
PARP14 Contributes to the Development of the Tumor-Associated Macrophage Phenotype



 , , , and
, , , and 
Abstract
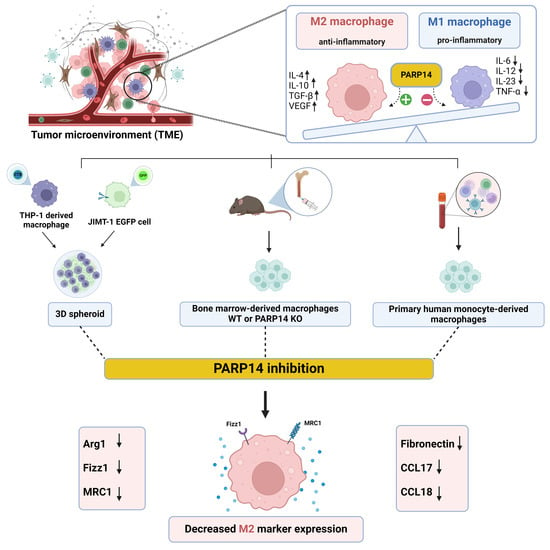
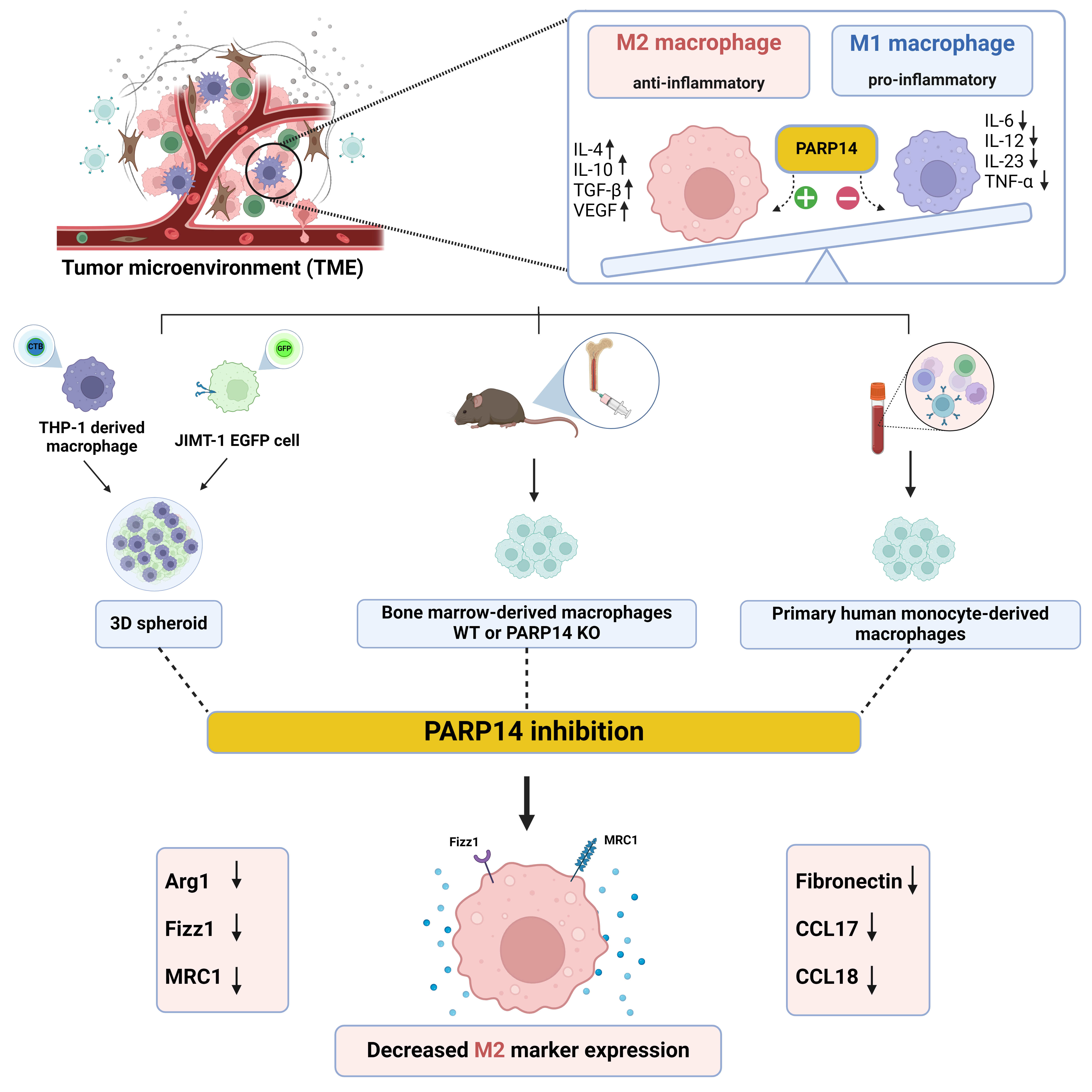{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
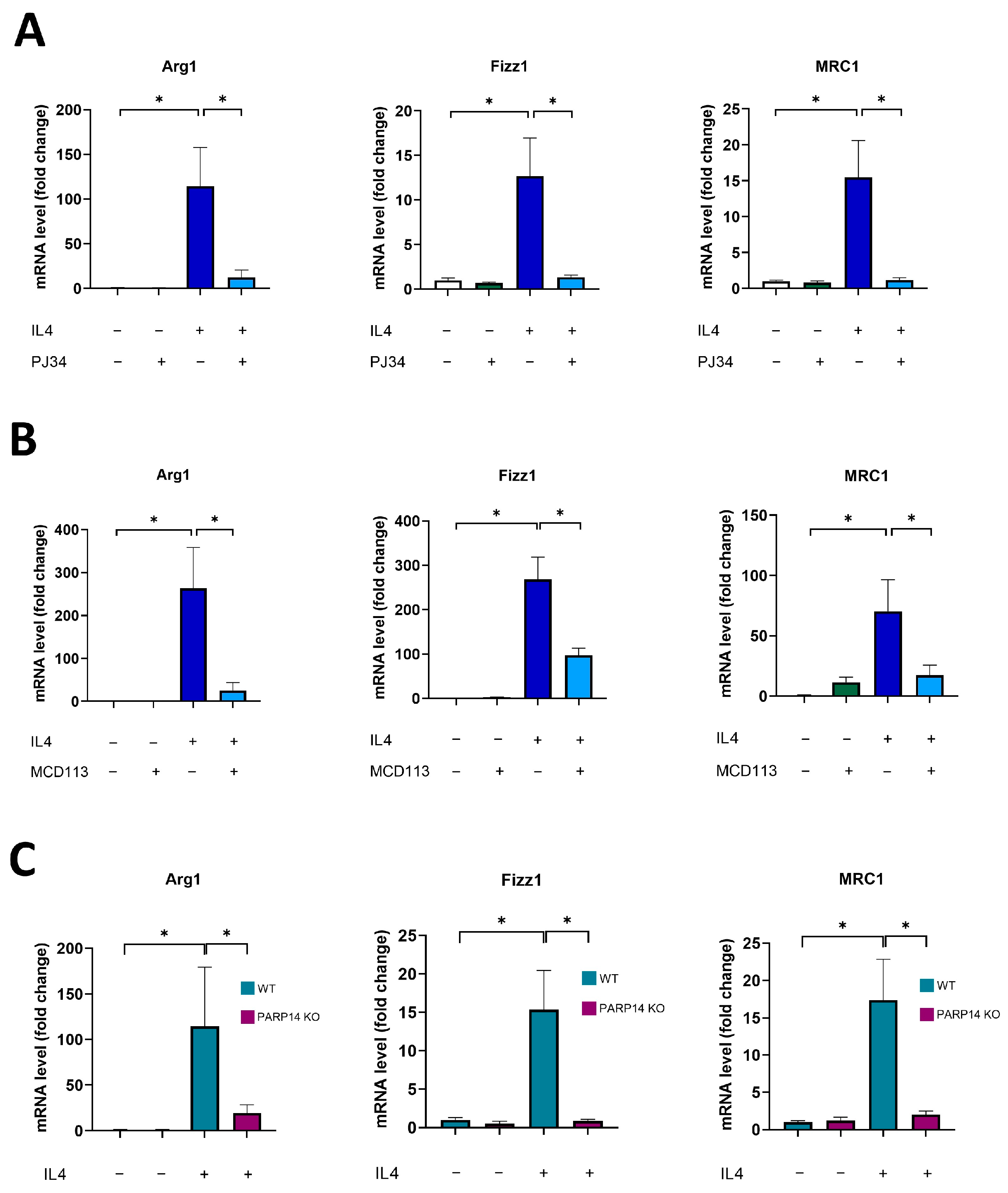
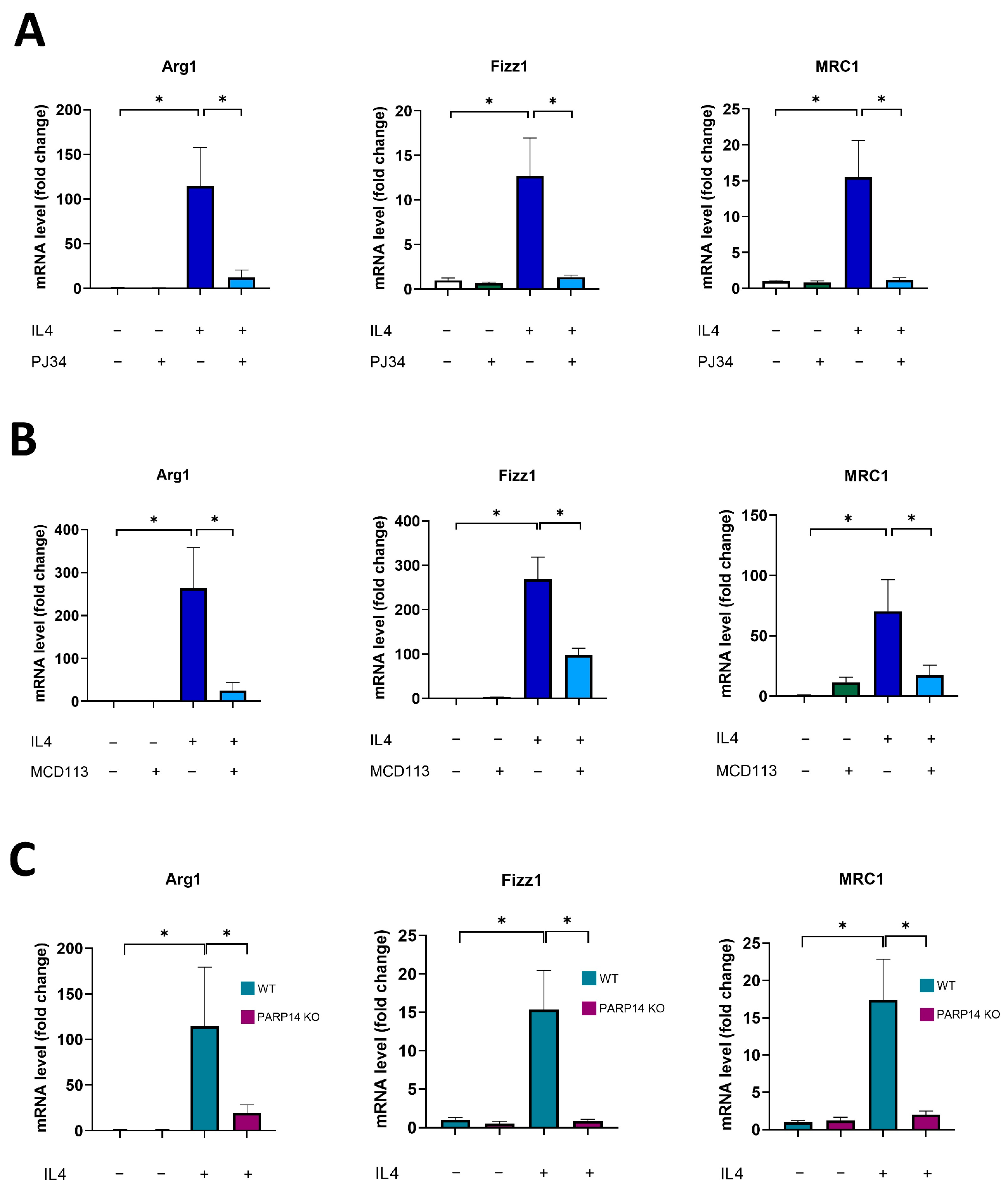
2.1. PARP14 Mediated M2 Polarization in Murine BMDMs
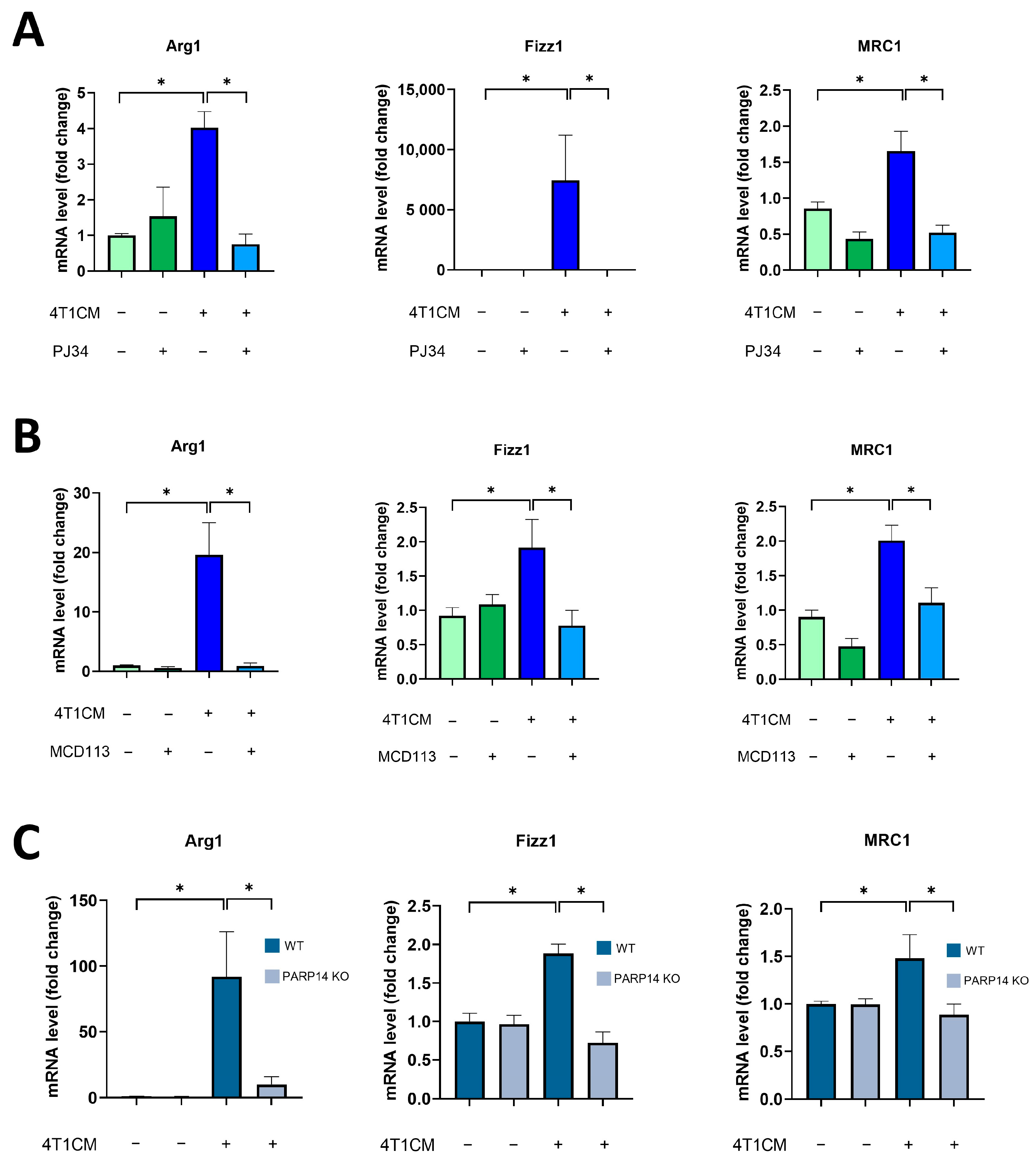
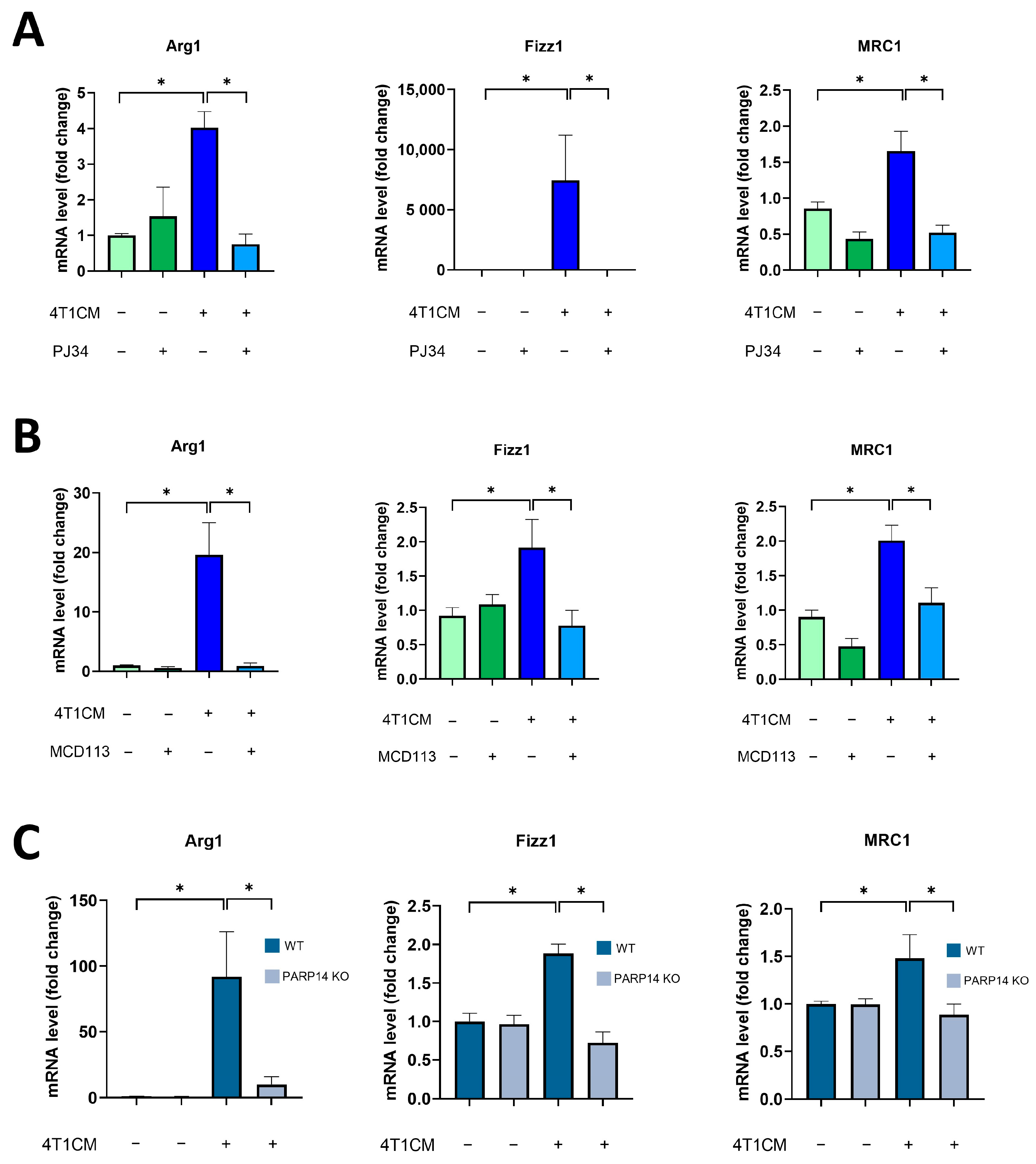
2.2. Soluble Factors Produced by 4T1 Murine Breast Cancer Cells Reprogrammed Murine BMDMs toward an M2-like TAM Phenotype in a PARP14-Dependent Manner
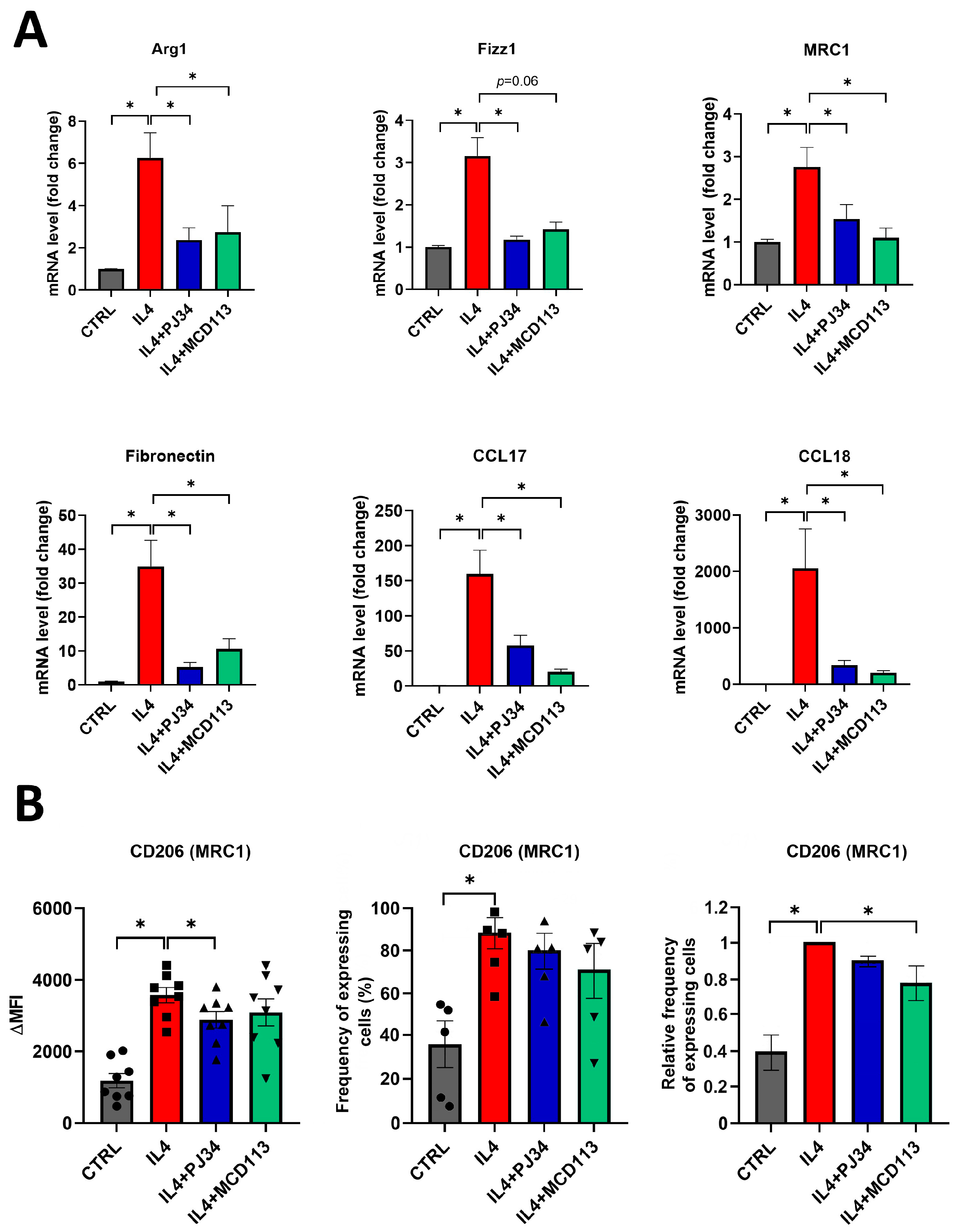
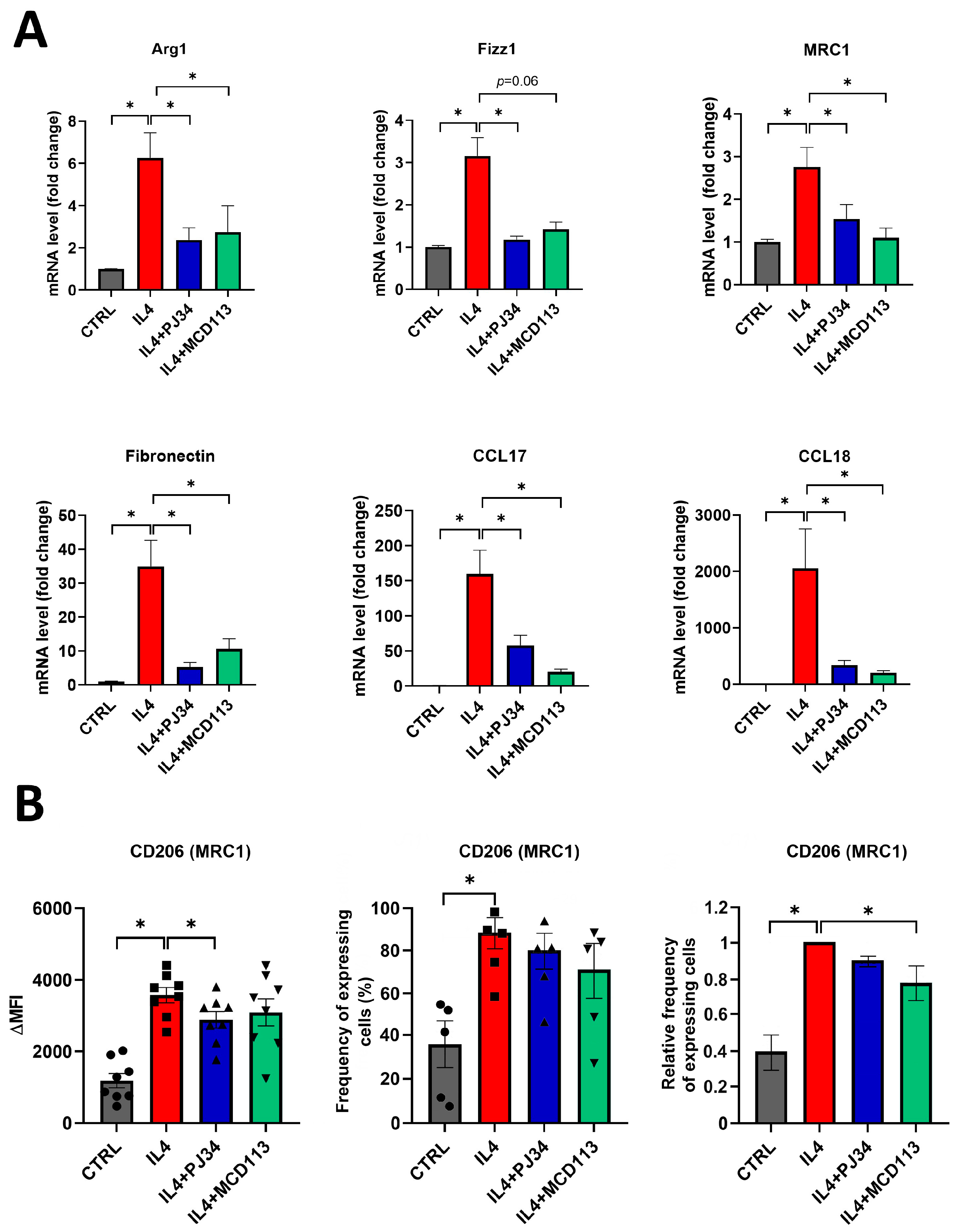
2.3. PARP14 Mediated M2 Polarization in Human PBMC-MΦs
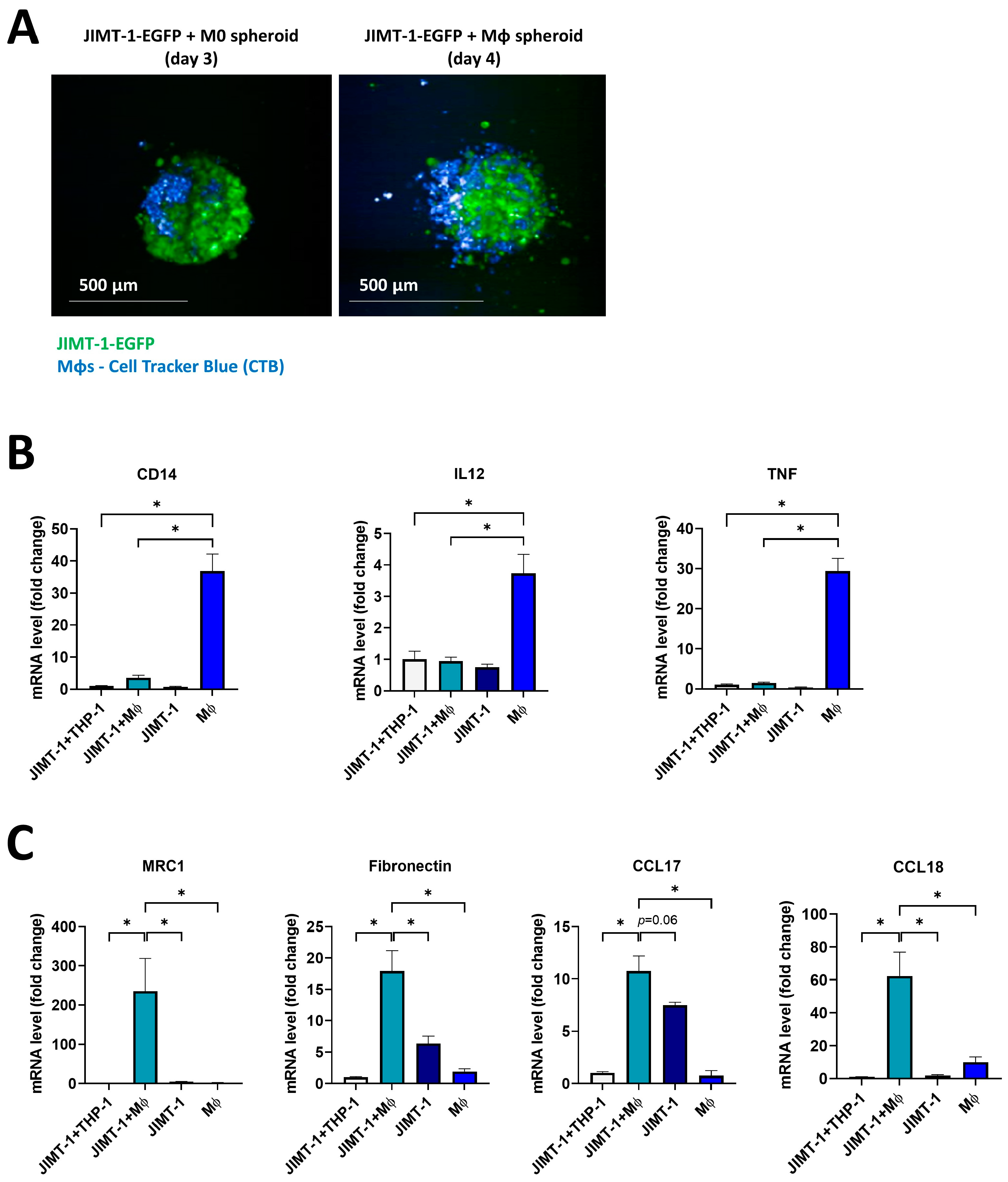
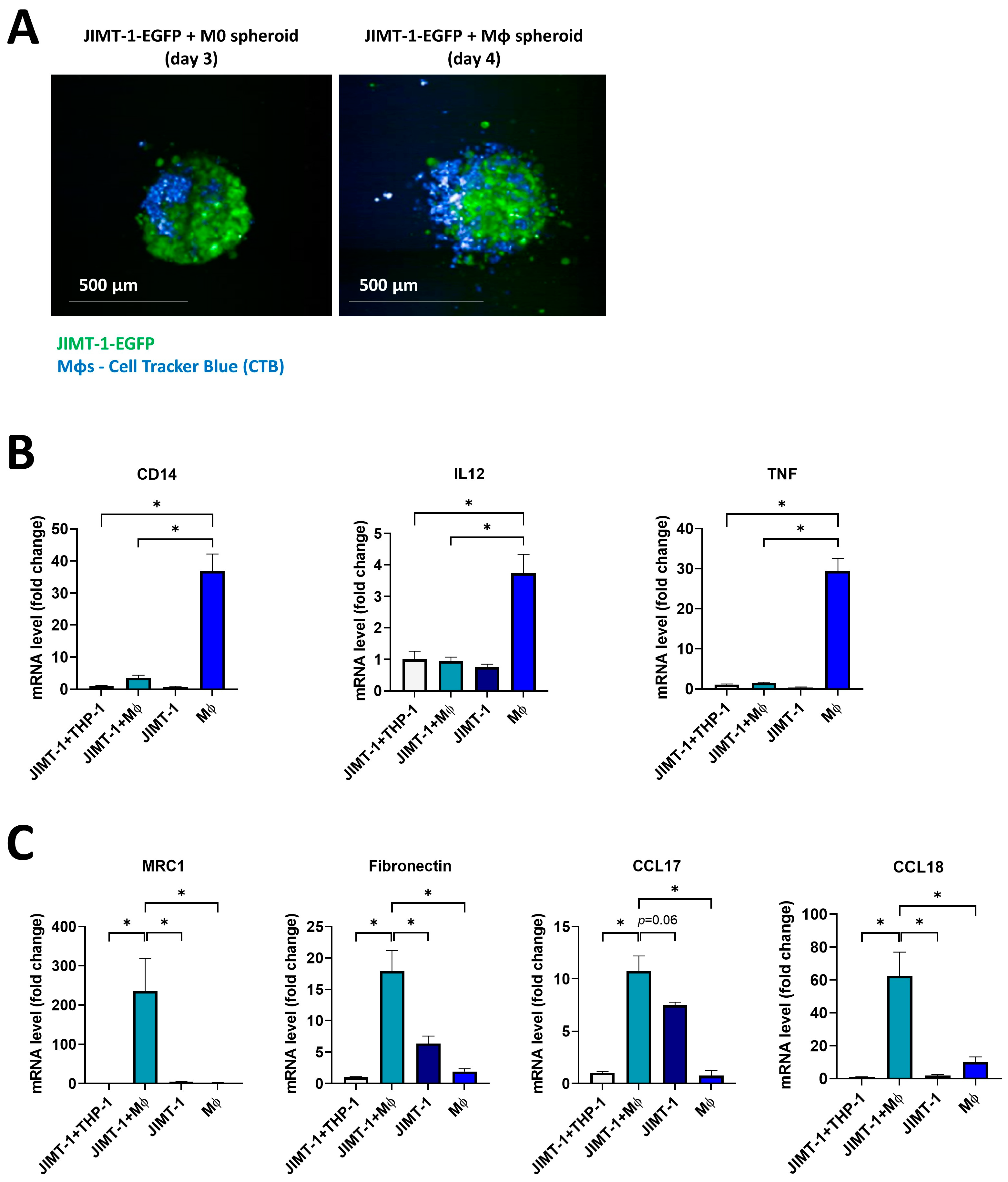
2.4. THP-1-Derived MΦs Incorporated into JIMT-1 Breast Cancer Cell Spheroids Underwent M2-like Polarization
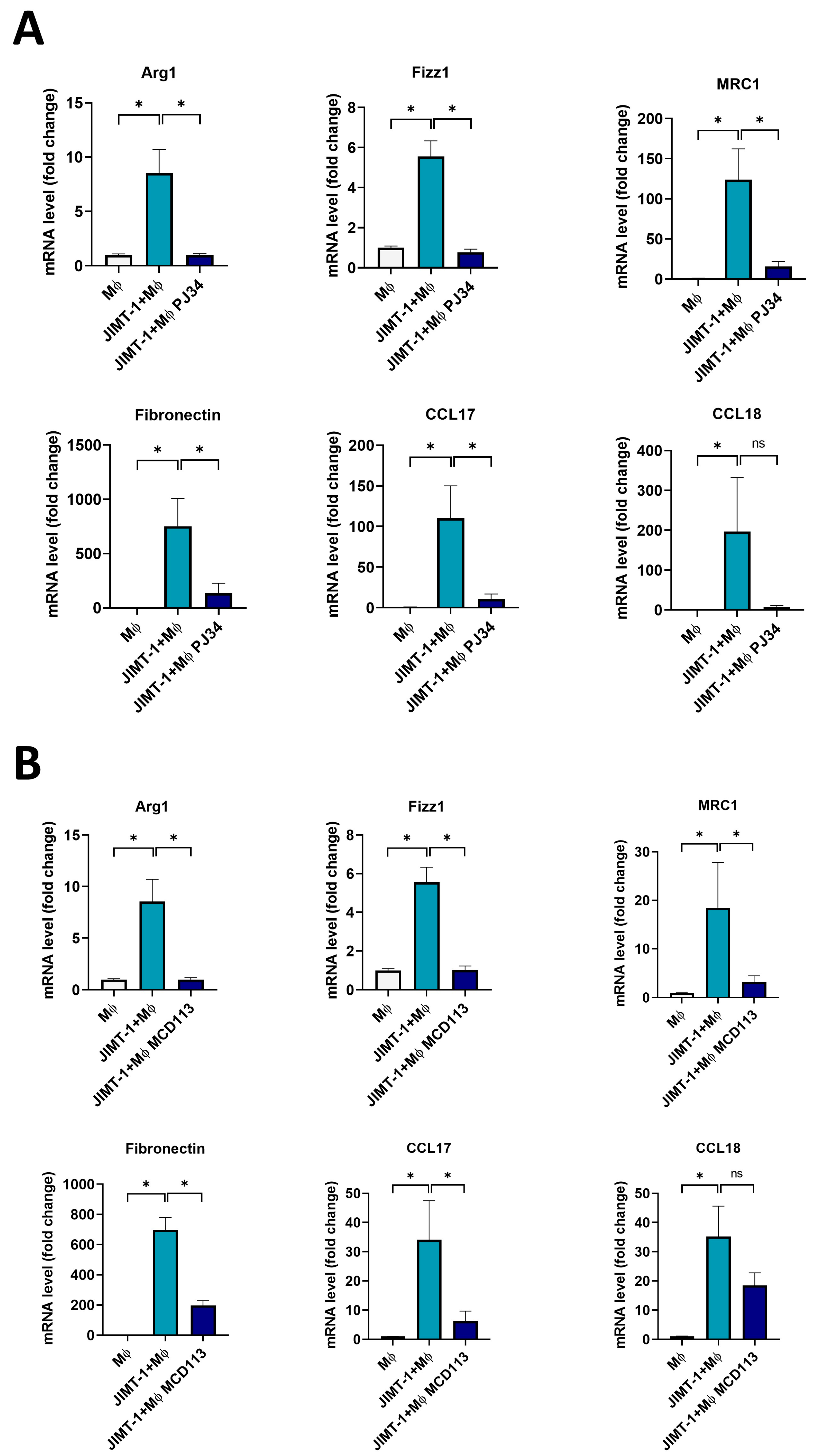
2.5. The PARPi PJ34 and the PARP14i MCD113 Decreased M2 Marker Gene Expression in JIMT-1-THP-1-Derived MΦ Co-Culture Spheroids
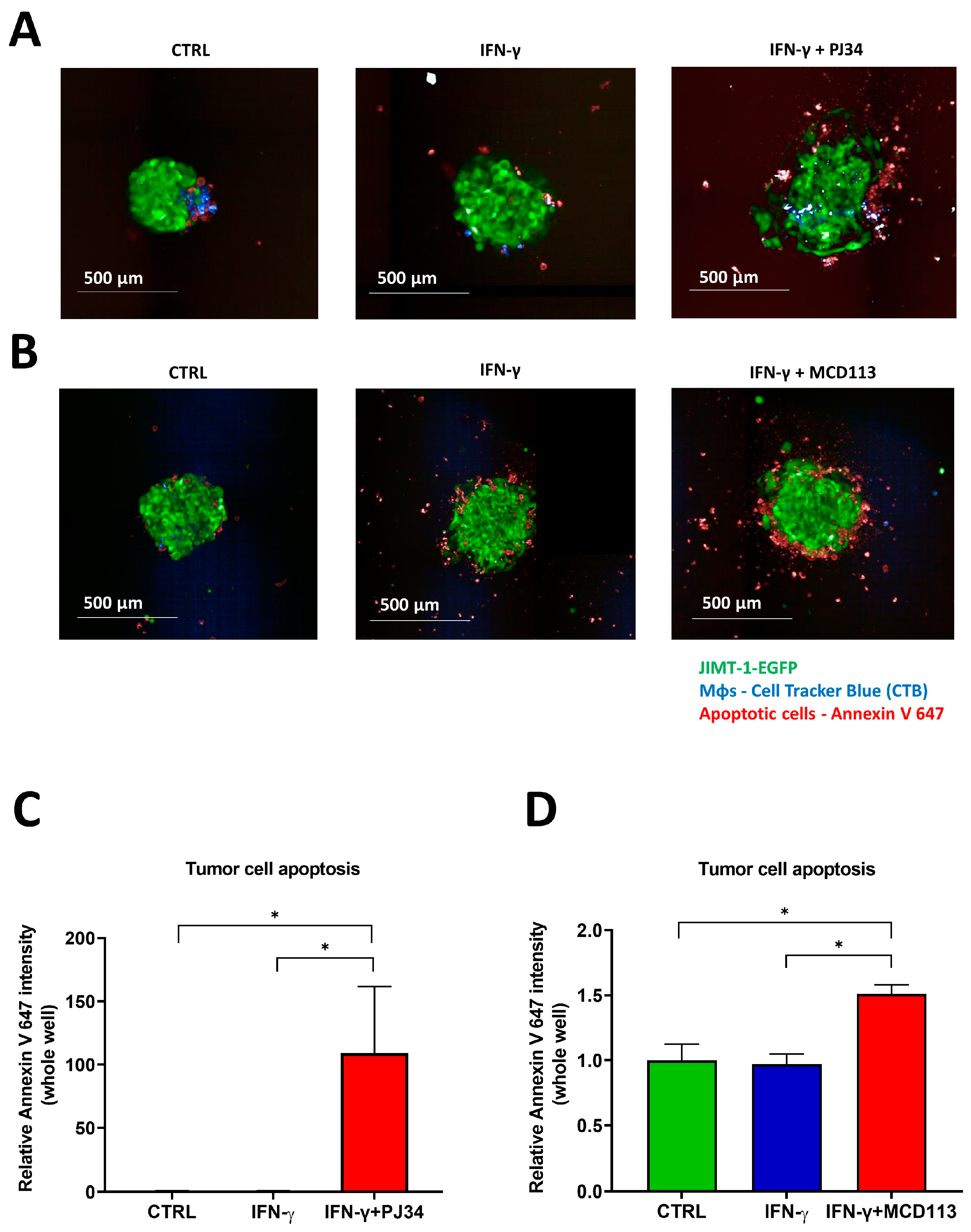
2.6. Effects of the PARP Inhibitors PJ34 and MCD113 on Morphological Changes and Cell Death of the Spheroid Co-Culture System
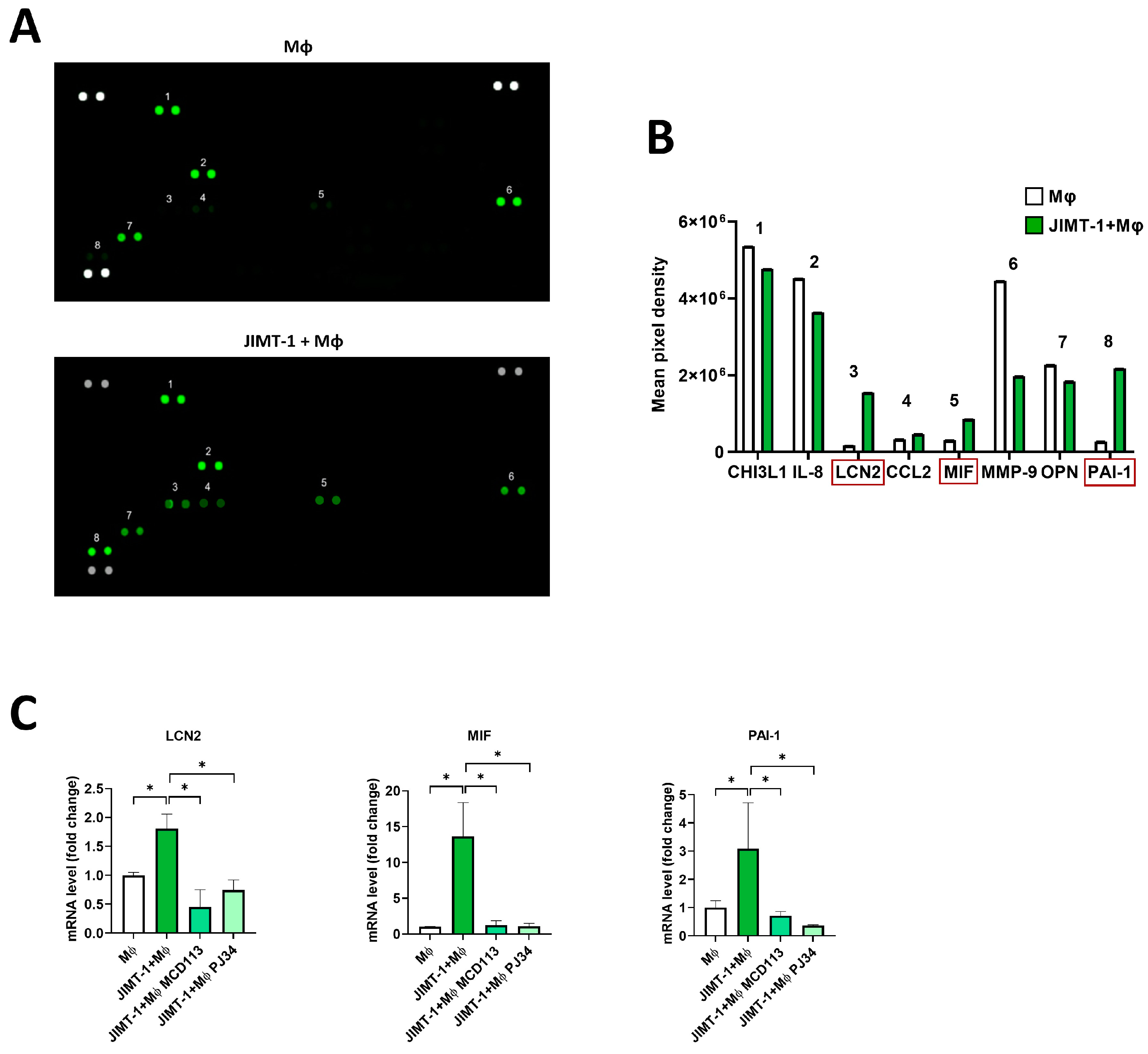
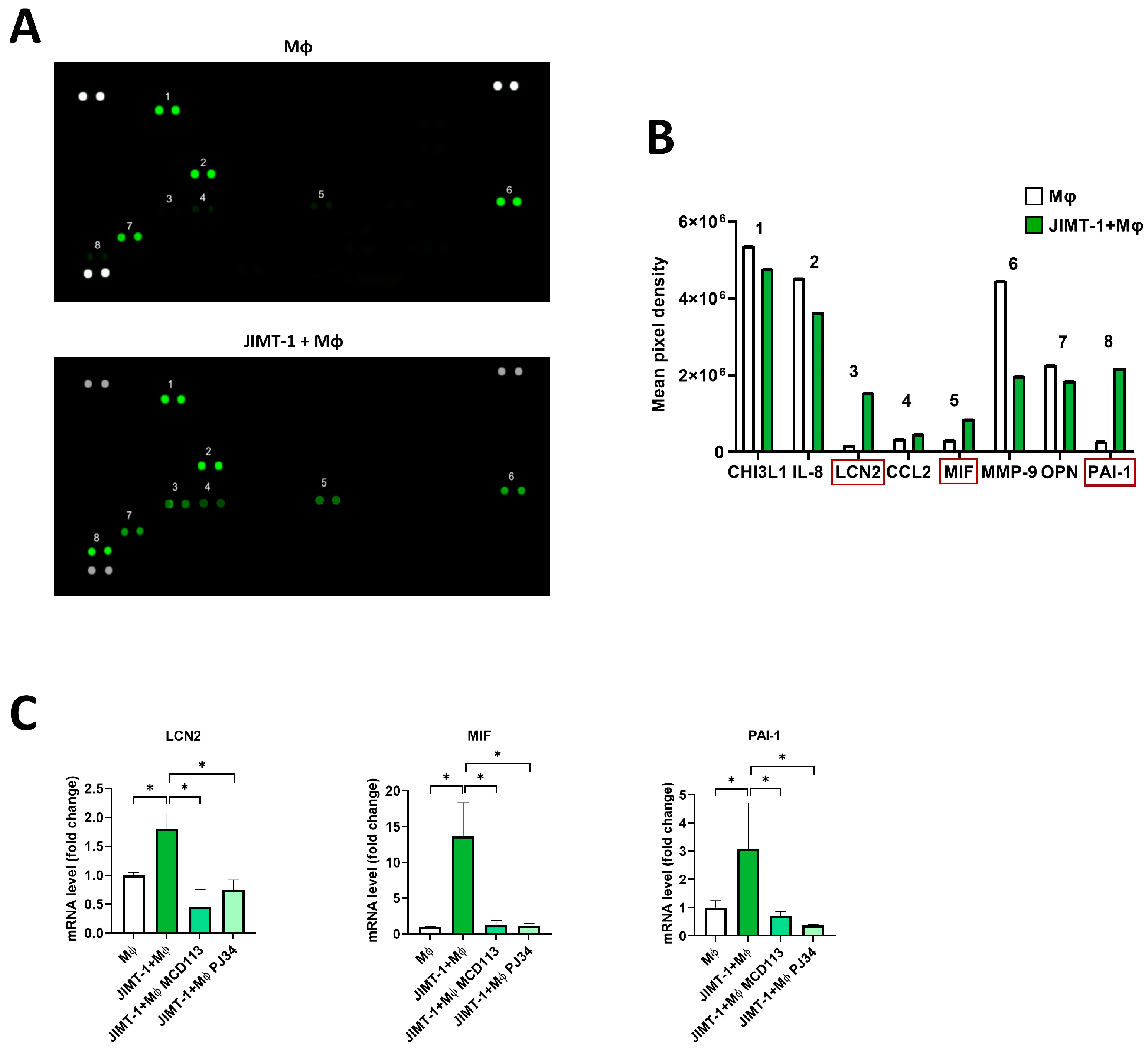
2.7. Proteome Profiling Revealed the Possible Roles of LCN2, MIF, and PAI-1 Cytokines in TAM Polarization
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines
4.3. Murine Bone-Marrow-Derived Macrophages (BMDMs)
4.4. Preparation of 4T1 Conditioned Media (4T1CM) and Induction of the TAM Phenotype
4.5. Human Peripheral Blood Mononuclear Cells (PBMCs)
4.6. THP-1 MΦ Differentiation
4.7. Generation of THP-1-Derived MΦ Spheroids, JIMT-1 Spheroids, and JIMT-1-THP-1 Co-Culture Spheroids
4.8. RNA Isolation, Reverse Transcription, and Quantitative Real-Time PCR
4.9. Flow Cytometry
4.10. High-Content Analysis (HCA) on Live Cells: Cell Death in Spheroids
4.11. Human Cytokine Array
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burguin, A.; Diorio, C.; Durocher, F. Breast Cancer Treatments: Updates and New Challenges. J. Pers. Med. 2021, 11, 808. [Google Scholar] [CrossRef]
- Musolino, A.; Gradishar, W.J.; Rugo, H.S.; Nordstrom, J.L.; Rock, E.P.; Arnaldez, F.; Pegram, M.D. Role of Fcgamma receptors in HER2-targeted breast cancer therapy. J. Immunother. Cancer 2022, 10, e003171. [Google Scholar] [CrossRef]
