
Biological Actions of Bile Acids via Cell Surface Receptors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Biosynthesis of Various Bile Acids
3. Bile Acid Membrane Receptors
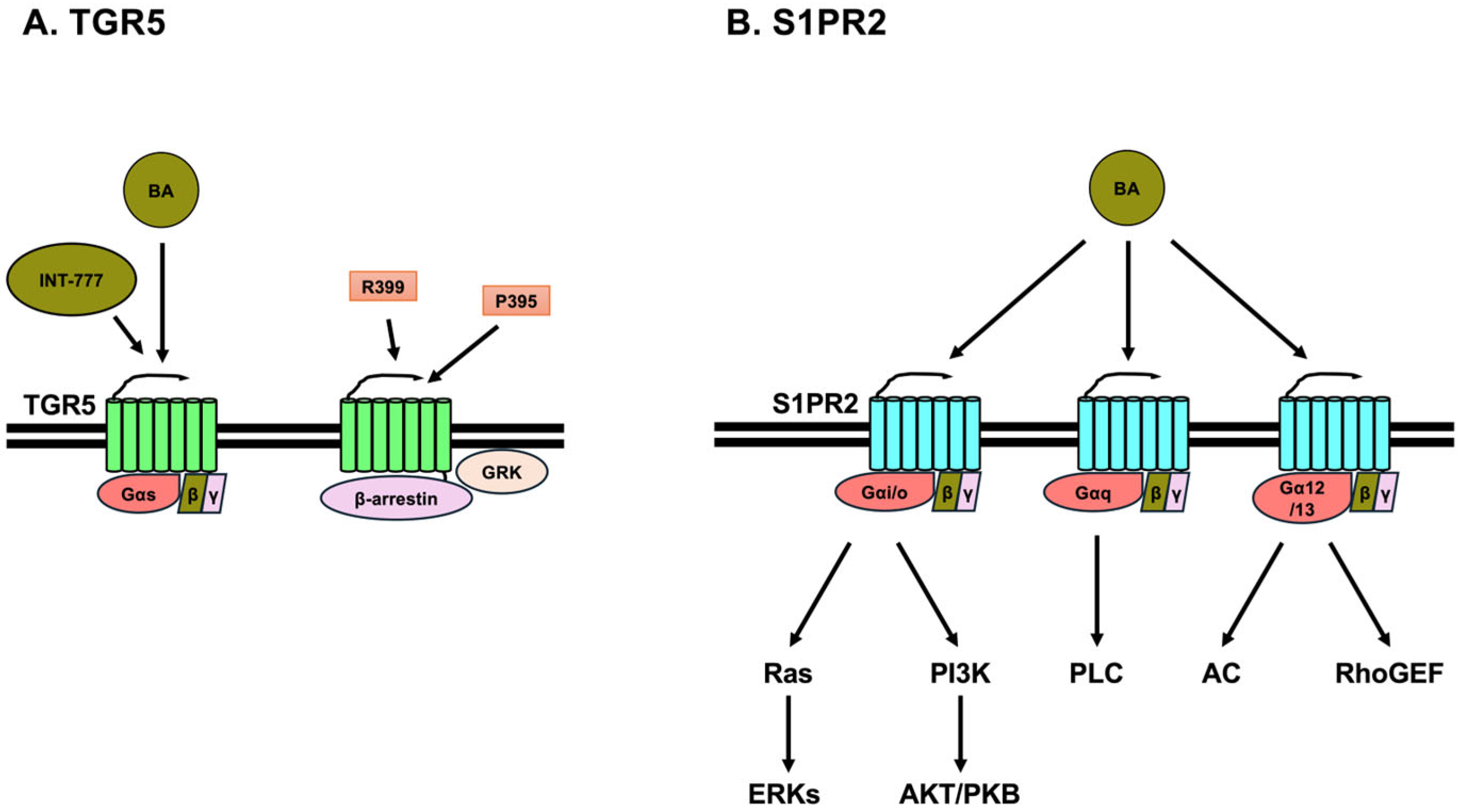
3.1. TGR5
3.2. Sphingosine-1 Phosphate Receptor 2 (S1PR2)
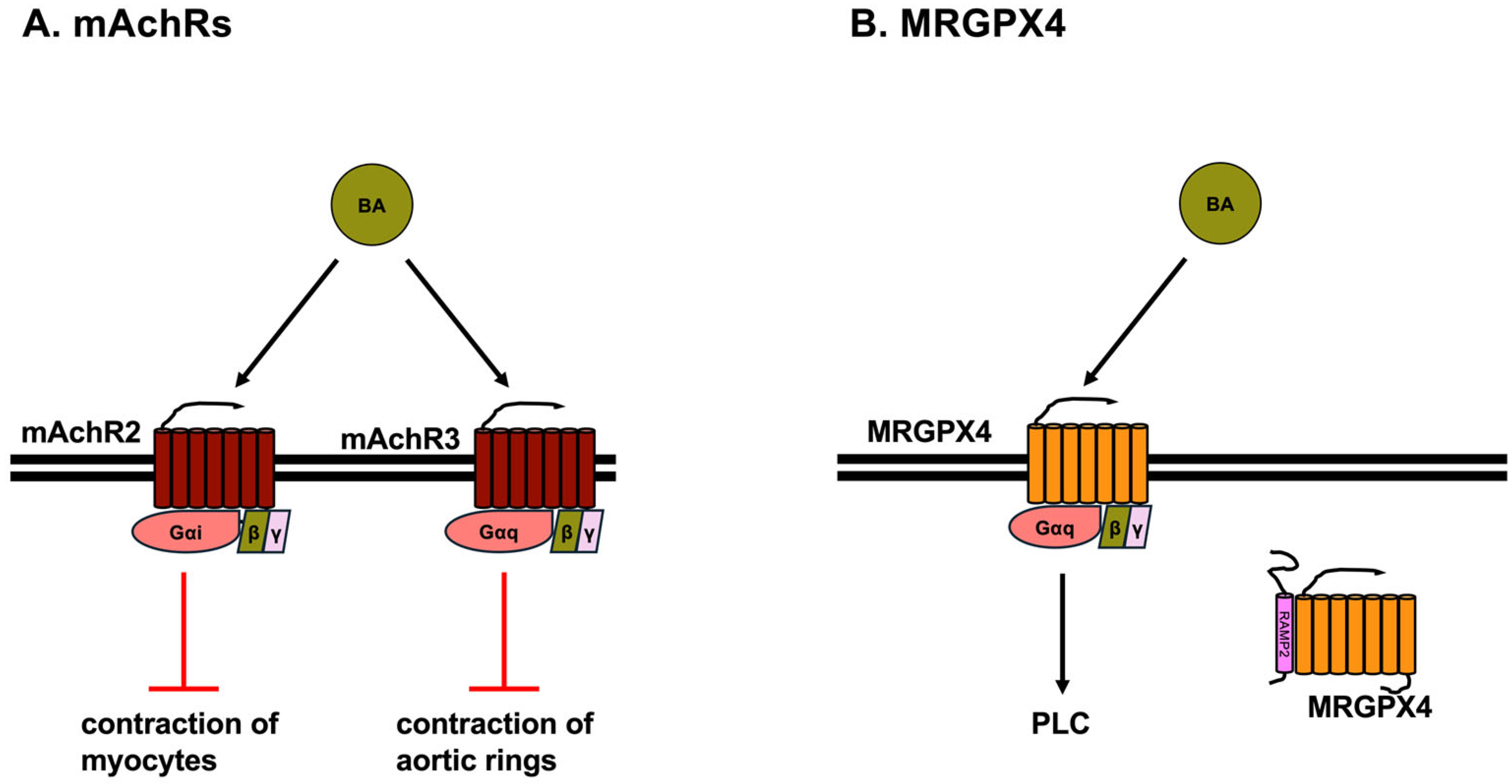
3.3. Muscarinic Acetylcholine Receptors (mAChRs)
3.4. MRGPRX4
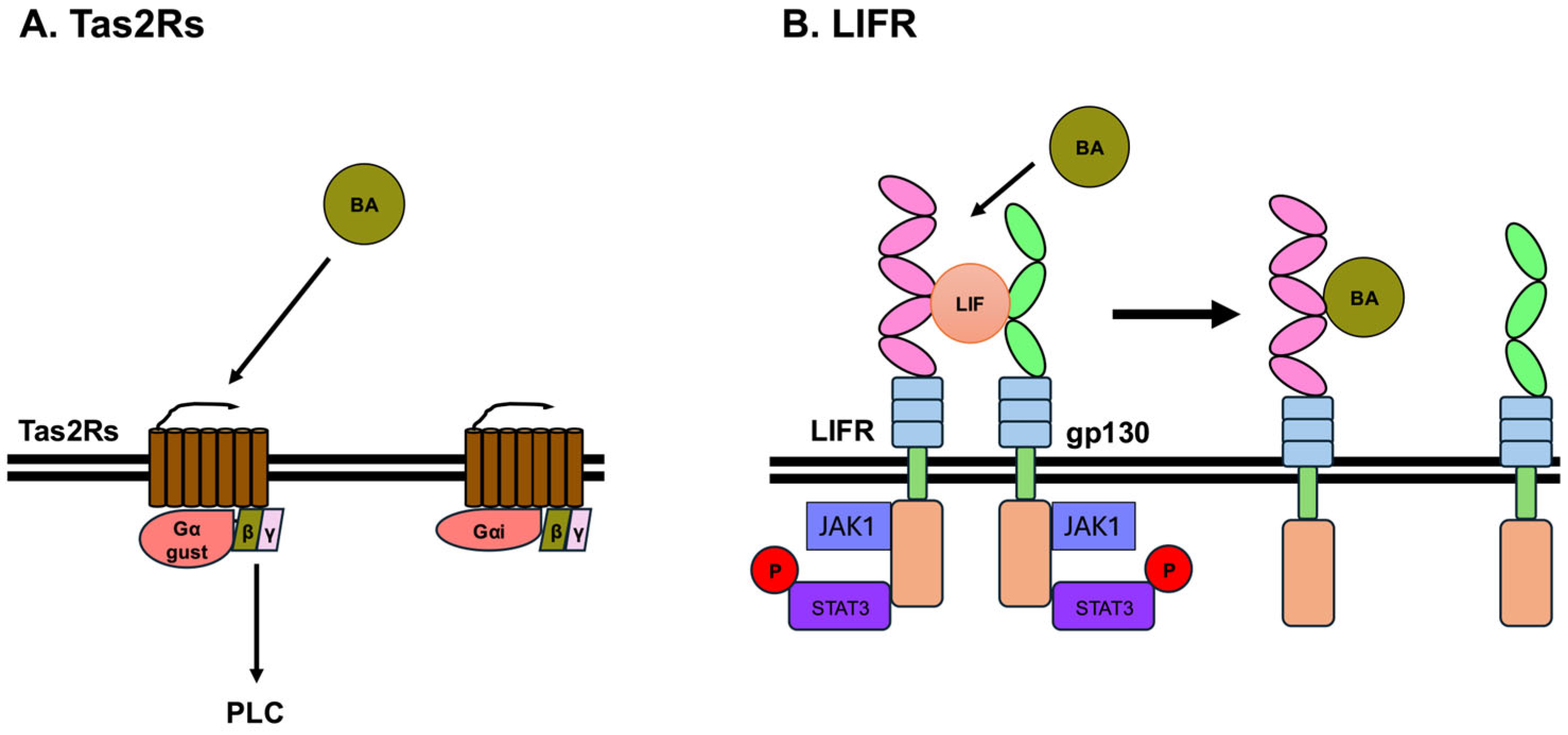
3.5. Bitter Taste Receptors
3.6. Leukemia Inhibitory Factor Receptor (LIFR)
4. Potential Medicines Targeting Cell Surface Receptors for Bile Acids
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Russell, D.W. Fifty years of advances in bile acid synthesis and metabolism. J. Lipid Res. 2009, 50, S120–S125. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic Roles of Bile Acids in Metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Gaskins, H.R. Another renaissance for bile acid gastrointestinal microbiology. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Demagny, H.; Velazquez-Villegas, L.; Schoonjans, K. Molecular Physiology of Bile Acid Signaling in Health, Disease, and Aging. Physiol. Rev. 2021, 101, 683–731. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. Role of Microbiota-Modified Bile Acids in the Regulation of Intracellular Organelles and Neurodegenerative Diseases. Genes 2023, 14, 825. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. The Biosynthesis, Signaling, and Neurological Functions of Bile Acids. Biomolecules 2019, 9, 232. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. Physiological Role of Bile Acids Modified by the Gut Microbiome. Microorganisms 2021, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. The Role of Gut Microbiota-Derived Lithocholic Acid, Deoxycholic Acid and Their Derivatives on the Function and Differentiation of Immune Cells. Microorganisms 2023, 11, 2730. [Google Scholar] [CrossRef]
- Pandak, W.M.; Kakiyama, G. The acidic pathway of bile acid synthesis: Not just an alternative pathway(☆). Liver Res. 2019, 3, 88–98. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Up to date on cholesterol 7 alpha-hydroxylase (CYP7A1) in bile acid synthesis. Liver Res. 2020, 4, 47–63. [Google Scholar] [CrossRef]
- Xue, R.; Su, L.; Lai, S.; Wang, Y.; Zhao, D.; Fan, J.; Chen, W.; Hylemon, P.B.; Zhou, H. Bile Acid Receptors and the Gut-Liver Axis in Nonalcoholic Fatty Liver Disease. Cells 2021, 10, 2806. [Google Scholar] [CrossRef] [PubMed]
- Styles, N.A.; Shonsey, E.M.; Falany, J.L.; Guidry, A.L.; Barnes, S.; Falany, C.N. Carboxy-terminal mutations of bile acid CoA:<em>N</em>-acyltransferase alter activity and substrate specificity. J. Lipid Res. 2016, 57, 1133–1143. [Google Scholar] [PubMed]
- Daly, J.W.; Keely, S.J.; Gahan, C.G.M. Functional and Phylogenetic Diversity of BSH and PVA Enzymes. Microorganisms 2021, 9, 732. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Cowley, E.S.; Wolf, P.G.; Doden, H.L.; Murai, T.; Caicedo, K.Y.O.; Ly, L.K.; Sun, F.; Takei, H.; Nittono, H.; et al. Formation of secondary allo-bile acids by novel enzymes from gut Firmicutes. Gut Microbes 2022, 14, 2132903. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Daniel, S.L.; Gaskins, H.R. The Hylemon-Björkhem pathway of bile acid 7-dehydroxylation: History, biochemistry, and microbiology. J. Lipid Res. 2023, 64, 100392. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, T.; Lu, X.; Lan, X.; Chen, Z.; Lu, S. G protein-coupled receptors (GPCRs): Advances in structures, mechanisms, and drug discovery. Signal Transduct. Target. Ther. 2024, 9, 88. [Google Scholar] [CrossRef]
- Scharf, M.M.; Humphrys, L.J.; Berndt, S.; Di Pizio, A.; Lehmann, J.; Liebscher, I.; Nicoli, A.; Niv, M.Y.; Peri, L.; Schihada, H.; et al. The dark sides of the GPCR tree—Research progress on understudied GPCRs. Br. J. Pharmacol. 2024. [Google Scholar] [CrossRef]
