
Targeting N-Methyl-d-Aspartate Receptors in Neurodegenerative Diseases

 ,
,  and
and
Abstract
1. Introduction
2. Physiology of NMDA Receptors
2.1. Structure, Composition, and Localization
2.2. NMDARs in the Glutamatergic Synapse

2.3. Regulation of NMDAR Activity
2.4. NMDARs Modulators

| Compounds | Modulator | Selectivity | Action | References |
|---|---|---|---|---|
D-AP5 | Competitive antagonist | GluN2A | Inhibits excitatory response. Impacts behavioral learning; blocks plasticity (LTP). | [95,96,97] |
Riluzole | Competitive antagonist | GluN1/GluN2B | Indirect block of NMDAR. Protects from motor deficit. | [103,104,105] |
Phencyclidine | Selective uncompetitive antagonist | GluN2B/D(PCP site) | Induces psychotic and dissociative schizophrenia-like symptoms. Impairs NMDAR neurotransmission in vivo. | [98] |
Ketamine | Uncompetitive antagonist | GluN2B/D(PCP site) | Applied in post-synapse: inhibits excitatory pyramidal neuron in extra-synaptic GluN2B. Applied in pre-synapse: inhibits GluN2D in interneuron (induces disinhibition of Glu release in post-synapse). Up-regulates hippocampal AMPARs (GluA1/GluA2). Antidepressant. | [100,106] |
Dizocilpine | Uncompetitive antagonist | GluN2A/B/D(PCP site) | Anticonvulsant, antidepressant. Induces memory impairments. | [102,107] |
Ifenprodil | Uncompetitive antagonist | GluN1/GluN2B(N-Terminal domain) | Blocks GluN2B (140-fold preference for NR2B over NR2A subunits). Induces inhibition of GluN2R receptor currents. Anti-Parkinsonian effect. | [108,109,110] |
Memantine | Uncompetitive antagonist | GluN1/GluN2B | Blocks GluN2B extra-synaptic and induces glutamatergic excitotoxicity. Used for moderate-to-severe AD. | [111,112] |
Amantadine | Uncompetitive antagonist | GluN1/GluN2B | Blocks GluN1/GluN2B by accelerating channel closure during channel block. Used as anti-Parkinsonian drug. | [113] |
Dextromethorphan | Uncompetitive antagonist | GluN2A | Blocks GluN2A subunit. Prevents neuronal damage and modulates pain sensation | [114,115,116,117,118] |
3. The Impact of NMDARs in Neurodegenerative Diseases
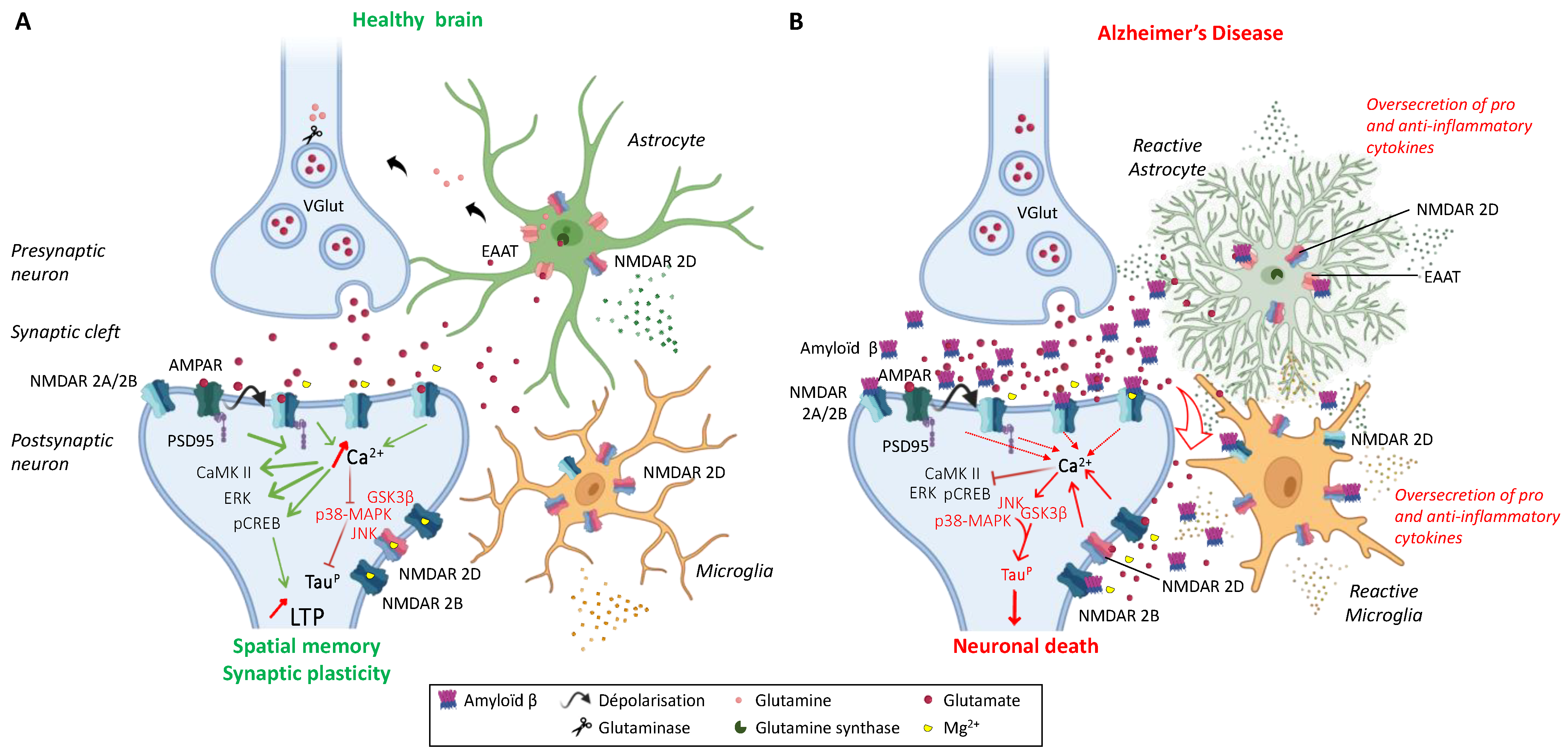
3.1. Alzheimer’s Disease
3.2. Huntington’s Disease
3.3. Parkinson’s Disease
4. Fluoroethylnormemantine (FENM): A New Generation NMDAR Uncompetitive Antagonist
4.1. 18F-FENM as a PET NMDAR Radiotracer
4.2. FENM as an Anxiolytic Agent in PTSD
4.3. FENM as a Neuroprotective Agent in AD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABD | agonist-binding domain |
| AD | Alzheimer’s disease |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| Aβ | amyloid-β |
| BDNF | brain-derived neurotrophic factor |
| CaMKII | camodulin kinase II |
| cAMP | 3′,5′-adenosine monophosphate |
| cGMP | 3′,5′-guanosine monophosphate |
| CNS | central nervous system |
| CREB | cAMP response element-binding protein |
| CTD | carboxyl C-terminal domain |
| DYRK1A | dual-specificity tyrosine-phosphorylation-regulated kinase 1 |
| EAAT | excitatory amino acid transporter |
| ERK1/2 | extracellular signal-regulated protein kinase 1/2 |
| FENM | fluoroethylnormemantine |
| GABA | γ-aminobutyric acid |
| HD | Huntington’s disease |
| HEK-293 | human embryonic kidney 293 cells |
| L-Dopa | L-3,4-dihydroxyphenylalanine |
| LTD | long-term depression |
| LTP | long-term potentiation |
| MAPK | mitogen-activated protein kinases |
| NMDAR | N-methyl-d-aspartate receptor |
| PCP | phencycline |
| PD | Parkinson’s disease |
| PET | positron emission tomography |
| PKA | protein kinase A |
| PSD-95 | post-synaptic density protein 95 |
| PTSD | post-traumatic stress disorder |
| RCPG | G protein-coupled receptor |
| SANT | sodium-coupled neutral amino acid transporter |
| tPA | tissue-type plasminogen activator |
References
- Greenamyre, J.T.; Maragos, W.F.; Albin, R.L.; Penney, J.B.; Young, A.B. Glutamate transmission and toxicity in Alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1988, 12, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Reis, H.J.; Guatimosim, C.; Paquet, M.; Santos, M.; Ribeiro, F.M.; Kummer, A.; Schenatto, G.; Salgado, J.V.; Vieira, L.B.; Teixeira, A.L.; et al. Neuro-transmitters in the central nervous system & their implication in learning and memory processes. Curr. Med. Chem. 2009, 16, 796–840. [Google Scholar] [CrossRef] [PubMed]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar]
- Volianskis, A.; France, G.; Jensen, M.S.; Bortolotto, Z.A.; Jane, D.E.; Collingridge, G.L. Long-term potentiation and the role of N-methyl-d-aspartate receptors. Brain Res. 2015, 1621, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Lynch, G.; Baudry, M. The biochemistry of memory: A new and specific hypothesis. Science 1984, 8, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Peng, R.-Y. Basic roles of key molecules connected with NMDAR signaling pathway on regulating learning and memory and synaptic plasticity. Mil. Med. Res. 2016, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E. NMDA receptor C-terminal signaling in development, plasticity, and disease. F1000Research 2019, 8, 1547. [Google Scholar] [CrossRef]
- Pol, A.N.V.D.; Hermans-Borgmeyer, I.; Hofer, M.; Ghosh, P.; Heinemann, S. Ionotropic glutamate-receptor gene expression in hypothalamus: Localization of AMPA, kainate, and NMDA receptor RNA with in situ hybridization. J. Comp. Neurol. 1994, 343, 428–444. [Google Scholar] [CrossRef] [PubMed]
- Tovar, K.R.; Westbrook, G.L. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J. Neurosci. 1999, 19, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, J.R.; Dubé, G.R.; Egles, C.; Liu, G. Distribution, density, and clustering of functional glutamate receptors before and after synaptogenesis in hippocampal neurons. J. Neurophysiol. 2000, 84, 1573–1587. [Google Scholar] [CrossRef] [PubMed]
- Rodenas-Ruano, A.; Chávez, A.E.; Cossio, M.J.; Castillo, P.E.; Zukin, R.S. REST-dependent epigenetic remodeling promotes the in vivo developmental switch in NMDA receptors. Nat. Neurosci. 2012, 15, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Ogawa, I.; Yamasaki, M.; Kiyama, Y.; Kassai, H.; Watabe, A.M.; Nakao, K.; Aiba, A.; Watanabe, M.; Manabe, T. The glutamate receptor GluN2 subunit regulates synaptic trafficking of AMPA receptors in the neonatal mouse brain. Eur. J. Neurosci. 2014, 40, 3136–3146. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.L.; Westbrook, G.L.; Guthrie, P.B. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 1984, 309, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef] [PubMed]
