Turner, A.M.; Beiko, T.; Spittle, D.A.; Maier, L.; Hamzeh, N.; Silverman, E.K.; et al. Unique and Shared Systemic Biomarkers for Emphysema in Alpha-1 Antitrypsin Deficiency and Chronic Obstructive Pulmonary Disease. eBioMedicine 2022, 84, 104262. [Google Scholar] [CrossRef] [PubMed]
- Candia, J.; Cheung, F.; Kotliarov, Y.; Fantoni, G.; Sellers, B.; Griesman, T.; Huang, J.; Stuccio, S.; Zingone, A.; Ryan, B.M.; et al. Assessment of Variability in the SOMAscan Assay. Sci. Rep. 2017, 7, 14248. [Google Scholar] [CrossRef] [PubMed]
- Beiko, T.; Janech, M.G.; Alekseyenko, A.V.; Atkinson, C.; Coxson, H.O.; Barth, J.L.; Stephenson, S.E.; Wilson, C.L.; Schnapp, L.M.; Barker, A.; et al. Serum Proteins Associated with Emphysema Progression in Severe Alpha-1 Antitrypsin Deficiency. Chronic Obstr. Pulm. Dis. 2017, 4, 204–216. [Google Scholar] [CrossRef]
- Karatas, E.; Raymond, A.-A.; Leon, C.; Dupuy, J.-W.; Di-Tommaso, S.; Senant, N.; Collardeau-Frachon, S.; Ruiz, M.; Lachaux, A.; Saltel, F.; et al. Hepatocyte Proteomes Reveal the Role of Protein Disulfide Isomerase 4 in Alpha 1-Antitrypsin Deficiency. JHEP Rep. 2021, 3, 100297. [Google Scholar] [CrossRef] [PubMed]
- Ohlmeier, S.; Nieminen, P.; Gao, J.; Kanerva, T.; Rönty, M.; Toljamo, T.; Bergmann, U.; Mazur, W.; Pulkkinen, V. Lung Tissue Proteomics Identifies Elevated Transglutaminase 2 Levels in Stable Chronic Obstructive Pulmonary Disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L1155–L1165. [Google Scholar] [CrossRef]
- Murphy, M.P.; McEnery, T.; McQuillan, K.; McElvaney, O.F.; McElvaney, O.J.; Landers, S.; Coleman, O.; Bussayajirapong, A.; Hawkins, P.; Henry, M.; et al. A1 Antitrypsin Therapy Modulates the Neutrophil Membrane Proteome and Secretome. Eur. Respir. J. 2020, 55, 1901678. [Google Scholar] [CrossRef] [PubMed]
- Bergin, D.A.; Reeves, E.P.; Meleady, P.; Henry, M.; McElvaney, O.J.; Carroll, T.P.; Condron, C.; Chotirmall, S.H.; Clynes, M.; O’Neill, S.J.; et al. α-1 Antitrypsin Regulates Human Neutrophil Chemotaxis Induced by Soluble Immune Complexes and IL-8. J. Clin. Investig. 2010, 120, 4236–4250. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Ferrari, F.; Luisetti, M.; Stolk, J.; Hiemstra, P.S.; Capuano, D.; Viglio, S.; Fregonese, L.; Cerveri, I.; Corana, F.; et al. Profiling the Proteome of Exhaled Breath Condensate in Healthy Smokers and COPD Patients by LC-MS/MS. Int. J. Mol. Sci. 2012, 13, 13894–13910. [Google Scholar] [CrossRef]
- Airoldi, C.; Ciaramelli, C.; Fumagalli, M.; Bussei, R.; Mazzoni, V.; Viglio, S.; Iadarola, P.; Stolk, J. 1H NMR To Explore the Metabolome of Exhaled Breath Condensate in A1-Antitrypsin Deficient Patients: A Pilot Study. J. Proteome Res. 2016, 15, 4569–4578. [Google Scholar] [CrossRef]
- de Serres, F.J. Alpha-1 Antitrypsin Deficiency Is Not a Rare Disease but a Disease That Is Rarely Diagnosed. Environ. Health Perspect. 2003, 111, 1851–1854. [Google Scholar] [CrossRef]
- Cortes-Lopez, R.; Barjaktarevic, I. Alpha-1 Antitrypsin Deficiency: A Rare Disease? Curr. Allergy Asthma Rep. 2020, 20, 51. [Google Scholar] [CrossRef]
- Smith, G.; Singh, K. Alpha-1 Antitrypsin Deficiency: Navigating Challenges Through Collaborative Innovation. Chest 2024, 166, 1288–1290. [Google Scholar] [CrossRef]
- Park, H.Y.; Churg, A.; Wright, J.L.; Li, Y.; Tam, S.; Man, S.F.P.; Tashkin, D.; Wise, R.A.; Connett, J.E.; Sin, D.D. Club Cell Protein 16 and Disease Progression in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2013, 188, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Spittle, D.A.; Mansfield, A.; Pye, A.; Turner, A.M.; Newnham, M. Predicting Lung Function Using Biomarkers in Alpha-1 Antitrypsin Deficiency. Biomedicines 2023, 11, 2001. [Google Scholar] [CrossRef]