- Foster, S.R.; Hauser, A.S.; Vedel, L.; Strachan, R.T.; Huang, X.-P.; Gavin, A.C.; Shah, S.D.; Nayak, A.P.; Haugaard-Kedström, L.M.; Penn, R.B.; et al. Discovery of Human Signaling Systems: Pairing Peptides to G Protein-Coupled Receptors. Cell 2019, 179, 895–908.e21. [Google Scholar] [CrossRef]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef]
- Maruyama, T.; Tanaka, K.; Suzuki, J.; Miyoshi, H.; Harada, N.; Nakamura, T.; Miyamoto, Y.; Kanatani, A.; Tamai, Y. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J. Endocrinol. 2006, 191, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Zuo, G.; Zhang, T.; Huang, L.; Araujo, C.; Peng, J.; Travis, Z.; Okada, T.; Ocak, U.; Zhang, G.; Tang, J.; et al. Activation of TGR5 with INT-777 attenuates oxidative stress and neuronal apoptosis via cAMP/PKCepsilon/ALDH2 pathway after subarachnoid hemorrhage in rats. Free Radic. Biol. Med. 2019, 143, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Keitel, V.; Gorg, B.; Bidmon, H.J.; Zemtsova, I.; Spomer, L.; Zilles, K.; Haussinger, D. The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia 2010, 58, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, X.; Ge, Y.; Ma, L.; Chen, Q.; Liu, H.; Du, Y.; Ye, R.D.; Hu, H.; Ren, R. Cryo-EM structure of activated bile acids receptor TGR5 in complex with stimulatory G protein. Signal Transduct. Target. Ther. 2020, 5, 142. [Google Scholar] [CrossRef]
- Ahmed, M.B.; Alghamdi, A.A.A.; Islam, S.U.; Lee, J.S.; Lee, Y.S. cAMP Signaling in Cancer: A PKA-CREB and EPAC-Centric Approach. Cells 2022, 11, 2020. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.; Liu, J.; Chen, J.; Wang, J.; Hua, H.; Jiang, Y. cAMP-PKA/EPAC signaling and cancer: The interplay in tumor microenvironment. J. Hematol. Oncol. 2024, 17, 5. [Google Scholar] [CrossRef]
- Hong, J.; Behar, J.; Wands, J.; Resnick, M.; Wang, L.J.; DeLellis, R.A.; Lambeth, D.; Souza, R.F.; Spechler, S.J.; Cao, W. Role of a novel bile acid receptor TGR5 in the development of oesophageal adenocarcinoma. Gut 2010, 59, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, A.I.; Huang, B.Q.; Radtke, B.N.; Gajdos, G.B.; Splinter, P.L.; Masyuk, T.V.; Gradilone, S.A.; LaRusso, N.F. Ciliary subcellular localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G1013–G1024. [Google Scholar] [CrossRef]
- Kumar, D.P.; Asgharpour, A.; Mirshahi, F.; Park, S.H.; Liu, S.; Imai, Y.; Nadler, J.L.; Grider, J.R.; Murthy, K.S.; Sanyal, A.J. Activation of Transmembrane Bile Acid Receptor TGR5 Modulates Pancreatic Islet alpha Cells to Promote Glucose Homeostasis. J. Biol. Chem. 2016, 291, 6626–6640. [Google Scholar] [CrossRef]
- Kida, T.; Tsubosaka, Y.; Hori, M.; Ozaki, H.; Murata, T. Bile acid receptor TGR5 agonism induces NO production and reduces monocyte adhesion in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1663–1669. [Google Scholar] [CrossRef]
- Perino, A.; Pols, T.W.; Nomura, M.; Stein, S.; Pellicciari, R.; Schoonjans, K. TGR5 reduces macrophage migration through mTOR-induced C/EBPbeta differential translation. J. Clin. Investig. 2014, 124, 5424–5436. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Villegas, L.A.; Perino, A.; Lemos, V.; Zietak, M.; Nomura, M.; Pols, T.W.H.; Schoonjans, K. TGR5 signalling promotes mitochondrial fission and beige remodelling of white adipose tissue. Nat. Commun. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.D.; Chen, W.D.; Yu, D.; Forman, B.M.; Huang, W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology 2011, 54, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, C.; Huang, X.; Yi, S.; Pan, S.; Zhang, Y.; Yuan, G.; Cao, Q.; Ye, X.; Li, H. Gut microbiota-mediated secondary bile acids regulate dendritic cells to attenuate autoimmune uveitis through TGR5 signaling. Cell Rep. 2021, 36, 109726. [Google Scholar] [CrossRef]