- Nowak, L.; Bregestovski, P.; Ascher, P.; Herbet, A.; Prochiantz, A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature 1984, 307, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.W.; Ascher, P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 1987, 325, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Manabe, T.; Takahashi, T. Presynaptic long-term depression at the hippocampal mossy fiber-CA3 synapse. Science 1996, 273, 648–650. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C.; Kauer, J.A.; Perkel, D.J.; Nicoll, R.A. The impact of postsynaptic calcium on synaptic transmission—Its role in long-term potentiation. Trends Neurosci. 1989, 12, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, X.; Tzingounis, A.; Danbolt, N.C.; Larsson, H.P. EAAT2 (GLT-1; slc1a2) glutamate transporters reconstituted in liposomes argues against heteroexchange being substantially faster than net uptake. J. Neurosci. 2014, 34, 13472–13485. [Google Scholar] [CrossRef]
- Dupuis, J.P.; Nicole, O.; Groc, L. NMDA receptor functions in health and disease: Old actor, new dimensions. Neuron 2023, 111, 2312–2328. [Google Scholar] [CrossRef]
- Geddes, J.W.; Cotman, C.W. Plasticity in hippocampal excitatory amino acid receptors in Alzheimer’s disease. Neurosci. Res. 1986, 3, 672–678. [Google Scholar] [CrossRef]
- Weiss, J.; Goldberg, M.P.; Choi, D.W. Ketamine protects cultured neocortical neurons from hypoxic injury. Brain Res. 1986, 380, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Etienne, P.; Baudry, M. Calcium dependent aspects of synaptic plasticity, excitatory amino acid neurotransmission, brain aging and schizophrenia: A unifying hypothesis. Neurobiol. Aging 1987, 8, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Adamec, R. Transmitter systems involved in neural plasticity underlying increased anxiety and defense-implications for understanding anxiety following traumatic stress. Neurosci. Biobehav. Rev. 1997, 21, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Zeevalk, G.D.; Nicklas, W.J. Evidence that the loss of the voltage-dependent Mg2+ block at the N-methyl-d-aspartate receptor underlies receptor activation during inhibition of neuronal metabolism. J. Neurochem. 1992, 59, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Chen, H.S.; Zhang, D.; Lipton, S.A. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J. Neurosci. 2010, 30, 11246–11250. [Google Scholar] [CrossRef]
- Stroebel, D.; Casado, M.; Paoletti, P. Triheteromeric NMDA receptors: From structure to synaptic physiology. Curr. Opin. Physiol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Snell, L.D.; Johnson, K.M. Characterization of the inhibition of excitatory amino acid-induced neurotransmitter release in the rat striatum by phencyclidine-like drugs. J. Pharmacol. Exp. Ther. 1986, 238, 938–946. [Google Scholar] [PubMed]
- Burnashev, N.; Schoepfer, R.; Monyer, H.; Ruppersberg, J.P.; Günther, W.; Seeburg, P.H.; Sakmann, B. Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science 1992, 257, 1415–1419. [Google Scholar] [CrossRef]
- Lee, E.; Williams, Z.; Goodman, C.B.; Oriaku, E.T.; Harris, C.; Thomas, M.; Soliman, K.F.A. Effects of NMDA receptor inhibition by phencyclidine on the neuronal differentiation of PC12 cells. Neurotoxicology 2006, 27, 558–566. [Google Scholar] [CrossRef]
- Paoletti, P.; Ascher, P.; Neyton, J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J. Neurosci. 1997, 17, 5711–5725. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Cummings, J.; Roldan, L.A.; Jan, Y.N.; Jan, L.Y. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 1994, 368, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Chazot, P.L.; Stephenson, F.A. Molecular dissection of native mammalian forebrain NMDA receptors containing the NR1 C2 exon: Direct demonstration of NMDA receptors comprising NR1, NR2A, and NR2B subunits within the same complex. J. Neurochem. 1997, 69, 2138–2144. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, Y.; Yasuda, R.P.; Dunah, A.W.; Wolfe, B.B. The majority of N-methyl-d-aspartate receptor complexes in adult rat cerebral cortex contain at least three different subunits (NR1/NR2A/NR2B). Mol. Pharmacol. 1997, 51, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Al-Hallaq, R.A.; Conrads, T.P.; Veenstra, T.D.; Wenthold, R.J. NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J. Neurosci. 2007, 27, 8334–8343. [Google Scholar] [CrossRef] [PubMed]
- Monyer, H.; Sprengel, R.; Schoepfer, R.; Herb, A.; Higuchi, M.; Lomeli, H.; Burnashev, N.; Sakmann, B.; Seeburg, P.H. Heteromeric NMDA receptors: Molecular and functional distinction of subtypes. Science 1992, 256, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Otaño, I.; Luján, R.; Tavalin, S.J.; Plomann, M.; Modregger, J.; Liu, X.-B.; Jones, E.G.; Heinemann, S.F.; Lo, D.C.; Ehlers, M.D. Endocytosis and synaptic removal of NR3A-containing NMDA receptors by PAC-SIN1/syndapin1. Nat. Neurosci. 2006, 9, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Wu, A.; Berglund, K.; Gu, X.; Jiang, M.Q.; Talati, J.; Zhao, J.; Wei, L.; Yu, S.P. Pathogenesis of sporadic Alzheimer’s disease by deficiency of NMDA receptor subunit GluN3A. Alzheimers Dement. 2022, 18, 222–239. [Google Scholar] [CrossRef] [PubMed]
- Grand, T.; Abi Gerges, S.; David, M.; Diana, M.A.; Paoletti, P. Unmasking GluN1/GluN3A excitatory glycine NMDA receptors. Nat. Commun. 2018, 9, 4769. [Google Scholar] [CrossRef]
- Harney, S.C.; Jane, D.E.; Anwyl, R. Extrasynaptic NR2D-containing NMDARs are recruited to the synapse during LTP of NMDAR-EPSCs. J. Neurosci. 2008, 28, 11685–11694. [Google Scholar] [CrossRef] [PubMed]
- Eapen, A.V.; Fernández-Fernández, D.; Georgiou, J.; Bortolotto, Z.A.; Lightman, S.; Jane, D.E.; Volianskis, A.; Collingridge, G.L. Multiple roles of GluN2D-containing NMDA receptors in short-term potentiation and long-term potentiation in mouse hippocampal slices. Neuropharmacology 2021, 201, 108833. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef]