- Lei, X.; Lei, Y.; Li, J.K.; Du, W.X.; Li, R.G.; Yang, J.; Li, J.; Li, F.; Tan, H.B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Retecki, K.; Seweryn, M.; Graczyk-Jarzynka, A.; Bajor, M. The Immune Landscape of Breast Cancer: Strategies for Overcoming Immunotherapy Resistance. Cancers 2021, 13, 6012. [Google Scholar] [CrossRef]
- Domblides, C.; Alizadeh, D.; Larmonier, N. Editorial: Tumor-promoting immune cells: Cancer immune escape and beyond. Front. Immunol. 2023, 14, 1168884. [Google Scholar] [CrossRef]
- Corti, C.; Venetis, K.; Sajjadi, E.; Zattoni, L.; Curigliano, G.; Fusco, N. CAR-T cell therapy for triple-negative breast cancer and other solid tumors: Preclinical and clinical progress. Expert Opin. Investig. Drugs 2022, 31, 593–605. [Google Scholar] [CrossRef]
- Simeone, E.; Ascierto, P.A. Immunomodulating antibodies in the treatment of metastatic melanoma: The experience with anti-CTLA-4, anti-CD137, and anti-PD1. J. Immunotoxicol. 2012, 9, 241–247. [Google Scholar] [CrossRef]
- Ramos, C.A.; Heslop, H.E.; Brenner, M.K. CAR-T Cell Therapy for Lymphoma. Annu. Rev. Med. 2016, 67, 165–183. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef]
- Molawi, K.; Sieweke, M.H. Transcriptional control of macrophage identity, self-renewal, and function. Adv. Immunol. 2013, 120, 269–300. [Google Scholar]
- Burkle, A.; Virag, L. Poly(ADP-ribose): PARadigms and PARadoxes. Mol. Aspects Med. 2013, 34, 1046–1065. [Google Scholar] [CrossRef]
- Crawford, K.; Bonfiglio, J.J.; Mikoc, A.; Matic, I.; Ahel, I. Specificity of reversible ADP-ribosylation and regulation of cellular processes. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.W.; Jijon, H.B.; Madsen, K.L. AMP-activated protein kinase is a positive regulator of poly(ADP-ribose) polymerase. Biochem. Biophys. Res. Commun. 2006, 342, 336–341. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Chan, W.Y.; Suh, S.W.; Wiggins, A.K.; Huang, E.J.; Swanson, R.A. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc. Natl. Acad. Sci. USA 2006, 103, 7136–7141. [Google Scholar] [CrossRef]
- Castri, P.; Lee, Y.J.; Ponzio, T.; Maric, D.; Spatz, M.; Bembry, J.; Hallenbeck, J. Poly(ADP-ribose) polymerase-1 and its cleavage products differentially modulate cellular protection through NF-kappaB-dependent signaling. Biochim. Biophys. Acta 2014, 1843, 640–651. [Google Scholar] [CrossRef]