- Jin, D.; Huang, K.; Xu, M.; Hua, H.; Ye, F.; Yan, J.; Zhang, G.; Wang, Y. Deoxycholic acid induces gastric intestinal metaplasia by activating STAT3 signaling and disturbing gastric bile acids metabolism and microbiota. Gut Microbes 2022, 14, 2120744. [Google Scholar] [CrossRef]
- Li, C.; Wang, L.; Xie, W.; Chen, E.; Chen, Y.; Li, H.; Can, D.; Lei, A.; Wang, Y.; Zhang, J. TGR5 deficiency in excitatory neurons ameliorates Alzheimer’s pathology by regulating APP processing. Sci. Adv. 2024, 10, eado1855. [Google Scholar] [CrossRef]
- Hu, M.M.; He, W.R.; Gao, P.; Yang, Q.; He, K.; Cao, L.B.; Li, S.; Feng, Y.Q.; Shu, H.B. Virus-induced accumulation of intracellular bile acids activates the TGR5-beta-arrestin-SRC axis to enable innate antiviral immunity. Cell Res. 2019, 29, 193–205. [Google Scholar] [CrossRef]
- Yang, F.; Mao, C.; Guo, L.; Lin, J.; Ming, Q.; Xiao, P.; Wu, X.; Shen, Q.; Guo, S.; Shen, D.D.; et al. Structural basis of GPBAR activation and bile acid recognition. Nature 2020, 587, 499–504. [Google Scholar] [CrossRef]
- Ma, L.; Yang, F.; Wu, X.; Mao, C.; Guo, L.; Miao, T.; Zang, S.K.; Jiang, X.; Shen, D.D.; Wei, T.; et al. Structural basis and molecular mechanism of biased GPBAR signaling in regulating NSCLC cell growth via YAP activity. Proc. Natl. Acad. Sci. USA 2022, 119, e2117054119. [Google Scholar] [CrossRef]
- Chen, H.; Wang, J.; Zhang, C.; Ding, P.; Tian, S.; Chen, J.; Ji, G.; Wu, T. Sphingosine 1-phosphate receptor, a new therapeutic direction in different diseases. Biomed. Pharmacother. 2022, 153, 113341. [Google Scholar] [CrossRef]
- Xu, X.; Han, Y.; Zhu, T.; Fan, F.; Wang, X.; Liu, Y.; Luo, D. The role of SphK/S1P/S1PR signaling pathway in bone metabolism. Biomed. Pharmacother. 2023, 169, 115838. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Luo, D.; Jiang, Y.; Wan, S.; Li, X. An overview of sphingosine-1-phosphate receptor 2: Structure, biological function, and small-molecule modulators. Med. Res. Rev. 2024, 44, 2331–2362. [Google Scholar] [CrossRef] [PubMed]
- Studer, E.; Zhou, X.; Zhao, R.; Wang, Y.; Takabe, K.; Nagahashi, M.; Pandak, W.M.; Dent, P.; Spiegel, S.; Shi, R.; et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 2012, 55, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Aoki, H.; Yang, J.; Peng, K.; Liu, R.; Li, X.; Qiang, X.; Sun, L.; Gurley, E.C.; Lai, G.; et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017, 65, 2005–2018. [Google Scholar] [CrossRef]
- Liu, L.; Panzitt, K.; Racedo, S.; Wagner, M.; Platzer, W.; Zaufel, A.; Theiler-Schwetz, V.; Obermayer-Pietsch, B.; Muller, H.; Hofler, G.; et al. Bile acids increase steroidogenesis in cholemic mice and induce cortisol secretion in adrenocortical H295R cells via S1PR2, ERK and SF-1. Liver Int. 2019, 39, 2112–2123. [Google Scholar] [CrossRef]
- Al Alawi, A.M.; Nordenstrom, A.; Falhammar, H. Clinical perspectives in congenital adrenal hyperplasia due to 3beta-hydroxysteroid dehydrogenase type 2 deficiency. Endocrine 2019, 63, 407–421. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, S.; Zaufel, A.; Xie, Z.; Racedo, S.; Wagner, M.; Zollner, G.; Fickert, P.; Zhang, Q. Bile acids regulate SF-1 to alter cholesterol balance in adrenocortical cells via S1PR2. Biochem. Biophys. Res. Commun. 2024, 692, 149342. [Google Scholar] [CrossRef]
- Yang, J.; Tang, X.; Liang, Z.; Chen, M.; Sun, L. Taurocholic acid promotes hepatic stellate cell activation via S1PR2/p38 MAPK/YAP signaling under cholestatic conditions. Clin. Mol. Hepatol. 2023, 29, 465–481. [Google Scholar] [CrossRef]
- Huang, K.; Wang, C.; Mei, B.; Li, J.; Ren, T.; Zhan, H.; Zhang, Y.; Zhang, B.; Lv, X.; Zhang, Q.; et al. Bile acids attenuate hepatic inflammation during ischemia/reperfusion injury. JHEP Rep. 2024, 6, 101101. [Google Scholar] [CrossRef]