- Yoshioka, A.; Yamaya, Y.; Saiki, S.; Kanemoto, M.; Hirose, G.; Beesley, J.; Pleasure, D. Non-N-methyl-d-aspartate glutamate receptors mediate oxygen-glucose deprivation-induced oligodendroglial injury. Brain Res. 2000, 854, 207–215. [Google Scholar] [CrossRef]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef]
- Palygin, O.; Lalo, U.; Pankratov, Y. Distinct pharmacological and functional properties of NMDA receptors in mouse cortical astrocytes. Br. J. Pharmacol. 2011, 163, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Adamsky, A.; Kol, A.; Kreisel, T.; Doron, A.; Ozeri-Engelhard, N.; Melcer, T.; Refaeli, R.; Horn, H.; Regev, L.; Groysman, M.; et al. Astrocytic activation generates de novo neuronal potentiation and memory enhancement. Cell 2018, 174, 59–71.e14. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Araque, A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science 2007, 317, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, C.; Papouin, T.; Oliet, S.H.R.; Rusakov, D.A. Long-term potentiation depends on release of D-serine from as-trocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Koh, W.; Lee, C.J.; Pankratov, Y. The tripartite glutamatergic synapse. Neuropharmacology 2021, 199, 108758. [Google Scholar] [CrossRef] [PubMed]
- Raghunatha, P.; Vosoughi, A.; Kauppinen, T.M.; Jackson, M.F. Microglial NMDA receptors drive pro-inflammatory responses via PARP-1/TRMP2 signaling. Glia 2020, 68, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.D.; Lester, R.A.; Tong, G.; Jahr, C.E.; Westbrook, G.L. The time course of glutamate in the synaptic cleft. Science 1992, 258, 1498–1501. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.; Defamie, N.; Zhang, X.; De Gois, S.; Shawki, A.; Mackenzie, B.; Chen, C.; Varoqui, H.; Erickson, J.D. SNAT2 amino acid transporter is regulated by amino acids of the SLC6 gamma-aminobutyric acid transporter subfamily in neocortical neurons and may play no role in delivering glutamine for glutamatergic transmission. J. Biol. Chem. 2009, 284, 11224–11236. [Google Scholar] [CrossRef]
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zheng, F.; Moon, C.; Schlüter, O.M.; Wang, H. Bi-directional regulation of CaMKIIα phosphorylation at Thr286 by NMDA receptors in cultured cortical neurons. J. Neurochem. 2012, 122, 295–307. [Google Scholar] [CrossRef]
- Kristensen, A.S.; Jenkins, M.A.; Banke, T.G.; Schousboe, A.; Makino, Y.; Johnson, R.C.; Huganir, R.; Traynelis, S.F. Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat. Neurosci. 2011, 14, 727–735. [Google Scholar] [CrossRef]
- Yang, X.; Gong, R.; Qin, L.; Bao, Y.; Fu, Y.; Gao, S.; Yang, H.; Ni, J.; Yuan, T.-F.; Lu, W. Trafficking of NMDA receptors is es-sential for hippocampal synaptic plasticity and memory consolidation. Cell Rep. 2022, 40, 111217. [Google Scholar] [CrossRef]
- Niethammer, M.; Kim, E.; Sheng, M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J. Neurosci. 1996, 16, 2157–2163. [Google Scholar] [CrossRef]
- El-Husseini, A.E.; Schnell, E.; Chetkovich, D.M.; Nicoll, R.A.; Bredt, D.S. PSD-95 involvement in maturation of excitatory synapses. Science 2000, 290, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Lim, I.A.; Hall, D.D.; Hell, J.W. Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and synapse-associated protein 102. J. Biol. Chem. 2002, 277, 21697–21711. [Google Scholar] [CrossRef]
- Barco, A.; Alarcon, J.M.; Kandel, E.R. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell 2002, 108, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Benito, E.; Barco, A. CREB’s control of intrinsic and synaptic plasticity: Implications for CREB-dependent memory models. Trends Neurosci. 2010, 33, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Fiumelli, H.; Jabaudon, D.; Magistretti, P.J.; Martin, J.L. BDNF stimulates expression, activity and release of tissue-type plasminogen activator in mouse cortical neurons. Eur. J. Neurosci. 1999, 11, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Pang, P.T.; Teng, H.K.; Zaitsev, E.; Woo, N.T.; Sakata, K.; Zhen, S.; Teng, K.K.; Yung, W.H.; Hempstead, B.L.; Lu, B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 2004, 306, 487–491. [Google Scholar] [CrossRef]
- Mulkey, R.M.; Herron, C.E.; Malenka, R.C. An essential role for protein phosphatases in hippocampal long-term depression. Science 1993, 261, 1051–1055. [Google Scholar] [CrossRef]
- Li, Z.; Jo, J.; Jia, J.-M.; Lo, S.-C.; Whitcomb, D.J.; Jiao, S.; Cho, K.; Sheng, M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 2010, 141, 859–871. [Google Scholar] [CrossRef]
- Bloodgood, B.L.; Giessel, A.J.; Sabatini, B.L. Biphasic synaptic Ca influx arising from compartmentalized elec-trical signals in dendritic spines. PLoS Biol. 2009, 7, e1000190. [Google Scholar] [CrossRef] [PubMed]
- Lanté, F.; Cavalier, M.; Cohen-Solal, C.; Guiramand, J.; Vignes, M. Developmental switch from LTD to LTP in low frequency-induced plasticity. Hippocampus 2006, 16, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Roche, K.W.; Standley, S.; McCallum, J.; Dune Ly, C.; Ehlers, M.D.; Wenthold, R.J. Molecular determinants of NMDA recep-tor internalization. Nat. Neurosci. 2001, 4, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Grosshans, D.R.; Clayton, D.A.; Coultrap, S.J.; Browning, M.D. LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat. Neurosci. 2002, 5, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, K.; Philpot, B.D. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and meta-plasticity. Neuropharmacology 2008, 55, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wong, T.P.; Pozza, M.F.; Lingenhoehl, K.; Wang, Y.; Sheng, M.; Auberson, Y.P.; Wang, Y.T. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 2004, 304, 1021–1024. [Google Scholar] [CrossRef] [PubMed]
