
Different Patterns of Platinum Resistance in Ovarian Cancer Cells with Homologous Recombination Proficient and Deficient Background

 , , and
, , and 
Abstract
1. Introduction
2. Results
2.1. Generation of DDP-Resistant Models
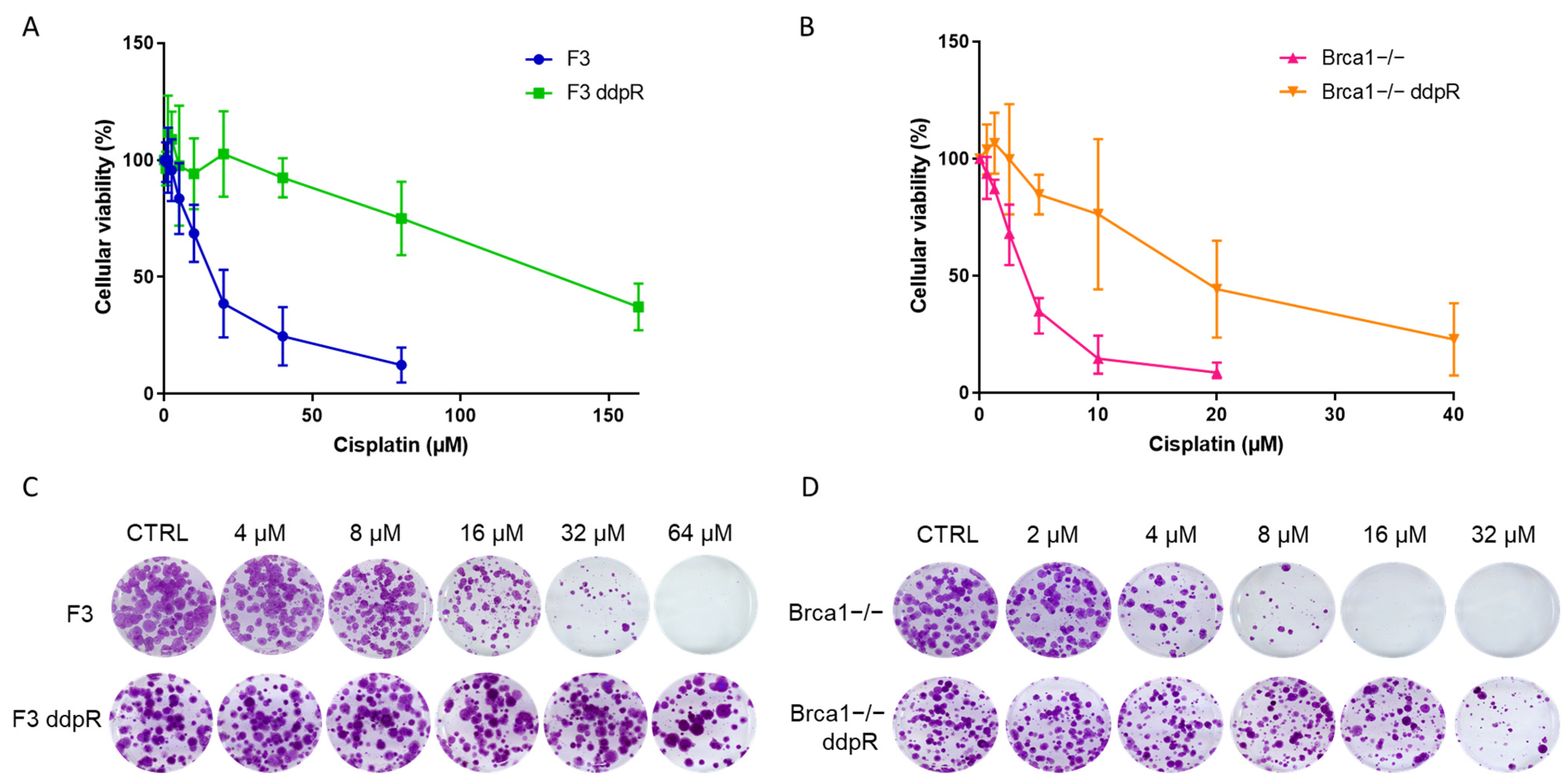
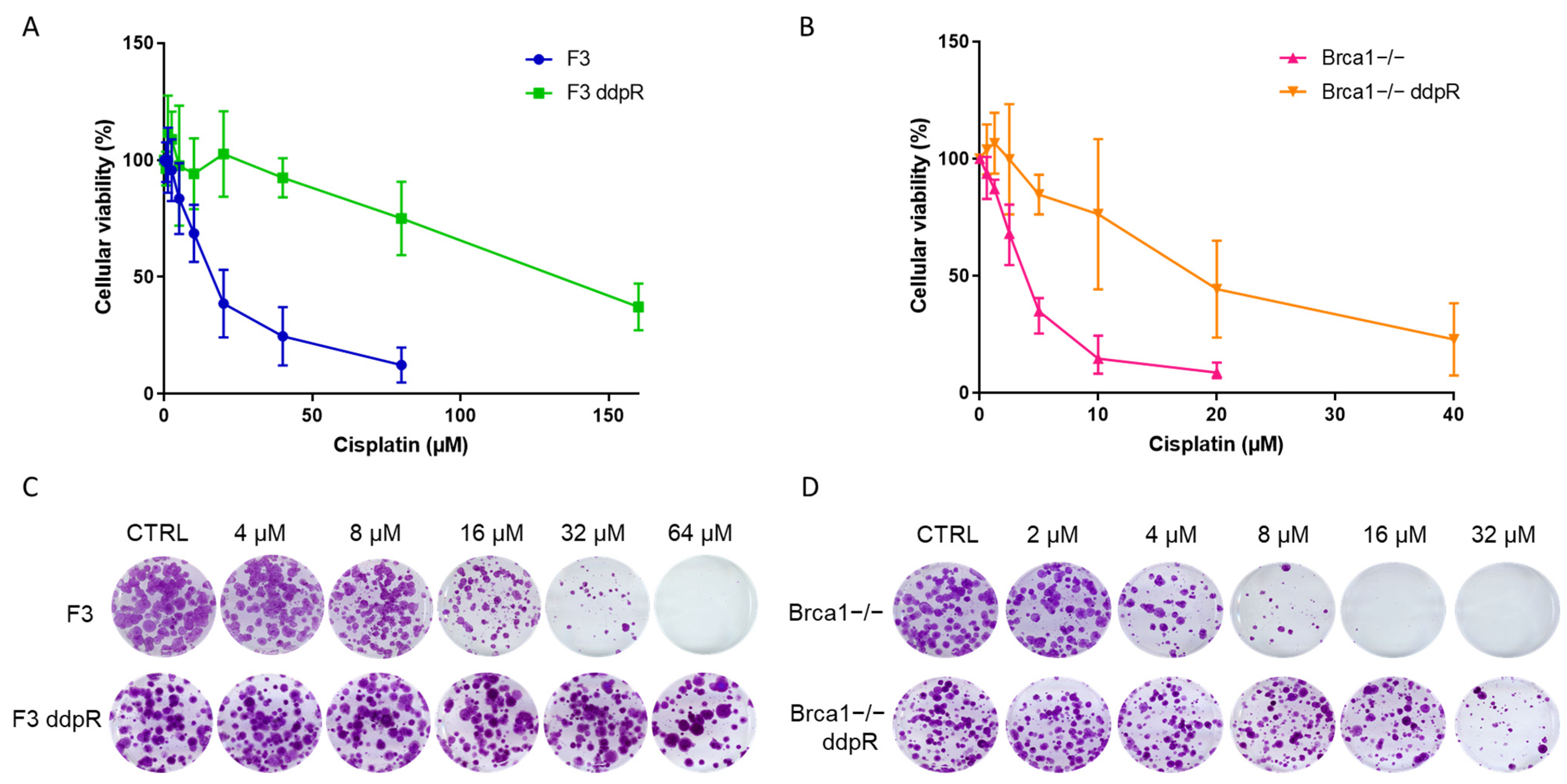
2.2. Pharmacological Characterization
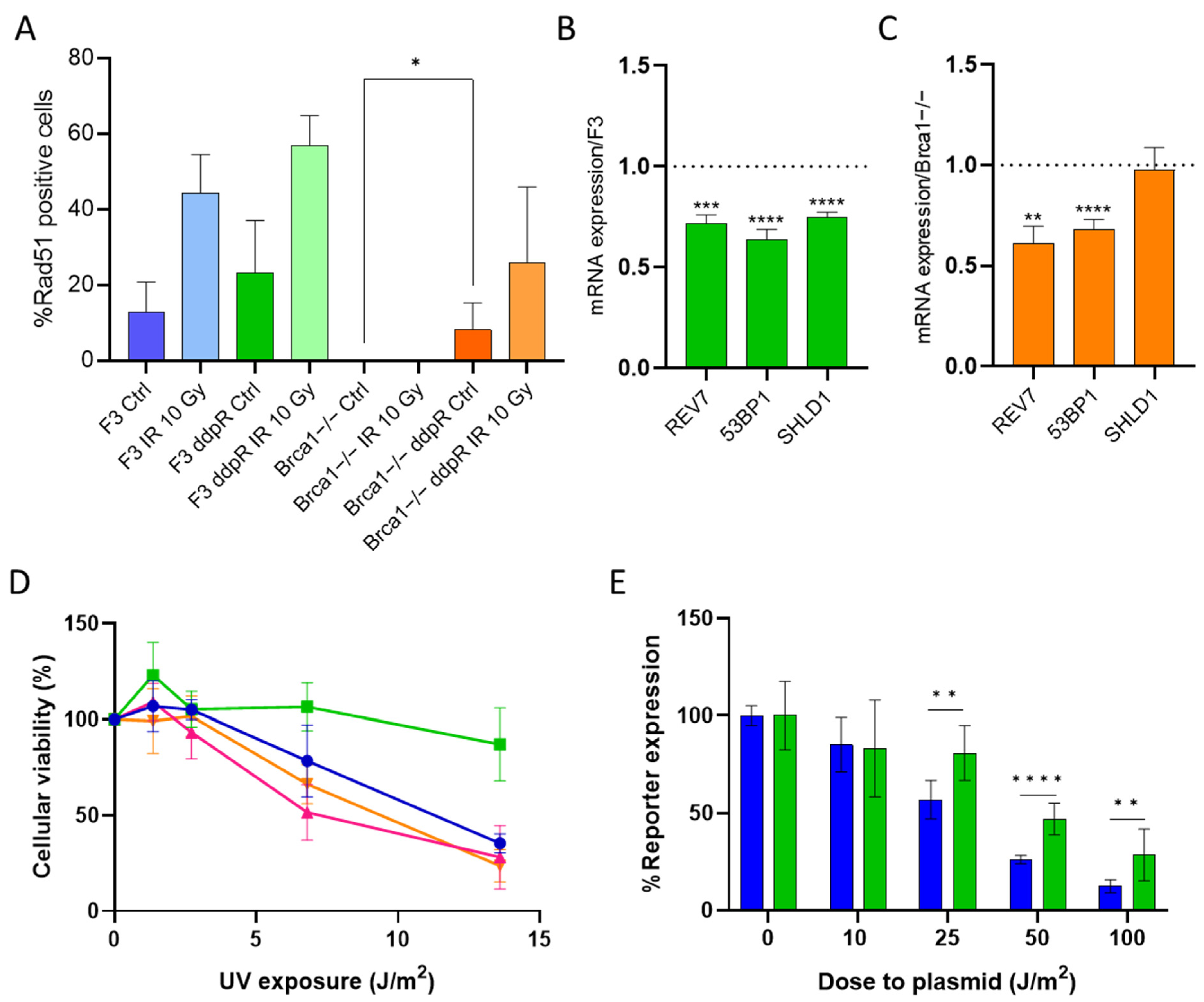
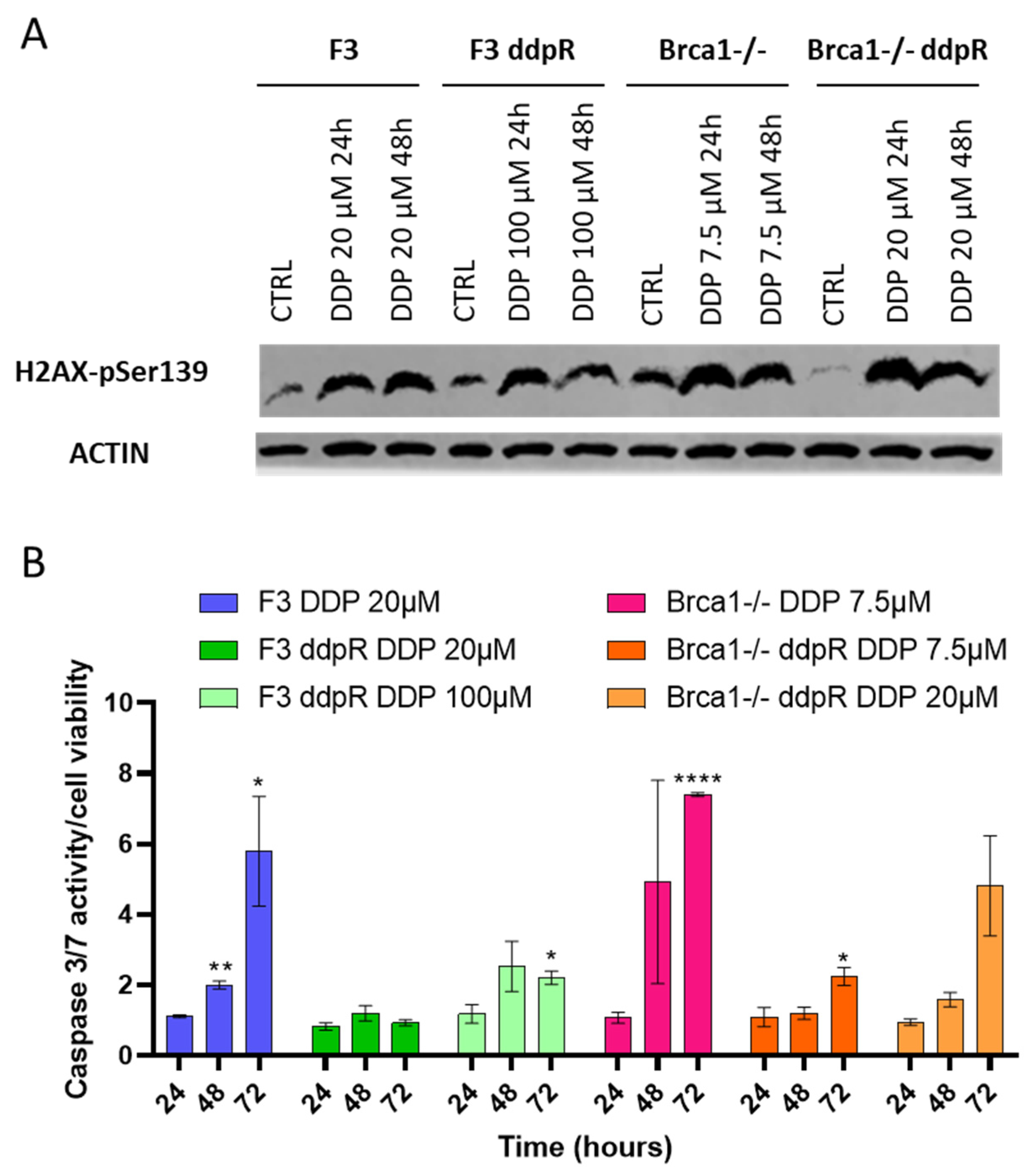
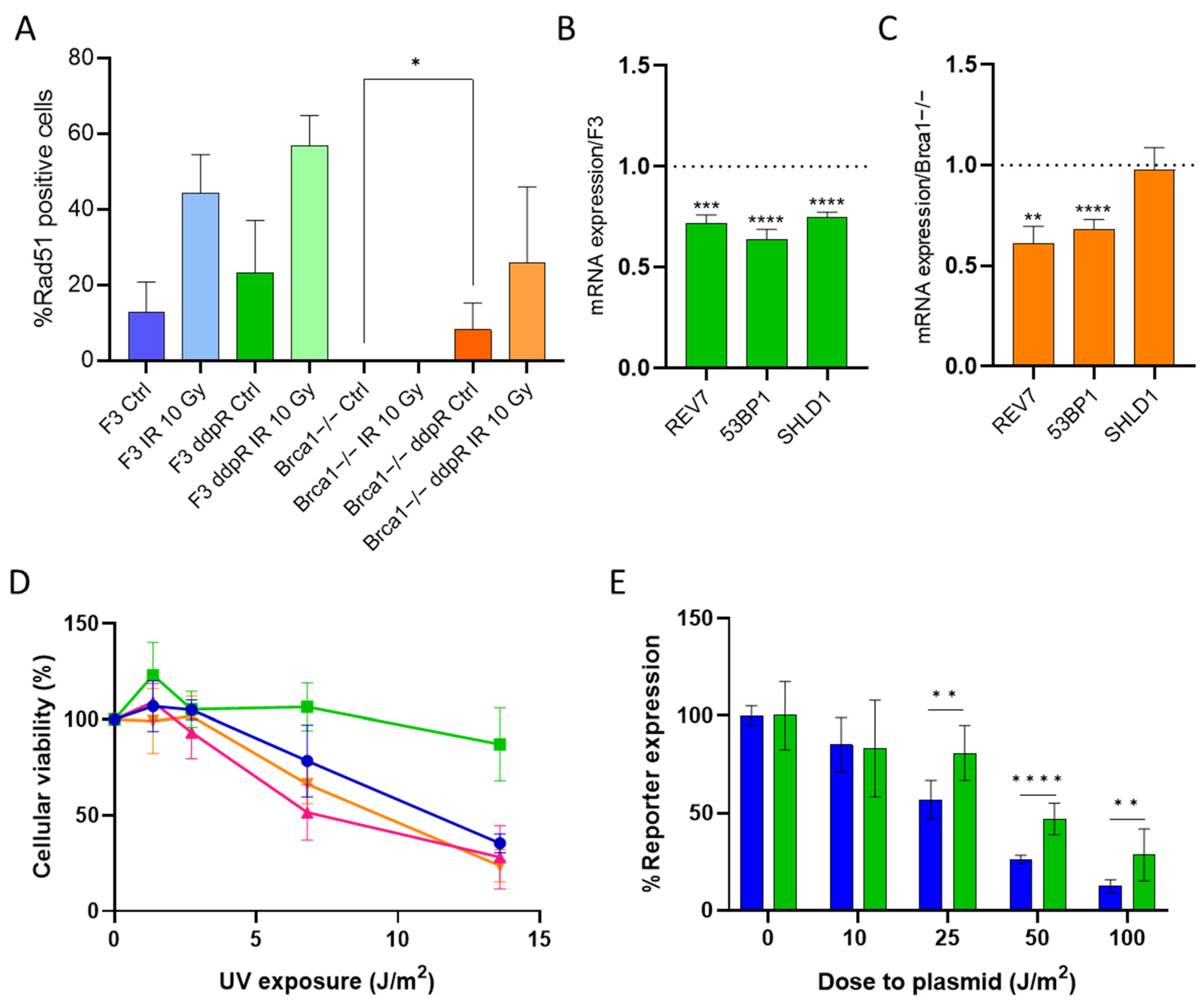
2.3. DDP Induces Damage, Apoptosis and Different Pattern of Repair in Sensitive and Resistant Cells
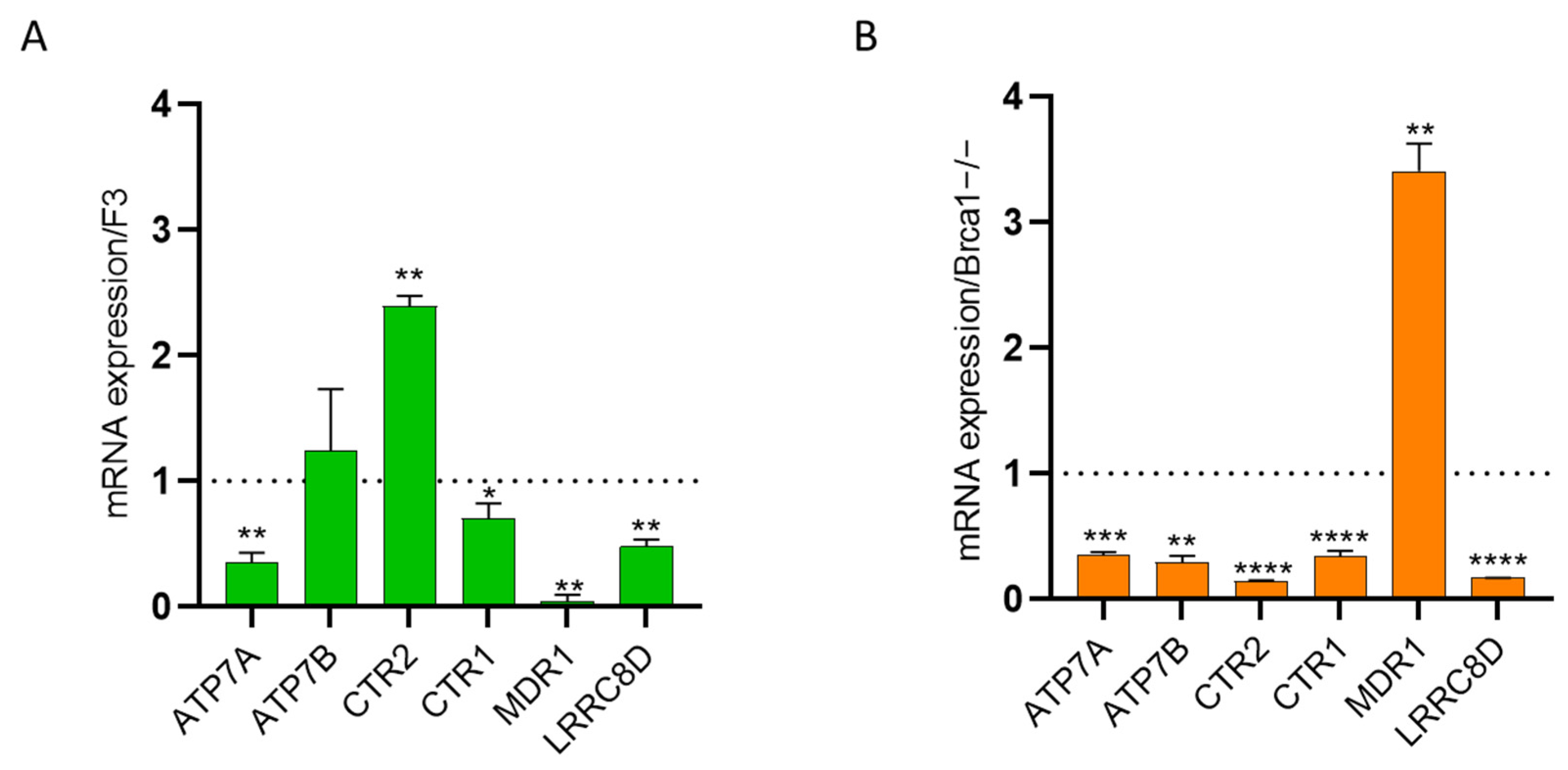
2.4. Intracellular Drug Levels
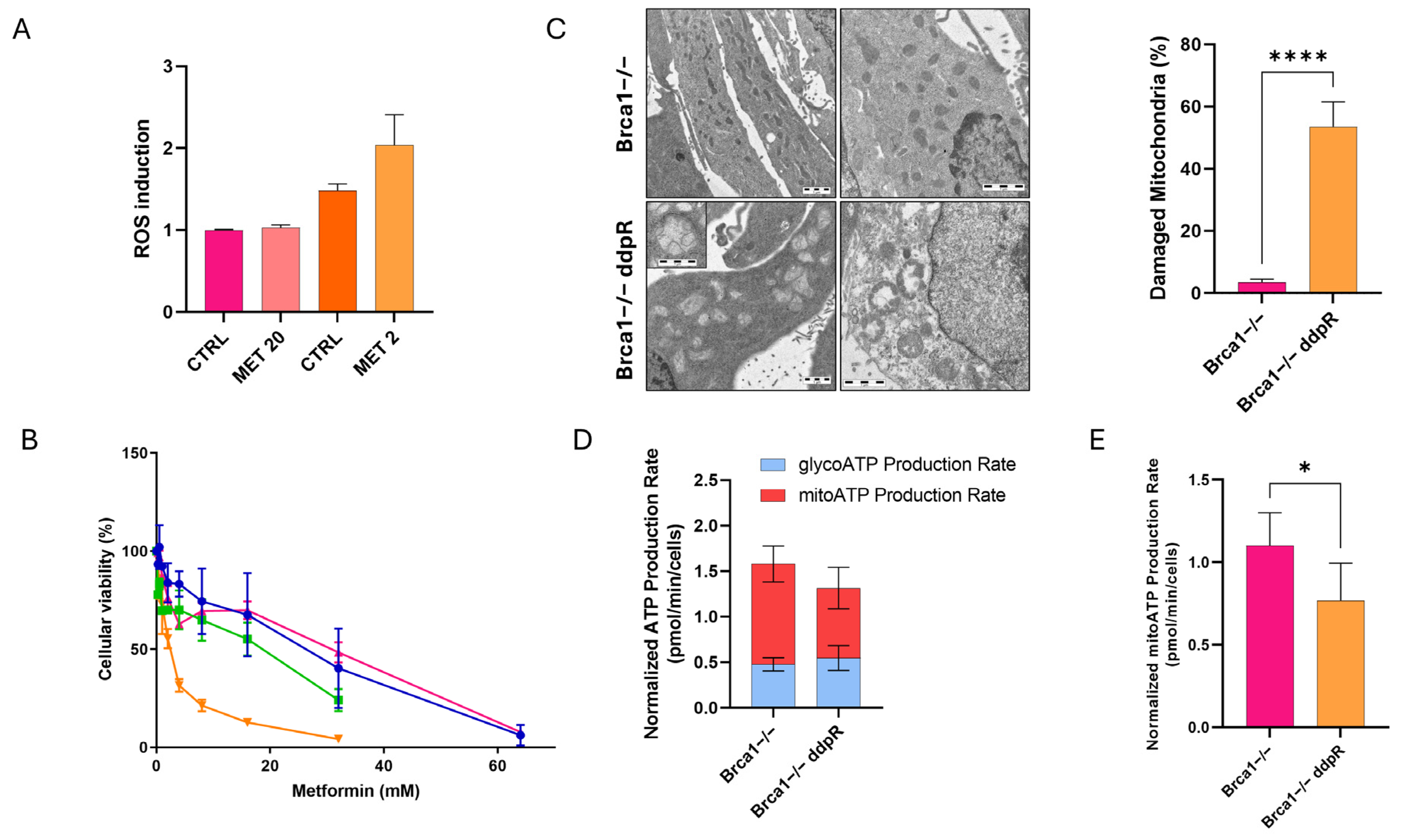
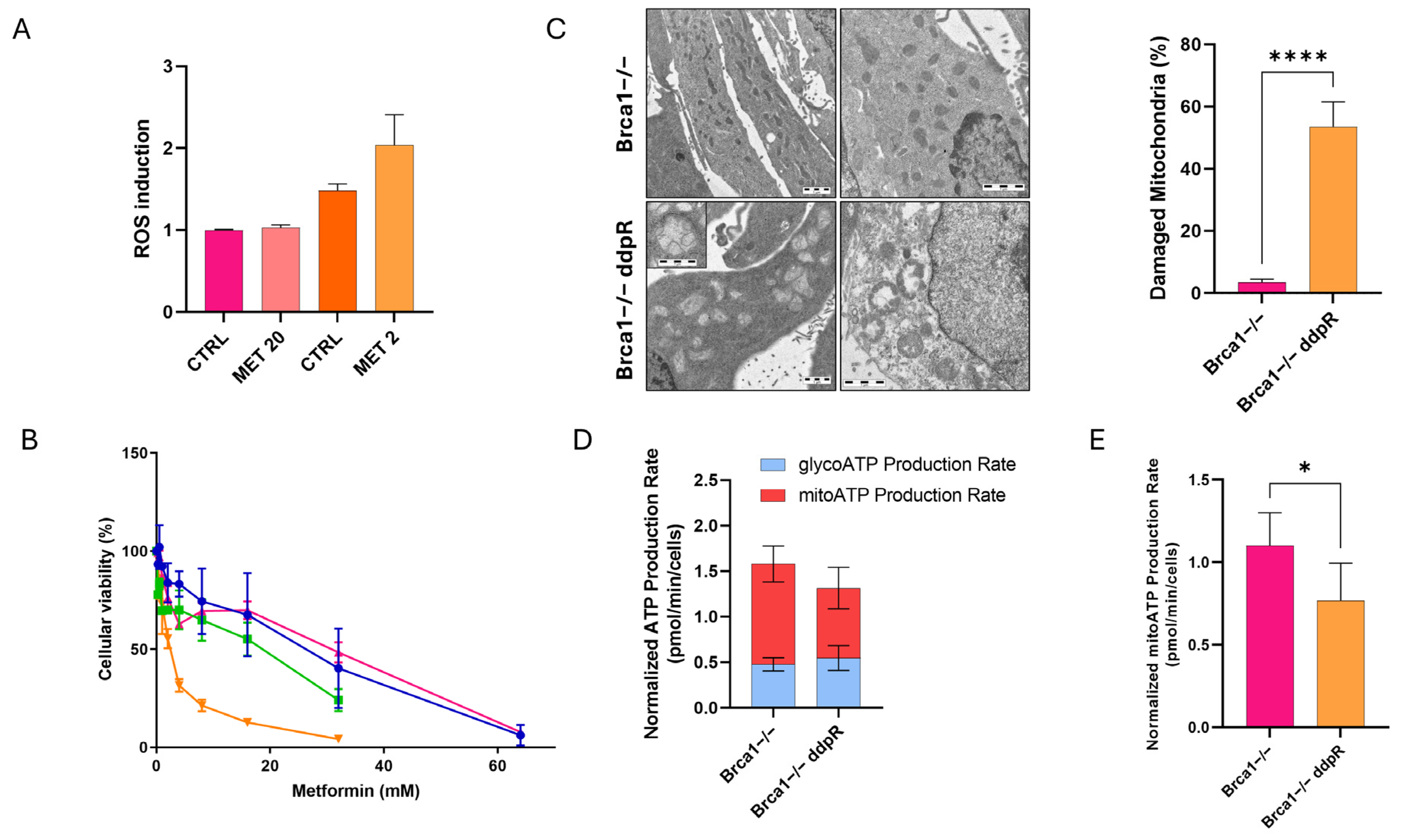
2.5. Metabolic Stress
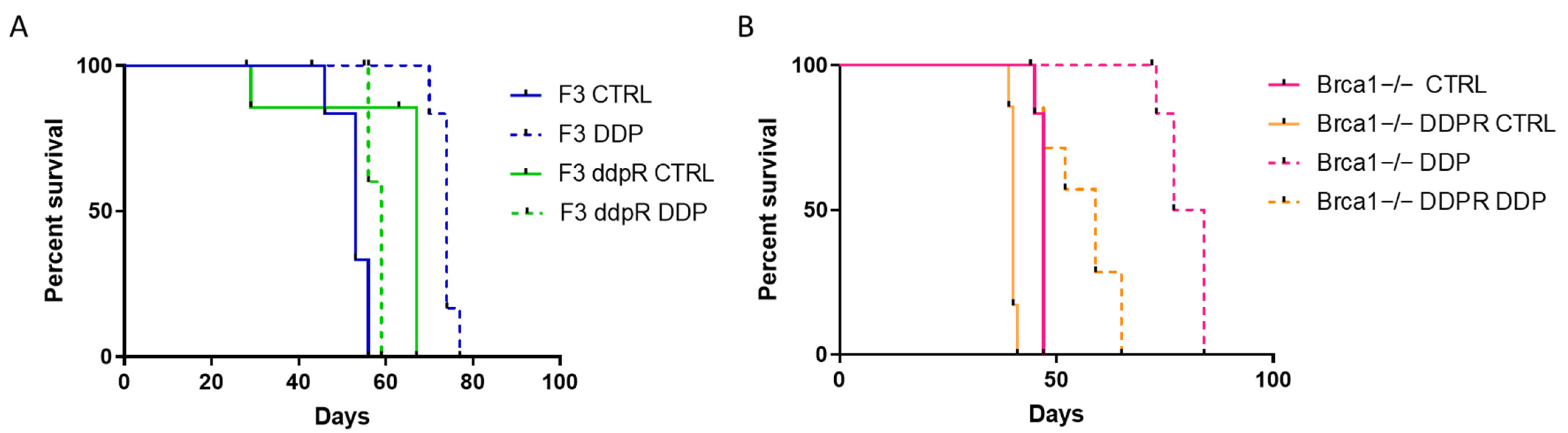
2.6. In Vivo Activity
3. Discussion
4. Materials and Methods
4.1. Cisplatin-Resistant Clones
4.2. Cell Growth
4.3. Drugs and Treatments
4.4. Western Blot
4.5. Caspase 3/7 Activity
4.6. Quantitative Reverse Transcription (RT)-PCR
4.7. RAD51 Immunofluorescence
4.8. Host Cell Reactivation Assay
4.9. ROS Detection
4.10. Seahorse Analyses
4.11. Transmission Electron Microscopy
4.12. Flow Cytometry
4.13. In Vivo Studies
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, H.; Lord, C.J.; Tutt, A.H.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Tew, W.P.; Lacchetti, C.; Ellis, A.; Maxian, K.; Banerjee, S.; Bookman, M.; Jones, M.B.; Lee, J.-M.; Lheureux, S.; Liu, J.F.; et al. PARP Inhibitors in the Management of Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3468–3493. [Google Scholar] [CrossRef]
- Rottenberg, S.; Disler, C.; Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef]