- Dai, Z.; Liu, W.C.; Chen, X.Y.; Wang, X.; Li, J.L.; Zhang, X. Gasdermin D-mediated pyroptosis: Mechanisms, diseases, and inhibitors. Front. Immunol. 2023, 14, 1178662. [Google Scholar] [CrossRef]
- Takezawa, T.; Uzu, M. HepG2-NIAS cells, a new subline of HepG2 cells that can enhance not only CYP3A4 activity but also expression of drug transporters and form bile canaliculus-like networks by the oxygenation culture via a collagen vitrigel membrane. J. Toxicol. Sci. 2022, 47, 39–50. [Google Scholar] [CrossRef]
- Miyagawa-Hayashino, A.; Imura, T.; Takezawa, T.; Hirai, M.; Shibata, S.; Ogi, H.; Tsujikawa, T.; Konishi, E. Activation of S1PR2 on macrophages and the hepatocyte S1PR2/RhoA/ROCK1/MLC2 pathway in vanishing bile duct syndrome. PLoS ONE 2025, 20, e0317568. [Google Scholar] [CrossRef] [PubMed]
- McMillin, M.; Frampton, G.; Grant, S.; Khan, S.; Diocares, J.; Petrescu, A.; Wyatt, A.; Kain, J.; Jefferson, B.; DeMorrow, S. Bile Acid-Mediated Sphingosine-1-Phosphate Receptor 2 Signaling Promotes Neuroinflammation during Hepatic Encephalopathy in Mice. Front. Cell. Neurosci. 2017, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yue, J.; Ha, F.; Wang, Y.; Wang, R.; Yang, X.; Zhang, J.; Liu, X.; Zhang, Y.; Han, T.; et al. Bile acid derivatives from gut microbiota promote GBPs-mediated activation of caspase-4/11 by LPS through lncRNA57RIK. Int. J. Biol. Sci. 2024, 20, 5831–5849. [Google Scholar] [CrossRef]
- Liu, X.; Yu, Y.; Zhang, H.; Zhang, M.; Liu, Y. The Role of Muscarinic Acetylcholine Receptor M(3) in Cardiovascular Diseases. Int. J. Mol. Sci. 2024, 25, 7560. [Google Scholar] [CrossRef]
- Sheikh Abdul Kadir, S.H.; Miragoli, M.; Abu-Hayyeh, S.; Moshkov, A.V.; Xie, Q.; Keitel, V.; Nikolaev, V.O.; Williamson, C.; Gorelik, J. Bile acid-induced arrhythmia is mediated by muscarinic M2 receptors in neonatal rat cardiomyocytes. PLoS ONE 2010, 5, e9689. [Google Scholar] [CrossRef]
- Ibrahim, E.; Diakonov, I.; Arunthavarajah, D.; Swift, T.; Goodwin, M.; McIlvride, S.; Nikolova, V.; Williamson, C.; Gorelik, J. Bile acids and their respective conjugates elicit different responses in neonatal cardiomyocytes: Role of Gi protein, muscarinic receptors and TGR5. Sci. Rep. 2018, 8, 7110. [Google Scholar] [CrossRef]
- Khurana, S.; Yamada, M.; Wess, J.; Kennedy, R.H.; Raufman, J.P. Deoxycholyltaurine-induced vasodilation of rodent aorta is nitric oxide- and muscarinic M(3) receptor-dependent. Eur. J. Pharmacol. 2005, 517, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Farhana, L.; Nangia-Makker, P.; Arbit, E.; Shango, K.; Sarkar, S.; Mahmud, H.; Hadden, T.; Yu, Y.; Majumdar, A.P. Bile acid: A potential inducer of colon cancer stem cells. Stem Cell Res. Ther. 2016, 7, 181. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Serhan, N.; Cenac, N.; Basso, L.; Gaudenzio, N. Mas-related G protein-coupled receptors (Mrgprs)—Key regulators of neuroimmune interactions. Neurosci. Lett. 2021, 749, 135724. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, T.; Fan, J.; Zou, H.; Lan, G.; Guo, F.; Shi, Y.; Ke, H.; Yu, H.; Yue, Z.; et al. Structure-guided discovery of bile acid derivatives for treating liver diseases without causing itch. Cell 2024, 187, 7164–7182 e18. [Google Scholar] [CrossRef]
- Yu, H.; Zhao, T.; Liu, S.; Wu, Q.; Johnson, O.; Wu, Z.; Zhuang, Z.; Shi, Y.; Peng, L.; He, R.; et al. MRGPRX4 is a bile acid receptor for human cholestatic itch. eLife 2019, 8, e48431. [Google Scholar] [CrossRef] [PubMed]
- Meixiong, J.; Vasavda, C.; Snyder, S.H.; Dong, X. MRGPRX4 is a G protein-coupled receptor activated by bile acids that may contribute to cholestatic pruritus. Proc. Natl. Acad. Sci. USA 2019, 116, 10525–10530. [Google Scholar] [CrossRef]