- Bellone, C.; Nicoll, R.A. Rapid bidirectional switching of synaptic NMDA receptors. Neuron 2007, 55, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J.P.; Ladépêche, L.; Seth, H.; Bard, L.; Varela, J.; Mikasova, L.; Bouchet, D.; Rogemond, V.; Honnorat, J.; Hanse, E.; et al. Surface dynamics of GluN2B-NMDA receptors controls plasticity of maturing glutamate synapses. EMBO J. 2014, 33, 842–861. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Clemente, A.; Matta, J.A.; Isaac, J.T.R.; Roche, K.W. Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron 2010, 67, 984–996. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Clemente, A.; Gray, J.A.; Ogilvie, K.A.; Nicoll, R.A.; Roche, K.W. Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep. 2013, 3, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Borgdorff, A.J.; Choquet, D. Regulation of AMPA receptor lateral movements. Nature 2002, 417, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Opazo, P.; Labrecque, S.; Tigaret, C.M.; Frouin, A.; Wiseman, P.W.; De Koninck, P.; Choquet, D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 2010, 67, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Sumioka, A.; Yan, D.; Tomita, S. TARP phosphorylation regulates synaptic AMPA receptors through lipid bilayers. Neuron 2010, 66, 755–767. [Google Scholar] [CrossRef]
- Penn, A.C.; Zhang, C.L.; Georges, F.; Royer, L.; Breillat, C.; Hosy, E.; Petersen, J.D.; Humeau, Y.; Choquet, D. Hippocampal LTP and contextual learning require surface diffusion of AMPA receptors. Nature 2017, 549, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Gardoni, F.; Schrama, L.H.; Kamal, A.; Gispen, W.H.; Cattabeni, F.; Di Luca, M. Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor. J. Neurosci. 2001, 21, 1501–1509. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gardoni, F.; Mauceri, D.; Fiorentini, C.; Bellone, C.; Missale, C.; Cattabeni, F.; Di Luca, M. CaMKII-dependent phosphoryla-tion regulates SAP97/NR2A interaction. J. Biol. Chem. 2003, 278, 44745–44752. [Google Scholar] [CrossRef]
- Mauceri, D.; Gardoni, F.; Marcello, E.; Di Luca, M. Dual role of CaMKII-dependent SAP97 phosphorylation in mediating trafficking and insertion of NMDA receptor subunit NR2A. J. Neurochem. 2007, 100, 1032–1046. [Google Scholar] [CrossRef] [PubMed]
- Tingley, W.G.; Ehlers, M.D.; Kameyama, K.; Doherty, C.; Ptak, J.B.; Riley, C.T.; Huganir, R.L. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-d-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J. Biol. Chem. 1997, 272, 5157–5166. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.B.; Blanpied, T.A.; Swanson, G.T.; Zhang, C.; Ehlers, M.D. An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J. Neurosci. 2001, 21, 3063–3072. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.B.; Blanpied, T.A.; Ehlers, M.D. Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER reten-tion and regulates the surface delivery of NMDA receptors. Neuropharmacology 2003, 45, 755–767. [Google Scholar] [CrossRef]
- Taniguchi, S.; Nakazawa, T.; Tanimura, A.; Kiyama, Y.; Tezuka, T.; Watabe, A.M.; Katayama, N.; Yokoyama, K.; Inoue, T.; Izumi-Nakaseko, H.; et al. Involvement of NMDAR2A tyrosine phosphorylation in depression-related behaviour. EMBO J. 2009, 28, 3717–3729. [Google Scholar] [CrossRef]
- Grau, C.; Arató, K.; Fernández-Fernández, J.M.; Valderrama, A.; Sindreu, C.; Fillat, C.; Ferrer, I.; de la Luna, S.; Altafaj, X. DYRK1A-mediated phosphorylation of GluN2A at Ser1048 regulates the surface expression and channel activity of GluN1/GluN2A receptors. Front. Cell Neurosci. 2014, 8, 331. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-C.; Chang, C.-P.; Lin, C.-J.; Lai, H.-L.; Kao, Y.-H.; Cheng, S.-J.; Chen, H.-M.; Liao, Y.-P.; Faivre, E.; Buée, L.; et al. Adenosine augmentation evoked by an ENT1 inhibitor improves memory impairment and neuronal plasticity in the APP/PS1 mouse model of Alzheimer’s disease. Mol. Neurobiol. 2018, 55, 8936–8952. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Komai, S.; Tezuka, T.; Hisatsune, C.; Umemori, H.; Semba, K.; Mishina, M.; Manabe, T.; Yamamoto, T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-d-aspartate receptor. J. Biol. Chem. 2001, 276, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, B.; Li, G.; Chen, L.; Chen, L. Simvastatin enhances NMDA receptor GluN2B expression and phosphorylation of GluN2B and GluN2A through increased histone acetylation and Src signaling in hippocampal CA1 neurons. Neuropharmacology 2016, 107, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Meng, Z.-X.; Chen, Y.-Z.; Li, Y.-P.; Zhou, H.-Y.; Yang, M.; Zhao, T.-T.; Gong, Y.-L.; Wu, Y.; Liu, T. Enriched environ-ment enhances histone acetylation of NMDA receptor in the hippocampus and improves cognitive dysfunction in aged mice. Neural Regen. Res. 2020, 15, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.G.; Anderson, E.; Lynch, G.S.; Baudry, M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature 1986, 319, 774–776. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Butcher, S.P.; Morris, R.G. The NMDA receptor antagonist D-2-amino-5-phosphonopentanoate (D-AP5) impairs spatial learning and LTP in vivo at intracerebral concentrations comparable to those that block LTP in vitro. J. Neurosci. 1992, 12, 21–34. [Google Scholar] [CrossRef]
- Lodge, D.; Watkins, J.C.; Bortolotto, Z.A.; Jane, D.E.; Volianskis, A. The 1980s: D-AP5, LTP and a decade of NMDA receptor discoveries. Neurochem. Res. 2019, 44, 516–530. [Google Scholar] [CrossRef] [PubMed]
- Gozzi, A.; Herdon, H.; Schwarz, A.; Bertani, S.; Crestan, V.; Turrini, G.; Bifone, A. Pharmacological stimulation of NMDA receptors via co-agonist site suppresses fMRI response to phencyclidine in the rat. Psychopharmacology 2008, 201, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-H.; Vicknasingam, B.; Cheung, Y.-W.; Zhou, W.; Nurhidayat, A.W.; Jarlais, D.C.D.; Schottenfeld, R. To use or not to use: An update on licit and illicit ketamine use. Subst. Abus. Rehabil. 2011, 2, 11–20. [Google Scholar] [CrossRef]