- Ortiz, M.; Wabel, E.; Mitchell, K.; Horibata, S. Mechanisms of chemotherapy resistance in ovarian cancer. Cancer Drug Resist. 2022, 5, 304–316. [Google Scholar] [CrossRef]
- Widmer, C.A.; Klebic, I.; Domanitskaya, N.; Decollogny, M.; Howald, D.; Siffert, M.; Essers, P.; Nowicka, Z.; Stokar-Regenscheit, N.; van de Ven, M.; et al. Loss of the Volume-regulated Anion Channel Components LRRC8A and LRRC8D Limits Platinum Drug Efficacy. Cancer Res. Commun. 2022, 2, 1266–1281. [Google Scholar] [CrossRef]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef]
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef]
- Walton, J.; Blagih, J.; Ennis, D.; Leung, E.; Dowson, S.; Farquharson, M.; Tookman, L.A.; Orange, C.; Athineos, D.; Mason, S.; et al. CRISPR/Cas9-Mediated Trp53 and Brca2 Knockout to Generate Improved Murine Models of Ovarian High-Grade Serous Carcinoma. Cancer Res. 2016, 76, 6118–6129. [Google Scholar] [CrossRef]
- Walton, J.B.; Farquharson, M.; Mason, S.; Port, J.; Kruspig, B.; Dowson, S.; Stevenson, D.; Murphy, D.; Matzuk, M.; Kim, J.; et al. CRISPR/Cas9-derived models of ovarian high grade serous carcinoma targeting Brca1, Pten and Nf1, and correlation with platinum sensitivity. Sci. Rep. 2017, 7, 16827. [Google Scholar] [CrossRef]
- Huntoon, C.J.; Flatten, K.S.; Wahner Hendrickson, A.E.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. ATR Inhibition Broadly Sensitizes Ovarian Cancer Cells to Chemotherapy Independent of BRCA Status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef]