- Kotliar, I.B.; Ceraudo, E.; Kemelmakher-Liben, K.; Oren, D.A.; Lorenzen, E.; Dodig-Crnković, T.; Horioka-Duplix, M.; Huber, T.; Schwenk, J.M.; Sakmar, T.P. Itch receptor MRGPRX4 interacts with the receptor activity–modifying proteins. J. Biol. Chem. 2023, 299, 104664. [Google Scholar]
- Song, M.H.; Shim, W.S. Lithocholic Acid Activates Mas-Related G Protein-Coupled Receptors, Contributing to Itch in Mice. Biomol. Ther. 2022, 30, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.; Garraway, S.M. A review of dorsal root ganglia and primary sensory neuron plasticity mediating inflammatory and chronic neuropathic pain. Neurobiol. Pain 2024, 15, 100151. [Google Scholar] [CrossRef]
- Kotliar, I.B.; Lorenzen, E.; Schwenk, J.M.; Hay, D.L.; Sakmar, T.P. Elucidating the Interactome of G Protein-Coupled Receptors and Receptor Activity-Modifying Proteins. Pharmacol. Rev. 2023, 75, 1–34. [Google Scholar] [CrossRef]
- McLatchie, L.M.; Fraser, N.J.; Main, M.J.; Wise, A.; Brown, J.; Thompson, N.; Solari, R.; Lee, M.G.; Foord, S.M. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 1998, 393, 333–339. [Google Scholar] [CrossRef]
- Armour, S.L.; Foord, S.; Kenakin, T.; Chen, W.J. Pharmacological characterization of receptor-activity-modifying proteins (RAMPs) and the human calcitonin receptor. J. Pharmacol. Toxicol. Methods 1999, 42, 217–224. [Google Scholar] [CrossRef]
- Aiyar, N.; Disa, J.; Pullen, M.; Nambi, P. Receptor activity modifying proteins interaction with human and porcine calcitonin receptor-like receptor (CRLR) in HEK-293 cells. Mol. Cell. Biochem. 2001, 224, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. Role and Cytotoxicity of Amylin and Protection of Pancreatic Islet beta-Cells from Amylin Cytotoxicity. Cells 2018, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. Procalcitonin and Adrenomedullin in Infectious Diseases. Microbiol. Res. 2023, 14, 190–204. [Google Scholar] [CrossRef]
- Pallante, L.; Malavolta, M.; Grasso, G.; Korfiati, A.; Mavroudi, S.; Mavkov, B.; Kalogeras, A.; Alexakos, C.; Martos, V.; Amoroso, D.; et al. On the human taste perception: Molecular-level understanding empowered by computational methods. Trends Food Sci. Technol. 2021, 116, 445–459. [Google Scholar] [CrossRef]
- Nishihara, H.; Toda, Y.; Kuramoto, T.; Kamohara, K.; Goto, A.; Hoshino, K.; Okada, S.; Kuraku, S.; Okabe, M.; Ishimaru, Y. A vertebrate-wide catalogue of T1R receptors reveals diversity in taste perception. Nat. Ecol. Evol. 2024, 8, 111–120. [Google Scholar] [CrossRef]
- Li, X.; Staszewski, L.; Xu, H.; Durick, K.; Zoller, M.; Adler, E. Human receptors for sweet and umami taste. Proc. Natl. Acad. Sci. USA 2002, 99, 4692–4696. [Google Scholar] [CrossRef]
- Taruno, A.; Nomura, K.; Kusakizako, T.; Ma, Z.; Nureki, O.; Foskett, J.K. Taste transduction and channel synapses in taste buds. Pflugers Arch. 2021, 473, 3–13. [Google Scholar] [CrossRef]
- Schaefer, S.; Ziegler, F.; Lang, T.; Steuer, A.; Di Pizio, A.; Behrens, M. Membrane-bound chemoreception of bitter bile acids and peptides is mediated by the same subset of bitter taste receptors. Cell. Mol. Life Sci. 2024, 81, 217. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, F.; Steuer, A.; Di Pizio, A.; Behrens, M. Physiological activation of human and mouse bitter taste receptors by bile acids. Commun. Biol. 2023, 6, 612. [Google Scholar] [CrossRef]
- Kim, Y.; Gumpper, R.H.; Liu, Y.; Kocak, D.D.; Xiong, Y.; Cao, C.; Deng, Z.; Krumm, B.E.; Jain, M.K.; Zhang, S.; et al. Bitter taste receptor activation by cholesterol and an intracellular tastant. Nature 2024, 628, 664–671. [Google Scholar] [CrossRef]
- Ziaikin, E.; David, M.; Uspenskaya, S.; Niv, M.Y. BitterDB: 2024 update on bitter ligands and taste receptors. Nucleic Acids Res. 2025, 53, D1645–D1650. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M. International Union of Basic and Clinical Pharmacology. CXVII: Taste 2 receptors-Structures, functions, activators, and blockers. Pharmacol. Rev. 2025, 77, 100001. [Google Scholar] [CrossRef] [PubMed]