- Moll, M.; Hobbs, B.D.; Pratte, K.A.; Zhang, C.; Ghosh, A.J.; Bowler, R.P.; Lomas, D.A.; Silverman, E.K.; DeMeo, D.L. Assessing Inflammatory Protein Biomarkers in COPD Subjects with and without Alpha-1 Antitrypsin Deficiency. Am. J. Respir. Crit. Care Med. 2025, 211, A3191. [Google Scholar] [CrossRef]
- Donato, L.J.; Karras, R.M.; Katzmann, J.A.; Murray, D.L.; Snyder, M.R. Quantitation of Circulating Wild-Type Alpha-1-Antitrypsin in Heterozygous Carriers of the S and Z Deficiency Alleles. Respir. Res. 2015, 16, 96. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Snyder, M.R.; Zhu, Y.; Tostrud, L.J.; Benson, L.M.; Katzmann, J.A.; Bergen, H.R. Simultaneous Phenotyping and Quantification of α-1-Antitrypsin by Liquid Chromatography-Tandem Mass Spectrometry. Clin. Chem. 2011, 57, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, N.; Oshins, R.; Gu, T.; Clark, V.; Lascano, J.; Assarzadegan, N.; Marek, G.; Brantly, M.; Khodayari, N. Liver Characterization of a Cohort of Alpha-1 Antitrypsin Deficiency Patients with and without Lung Disease. J. Clin. Transl. Hepatol. 2024, 12, 845–856. [Google Scholar] [CrossRef]
- Pérez-Luz, S.; Lalchandani, J.; Matamala, N.; Barrero, M.J.; Gil-Martín, S.; Saz, S.R.; Varona, S.; Monzón, S.; Cuesta, I.; Justo, I.; et al. Quantitative Lipid Profiling Reveals Major Differences between Liver Organoids with Normal Pi*M and Deficient Pi*Z Variants of Alpha-1-antitrypsin. Int. J. Mol. Sci. 2023, 24, 12472. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.A.; Chilet-Rosell, E.; Hernández-Aguado, I.; Pastor-Valero, M.; Gea, S.; Lumbreras, B. Diagnostic Biomarkers: Are We Moving from Discovery to Clinical Application? Clin. Chem. 2018, 64, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Rifai, N.; Gillette, M.A.; Carr, S.A. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 24, 971–983. [Google Scholar] [CrossRef]
- Jardim, J.R.; Casas-Maldonado, F.; Fernandes, F.L.A.; de Castellano, M.V.C.O.; Torres-Durán, M.; Miravitlles, M. Update on and Future Perspectives for the Diagnosis of Alpha-1 Antitrypsin Deficiency in Brazil. J. Bras. Pneumol. 2021, 47, e20200380. [Google Scholar] [CrossRef]
- Erion, D.M.; Liu, L.Y.; Brown, C.R.; Rennard, S.; Farah, H. Editing approaches to treat alpha1-antitrypsin deficiency. Chest 2025, 167, 444–452. [Google Scholar] [CrossRef]
- Abreu, N.; Pereira, V.M.; Pestana, M.; Jasmins, L. Future Perspectives in the Diagnosis and Treatment of Liver Disease Associated with Alpha-1 Antitrypsin Deficiency. GE Port. J. Gastroenterol. 2023, 30, 327–335. [Google Scholar] [CrossRef]
- McCarthy, C.; Saldova, R.; O’Brien, M.E.; Bergin, D.A.; Carroll, T.P.; Keenan, J.; Meleady, P.; Henry, M.; Clynes, M.; Rudd, P.M.; et al. Increased Outer Arm and Core Fucose Residues on the N-Glycans of Mutated Alpha-1 Antitrypsin Protein from Alpha-1 Antitrypsin Deficient Individuals. J. Proteome Res. 2014, 13, 596–605. [Google Scholar] [CrossRef]
- He, N.; Liu, X.; Vegter, A.R.; Evans, T.I.A.; Gray, J.S.; Guo, J.; Moll, S.R.; Guo, L.J.; Luo, M.; Ma, N.; et al. Ferret Models of Alpha-1 Antitrypsin Deficiency Develop Lung and Liver Disease. JCI Insight 2022, 7, e143004. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Subjects Investigated * | Source | Proteomic Technique | Main Findings |
|---|---|---|---|---|
| [21] | P = 5 | Serum | LC-ESI-Triple TOF-MS | MS can easily identify S/Z mutations by detecting mass differences between S/Z and non-S/non-Z peptides. Combining peptide pattern analysis with AAT quantification via immunoassay ensures an accurate assessment of deficiency alleles in most patients. |
| [27] | C = 5607 (COPD) P = 317 (GRADS = 133 QUANTUM-1 = 38 Birmingham = 146) | Plasma | SomaScan v4.0 | Common plasma biomarkers have been identified in both AATD and COPD patients, along with proteins associated with emphysema. PiZZ patients also exhibited biomarkers related to DLCO and emphysema. Additionally, PiZZ individuals undergoing augmentation therapy showed near-normal AAT levels. |