- Tavecchio, M.; Simone, M.; Erba, E.; Chiolo, I.; Liberi, G.; Foiani, M.; D’Incalci, M.; Damia, G. Role of homologous recombination in trabectedin-induced DNA damage. Eur. J. Cancer 2008, 44, 609–618. [Google Scholar] [CrossRef]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Tumiati, M.; Hietanen, S.; Hynninen, J.; Pietilä, E.; Färkkilä, A.; Kaipio, K.; Roering, P.; Huhtinen, K.; Alkodsi, A.; Li, Y.; et al. A Functional Homologous Recombination Assay Predicts Primary Chemotherapy Response and Long-Term Survival in Ovarian Cancer Patients. Clin. Cancer Res. 2018, 24, 4482–4493. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, J.S.; Latimer, J.J. Host Cell Reactivation: Assay for Actively Transcribed DNA (Nucleotide Excision) Repair Using Luciferase Family Expression Vectors. In Molecular Toxicology Protocols; Springer: New York, NY, USA, 2020; pp. 509–528. [Google Scholar]
- Kilari, D.; Guancial, E.; Kim, E.S. Role of copper transporters in platinum resistance. World J. Clin. Oncol. 2016, 7, 106–113. [Google Scholar] [CrossRef]
- Ricci, F.; Brunelli, L.; Affatato, R.; Chilà, R.; Verza, M.; Indraccolo, S.; Falcetta, F.; Fratelli, M.; Fruscio, R.; Pastorelli, R.; et al. Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts. Ther. Adv. Med. Oncol. 2019, 11, 1758835919839543. [Google Scholar] [CrossRef] [PubMed]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS production by mitochondria: Function or dysfunction? Oncogene 2024, 43, 295–303. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2020, 42, 77–96. [Google Scholar] [CrossRef]
- Gadducci, A.; Guarneri, V.; Peccatori, F.A.; Ronzino, G.; Scandurra, G.; Zamagni, C.; Zola, P.; Salutari, V. Current strategies for the targeted treatment of high-grade serous epithelial ovarian cancer and relevance of BRCA mutational status. J. Ovarian. Res. 2019, 12, 9. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef]