- Descamps-Sola, M.; Vilalta, A.; Jalsevac, F.; Blay, M.T.; Rodriguez-Gallego, E.; Pinent, M.; Beltran-Debon, R.; Terra, X.; Ardevol, A. Bitter taste receptors along the gastrointestinal tract: Comparison between humans and rodents. Front. Nutr. 2023, 10, 1215889. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.; Yang, H.; Yu, X.; Wang, D.; Guan, J.; Zhao, M.; Li, J. Mechanisms and novel therapeutic roles of bitter taste receptors in diseases. Theranostics 2025, 15, 3961–3978. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Ugawa, S.; Yamamura, H.; Imaizumi, Y.; Shimada, S. Functional interaction between T2R taste receptors and G-protein alpha subunits expressed in taste receptor cells. J. Neurosci. 2003, 23, 7376–7380. [Google Scholar] [CrossRef]
- Lossow, K.; Hubner, S.; Roudnitzky, N.; Slack, J.P.; Pollastro, F.; Behrens, M.; Meyerhof, W. Comprehensive Analysis of Mouse Bitter Taste Receptors Reveals Different Molecular Receptive Ranges for Orthologous Receptors in Mice and Humans. J. Biol. Chem. 2016, 291, 15358–15377. [Google Scholar] [CrossRef]
- Hu, X.; Ao, W.; Gao, M.; Wu, L.; Pei, Y.; Liu, S.; Wu, Y.; Zhao, F.; Sun, Q.; Liu, J.; et al. Bitter taste TAS2R14 activation by intracellular tastants and cholesterol. Nature 2024, 631, 459–466. [Google Scholar] [CrossRef]
- Kang, S.; Narazaki, M.; Metwally, H.; Kishimoto, T. Historical overview of the interleukin-6 family cytokine. J. Exp. Med. 2020, 217, e20190347. [Google Scholar] [CrossRef]
- Di Giorgio, C.; Morretta, E.; Lupia, A.; Bellini, R.; Massa, C.; Urbani, G.; Bordoni, M.; Marchiano, S.; Lachi, G.; Rapacciuolo, P.; et al. Bile acids serve as endogenous antagonists of the Leukemia inhibitory factor (LIF) receptor in oncogenesis. Biochem. Pharmacol. 2024, 223, 116134. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, M.M.; de la Puente, P. Leukemia Inhibitory Factor: An Important Cytokine in Pathologies and Cancer. Biomolecules 2022, 12, 217. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Stevis, P.E.; Cao, J.; Saotome, K.; Wu, J.; Glatman Zaretsky, A.; Haxhinasto, S.; Yancopoulos, G.D.; Murphy, A.J.; Sleeman, M.W.; et al. Structural insights into the assembly of gp130 family cytokine signaling complexes. Sci. Adv. 2023, 9, eade4395. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Liu, H.; Zhu, M.; Zhang, S.; Wang, M.; Ruan, Y.; Zheng, Y. Molecular dynamics simulations reveal key roles of the LIF receptor in the assembly of human LIF signaling complex. Comput. Struct. Biotechnol. J. 2025, 27, 585–594. [Google Scholar] [CrossRef]
- Viswanadhapalli, S.; Dileep, K.V.; Zhang, K.Y.J.; Nair, H.B.; Vadlamudi, R.K. Targeting LIF/LIFR signaling in cancer. Genes. Dis. 2022, 9, 973–980. [Google Scholar] [CrossRef]
- Bian, S.B.; Yang, Y.; Liang, W.Q.; Zhang, K.C.; Chen, L.; Zhang, Z.T. Leukemia inhibitory factor promotes gastric cancer cell proliferation, migration, and invasion via the LIFR-Hippo-YAP pathway. Ann. N. Y. Acad. Sci. 2021, 1484, 74–89. [Google Scholar] [CrossRef]
- Sato, H.; Macchiarulo, A.; Thomas, C.; Gioiello, A.; Une, M.; Hofmann, A.F.; Saladin, R.; Schoonjans, K.; Pellicciari, R.; Auwerx, J. Novel Potent and Selective Bile Acid Derivatives as TGR5 Agonists: Biological Screening, Structure−Activity Relationships, and Molecular Modeling Studies. J. Med. Chem. 2008, 51, 1831–1841. [Google Scholar] [CrossRef]
- Sato, H.; Genet, C.; Strehle, A.; Thomas, C.; Lobstein, A.; Wagner, A.; Mioskowski, C.; Auwerx, J.; Saladin, R. Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem. Biophys. Res. Commun. 2007, 362, 793–798. [Google Scholar] [CrossRef]
