

Maximizing the Therapeutic Effect of Endothelin Receptor Antagonists in Pulmonary Fibrosis: A Paradigm for Treating the Disease
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. ERAs in Animal Models of Pulmonary Fibrosis
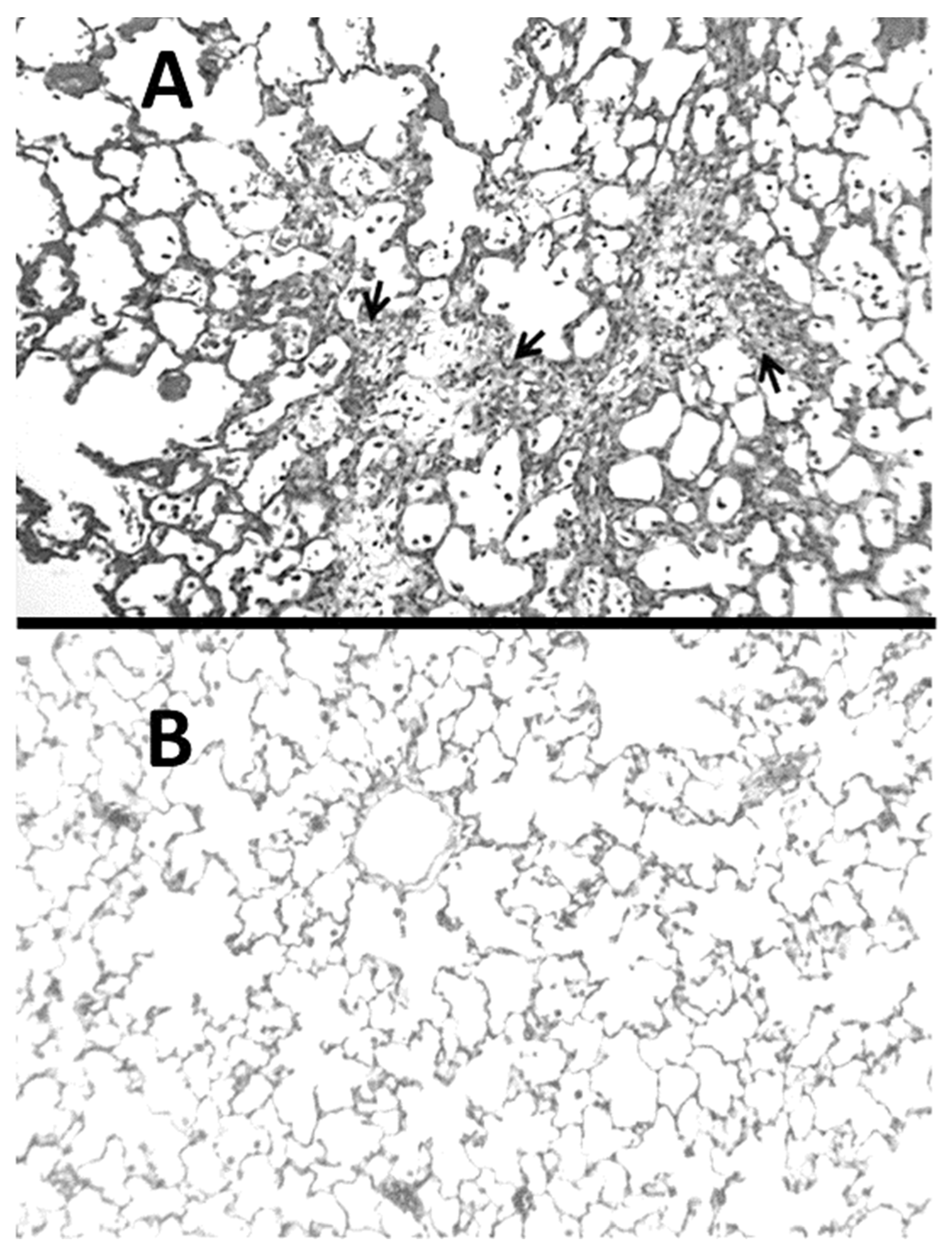
2.1. BLM-Induced Pulmonary Fibrosis
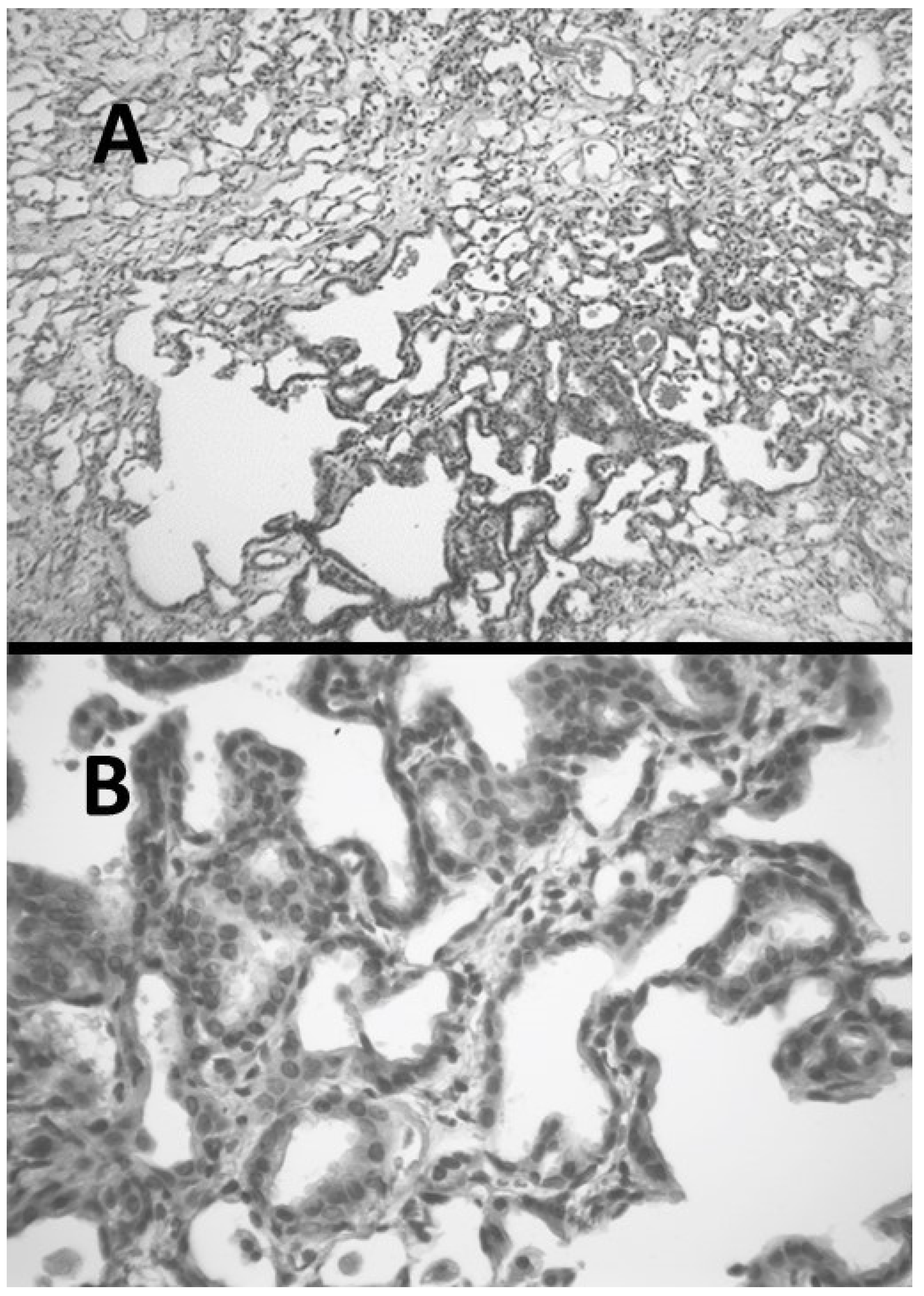
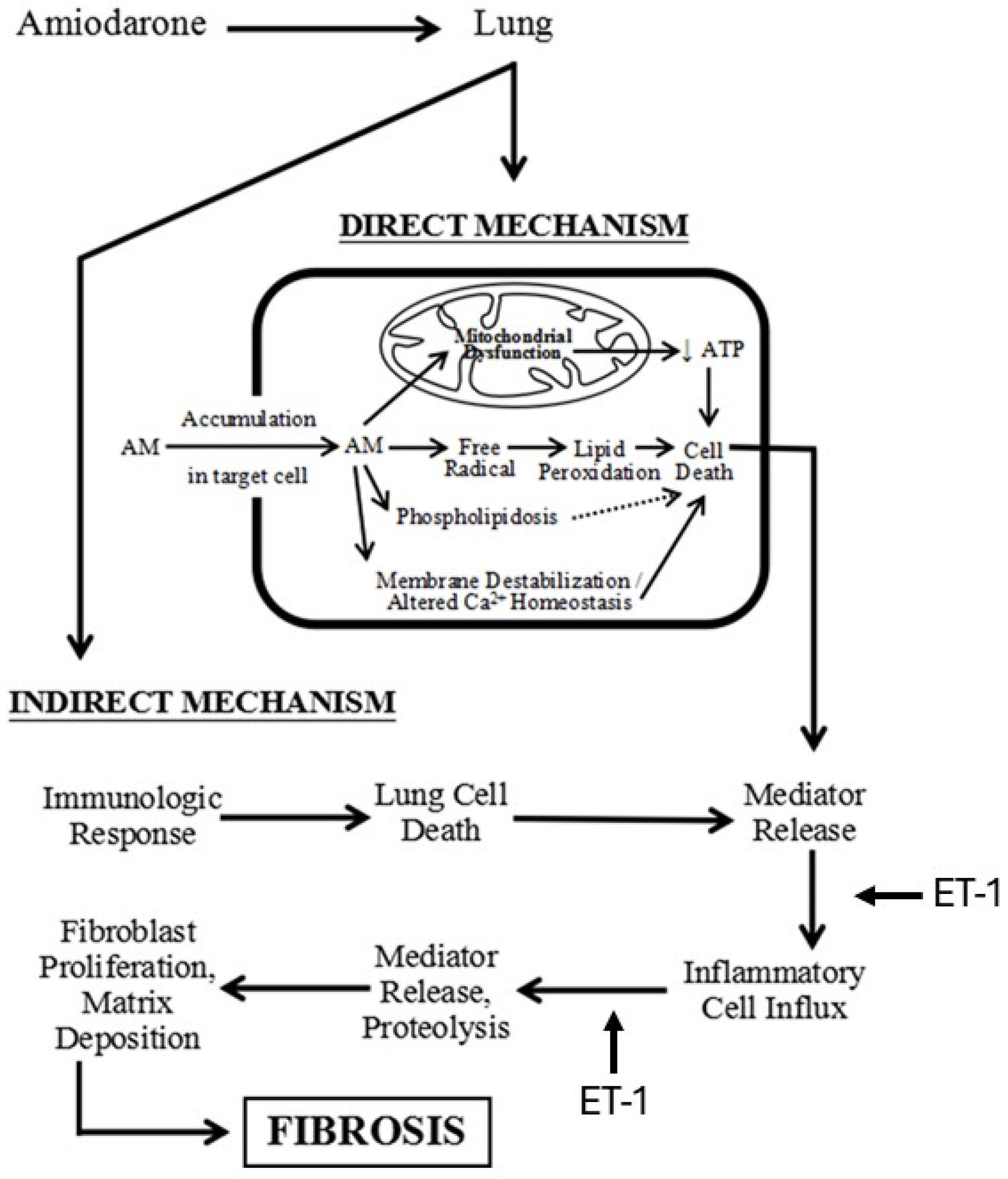
2.2. AM-Induced Pulmonary Fibrosis
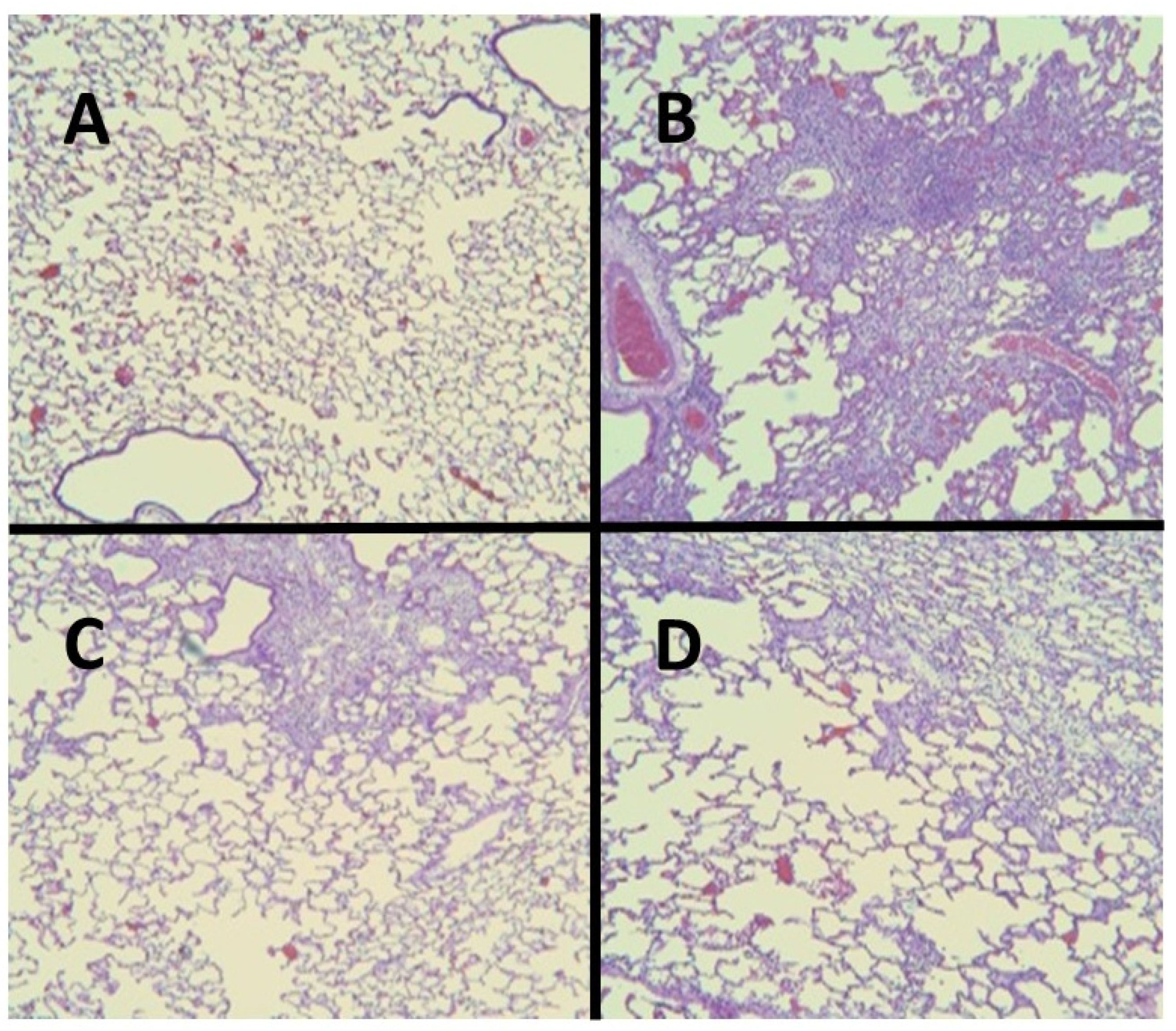
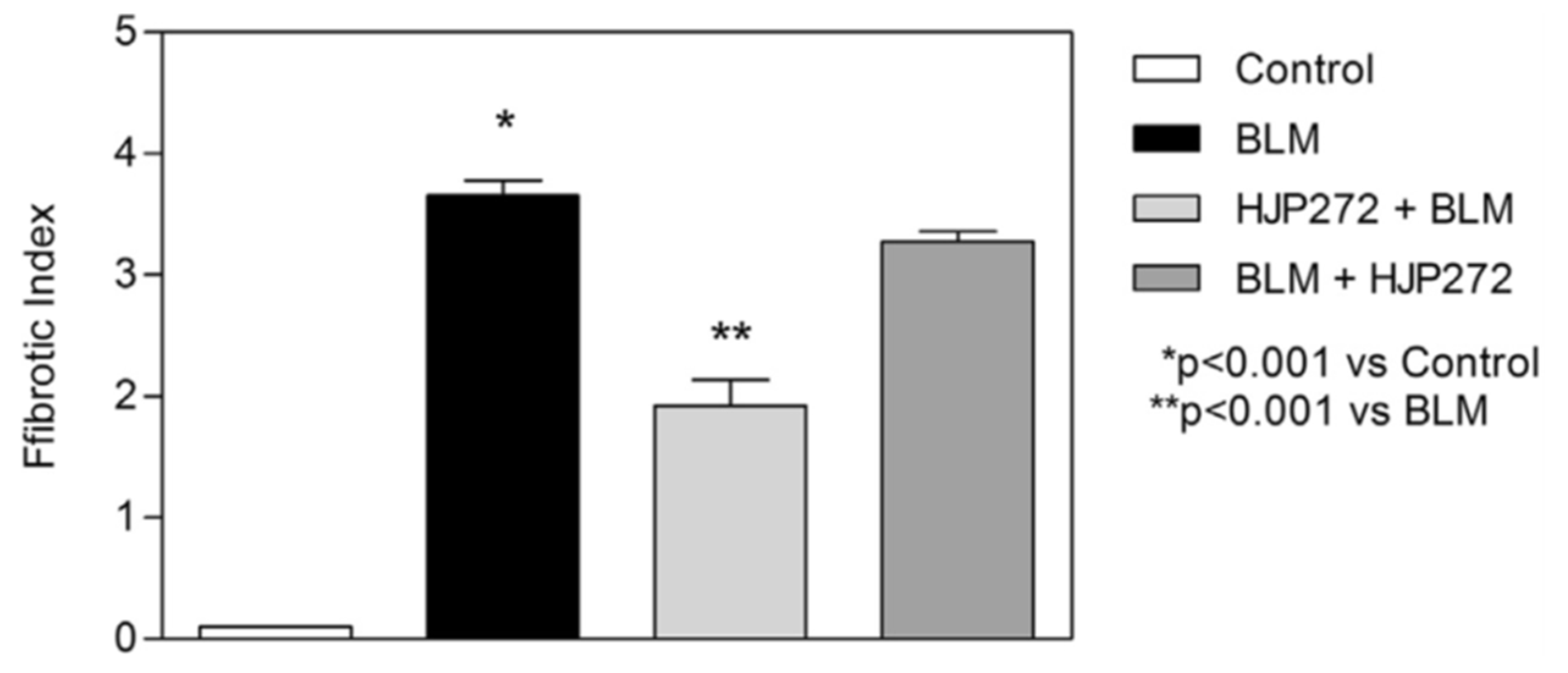
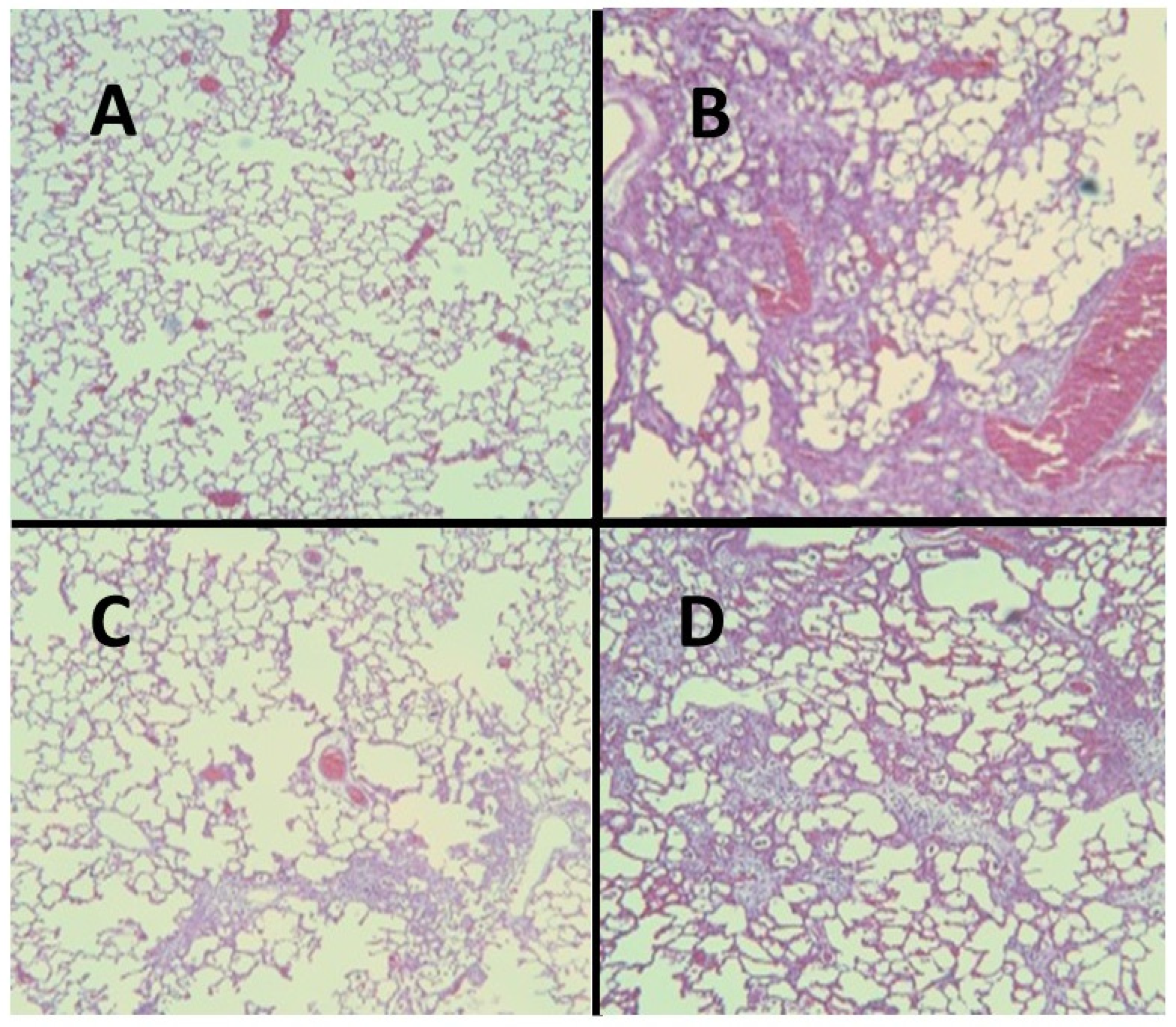
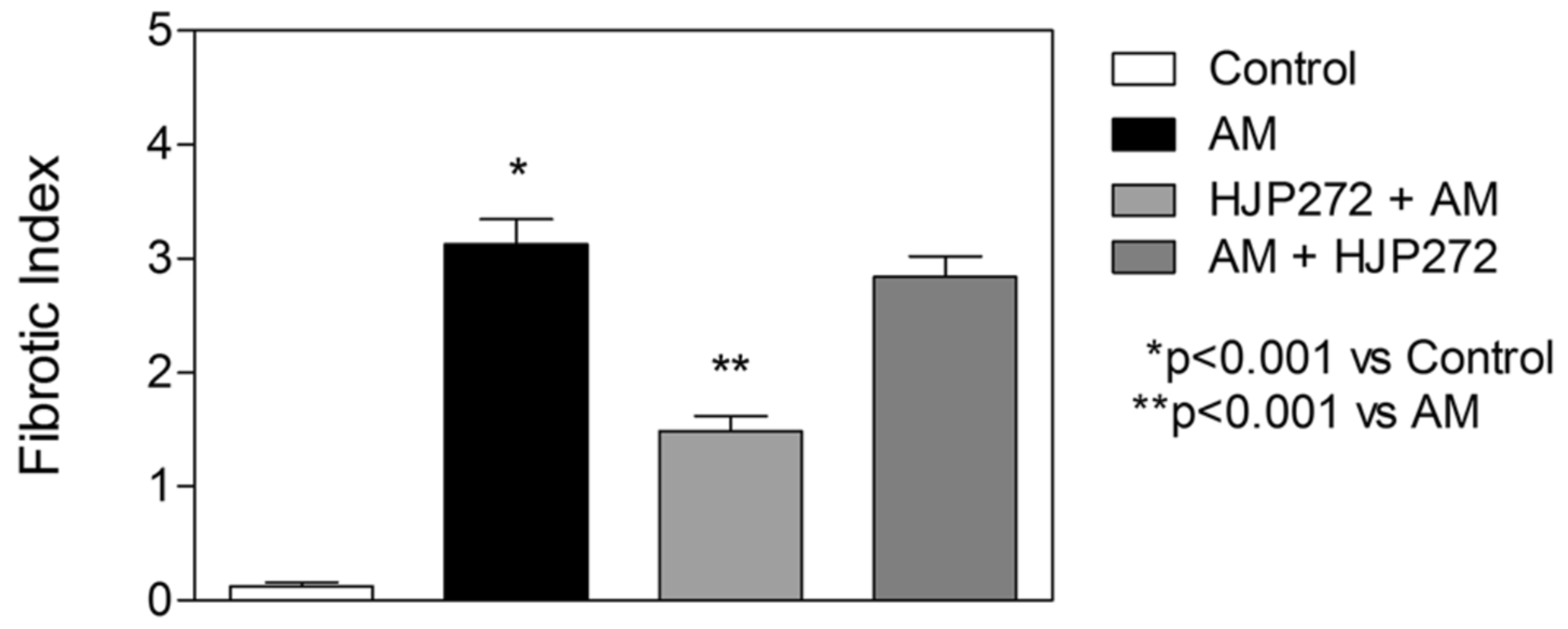
2.3. ERA Modulation of Fibrosis in the BLM and AM Models
3. Therapeutic Considerations
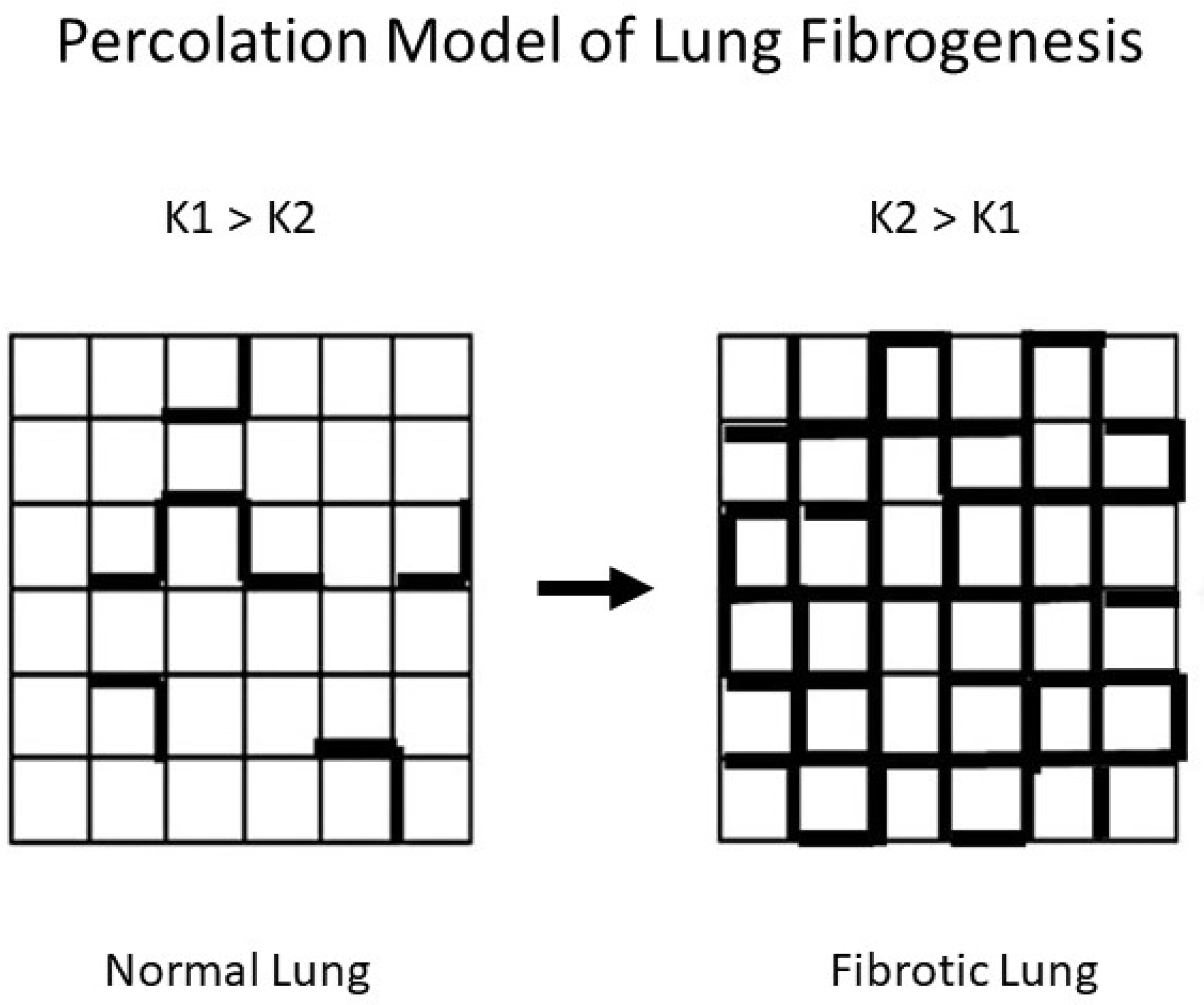
3.1. The Potential Role of Disease Emergence in Pulmonary Fibrosis
3.2. Developing a More Effective Treatment for Pulmonary Fibrosis
4. Elastin Crosslinks as a Potential Biomarker for Pulmonary Fibrosis
4.1. The Ratio of Peptide-Free to Peptide-Bound Elastin Crosslinks
4.2. Modeling the Effects of Pulmonary Fibrosis on DID Crosslinking
4.3. Potential Limitations of the DID Biomarker
4.4. Future Directions of Biomarker Research
5. Conclusions
Funding
Conflicts of Interest
References
- Hardie, W.D.; Glasser, S.W.; Hagood, J.S. Emerging concepts in the pathogenesis of lung fibrosis. Am. J. Pathol. 2009, 175, 3–16. [Google Scholar] [CrossRef]
- Lüscher, T.F.; Barton, M. Endothelins and endothelin receptor antagonists: Therapeutic considerations for a novel class of cardiovascular drugs. Circulation 2000, 102, 2434–2440. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Yanagisawa, M.; Kimura, S.; Kasuya, Y.; Miyauchi, T.; Goto, K.; Masaki, T. The human endothelin family: Three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc. Natl. Acad. Sci. USA 1989, 86, 2863–2867. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Olgun, N.; Patel, H.J.; Stephani, R.; Wang, W.; Yen, H.; Reznik, S.E. Effect of the putative novel selective ETA-receptor antagonist HJP272, a 1,3,6-trisubstituted-2-carboxy-quinol-4-one, on infection-mediated premature delivery. Can. J. Physiol. Pharmacol. 2008, 86, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Liu, X.; Liu, M.; Stephani, R.; Patel, H.; Cantor, J. HJP272, a novel endothelin receptor antagonist, attenuates lipopolysaccharide-induced acute lung injury in hamsters. Lung 2014, 192, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, T.; Liu, X.J.; Patel, H.; Stephani, R.; Cantor, J.O. Preferential recruitment of neutrophils by endothelin-1 in acute lung inflammation induced by lipopolysaccharide or cigarette smoke. Int. J. Chron. Obstruct. Pulmon. Dis. 2008, 3, 477–481. [Google Scholar] [PubMed]
- DiVietro, J.A.; Smith, M.J.; Smith, B.R.E.; Petruzzelli, L.; Larson, R.S.; Lawrence, M.B. Immobilized IL-8 triggers progressive activation of neutrophils rolling in vitro on P-selectin and intercellular adhesion molecule-1. J. Immunol. 2001, 167, 4017–4025. [Google Scholar] [CrossRef] [PubMed]
- Reutershan, J.; Morris, M.A.; Burcin, T.L.; Smith, D.F.; Chang, D.; Saprito, M.S.; Ley, K. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J. Clin. Investig. 2006, 116, 695–702. [Google Scholar] [CrossRef]
- Sato, Y.; Hogg, J.C.; English, D.; van Eeden, S.F. Endothelin-1 changes polymorphonuclear leukocytes’ deformability and CD11b expression and promotes their retention in the lun. Am. J. Respir. Cell Mol. Biol. 2000, 23, 404–410. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Hoidal, J.R.; Mukherjee, T.K. Role of TNFalpha in pulmonary pathophysiology. Respir. Res. 2006, 7, 125. [Google Scholar] [CrossRef]