- Stordal, B.; Pavlakis, N.; Davey, R. Oxaliplatin for the treatment of cisplatin-resistant cancer: A systematic review. Cancer Treat. Rev. 2007, 33, 347–357. [Google Scholar] [CrossRef]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Damia, G.; D’Incalci, M. Targeting DNA repair as a promising approach in cancer therapy. Eur. J. Cancer 2007, 43, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; O’Connor, K.W.; Mouw, K.W.; Li, A.Y.; Matulonis, U.A.; D’Andrea, A.D.; Konstantinopoulos, P.A. A Unique Subset of Epithelial Ovarian Cancers with Platinum Sensitivity and PARP Inhibitor Resistance. Cancer Res. 2015, 75, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Damia, G.; Imperatori, L.; Stefanini, M.; D’Incalci, M. Sensitivity of CHO mutant cell lines with specific defects in nucleotide excision repair to different anti-cancer agents. Int. J. Cancer 1996, 66, 779–783. [Google Scholar] [CrossRef]
- Muallem, M.Z.; Braicu, I.; Nassir, M.; Richter, R.; Sehouli, J.; Arsenic, R. ERCC1 Expression as a Predictor of Resistance to Platinum-based Chemotherapy in Primary Ovarian Cancer. Anticancer. Res. 2014, 34, 393–399. [Google Scholar] [PubMed]
- Zhang, C.; Gao, S.; Hou, J. ERCC1 expression and platinum chemosensitivity in patients with ovarian cancer: A meta-analysis. Int. J. Biol. Markers. 2020, 35, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Chiappa, M.; Guffanti, F.; Bertoni, F.; Colombo, I.; Damia, G. Overcoming PARPi resistance: Preclinical and clinical evidence in ovarian cancer. Drug Resist. Updat. 2021, 55, 100744. [Google Scholar] [CrossRef] [PubMed]