- Iwata, H.; Goettsch, C.; Sharma, A.; Ricchiuto, P.; Goh, W.W.; Halu, A.; Yamada, I.; Yoshida, H.; Hara, T.; Wei, M.; et al. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat. Commun. 2016, 7, 12849. [Google Scholar] [CrossRef]
- Goenka, S.; Cho, S.H.; Boothby, M. Collaborator of Stat6 (CoaSt6)-associated poly(ADP-ribose) polymerase activity modulates Stat6-dependent gene transcription. J. Biol. Chem. 2007, 282, 18732–18739. [Google Scholar] [CrossRef]
- Mehrotra, P.; Hollenbeck, A.; Riley, J.P.; Li, F.; Patel, R.J.; Akhtar, N.; Goenka, S. Poly (ADP-ribose) polymerase 14 and its enzyme activity regulates T(H)2 differentiation and allergic airway disease. J. Allergy Clin. Immunol. 2013, 131, 521–531.e12. [Google Scholar] [CrossRef]
- Wong, C.W.; Evangelou, C.; Sefton, K.N.; Leshem, R.; Zhang, W.; Gopalan, V.; Chattrakarn, S.; Fernandez Carro, M.L.; Uzuner, E.; Mole, H.; et al. PARP14 inhibition restores PD-1 immune checkpoint inhibitor response following IFNgamma-driven acquired resistance in preclinical cancer models. Nat. Commun. 2023, 14, 5983. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, Z.; Wan, Y.; Zou, L.; Liu, L.; Ding, S.; Lu, C.; Qiu, F. PARP14 promotes the growth and glycolysis of acute myeloid leukemia cells by regulating HIF-1alpha expression. Clin. Immunol. 2022, 242, 109094. [Google Scholar] [CrossRef] [PubMed]
- Casella, G.; Garzetti, L.; Gatta, A.T.; Finardi, A.; Maiorino, C.; Ruffini, F.; Martino, G.; Muzio, L.; Furlan, R. IL4 induces IL6-producing M2 macrophages associated to inhibition of neuroinflammation in vitro and in vivo. J. Neuroinflamm. 2016, 13, 139. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, D.; Sonzogni, O.; Ke, S.; Wang, Q.; Thavamani, A.; Batalini, F.; Stopka, S.A.; Regan, M.S.; Vandal, S.; et al. PARP-inhibition reprograms macrophages toward an anti-tumor phenotype. Cell Rep. 2022, 41, 111462. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, E.; Karlberg, T.; Kouznetsova, E.; Markova, N.; Macchiarulo, A.; Thorsell, A.G.; Pol, E.; Frostell, A.; Ekblad, T.; Oncu, D.; et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat. Biotechnol. 2012, 30, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Singh, S.A.; Kerr, C.M.; Mukai, S.; Higashi, H.; Aikawa, M. The impact of PARPs and ADP-ribosylation on inflammation and host-pathogen interactions. Genes Dev. 2020, 34, 341–359. [Google Scholar] [CrossRef]