- Calkins, C.M.; Heimbach, J.K.; Bensard, D.D.; Song, Y.; Raeburn, C.D.; Meng, X.; McIntyre, R.C., Jr. TNF receptor I mediates chemokine production and neutrophil accumulation in the lung following systemic lipopolysaccharide. J. Surg. Res. 2001, 101, 232–237. [Google Scholar] [CrossRef]
- Manitsopoulos, N.; Nikitopoulou, I.; Maniatis, N.A.; Magkou, C.; Kotanidou, A.; Orfanos, S.E. Highly Selective Endothelin-1 Receptor A Inhibition Prevents Bleomycin-Induced Pulmonary Inflammation and Fibrosis in Mice. Respiration 2017, 95, 122–136. [Google Scholar] [CrossRef]
- Park, S.-H.; Saleh, D.; Giaid, A.; Michel, R.P. Increased endothelin-1 in bleomycin-induced pulmonary fibrosis and the effect of an endothelin receptor antagonist. Am. J. Respir. Crit. Care Med. 1997, 156 Pt 1, 600–608. [Google Scholar] [CrossRef]
- Burger, R.; Peisach, J.; Horwitz, S. Activated bleomycin. A transient complex of drug, iron, and oxygen that degrades DNA. J. Biol. Chem. 1981, 256, 11636–11644. [Google Scholar] [CrossRef]
- Hirano, S. Quantitative time-course profiles of bronchoalveolar lavage cells following intratracheal instillation of lipopolysaccharide in mice. Ind. Health 1997, 35, 353–358. [Google Scholar] [CrossRef]
- Izbicki, G.; Segel, M.; Christensen, T.; Conner, M.; Breuer, R. Time course of bleomycin-induced lung fibrosis. Int. J. Exp. Pathol. 2002, 83, 111–119. [Google Scholar] [CrossRef]
- Brass, D.M.; Hollingsworth, J.W.; Cinque, M.; Li, Z.; Potts, E.; Toloza, E.; Foster, W.M.; Schwartz, D.A. Chronic LPS inhalation causes emphysema-like changes in mouse lung that are associated with apoptosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 584–590. [Google Scholar] [CrossRef]