- Clairmont, C.S.; D’Andrea, A.D. REV7 directs DNA repair pathway choice. Trends Cell Biol. 2021, 31, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7–shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sarangi, P.; Iyer, D.R.; Feng, H.; Moreau, L.; Nguyen, H.; Clairmont, C.; D’Andrea, A.D. CHAMP1 binds to REV7/FANCV and promotes homologous recombination repair. Cell Rep. 2022, 40, 111297. [Google Scholar] [CrossRef]
- Cheng, B.; Pan, W.; Xing, Y.; Xiao, Y.; Chen, J.; Xu, Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur. J. Med. Chem. 2022, 230, 114109. [Google Scholar] [CrossRef]
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine ± cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 510–519. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Wesolowski, R.; Devoe, C.; Lord, S.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R.; Yap, T.A. Phase 1 study of the ATR inhibitor berzosertib in combination with cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 520–527. [Google Scholar] [CrossRef]
- Telli, M.L.; Tolaney, S.M.; Shapiro, G.I.; Middleton, M.; Lord, S.R.; Arkenau, H.T.; Tutt, A.; Abramson, V.; Dean, E.; Haddad, T.C.; et al. Phase 1b study of berzosertib and cisplatin in patients with advanced triple-negative breast cancer. NPJ Breast Cancer 2022, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; van Geel, R.M.J.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.A.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef]