| [29] | P = 31 (QUANTUM-1) | Serum | Multiplexed immunoassay | Proteome analyses revealed that C-reactive protein, adipocyte fatty acid-binding protein, and tissue plasminogen activator were linked to emphysema progression, highlighting them as potential therapeutic targets for COPD. |
| [30] | C = 7 P = 8 | FFPE liver tissues | nanoLC-ESI-Q-Exactive hybrid quadrupole-Orbitrap-MS | Among the 65 proteins upregulated exclusively in adult PiZZ samples, protein disulfide isomerase A4 (PDIA4) emerged as a promising therapeutic target. Its inhibition by cysteamine reduced Z-aggregate formation, while its silencing decreased oxidative stress, a hallmark of AATD-related liver disease. |
| [31] | C = 43 (NS = 9 SM without COPD = 9 SM with COPD stage I–II= 8, SM with COPD stage III–IV = 8 IPF = 9) P = 8 | Lung tissue | 2D-DIGE MALDI-TOF/TOF | The proteome analyses showed increased transglutaminase 2 (TGM2) across all sample types, reinforcing its potential as a diagnostic and therapeutic target for AATD-associated COPD. |
| [32] | C = 37 (HC = 30 COPD = 7) P = 31 (AATD = 6 COPD-AATD = 25) | Neutrophils Plasma | LC-MS/MS | AAT augmentation therapy influences the neutrophil membrane proteome by altering the levels of membrane-associated proteins in circulating neutrophils of AATD-COPD patients. |
| [34] | C = 60 (NS = 25 SM = 20 COPD = 15) AATD = 23 | EBC | ESI-LTQ-Orbitrap-MS SELDI-TOF | Several inflammatory cytokines, type I and II cytokeratins, two isoforms of surfactant protein A (SP-A), calgranulins A and B, and AAT have been identified in the COPD and AATD groups. |
| [35] | HC = 11 P = 11 | EBC | NMR | The analyses revealed that pyruvate metabolism is the most prominently involved pathway, with most metabolites originating from pyruvate. |
| Category | Unmet Need | Rationale |
|---|---|---|
| Biomarkers | Pediatric biomarkers | Lack of validated biomarkers to enable early diagnosis in infants and young children. |
| Prognostic biomarkers | Identification of markers that can predict disease progression and complications. | |
| Biomarkers to guide therapy | Lack of markers that inform therapeutic decision-making and response assessment | |
| Standardization/Screening | Robust liver-disease correlates | Need for non-invasive, reliable markers that correlate with liver disease severity and progression. |
| Pre-analytical protocols | Absence of standardized procedures for sample collection and handling in proteomic studies on AATD | |
| Broad implementation of screening strategies | Insufficient screening, particularly in asymptomatic individuals and family members |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grignano, M.A.; D’Amato, M.; Gregorini, M.; Rampino, T.; Iadarola, P.; Viglio, S. Can Proteomics Play a Significant Role in the Identification of Biomarkers for Alpha1-Antitrypsin Deficiency? Int. J. Mol. Sci. 2025, 26, 5085. https://doi.org/10.3390/ijms26115085
Grignano MA, D’Amato M, Gregorini M, Rampino T, Iadarola P, Viglio S. Can Proteomics Play a Significant Role in the Identification of Biomarkers for Alpha1-Antitrypsin Deficiency? International Journal of Molecular Sciences. 2025; 26(11):5085. https://doi.org/10.3390/ijms26115085
Chicago/Turabian StyleGrignano, Maria Antonietta, Maura D’Amato, Marilena Gregorini, Teresa Rampino, Paolo Iadarola, and Simona Viglio. 2025. "Can Proteomics Play a Significant Role in the Identification of Biomarkers for Alpha1-Antitrypsin Deficiency?" International Journal of Molecular Sciences 26, no. 11: 5085. https://doi.org/10.3390/ijms26115085
APA StyleGrignano, M. A., D’Amato, M., Gregorini, M., Rampino, T., Iadarola, P., & Viglio, S. (2025). Can Proteomics Play a Significant Role in the Identification of Biomarkers for Alpha1-Antitrypsin Deficiency? International Journal of Molecular Sciences, 26(11), 5085. https://doi.org/10.3390/ijms26115085