- Liu, X.; Khadtare, N.; Patel, H.; Stephani, R.; Cantor, J. Time-dependent effects of HJP272, an endothelin receptor antagonist, in bleomycin-induced pulmonary fibrosis. Pulm. Pharmacol. Ther. 2017, 45, 164–169. [Google Scholar] [CrossRef]
- Ashcroft, T.; Simpson, M.J.; Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1988, 41, 467–470. [Google Scholar] [CrossRef]
- Heath, M.F.; Costa-Jussà, F.R.; Jacobs, J.M.; Jacobson, W. The induction of pulmonary phospholipidosis and the inhibition of lysosomal phospholipases by amiodarone. Br. J. Exp. Pathol. 1985, 66, 391–397. [Google Scholar] [PubMed]
- Blake, T.; Reasor, M. Pulmonary responses to amiodarone in hamsters: Comparison of intratracheal and oral administrations. Toxicol. Appl. Pharmacol. 1995, 131, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Vereckei, A.; Blazovics, A.; Gyorgy, I.; Feher, E.; Toth, M.; Szenasi, G.; Zsinka, A.; Foldiak, G.; Feher, J. The role of free radicals in the pathogenesis of amiodarone toxicity. J. Cardiovasc. Electrophysiol. 1993, 4, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.P.; Gordon, G.B.; Paky, A.; McShane, A.; Adkinson, N.F., Jr.; Peters, S.P.; Friday, K.; Jackman, W.; Sciuto, A.M.; Gurtner, G.H. Amiodarone causes acute oxidant lung injury in ventilated and perfused rabbit lungs. J. Cardiovasc. Pharmacol. 1988, 12, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Larsen, B.T.; Vaszar, L.T.; Colby, T.V.; Tazelaar, H.D. Lymphoid hyperplasia and eosinophilic pneumonia as histologic manifestations of amiodarone-induced lung toxicity. Am. J. Surg. Pathol. 2012, 36, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Khadtare, N.; Patel, H.; Stephani, R.; Cantor, J. Transient Blockade of Endothelin-1 Mitigates Amiodarone-Induced Pulmonary Fibrosis. Lung 2018, 196, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Zagai, U.; Dadfar, E.; Lundahl, J.; Venge, P.; Skold, C.M. Eosinophil