- Moore, K.N.; Hong, D.S.; Patel, M.R.; Pant, S.; Ulahannan, S.V.; Jones, S.; Meric-Bernstam, F.; Wang, J.S.; Aljumaily, R.; Hamilton, E.P.; et al. A Phase 1b Trial of Prexasertib in Combination with Standard-of-Care Agents in Advanced or Metastatic Cancer. Target. Oncol. 2021, 16, 569–589. [Google Scholar] [CrossRef] [PubMed]
- Wehler, T.; Thomas, M.; Schumann, C.; Bosch-Barrera, J.; Viñolas Segarra, N.; Dickgreber, N.J.; Dalhoff, K.; Sebastian, M.; Jaime, J.C.; Alonso, M.; et al. A randomized, phase 2 evaluation of the CHK1 inhibitor, LY2603618, administered in combination with pemetrexed and cisplatin in patients with advanced nonsquamous non-small cell lung cancer. Lung Cancer 2017, 108, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Januchowski, R.; Sterzyńska, K.; Zaorska, K.; Sosińska, P.; Klejewski, A.; Brązert, M.; Nowicki, M.; Zabel, M. Analysis of MDR genes expression and cross-resistance in eight drug resistant ovarian cancer cell lines. J. Ovarian Res. 2016, 9, 65. [Google Scholar] [CrossRef]
- Blair, B.G.; Larson, C.A.; Safaei, R.; Howell, S.B. Copper Transporter 2 Regulates the Cellular Accumulation and Cytotoxicity of Cisplatin and Carboplatin. Clin. Cancer Res. 2009, 15, 4312–4321. [Google Scholar] [CrossRef]
- Zisowsky, J.; Koegel, S.; Leyers, S.; Devarakonda, K.; Kassack, M.U.; Osmak, M.; Jaehde, U. Relevance of drug uptake and efflux for cisplatin sensitivity of tumor cells. Biochem. Pharmacol. 2007, 73, 298–307. [Google Scholar] [CrossRef]