- Upton, K.; Meyers, M.; Thorsell, A.G.; Karlberg, T.; Holechek, J.; Lease, R.; Schey, G.; Wolf, E.; Lucente, A.; Schuler, H.; et al. Design and synthesis of potent inhibitors of the mono(ADP-ribosyl)transferase, PARP14. Bioorg. Med. Chem. Lett. 2017, 27, 2907–2911. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Goenka, S.; Henttinen, T.; Gudapati, P.; Reinikainen, A.; Eischen, C.M.; Lahesmaa, R.; Boothby, M. PARP-14, a member of the B aggressive lymphoma family, transduces survival signals in primary B cells. Blood 2009, 113, 2416–2425. [Google Scholar] [CrossRef]
- Benner, B.; Scarberry, L.; Suarez-Kelly, L.P.; Duggan, M.C.; Campbell, A.R.; Smith, E.; Lapurga, G.; Jiang, K.; Butchar, J.P.; Tridandapani, S.; et al. Generation of monocyte-derived tumor-associated macrophages using tumor-conditioned media provides a novel method to study tumor-associated macrophages in vitro. J. Immunother. Cancer 2019, 7, 140. [Google Scholar] [CrossRef] [PubMed]
- Muller-Quernheim, U.C.; Potthast, L.; Muller-Quernheim, J.; Zissel, G. Tumor-cell co-culture induced alternative activation of macrophages is modulated by interferons in vitro. J. Interferon Cytokine Res. 2012, 32, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Chanput, W.; Mes, J.J.; Wichers, H.J. THP-1 cell line: An in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014, 23, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.P.; Young, H.; Hurlstone, A.; Wellbrock, C. Differentiation of THP1 Cells into Macrophages for Transwell Co-culture Assay with Melanoma Cells. Bio. Protoc. 2015, 5, e1638. [Google Scholar] [CrossRef] [PubMed]
- Guti, E.; Regdon, Z.; Sturniolo, I.; Kiss, A.; Kovacs, K.; Demeny, M.; Szoor, A.; Vereb, G.; Szollosi, J.; Hegedus, C.; et al. The multitargeted receptor tyrosine kinase inhibitor sunitinib induces resistance of HER2 positive breast cancer cells to trastuzumab-mediated ADCC. Cancer Immunol. Immunother. 2022, 71, 2151–2168. [Google Scholar] [CrossRef] [PubMed]
- Khabipov, A.; Kading, A.; Liedtke, K.R.; Freund, E.; Partecke, L.I.; Bekeschus, S. RAW 264.7 Macrophage Polarization by Pancreatic Cancer Cells—A Model for Studying Tumour-promoting Macrophages. Anticancer Res. 2019, 39, 2871–2882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, M.; Hong, Z.; Chen, C. Co-culturing polarized M2 Thp-1-derived macrophages enhance stemness of lung adenocarcinoma A549 cells. Ann. Transl. Med. 2021, 9, 709. [Google Scholar] [CrossRef]