cationic protein stimulates TGF-beta1 release by human lung fibroblasts in vitro. Inflammation 2007, 30, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Guarino, M.; Tosoni, A.; Nebuloni, M. Direct contribution of epithelium to organ fibrosis: Epithelial-mesenchymal transition. Hum. Pathol. 2009, 40, 1365–1376. [Google Scholar] [CrossRef]
- Lasky, J.A.; Brody, A.R. Interstitial fibrosis and growth factors. Environ. Health Perspect. 2000, 108, 751–762. [Google Scholar]
- Andersson-Sjöland, A.; de Alba, C.G.; Nihlberg, K.; Becerril, C.; Ramírez, R.; Pardo, A.; Westergren-Thorsson, G.; Selman, M. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.J.; Burdick, M.D.; Hong, K.; Lutz, M.A.; Murray, L.A.; Xue, Y.Y.; Belperio, J.A.; Keane, M.P.; Strieter, R.M. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Investig. 2004, 114, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.-M.; Yu, C.-C.; Kuo, M.-L.; Chen, B.-C.; Lin, C.-H. Endothelin-1 induces connective tissue growth factor expression in human lung fibroblasts by ETAR-dependent JNK/AP-1 pathway. Biochem. Pharmacol. 2014, 88, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Pan, Y.-L.; Xin, W.; Yan, C. The potential benefit of endothelin receptor antagonists’ therapy in idiopathic pulmonary fibrosis: A meta-analysis of results from randomized controlled trials. Medicine 2022, 101, e29981. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Behr, J.; Brown, K.K.; Egan, J.J.; Kawut, S.M.; Flaherty, K.R.; Martinez, F.J.; Nathan, S.D.; Wells, A.U.; Collard, H.R.; et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: A parallel, randomized trial. Ann. Intern. Med. 2013, 158, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Furuya, Y.; Kuwana, M. Effect of Bosentan on systemic sclerosis-associated interstitial lung disease ineligible for cyclophosphamide therapy: A prospective open-label study. J. Rheumatol. 