- Ricci, F.; Corbelli, A.; Affatato, R.; Chilà, R.; Chiappa, M.; Brunelli, L.; Fruscio, R.; Pastorelli, R.; Fiordaliso, F.; Damia, G. Mitochondrial structural alterations in ovarian cancer patient-derived xenografts resistant to cisplatin. Am. J. Cancer Res. 2021, 11, 2303–2311. [Google Scholar]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II clinical trial of metformin as a cancer stem cell–targeting agent in ovarian cancer. JCI Insight 2020, 5, e133247. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DRUG | F3 | F3 ddpR | FI | p Value * | Brca1−/− | Brca1−/− ddpR | FI | p Value * |
|---|---|---|---|---|---|---|---|---|
| CISPLATIN (µM) | 16.94 ± 8.26 | >150 | >10 | p = 0.003 | 3.88 ± 0.86 | 22.05 ± 11.63 | 5.7 | p = 0.02 |
| CARBOPLATIN (µM) | 60.67 ± 9.08 | 1060 ± 176.64 | 17.5 | p = 0.015 | 25.2 ± 0.35 | 86.7 ± 1.27 | 3.4 | p = 0.002 |
| OXALIPLATIN (µM) | 79.08 ± 17.92 | 212 ± 82.72 | 2.7 | p = 0.05 | 14.41 ± 2.18 | 32.55 ± 3.56 | 2.1 | p = 0.0078 |
| OLAPARIB (µM) | 6.44 ± 0.5 | 7.53 ± 3.98 | 1.2 | p = 0.63 | 0.08 ± 0.002 | 2.05 ± 0.7 | 24.5 | p = 0.05 |
| RUCAPARIB (µM) | 13.68 ± 2.86 | 5.24 ± 2.63 | 0.4 | p = 0.0197 | 0.21 ± 0.1 | 3.58 ± 1.71 | 17.3 | p = 0.027 |
| NIRAPARIB (µM) | 0.35 ± 0.04 | 0.33 ± 0.3 | 0.9 | p = 0.93 | 0.04 ± 0.01 | 0.26 ± 0.07 | 7.5 | p = 0.043 |
| YONDELIS (nM) | 0.8 ± 0.36 | 1.95 ± 0.74 | 2.5 | p = 0.18 | 0.11 ± 0.02 | 0.57 ± 0.04 | 5.2 | p = 0.0056 |
| PACLITAXEL (nM) | 19.72 ± 2.31 | 4.18 ± 1.8 | 0.2 | p = 0.017 | 22.27 ± 1.9 | 14.8 ± 3.1 | 0.7 | p = 0.1 |
| DOXORUBICIN (nM) | 84.62 ± 7.01 | 44.2 ± 0.49 | 0.5 | p = 0.01 | 34.8 ± 10.7 | 27.65 ± 0.2 | 0.8 | p = 0.44 |
| KU55933 (µM) | 4.06 ± 0.59 | 2.42 ± 0.16 | 0.6 | p = 0.06 | 1.66 ± 0.77 | 1.11 ± 0.18 | 0.7 | p = 0.43 |
| AZD6738 (µM) | 0.26 ± 0.07 | 0.83 ± 0.65 | 3.2 | p = 0.2 | 0.2 ± 0.04 | 0.23 ± 0.11 | 1.2 | p = 0.64 |