- Pothula, S.; Kato, T.; Liu, R.-J.; Wu, M.; Gerhard, D.; Shinohara, R.; Sliby, A.-N.; Chowdhury, G.M.I.; Behar, K.L.; Sanacora, G.; et al. Cell-type specific modulation of NMDA receptors triggers antidepressant actions. Mol. Psychiatr. 2021, 26, 5097–5111. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Brown, K.A.; Georgiou, P.; Yuan, P.; Zarate, C.A.; Thompson, S.M.; Gould, T.D. NMDA receptor activation-dependent antidepressant-relevant behavioral and synaptic actions of ketamine. J. Neurosci. 2023, 43, 1038–1050. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Su, T.P.; Parish, D.W.; Nabeshima, T.; Privat, A. PRE-084, a sigma selective PCP derivative, attenuates MK-801-induced impairment of learning in mice. Pharmacol. Biochem. Behav. 1994, 49, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Hubert, J.P.; Delumeau, J.C.; Glowinski, J.; Prémont, J.; Doble, A. Antagonism by riluzole of entry of calcium evoked by NMDA and veratridine in rat cultured granule cells: Evidence for a dual mechanism of action. Br. J. Pharmacol. 1994, 113, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, B.D.; Kratzer, U.; Schmidt, W.J. Riluzole, a glutamate release inhibitor, and motor behavior. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1998, 358, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Lamanauskas, N.; Nistri, A. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via pre-synaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur. J. Neurosci. 2008, 27, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Sircar, R.; Rappaport, M.; Nichtenhauser, R.; Zukin, S.R. The novel anticonvulsant MK-801: A potent and specific ligand of the brain phencyclidine/sigma-receptor. Brain Res. 1987, 435, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, I.J.; Hughes, N.; Carroll, C.B.; Brotchie, J.M. Reversal of parkinsonian symptoms by intrastriatal and systemic manipulations of excitatory amino acid and dopamine transmission in the bilateral 6-OHDA lesioned marmoset. Behav. Pharmacol. 1995, 6, 492–507. [Google Scholar] [CrossRef]
- Gallagher, M.J.; Huang, H.; Pritchett, D.B.; Lynch, D.R. Interactions between ifenprodil and the NR2B subunit of the N-Methyl-d-aspartate receptor. J. Biol. Chem. 1996, 271, 9603–9611. [Google Scholar] [CrossRef]
- Sarre, S.; Lanza, M.; Makovec, F.; Artusi, R.; Caselli, G.; Michotte, Y. In vivo neurochemical effects of the NR2B selective NMDA receptor antagonist CR 3394 in 6-hydroxydopamine lesioned rats. Eur. J. Pharmacol. 2008, 584, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G. The NMDA receptor antagonist memantine as a symptomatological and neuro-protective treatment for Alzheimer’s disease: Preclinical evidence. Int. J. Geriatr. Psychiatry 2003, 18, S23–S32. [Google Scholar] [CrossRef] [PubMed]
- Murakawa-Hirachi, T.; Mizoguchi, Y.; Ohgidani, M.; Haraguchi, Y.; Monji, A. Effect of memantine, an anti-Alzheimer’s drug, on rodent microglial cells in vitro. Sci. Rep. 2021, 11, 6151. [Google Scholar] [CrossRef] [PubMed]
- Blanpied, T.A.; Clarke, R.J.; Johnson, J.W. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J. Neurosci. 2005, 25, 3312–3322. [Google Scholar] [CrossRef]
- Block, F.; Schwarz, M. Dextromethorphan reduces functional deficits and neuronal damage after global ischemia in rats. Brain Res. 1996, 741, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Dere, E.; Topic, B.; De Souza Silva, M.A.; Fink, H.; Buddenberg, T.; Huston, J.P. NMDA-receptor antagonism via dextromethorphan and ifenprodil modulates graded anxiety test performance of C57BL/6 mice. Behav. Pharmacol. 2003, 14, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Kemppainen, P.; Waltimo, A.; Waltimo, T.; Könönen, M.; Pertovaara, A. Differential effects of noxious conditioning stimulation of the cheek by capsaicin on human sensory and inhibitory masseter reflex responses evoked by tooth pulp stimulation. J. Dent. Res. 1997, 76, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Weinbroum, A.A.; Gorodetzky, A.; Nirkin, A.; Kollender, Y.; Bickels, J.; Marouani, N.; Rudick, V.; Meller, I. Dextrome-thorphan for the reduction of immediate and late postoperative pain and morphine consumption in orthopedic oncology patients: A randomized, placebo-controlled, double-blind study. Cancer 2002, 95, 1164–1170. [Google Scholar] [CrossRef]
- Weinbroum, A.A.; Bender, B.; Nirkin, A.; Chazan, S.; Meller, I.; Kollender, Y. Dextromethorphan-associated epidural patient-controlled analgesia provides better pain- and analgesics-sparing effects than dextromethorphan-associated intravenous patient-controlled analgesia after bone-malignancy resection: A randomized, placebo-controlled, double-blinded study. Anesth. Analg. 2004, 98, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Williams, K. Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptor: Selectivity and mechanisms at recombinant heteromeric receptors. Mol. Pharmacol. 1993, 44, 851–859. [Google Scholar] [PubMed]
- Tajima, N.; Karakas, E.; Grant, T.; Simorowski, N.; Diaz-Avalos, R.; Grigorieff, N.; Furukawa, H. Activation of NMDA receptors and the mechanism of inhibition by ifenprodil. Nature 2016, 534, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, S.; Kishi, T.; Nomura, I.; Sakuma, K.; Okuya, M.; Ikuta, T.; Iwata, N. The efficacy and safety of memantine for the treatment of Alzheimer’s disease. Exp. Opin. Drug Saf. 2018, 17, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.R.; Pizon, A.F.; Brooks, D.E. Dextromethorphan-induced serotonin syndrome. Clin. Toxicol. 2008, 46, 771–773. [Google Scholar] [CrossRef] [PubMed]
- Guyot, M.C.; Hantraye, P.; Dolan, R.; Palfi, S.; Maziére, M.; Brouillet, E. Quantifiable bradykinesia, gait abnormalities and Huntington’s disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience 1997, 79, 45–56. [Google Scholar] [CrossRef]
- Albin, R.L.; Greenamyre, J.T. Alternative excitotoxic hypotheses. Neurology 1992, 42, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, M.; Fukumoto, H.; Newell, K.L.; Growdon, J.H.; Hedley-Whyte, E.T.; Frosch, M.P.; Albert, M.S.; Hyman, B.T.; Irizarry, M.C. Early Ab accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 2004, 62, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef]
- Fonte, C.; Smania, N.; Pedrinolla, A.; Munari, D.; Gandolfi, M.; Picelli, A.; Varalta, V.; Benetti, M.V.; Brugnera, A.; Federico, A.; et al. Comparison between physical and cognitive treatment in patients with MIC and Alzheimer’s disease. Aging 2019, 11, 3138–3155. [Google Scholar] [CrossRef] [PubMed]
- Blinkouskaya, Y.; Weickenmeier, J. Brain shape changes associated with cerebral atrophy in healthy aging and Alzheimer’s disease. Front. Mech. Eng. 2021, 7, 705653. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Xu, H.; Li, Z.; Wang, Z.; O’Malley, T.T.; Zhang, D.; Walsh, D.M.; Xu, P.; Selkoe, D.J.; Li, S. Soluble Aβ oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol. Dis. 2016, 85, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.E.; Ince, P.G.; Lace, G.; Forster, G.; Shaw, P.J.; Matthews, F.; Savva, G.; Brayne, C.; Wharton, S.B. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 2010, 31, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Nguyen, L.N.; Kessels, H.W.; Hagiwara, H.; Sisodia, S.; Malinow, R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 2010, 13, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble Aβ oligomers inhibit long-term poten-tiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef]