- Genet, C.; Strehle, A.; Schmidt, C.; Boudjelal, G.; Lobstein, A.; Schoonjans, K.; Souchet, M.; Auwerx, J.; Saladin, R.; Wagner, A. Structure−Activity Relationship Study of Betulinic Acid, A Novel and Selective TGR5 Agonist, and Its Synthetic Derivatives: Potential Impact in Diabetes. J. Med. Chem. 2010, 53, 178–190. [Google Scholar] [CrossRef]
- Ladurner, A.; Zehl, M.; Grienke, U.; Hofstadler, C.; Faur, N.; Pereira, F.C.; Berry, D.; Dirsch, V.M.; Rollinger, J.M. Allspice and Clove As Source of Triterpene Acids Activating the G Protein-Coupled Bile Acid Receptor TGR5. Front. Pharmacol. 2017, 8, 468. [Google Scholar] [CrossRef]
- Pellicciari, R.; Gioiello, A.; Macchiarulo, A.; Thomas, C.; Rosatelli, E.; Natalini, B.; Sardella, R.; Pruzanski, M.; Roda, A.; Pastorini, E.; et al. Discovery of 6alpha-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J. Med. Chem. 2009, 52, 7958–7961. [Google Scholar] [CrossRef]
- Renga, B.; Cipriani, S.; Carino, A.; Simonetti, M.; Zampella, A.; Fiorucci, S. Reversal of Endothelial Dysfunction by GPBAR1 Agonism in Portal Hypertension Involves a AKT/FOXOA1 Dependent Regulation of H2S Generation and Endothelin-1. PLoS ONE 2015, 10, e0141082. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, J.; Wang, X.; Luo, Z.; Shao, X.; Li, Y.; Cao, Q.; Zhao, S.; Qian, M.; Chen, X. Discovery and biological evaluation of cholic acid derivatives as potent TGR5 positive allosteric modulators. Bioorganic Med. Chem. 2023, 92, 117418. [Google Scholar] [CrossRef] [PubMed]
- Nakhi, A.; McDermott, C.M.; Stoltz, K.L.; John, K.; Hawkinson, J.E.; Ambrose, E.A.; Khoruts, A.; Sadowsky, M.J.; Dosa, P.I. 7-Methylation of Chenodeoxycholic Acid Derivatives Yields a Substantial Increase in TGR5 Receptor Potency. J. Med. Chem. 2019, 62, 6824–6830. [Google Scholar] [CrossRef]
- Yang, W.J.; Han, F.H.; Gu, Y.P.; Qu, H.; Liu, J.; Shen, J.H.; Leng, Y. TGR5 agonist inhibits intestinal epithelial cell apoptosis via cAMP/PKA/c-FLIP/JNK signaling pathway and ameliorates dextran sulfate sodium-induced ulcerative colitis. Acta Pharmacol. Sin. 2023, 44, 1649–1664. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Ning, M.; Chen, X.; Zou, Q.; Zhang, L.; Feng, Y.; Zhang, L.; Leng, Y.; Shen, J. Design, Synthesis, and Antidiabetic Activity of 4-Phenoxynicotinamide and 4-Phenoxypyrimidine-5-carboxamide Derivatives as Potent and Orally Efficacious TGR5 Agonists. J. Med. Chem. 2012, 55, 10475–10489. [Google Scholar] [CrossRef]
- Yu, D.D.; Sousa, K.M.; Mattern, D.L.; Wagner, J.; Fu, X.; Vaidehi, N.; Forman, B.M.; Huang, W. Stereoselective synthesis, biological evaluation, and modeling of novel bile acid-derived G-protein coupled bile acid receptor 1 (GP-BAR1, TGR5) agonists. Bioorganic Med. Chem. 2015, 23, 1613–1628. [Google Scholar] [CrossRef]
- Han, F.; Ning, M.; Cao, H.; Ye, Y.; Feng, Y.; Leng, Y.; Shen, J. Design of G-protein-coupled bile acid receptor 1 (GPBAR1, TGR5) soft drugs with reduced gallbladder-filling effects. Eur. J. Med. Chem. 2020, 203, 112619. [Google Scholar] [CrossRef]
- Fiorucci, S.; Rapacciuolo, P.; Fiorillo, B.; Roselli, R.; Marchiano, S.; Di Giorgio, C.; Bordoni, M.; Bellini, R.; Cassiano, C.; Conflitti, P.; et al. Discovery of a Potent and Orally Active Dual GPBAR1/CysLT(1)R Modulator for the Treatment of Metabolic Fatty Liver Disease. Front. Pharmacol. 2022, 13, 858137. [Google Scholar] [CrossRef]
- Briere, D.A.; Ruan, X.; Cheng, C.C.; Siesky, A.M.; Fitch, T.E.; Dominguez, C.; Sanfeliciano, S.G.; Montero, C.; Suen, C.S.; Xu, Y.; et al. Novel Small Molecule Agonist of TGR5 Possesses Anti-Diabetic Effects but Causes Gallbladder Filling in Mice. PLoS ONE 2015, 10, e0136873. [Google Scholar] [CrossRef]