| AZD1775 (µM) | 0.42 ± 0.2 | 1.08 ± 0.69 | 2.6 | p = 0.11 | 0.2 ± 0.09 | 0.2 ± 0.14 | 0.9 | p = 0.76 |
| AZD7762 (µM) | 0.19 ± 0.08 | 0.38 ± 0.17 | 2 | p = 0.08 | 0.13 ± 0.06 | 0.22 ± 0.15 | 1.7 | p = 0.28 |
| NU7741 (µM) | 1.72 ± 0.11 | 1.34 ± 0.07 | 0.8 | p = 0.06 | 2.32 ± 0.11 | 1.02 ± 0.29 | 0.4 | p = 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiappa, M.; Guffanti, F.; Grasselli, C.; Panini, N.; Corbelli, A.; Fiordaliso, F.; Damia, G. Different Patterns of Platinum Resistance in Ovarian Cancer Cells with Homologous Recombination Proficient and Deficient Background. Int. J. Mol. Sci. 2024, 25, 3049. https://doi.org/10.3390/ijms25053049
Chiappa M, Guffanti F, Grasselli C, Panini N, Corbelli A, Fiordaliso F, Damia G. Different Patterns of Platinum Resistance in Ovarian Cancer Cells with Homologous Recombination Proficient and Deficient Background. International Journal of Molecular Sciences. 2024; 25(5):3049. https://doi.org/10.3390/ijms25053049
Chicago/Turabian StyleChiappa, Michela, Federica Guffanti, Chiara Grasselli, Nicolò Panini, Alessandro Corbelli, Fabio Fiordaliso, and Giovanna Damia. 2024. "Different Patterns of Platinum Resistance in Ovarian Cancer Cells with Homologous Recombination Proficient and Deficient Background" International Journal of Molecular Sciences 25, no. 5: 3049. https://doi.org/10.3390/ijms25053049
APA StyleChiappa, M., Guffanti, F., Grasselli, C., Panini, N., Corbelli, A., Fiordaliso, F., & Damia, G. (2024). Different Patterns of Platinum Resistance in Ovarian Cancer Cells with Homologous Recombination Proficient and Deficient Background. International Journal of Molecular Sciences, 25(5), 3049. https://doi.org/10.3390/ijms25053049