- Companys-Alemany, J.; Turcu, A.L.; Vázquez, S.; Pallàs, M.; Griñán-Ferré, C. Glial cell reactivity and oxidative stress prevention in Alzheimer’s disease mice model by an optimized NMDA receptor antagonist. Sci. Rep. 2022, 12, 17908. [Google Scholar] [CrossRef] [PubMed]
- Esposito, Z.; Belli, L.; Toniolo, S.; Sancesario, G.; Bianconi, C.; Martorana, A. Amyloid β, Glutamate, Excitotoxicity in Alzheimer’s Disease: Are We on the Right Track? CNS Neurosci. Ther. 2013, 19, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-b toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Vitolo, O.V.; Sant’Angelo, A.; Costanzo, V.; Battaglia, F.; Arancio, O.; Shelanski, M. Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: Reversibility by drugs that enhance cAMP signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13217–13221. [Google Scholar] [CrossRef] [PubMed]
- Rosa, E.; Fahnestock, M. CREB expression mediates amyloid β-induced basal BDNF downregulation. Neurobiol. Aging 2015, 36, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.B.C.; Emptage, N.J.; Jeans, A.F. Long-term depression links amyloid-β to the pathological hyperphosphorylation of tau. Cell Rep. 2021, 36, 109638. [Google Scholar] [CrossRef] [PubMed]
- Bordji, K.; Becerril-Ortega, J.; Nicole, O.; Buisson, A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-ß production. J. Neurosci. 2010, 30, 15927–15942. [Google Scholar] [CrossRef] [PubMed]
- Sze, S.C.; Wong, C.K.; Yung, K.K. Modulation of the gene expression of N-methyl-d-aspartate receptor NR2B subunit in the rat neostriatum by a single dose of specific antisense oligodeoxynucleotide. Neurochem. Int. 2001, 39, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Sze, C.-I. N-Methyl-d-aspartate receptor subunit NR2A and NR2B messenger RNA levels are altered in the hippo-campus and entorhinal cortex in Alzheimer’s disease. J. Neurol. Sci. 2002, 200, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Differential expression of N-methyl-d-aspartate receptor NR2 isoforms in Alzheimer’s disease. J. Neurochem. 2004, 90, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Mishizen-Eberz, A.J.; Rissman, R.A.; Carter, T.L.; Ikonomovic, M.D.; Wolfe, B.B.; Armstrong, D.M. Biochemical and molecular studies of NMDA receptor subunits NR1/2A/2B in hippocampal subregions throughout progression of Alzheimer’s disease pathology. Neurobiol. Dis. 2004, 15, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.W.Y.; Vinters, H.V.; Cummings, J.L.; Wong, P.T.-H.; Chen, C.P.L.-H.; Lai, M.K.P. Alterations in NMDA receptor subunit densities and ligand binding to glycine recognition sites are associated with chronic anxiety in Alzheimer’s disease. Neurobiol. Aging 2008, 29, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Aggarwal, N.T.; Vila-Castelar, C.; Agarwal, P.; Arenaza-Urquijo, E.M.; Brett, B.; Brugulat-Serrat, A.; DuBose, L.E.; Eikelboom, W.S.; Flatt, J.; et al. Diversity and Disparity Professional Interest Area Sex and Gender Special Interest Group. Consideration of sex and gender in Alzheimer’s disease and related disorders from a global perspective. Alzheimers Dement. 2022, 18, 2707–2724. [Google Scholar] [CrossRef] [PubMed]
- Snyder, H.M.; Asthana, S.; Bain, L.; Brinton, R.; Craft, S.; Dubal, D.B.; Espeland, M.A.; Gatz, M.; Mielke, M.M.; Raber, J.; et al. Sex biology contributions to vulnerability to Alzheimer’s disease: A think tank convened by the women’s Alzheimer’s research initiative. Alzheimers Dement. 2016, 12, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Maffioli, E.; Murtas, G.; Rabattoni, V.; Badone, B.; Tripodi, F.; Iannuzzi, F.; Licastro, D.; Nonnis, S.; Rinaldi, A.M.; Motta, Z.; et al. Insulin and serine metabolism as sex-specific hallmarks of Alzheimer’s disease in the human hippocampus. Cell Rep. 2022, 40, 111271. [Google Scholar] [CrossRef] [PubMed]
- Papouin, T.; Ladépêche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.P.; Oliet, S.H. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Baier, M.P.; Nagaraja, R.Y.; Yarbrough, H.P.; Owen, D.B.; Masingale, A.M.; Ranjit, R.; Stiles, M.A.; Murphy, A.; Agbaga, M.P.; Ahmad, M.; et al. Selective ablation of Sod2 in astrocytes induces sex-specific effects on cognitive function, d-serine availability, and astrogliosis. J. Neurosci. 2022, 42, 5992–6006. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.A.; Ashford, W.; Ernesto, C.; Saxton, J.; Schneider, L.S.; Clark, C.M.; Ferris, S.H.; Mackell, J.A.; Schafer, K.; Thal, L.J. The severe impairment battery: Concurrent validity and the assessment of longitudinal change in Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis. Assoc. Disord. 1997, 11 (Suppl. 2), S51–S56. [Google Scholar]
- Tariot, P.N.; Farlow, M.R.; Grossberg, G.T.; Graham, S.M.; McDonald, S.; Gergel, I.; For the Memantine Study Group. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil. A randomized controlled trial. J. Am. Med. Assoc. 2004, 291, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Mimica, N.; Presecki, P. Side effects of approved antidementives. Psychiatr. Danub. 2009, 21, 108–113. [Google Scholar] [PubMed]
- Medina, A.; Mahjoub, Y.; Shaver, L.; Pringsheim, T. Prevalence and incidence of Huntington’s disease: An updated systematic review and meta-analysis. Mov. Disord. 2022, 37, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Albin, R.L.; Anderson, K.D.; D’Amato, C.J.; Penney, J.B.; Young, A.B. Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. USA 1988, 85, 5733–5737. [Google Scholar] [CrossRef] [PubMed]
- Foroud, T.; Gray, J.; Ivashina, J.; Conneally, P.M. Differences in duration of Huntington’s disease based on age at onset. J. Neurol. Neurosurg. Psychiatr. 1999, 66, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Beck, C.A.; Darwin, K.; Nichols, P.; Brocht, A.F.D.; Biglan, K.M.; Shoulson, I.; Huntington Study Group CO-HORT Investigators. Natural history of Huntington disease. JAMA Neurol. 2013, 70, 1520–1530. [Google Scholar] [CrossRef][Green Version]