- Guo, H.; Jin, D.; Chen, X. Lipocalin 2 is a regulator of macrophage polarization and NF-kappaB/STAT3 pathway activation. Mol. Endocrinol. 2014, 28, 1616–1628. [Google Scholar] [CrossRef]
- Kai, K.; Moriyama, M.; Haque, A.; Hattori, T.; Chinju, A.; Hu, C.; Kubota, K.; Miyahara, Y.; Kakizoe-Ishiguro, N.; Kawano, S.; et al. Oral Squamous Cell Carcinoma Contributes to Differentiation of Monocyte-Derived Tumor-Associated Macrophages via PAI-1 and IL-8 Production. Int. J. Mol. Sci. 2021, 22, 9475. [Google Scholar] [CrossRef]
- Tugal, D.; Liao, X.; Jain, M.K. Transcriptional control of macrophage polarization. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1135–1144. [Google Scholar] [CrossRef]
- Higashi, H.; Maejima, T.; Lee, L.H.; Yamazaki, Y.; Hottiger, M.O.; Singh, S.A.; Aikawa, M. A Study into the ADP-Ribosylome of IFN-gamma-Stimulated THP-1 Human Macrophage-like Cells Identifies ARTD8/PARP14 and ARTD9/PARP9 ADP-Ribosylation. J. Proteome Res. 2019, 18, 1607–1622. [Google Scholar] [CrossRef]
- Sobczak, M.; Zyma, M.; Robaszkiewicz, A. The Role of PARP1 in Monocyte and Macrophage Commitment and Specification: Future Perspectives and Limitations for the Treatment of Monocyte and Macrophage Relevant Diseases with PARP Inhibitors. Cells 2020, 9, 2040. [Google Scholar] [CrossRef]
- Mehta, A.K.; Cheney, E.M.; Hartl, C.A.; Pantelidou, C.; Oliwa, M.; Castrillon, J.A.; Lin, J.R.; Hurst, K.E.; de Oliveira Taveira, M.; Johnson, N.T.; et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat. Cancer 2021, 2, 66–82. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, X.; Li, Z.; Zhu, B. Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages. Theranostics 2021, 11, 1016–1030. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Soldani, C.; Morabito, A.; Franceschini, B.; Ferlan, F.; Costa, G.; Pastorelli, R.; Donadon, M.; Brunelli, L. Implication of metabolism in the polarization of tumor-associated-macrophages: The mass spectrometry-based point of view. Front. Immunol. 2023, 14, 1193235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Meng, Y.; Xu, X.; Zuo, D. The role of glycolysis and lactate in the induction of tumor-associated macrophages immunosuppressive phenotype. Int. Immunopharmacol. 2022, 110, 108994. [Google Scholar] [CrossRef] [PubMed]