- Li, T.; Holmstrom, S.R.; Kir, S.; Umetani, M.; Schmidt, D.R.; Kliewer, S.A.; Mangelsdorf, D.J. The G protein-coupled bile acid receptor, TGR5, stimulates gallbladder filling. Mol. Endocrinol. 2011, 25, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, B.; Balemba, O.B.; Godfrey, C.; Watson, C.A.; Vassileva, G.; Corvera, C.U.; Nelson, M.T.; Mawe, G.M. Hydrophobic bile salts inhibit gallbladder smooth muscle function via stimulation of GPBAR1 receptors and activation of KATP channels. J. Physiol. 2010, 588, 3295–3305. [Google Scholar] [CrossRef] [PubMed]
- Satsu, H.; Schaeffer, M.T.; Guerrero, M.; Saldana, A.; Eberhart, C.; Hodder, P.; Cayanan, C.; Schurer, S.; Bhhatarai, B.; Roberts, E.; et al. A sphingosine 1-phosphate receptor 2 selective allosteric agonist. Bioorganic Med. Chem. 2013, 21, 5373–5382. [Google Scholar] [CrossRef] [PubMed]
- Chien, D.C.; Limjunyawong, N.; Cao, C.; Meixiong, J.; Peng, Q.; Ho, C.Y.; Fay, J.F.; Roth, B.L.; Dong, X. MRGPRX4 mediates phospho-drug-associated pruritus in a humanized mouse model. Sci. Transl. Med. 2024, 16, eadk8198. [Google Scholar] [CrossRef]
- Cao, C.; Kang, H.J.; Singh, I.; Chen, H.; Zhang, C.; Ye, W.; Hayes, B.W.; Liu, J.; Gumpper, R.H.; Bender, B.J.; et al. Structure, function and pharmacology of human itch GPCRs. Nature 2021, 600, 170–175. [Google Scholar] [CrossRef]
- Fiorucci, S.; Urbani, G.; Di Giorgio, C.; Biagioli, M.; Distrutti, E. Bile Acids-Based Therapies for Primary Sclerosing Cholangitis: Current Landscape and Future Developments. Cells 2024, 13, 1650. [Google Scholar] [CrossRef]
- Wang, M.; Zan, T.; Fan, C.; Li, Z.; Wang, D.; Li, Q.; Zhang, C. Advances in GPCR-targeted drug development in dermatology. Trends Pharmacol. Sci. 2024, 45, 678–690. [Google Scholar] [CrossRef]
- Tokmakova, A.; Kim, D.; Guthrie, B.; Kim, S.K.; Goddard, W.A., 3rd; Liggett, S.B. Predicted structure and cell signaling of TAS2R14 reveal receptor hyper-flexibility for detecting diverse bitter tastes. iScience 2023, 26, 106422. [Google Scholar] [CrossRef]
- Quinn, R.A.; Melnik, A.V.; Vrbanac, A.; Fu, T.; Patras, K.A.; Christy, M.P.; Bodai, Z.; Belda-Ferre, P.; Tripathi, A.; Chung, L.K.; et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 2020, 579, 123–129. [Google Scholar] [CrossRef]
- Lucas, L.N.; Barrett, K.; Kerby, R.L.; Zhang, Q.; Cattaneo, L.E.; Stevenson, D.; Rey, F.E.; Amador-Noguez, D. Dominant Bacterial Phyla from the Human Gut Show Widespread Ability To Transform and Conjugate Bile Acids. mSystems 2021, 6, e0080521. [Google Scholar] [CrossRef]
- Rimal, B.; Collins, S.L.; Tanes, C.E.; Rocha, E.R.; Granda, M.A.; Solanki, S.; Hoque, N.J.; Gentry, E.C.; Koo, I.; Reilly, E.R.; et al. Bile salt hydrolase catalyses formation of amine-conjugated bile acids. Nature 2024, 626, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, I.; Allaband, C.; Mannochio-Russo, H.; El Abiead, Y.; Hagey, L.R.; Knight, R.; Dorrestein, P.C. The changing metabolic landscape of bile acids—Keys to metabolism and immune regulation. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 493–516. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiriyama, Y.; Tokumaru, H.; Sadamoto, H.; Nochi, H. Biological Actions of Bile Acids via Cell Surface Receptors. Int. J. Mol. Sci. 2025, 26, 5004. https://doi.org/10.3390/ijms26115004
Kiriyama Y, Tokumaru H, Sadamoto H, Nochi H. Biological Actions of Bile Acids via Cell Surface Receptors. International Journal of Molecular Sciences. 2025; 26(11):5004. https://doi.org/10.3390/ijms26115004
Chicago/Turabian StyleKiriyama, Yoshimitsu, Hiroshi Tokumaru, Hisayo Sadamoto, and Hiromi Nochi. 2025. "Biological Actions of Bile Acids via Cell Surface Receptors" International Journal of Molecular Sciences 26, no. 11: 5004. https://doi.org/10.3390/ijms26115004
APA StyleKiriyama, Y., Tokumaru, H., Sadamoto, H., & Nochi, H. (2025). Biological Actions of Bile Acids via Cell Surface Receptors. International Journal of Molecular Sciences, 26(11), 5004. https://doi.org/10.3390/ijms26115004