- Duyao, M.; Ambrose, C.; Myers, R.; Novelletto, A.; Persichetti, F.; Frontali, M.; Folstein, S.; Ross, C.; Franz, M.; Abbott, M. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat. Genet. 1993, 4, 387–392. [Google Scholar] [CrossRef] [PubMed]
- McKinstry, S.U.; Karadeniz, Y.B.; Worthington, A.K.; Hayrapetyan, V.Y.; Ozlu, M.I.; Serafin-Molina, K.; Risher, W.C.; Ustunkaya, T.; Dragatsis, I.; Zeitlin, S.; et al. Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J. Neurosci. 2014, 34, 9455–9472. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s disease: Mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef]
- Sun, Y.; Savanenin, A.; Reddy, P.H.; Liu, Y.F. Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-d-aspartate receptors via post-synaptic density 95. J. Biol. Chem. 2001, 276, 24713–24718. [Google Scholar] [CrossRef]
- Young, A.B.; Greenamyre, J.T.; Hollingsworth, Z.; Albin, R.; D’Amato, C.; Shoulson, I.; Penney, J.B. NMDA receptor losses in putamen from patients with Huntington’s disease. Science 1988, 241, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Young, A.B.; Penney, J.B.; Handelin, B.; Balfour, R.; Anderson, K.D.; Markel, D.S.; Tourtellotte, W.W.; Reiner, A. Abnormalities of striatal projection neurons and N-methyl-d-aspartate receptors in presymptomatic Huntington’s disease. N. Engl. J. Med. 1990, 322, 1293–1298. [Google Scholar] [CrossRef]
- Matsushima, A.; Pineda, S.S.; Crittenden, J.R.; Lee, H.; Galani, K.; Mantero, J.; Tombaugh, G.; Kellis, M.; Heiman, M.; Graybiel, A.M. Transcriptional vulnerabilities of striatal neurons in human and rodent models of Huntington’s disease. Nat. Commun. 2023, 14, 282. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, C.; Ariano, M.A.; Calvert, C.R.; Flores-Hernández, J.; Chandler, S.H.; Leavitt, B.R.; Hayden, M.R.; Levine, M.S. NMDA receptor function in mouse models of Huntington disease. J. Neurosci. Res. 2001, 66, 525–539. [Google Scholar] [CrossRef]
- Ali, N.J.; Levine, M.S. Changes in expression of N-methyl-d-aspartate receptor subunits occur early in the R6/2 mouse model of Huntington’s disease. Dev. Neurosci. 2006, 28, 230–238. [Google Scholar] [CrossRef]
- Jarabek, B.R.; Yasuda, R.P.; Wolfe, B.B. Regulation of proteins affecting NMDA receptor-induced excitotoxicity in a Huntington’s mouse model. Brain 2004, 127, 505–516. [Google Scholar] [CrossRef]
- Faideau, M.; Kim, J.; Cormier, K.; Gilmore, R.; Welch, M.; Auregan, G.; Dufour, N.; Guillermier, M.; Brouillet, E.; Hantraye, P.; et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: A correlation with Huntington’s disease subjects. Hum. Mol. Genet. 2010, 19, 3053–3067. [Google Scholar] [CrossRef] [PubMed]
- Heng, M.Y.; Detloff, P.J.; Wang, P.L.; Tsien, J.Z.; Albin, R.L. In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J. Neurosci. 2009, 29, 3200–3205. [Google Scholar] [CrossRef]
- Lujan, B.; Liu, X.; Wan, Q. Differential roles of GluN2A- and GluN2B-containing NMDA receptors in neuronal survival and death. Int. J. Physiol. Pathophysiol. Pharmacol. 2012, 4, 211–218. [Google Scholar]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef]
- Jiang, H.; Poirier, M.A.; Liang, Y.; Pei, Z.; Weiskittel, C.E.; Smith, W.W.; DeFranco, D.B.; Ross, C.A. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 2006, 23, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Saft, C.; Burgunder, J.-M.; Dose, M.; Jung, H.H.; Katzenschlager, R.; Priller, J.; Nguyen, H.P.; Reetz, K.; Reilmann, R.; Seppi, K.; et al. Symptomatic treatment options for Huntington’s disease (guidelines of the German Neurological Society). Neurol. Res. Pract. 2023, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Neuropathology of Parkinson disease. Park. Relat. Disord. 2018, 46, S30–S33. [Google Scholar] [CrossRef] [PubMed]
- De Pablo-Fernández, E.; Lees, A.J.; Holton, J.L.; Warner, T.T. Prognosis and neuropathologic correlation of clinical subtypes of Parkinson disease. JAMA Neurol. 2019, 76, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Nagatsua, T.; Sawadab, M. L-Dopa therapy for Parkinson’s disease: Past, present, and future. Park. Relat. Disord. 2009, 15, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Bastide, M.F.; Meissner, W.G.; Picconi, B.; Fasano, S.; Fernagut, P.-O.; Feyder, M.; Francardo, V.; Alcacer, C.; Ding, Y.; Bram-billa, R.; et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog. Neurobiol. 2015, 132, 96–168. [Google Scholar] [CrossRef] [PubMed]
- Marti, M.; Paganini, F.; Stocchi, S.; Mela, F.; Beani, L.; Bianchi, C.; Morari, M. Plasticity of glutamatergic control of striatal acetylcholine release in experimental parkinsonism: Opposite changes at group-II metabotropic and NMDA receptors. J. Neurochem. 2003, 84, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.C.; Gunasekar, P.G.; Borowitz, J.L.; Isom, G.E. Dopamine-induced apoptosis is mediated by oxidative stress and is enhanced by cyanide in differentiated PC12 cells. J. Neurochem. 2000, 74, 2296–2304. [Google Scholar] [CrossRef] [PubMed]
- Jourdain, V.A.; Morin, N.; Grégoire, L.; Morissette, M.; Di Paolo, T. Changes in glutamate receptors in dyskinetic parkinsonian monkeys after unilateral subthalamotomy. J. Neurosurg. 2015, 123, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Verhagen Metman, L.; Blanchet, P.J.; van den Munckhof, P.; Del Dotto, P.; Natté, R.; Chase, T.N. A trial of dextromethorphan in parkinsonian patients with motor response complications. Mov. Disord. 1998, 13, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Rascol, O.; Fabbri, M.; Poewe, W. Amantadine in the treatment of Parkinson’s disease and other movement disorders. Lancet Neurol. 2021, 20, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Uitti, R.J.; Rajput, A.H.; Ahlskog, J.E.; Offord, K.P.; Schroeder, D.R.; Ho, M.M.; Prasad, M.; Rajput, A.; Basran, P. Amantadine treatment is an independent predictor of improved survival in Parkinson’s disease. Neurology 1996, 46, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Merello, M.; Nouzeilles, M.I.; Cammarota, A.; Leiguarda, R. Effect of memantine (NMDA antagonist) on Parkinson’s disease: A double-blind crossover randomized study. Clin. Neuropharmacol. 1999, 22, 273–276. [Google Scholar] [PubMed]