2011, 38, 2186–2192. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Brown, K.K.; Raghu, G.; du Bois, R.M.; Lynch, D.A.; Martinez, F.; Valeyre, D.; Leconte, I.; Morganti, A.; Roux, S.; et al. BUILD-3: A randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.-Q.; Jusup, M.; Jin, Z.; Wang, Y.; Wang, Z. Pattern transitions in spatial epidemics: Mechanisms and emergent properties. Phys. Life Rev. 2016, 19, 43–73. [Google Scholar] [CrossRef]
- Bunde, A.; Kantelhart, J.W. Diffusion and conduction in percolation systems. In Diffusion in Condensed Matter; Heitjans, P., Karger, J., Eds.; Springer: Berlin, Germany, 2005; pp. 895–914. [Google Scholar]
- Suki, B.; Bates, J.H.T.; Eskandari, M.; Arvayo, A.L.; Levenston, M.E.; Peták, F.; Fodor, G.H.; Babik, B.; Habre, W.; Prakash, Y.S.; et al. Lung tissue mechanics as an emergent phenomenon. J. Appl. Physiol. 2011, 110, 1111–1118. [Google Scholar] [CrossRef]
- Bates, J.H.T.; Davis, G.S.; Majumdar, A.; Butnor, K.J.; Suki, B. Linking parenchymal disease progression to changes in lung mechanical function by percolation. Am. J. Respir. Crit. Care Med. 2007, 176, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Stainer, A.; Faverio, P.; Busnelli, S.; Catalano, M.; Della Zoppa, M.; Marruchella, A.; Pesci, A.; Luppi, F. Molecular Biomarkers in Idiopathic Pulmonary Fibrosis: State of the Art and Future Directions. Int. J. Mol. Sci. 2021, 22, 6255. [Google Scholar] [CrossRef] [PubMed]