- Holbein, B.E.; Lehmann, C. Dysregulated Iron Homeostasis as Common Disease Etiology and Promising Therapeutic Target. Antioxidants 2023, 12, 671. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Duan, F.; Huang, J.; Bi, X.; Xia, W.; Song, C.; Wang, L.; Jiang, C.; Yuan, Z. A Novel Prognostic Model Based on the Serum Iron Level for Patients With Early-Stage Triple-Negative Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 777215. [Google Scholar] [CrossRef]
- Salnikow, K. Role of iron in cancer. Semin. Cancer Biol. 2021, 76, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Eickhoff, J.C.; Gould, M.N.; Mundhenke, C.; Maass, N.; Friedl, A. Neutrophil gelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res. Treat. 2008, 108, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Wenners, A.S.; Mehta, K.; Loibl, S.; Park, H.; Mueller, B.; Arnold, N.; Hamann, S.; Weimer, J.; Ataseven, B.; Darb-Esfahani, S.; et al. Neutrophil gelatinase-associated lipocalin (NGAL) predicts response to neoadjuvant chemotherapy and clinical outcome in primary human breast cancer. PLoS ONE 2012, 7, e45826. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bielenberg, D.R.; Rodig, S.J.; Doiron, R.; Clifton, M.C.; Kung, A.L.; Strong, R.K.; Zurakowski, D.; Moses, M.A. Lipocalin 2 promotes breast cancer progression. Proc. Natl. Acad. Sci. USA 2009, 106, 3913–3918. [Google Scholar] [CrossRef] [PubMed]
- Candido, S.; Abrams, S.L.; Steelman, L.S.; Lertpiriyapong, K.; Fitzgerald, T.L.; Martelli, A.M.; Cocco, L.; Montalto, G.; Cervello, M.; Polesel, J.; et al. Roles of NGAL and MMP-9 in the tumor microenvironment and sensitivity to targeted therapy. Biochim. Biophys. Acta 2016, 1863, 438–448. [Google Scholar] [CrossRef]
- Barbosa de Souza Rizzo, M.; Brasilino de Carvalho, M.; Kim, E.J.; Rendon, B.E.; Noe, J.T.; Darlene Wise, A.; Mitchell, R.A. Oral squamous carcinoma cells promote macrophage polarization in an MIF-dependent manner. QJM 2018, 111, 769–778. [Google Scholar] [CrossRef]
- Huang, W.C.; Kuo, K.T.; Wang, C.H.; Yeh, C.T.; Wang, Y. Cisplatin resistant lung cancer cells promoted M2 polarization of tumor-associated macrophages via the Src/CD155/MIF functional pathway. J. Exp. Clin. Cancer Res. 2019, 38, 180. [Google Scholar] [CrossRef]