- Dembitsky, V.M.; Gloriozova, T.A.; Poroikov, V.V. Pharmacological profile of natural and synthetic compounds with rigid adamantane-based scaffolds as potential agents for the treatment of neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2020, 529, 1225–1241. [Google Scholar] [CrossRef]
- Samnick, S.; Ametamey, S.; Leenders, K.L.; Vontobel, P.; Quack, G.; Parsons, C.G.; Neu, H.; Schubiger, P.A. Electrophysiolog-ical study, biodistribution in mice, and preliminary PET evaluation in a rhesus monkey of 1-amino-3-[18F]fluoromethyl-5-methyl-adamantane (18F-MEM): A potential radioligand for mapping the NMDA-receptor complex. Nucl. Med. Biol. 1998, 25, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Ametamey, S.M.; Samnick, S.; Leenders, K.L.; Vontobel, P.; Quack, G.; Parsons, C.G.; Schubiger, P.A. Fluorine-18 radiolabel-ling, biodistribution studies and preliminary PET evaluation of a new memantine derivative for imaging the NMDA receptor. J. Recept. Signal Transduct. Res. 1999, 19, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Ametamey, S.M.; Bruehlmeier, M.; Kneifel, S.; Kokic, M.; Honer, M.; Arigoni, M.; Buck, A.; Burger, C.; Samnick, S.; Quack, G.; et al. PET studies of 18F-memantine in healthy volunteers. Nucl. Med. Biol. 2002, 29, 227–231. [Google Scholar] [CrossRef]
- Weissman, A.D.; Casanova, M.F.; Kleinman, J.E.; De Souza, E.B. PCP and sigma receptors in brain are not altered after repeated exposure to PCP in humans. Neuropsychopharmacology 1991, 4, 95–102. [Google Scholar] [PubMed]
- Salabert, A.-S.; Fonta, C.; Fontan, C.; Adel, D.; Alonso, M.; Pestourie, C.; Belhadj-Tahar, H.; Tafani, M.; Payoux, P. Radio-labeling of [18F]-fluoroethylnormemantine and initial in vivo evaluation of this innovative PET tracer for imaging the PCP sites of NMDA receptors. Nucl. Med. Biol. 2015, 42, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Salabert, A.-S.; Mora-Ramirez, E.; Beaurain, M.; Alonso, M.; Fontan, C.; Tahar, H.B.; Boizeau, M.L.; Tafani, M.; Bardiès, M.; Payoux, P. Evaluation of [18F]FNM biodistribution and dosimetry based on whole-body PET imaging of rats. Nucl. Med. Biol. 2018, 59, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Beaurain, M.; Talmont, F.; Pierre, D.; Péran, P.; Boucher, S.; Hitzel, A.; Rols, M.-P.; Cuvillier, O.; Payoux, P.; Salabert, A.-S. Pharmacological characterization of [18F]-FNM and evaluation of NMDA receptors activation in a rat brain injury model. Mol. Imaging Biol. 2023, 25, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef] [PubMed]
- aan het Rot, M.; Collins, K.A.; Murrough, J.W.; Perez, A.M.; Reich, D.L.; Charney, D.S.; Mathew, S.J. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol. Psychiatry 2010, 67, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Amat, J.; Dolzani, S.D.; Tilden, S.; Christianson, J.P.; Kubala, K.H.; Bartholomay, K.; Sperr, K.; Ciancio, N.; Watkins, L.R.; Maier, S.F. Previous ketamine produces an enduring blockade of neurochemical and behavioral effects of uncontrollable stress. J. Neurosci. 2016, 36, 153–161. [Google Scholar] [CrossRef]
- Brachman, R.A.; McGowan, J.C.; Perusini, J.N.; Lim, S.C.; Pham, T.H.; Faye, C.; Gardier, A.M.; Mendez-David, I.; David, D.J.; Hen, R.; et al. Ketamine as a prophylactic against stress-induced depressive-like behavior. Biol. Psychiatr. 2016, 79, 776–786. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.C.; LaGamma, C.T.; Lim, S.C.; Tsitsiklis, M.; Neria, Y.; Brachman, R.A.; Denny, C.A. Prophylactic ketamine attenuates learned fear. Neuropsychopharmacology 2017, 42, 1577–1589. [Google Scholar] [CrossRef]
- Chen, B.K.; Le Pen, G.; Eckmier, A.; Rubinstenn, G.; Jay, T.M.; Denny, C.A. Fluoroethylnormemantine, a novel derivative of memantine, facilitates extinction learning without sensorimotor deficits. Int. J. Neuropychopharmacol. 2021, 24, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.K.; Luna, V.M.; Shannon, M.E.; Hunsberger, H.C.; Mastrodonato, A.; Stackmann, M.; McGowan, J.C.; Rubinstenn, G.; Denny, C.A. Fluoroethylnormemantine, a novel NMDA receptor antagonist, for the prevention and treatment of stress-induced maladaptive behavior. Biol. Psychiatr. 2021, 90, 458–472. [Google Scholar] [CrossRef]
- Maurice, T.; Phan, V.-L.; Privat, A. The anti-amnesic effects of sigma1 (σ1) receptor agonists confirmed by in vivo antisense strategy in the mouse. Brain Res. 2001, 898, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Couly, S.; Denus, M.; Bouchet, M.; Rubinstenn, G.; Maurice, T. Anti-amnesic and neuroprotective effects of fluoroethyl-normemantine in a pharmacological mouse model of Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2020, 24, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Slunt, H.H.; Ratovitski, T.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. Co-expression of multiple transgenes in mouse CNS: A comparison of strategies. Biomol. Eng. 2001, 17, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Carles, A.; Freyssin, A.; Guehairia, S.; Reguero, T.; Rubinstenn, G.; Maurice, T. Neuroprotective effects of Fluoroethylnormemantine (FENM) after chronic infusion by Alzet pumps in the Aß25-35 mouse model of Alzheimer’s disease. Neuroscience Meeting Planner; Society for Neuroscience: San Diego, CA, USA, 2022; Program No. 197.20. Online. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carles, A.; Freyssin, A.; Perin-Dureau, F.; Rubinstenn, G.; Maurice, T. Targeting N-Methyl-d-Aspartate Receptors in Neurodegenerative Diseases. Int. J. Mol. Sci. 2024, 25, 3733. https://doi.org/10.3390/ijms25073733
Carles A, Freyssin A, Perin-Dureau F, Rubinstenn G, Maurice T. Targeting N-Methyl-d-Aspartate Receptors in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2024; 25(7):3733. https://doi.org/10.3390/ijms25073733
Chicago/Turabian StyleCarles, Allison, Aline Freyssin, Florent Perin-Dureau, Gilles Rubinstenn, and Tangui Maurice. 2024. "Targeting N-Methyl-d-Aspartate Receptors in Neurodegenerative Diseases" International Journal of Molecular Sciences 25, no. 7: 3733. https://doi.org/10.3390/ijms25073733
APA StyleCarles, A., Freyssin, A., Perin-Dureau, F., Rubinstenn, G., & Maurice, T. (2024). Targeting N-Methyl-d-Aspartate Receptors in Neurodegenerative Diseases. International Journal of Molecular Sciences, 25(7), 3733. https://doi.org/10.3390/ijms25073733