- de Brouwer, B.; Drent, M.; van den Ouweland, J.M.W.; Wijnen, P.A.; van Moorsel, C.H.M.; Bekers, O.; Grutters, J.C.; White, E.S.; Janssen, R. Increased circulating desmosine and age-dependent elastinolysis in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 45. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ma, S.; Turino, G.; Cantor, J. The Pattern of Elastic Fiber Breakdown in Bleomycin-Induced Pulmonary Fibrosis May Reflect Microarchitectural Changes. Lung 2017, 195, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ma, S.; Liu, S.; Liu, M.; Turino, G.; Cantor, J. The Ratio of Free to Bound Desmosine and Isodesmosine May Reflect Emphysematous Changes in COPD. Lung 2015, 193, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.O.; Ma, S.; Liu, X.; Campos, M.A.; Strange, C.; Stocks, J.M.; Devine, M.S.; El Bayadi, S.G.; Lipchik, R.J.; Sandhaus, R.A.; et al. A 28-day clinical trial of aerosolized hyaluronan in alpha-1 antiprotease deficiency COPD using desmosine as a surrogate marker for drug efficacy. Respir. Med. 2021, 182, 106402. [Google Scholar] [CrossRef]
- Cantor, J.; Ma, S.; Turino, G. What percolation theory can tell us about COPD. Med. Hypotheses 2013, 81, 152–155. [Google Scholar] [CrossRef]
- Murphy, K.D.; Hunt, G.W.; Almond, D.P. Evidence of emergent scaling in mechanical systems. Philos. Mag. 2006, 86, 3325–3338. [Google Scholar] [CrossRef]
- Counts, D.F.; Evans, J.N.; DiPetrillo, T.A.; Sterling, K.M., Jr.; Kelley, J. Collagen lysyl oxidase activity in the lung increases during bleomycin-induced lung fibrosis. J. Pharmacol. Exp. Ther. 1981, 219, 675–678. [Google Scholar]
- Osman, M.; Kaldany RR, J.; Cantor, J.O.; Turino, G.M.; Mandl, I. Stimulation of lung lysyl oxidase activity in hamsters with elastase-induced emphysema. Am. Rev. Respir. Dis. 1985, 131, 169–170. [Google Scholar]