- Yang, Y.P.; Chien, C.S.; Yarmishyn, A.A.; Chan, M.S.; Lee, A.C.; Chen, Y.W.; Huang, P.I.; Ma, H.I.; Lo, W.L.; Chien, Y.; et al. Musashi-1 Regulates MIF1-Mediated M2 Macrophage Polarization in Promoting Glioblastoma Progression. Cancers 2021, 13, 1799. [Google Scholar] [CrossRef]
- Suzuki, F.; Nakamaru, Y.; Oridate, N.; Homma, A.; Nagahashi, T.; Yamaguchi, S.; Nishihira, J.; Furuta, Y.; Fukuda, S. Prognostic significance of cytoplasmic macrophage migration inhibitory factor expression in patients with squamous cell carcinoma of the head and neck treated with concurrent chemoradiotherapy. Oncol. Rep. 2005, 13, 59–64. [Google Scholar] [CrossRef]
- Kubala, M.H.; Punj, V.; Placencio-Hickok, V.R.; Fang, H.; Fernandez, G.E.; Sposto, R.; DeClerck, Y.A. Plasminogen Activator Inhibitor-1 Promotes the Recruitment and Polarization of Macrophages in Cancer. Cell Rep. 2018, 25, 2177–2191.e7. [Google Scholar] [CrossRef]
- Chen, S.; Morine, Y.; Tokuda, K.; Yamada, S.; Saito, Y.; Nishi, M.; Ikemoto, T.; Shimada, M. Cancer-associated fibroblast-induced M2-polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitor-1 pathway. Int. J. Oncol. 2021, 59, 1–14. [Google Scholar] [CrossRef]
- Li, Y.; Shi, Y.; Zhang, X.; Li, P.; Ma, L.; Hu, P.; Xu, L.; Dai, Y.; Xia, S.; Qiu, H. FGFR2 upregulates PAI-1 via JAK2/STAT3 signaling to induce M2 polarization of macrophages in colorectal cancer. Biochim. Biophys. Acta. Mol. Basis Dis. 2023, 1869, 166665. [Google Scholar] [CrossRef]
- Park, H.J.; Chi, G.Y.; Choi, Y.H.; Park, S.H. Lupeol suppresses plasminogen activator inhibitor-1-mediated macrophage recruitment and attenuates M2 macrophage polarization. Biochem. Biophys. Res. Commun. 2020, 527, 889–895. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Z.; Cui, S.; Nagy, T.; Xiong, M.P. Synthesis and evaluation of an amphiphilic deferoxamine:gallium-conjugated cationic random copolymer against a murine wound healing infection model of Pseudomonas aeruginosa. Acta. Biomater. 2021, 126, 384–393. [Google Scholar] [CrossRef]
- Liu, Y.; Xue, R.; Duan, X.; Shang, X.; Wang, M.; Wang, F.; Zhu, L.; Zhang, L.; Ge, X.; Zhao, X.; et al. PARP inhibition synergizes with CD47 blockade to promote phagocytosis by tumor-associated macrophages in homologous recombination-proficient tumors. Life Sci. 2023, 326, 121790. [Google Scholar] [CrossRef]
- Wang, Q.; Bergholz, J.S.; Ding, L.; Lin, Z.; Kabraji, S.K.; Hughes, M.E.; He, X.; Xie, S.; Jiang, T.; Wang, W.; et al. STING agonism reprograms tumor-associated macrophages and overcomes resistance to PARP inhibition in BRCA1-deficient models of breast cancer. Nat. Commun. 2022, 13, 3022. [Google Scholar] [CrossRef]
- Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; Tutt, A.N.J.; Lord, C.J. Opinion: PARP inhibitors in cancer-what do we still need to know? Open Biol. 2022, 12, 220118. [Google Scholar] [CrossRef] [PubMed]
- Pineda-Torra, I.; Gage, M.; de Juan, A.; Pello, O.M. Isolation, Culture, and Polarization of Murine Bone Marrow-Derived and Peritoneal Macrophages. Methods Mol. Biol. 2015, 1339, 101–109. [Google Scholar] [PubMed]
- Lakatos, P.; Hegedus, C.; Salazar Ayestaran, N.; Juarranz, A.; Kover, K.E.; Szabo, E.; Virag, L. The PARP inhibitor PJ-34 sensitizes cells to UVA-induced phototoxicity by a PARP independent mechanism. Mutat. Res. 2016, 790, 31–40. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sturniolo, I.; Váróczy, C.; Regdon, Z.; Mázló, A.; Muzsai, S.; Bácsi, A.; Intili, G.; Hegedűs, C.; Boothby, M.R.; Holechek, J.; et al. PARP14 Contributes to the Development of the Tumor-Associated Macrophage Phenotype. Int. J. Mol. Sci. 2024, 25, 3601. https://doi.org/10.3390/ijms25073601
Sturniolo I, Váróczy C, Regdon Z, Mázló A, Muzsai S, Bácsi A, Intili G, Hegedűs C, Boothby MR, Holechek J, et al. PARP14 Contributes to the Development of the Tumor-Associated Macrophage Phenotype. International Journal of Molecular Sciences. 2024; 25(7):3601. https://doi.org/10.3390/ijms25073601
Chicago/Turabian StyleSturniolo, Isotta, Csongor Váróczy, Zsolt Regdon, Anett Mázló, Szabolcs Muzsai, Attila Bácsi, Giorgia Intili, Csaba Hegedűs, Mark R. Boothby, Jacob Holechek, and et al. 2024. "PARP14 Contributes to the Development of the Tumor-Associated Macrophage Phenotype" International Journal of Molecular Sciences 25, no. 7: 3601. https://doi.org/10.3390/ijms25073601
APA StyleSturniolo, I., Váróczy, C., Regdon, Z., Mázló, A., Muzsai, S., Bácsi, A., Intili, G., Hegedűs, C., Boothby, M. R., Holechek, J., Ferraris, D., Schüler, H., & Virág, L. (2024). PARP14 Contributes to the Development of the Tumor-Associated Macrophage Phenotype. International Journal of Molecular Sciences, 25(7), 3601. https://doi.org/10.3390/ijms25073601