- Niewoehner, D.E.; Hoidal, J.R. Lung fibrosis and emphysema: Divergent responses to a common injury? Science 1982, 217, 359–360. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, R.A.; Miller, B.E.; Wrobel, K.; Ranjit, K.; Williams, M.C.; Drost, E.; Edwards, L.D.; Lomas, D.A.; Rennard, S.I.; Agustí, A.; et al. Circulating desmosine levels do not predict emphysema progression but are associated with cardiovascular risk and mortality in COPD. Eur. Respir. J. 2016, 47, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Ongay, S.; Sikma, M.; Horvatovich, P.; Hermans, J.; Miller, B.E.; Ten Hacken, N.H.T.; Bischoff, R. Free Urinary Desmosine and Isodesmosine as COPD Biomarkers: The Relevance of Confounding Factors. Chronic Obstr. Pulm. Dis. 2016, 3, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Luisetti, M.; Stolk, J.; Iadarola, P. Desmosine, a biomarker for COPD: Old and in the way. Eur. Respir. J. 2012, 39, 797–798. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantor, J. Maximizing the Therapeutic Effect of Endothelin Receptor Antagonists in Pulmonary Fibrosis: A Paradigm for Treating the Disease. Int. J. Mol. Sci. 2024, 25, 4184. https://doi.org/10.3390/ijms25084184
Cantor J. Maximizing the Therapeutic Effect of Endothelin Receptor Antagonists in Pulmonary Fibrosis: A Paradigm for Treating the Disease. International Journal of Molecular Sciences. 2024; 25(8):4184. https://doi.org/10.3390/ijms25084184
Chicago/Turabian StyleCantor, Jerome. 2024. "Maximizing the Therapeutic Effect of Endothelin Receptor Antagonists in Pulmonary Fibrosis: A Paradigm for Treating the Disease" International Journal of Molecular Sciences 25, no. 8: 4184. https://doi.org/10.3390/ijms25084184
APA StyleCantor, J. (2024). Maximizing the Therapeutic Effect of Endothelin Receptor Antagonists in Pulmonary Fibrosis: A Paradigm for Treating the Disease. International Journal of Molecular Sciences, 25(8), 4184. https://doi.org/10.3390/ijms25084184