
Inflammasomes in Intestinal Disease: Mechanisms of Activation and Therapeutic Strategies




Abstract

1. Introduction
2. Inflammasome Sensors
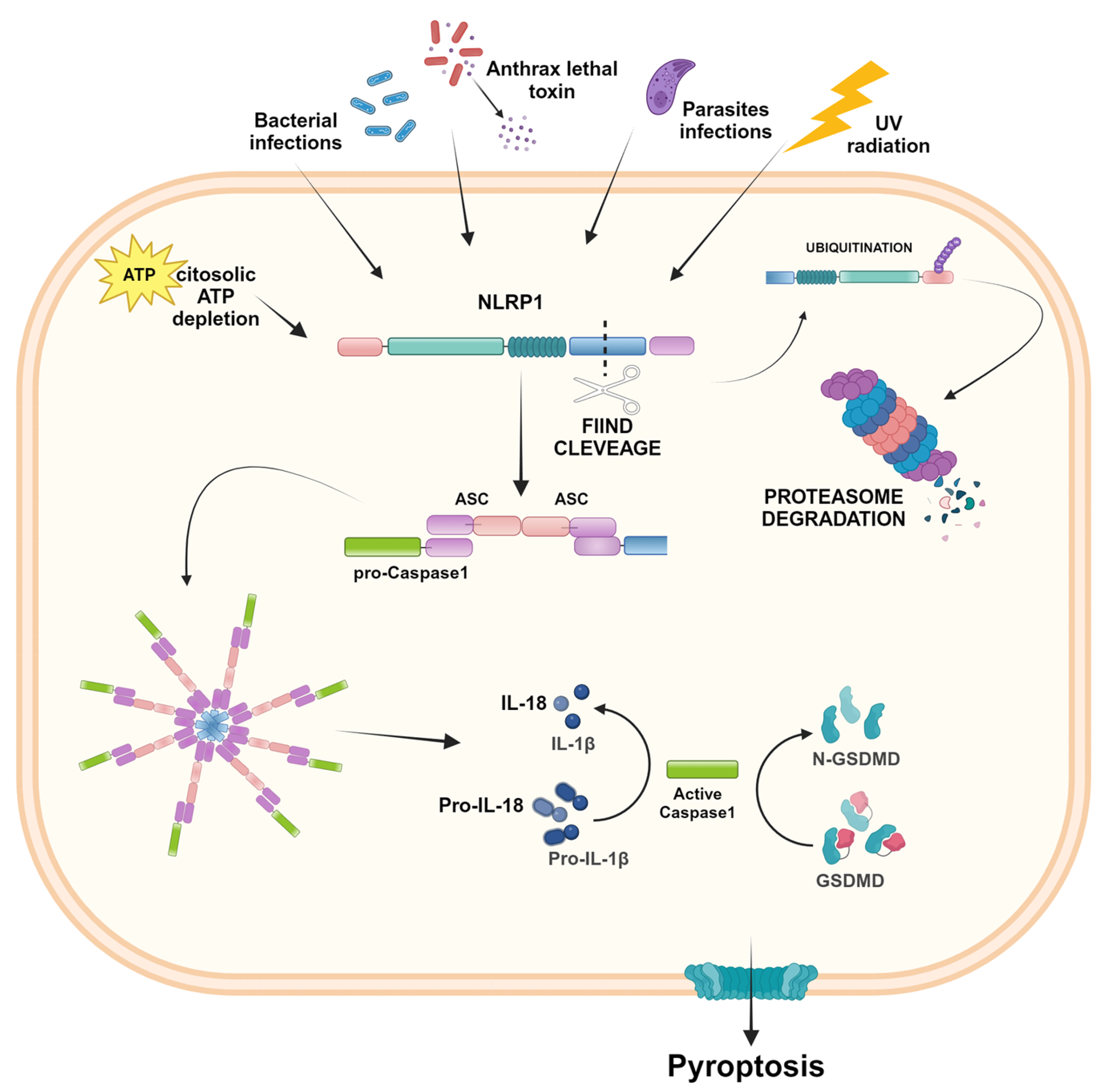
2.1. NLRP1
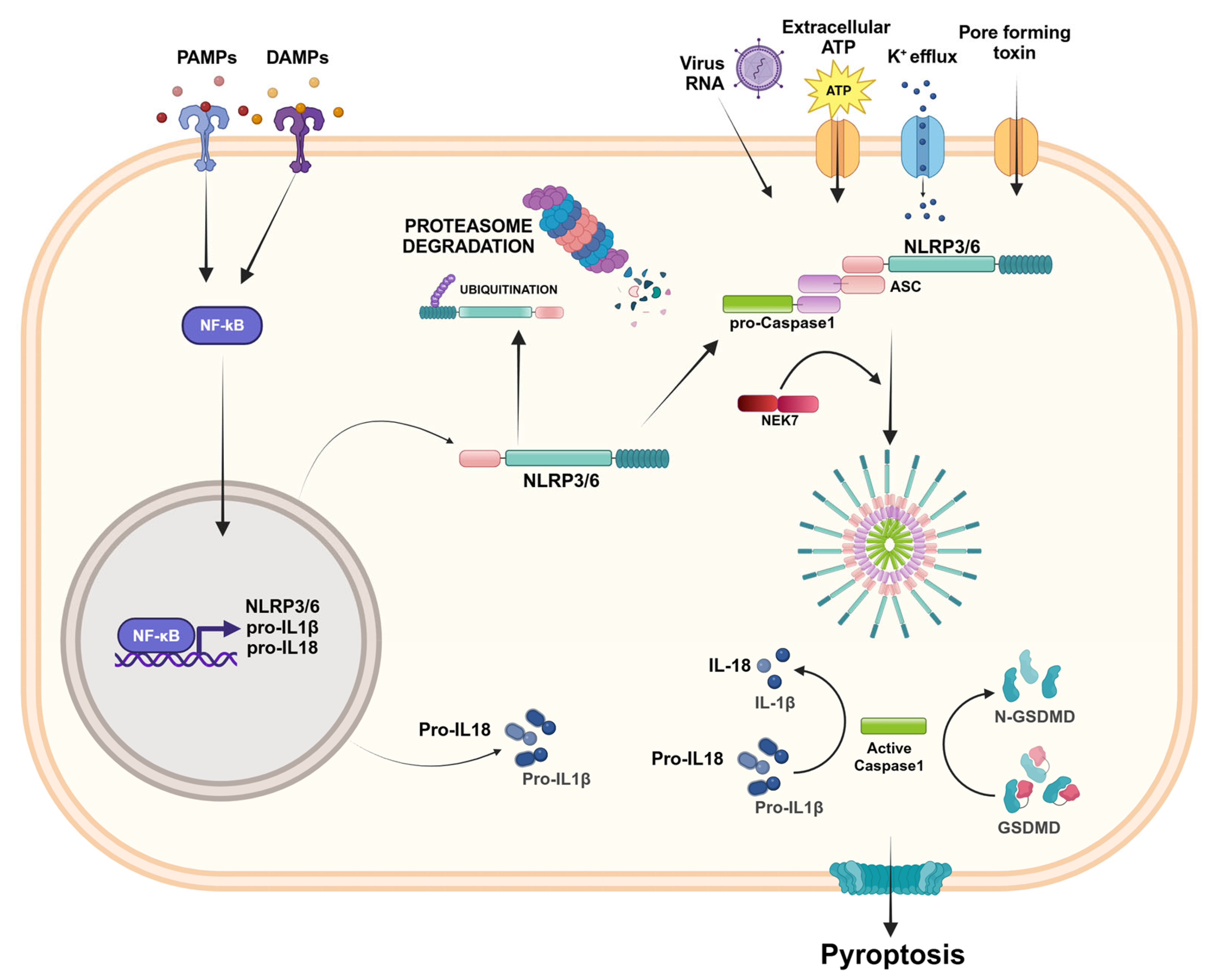
2.2. NLRP3
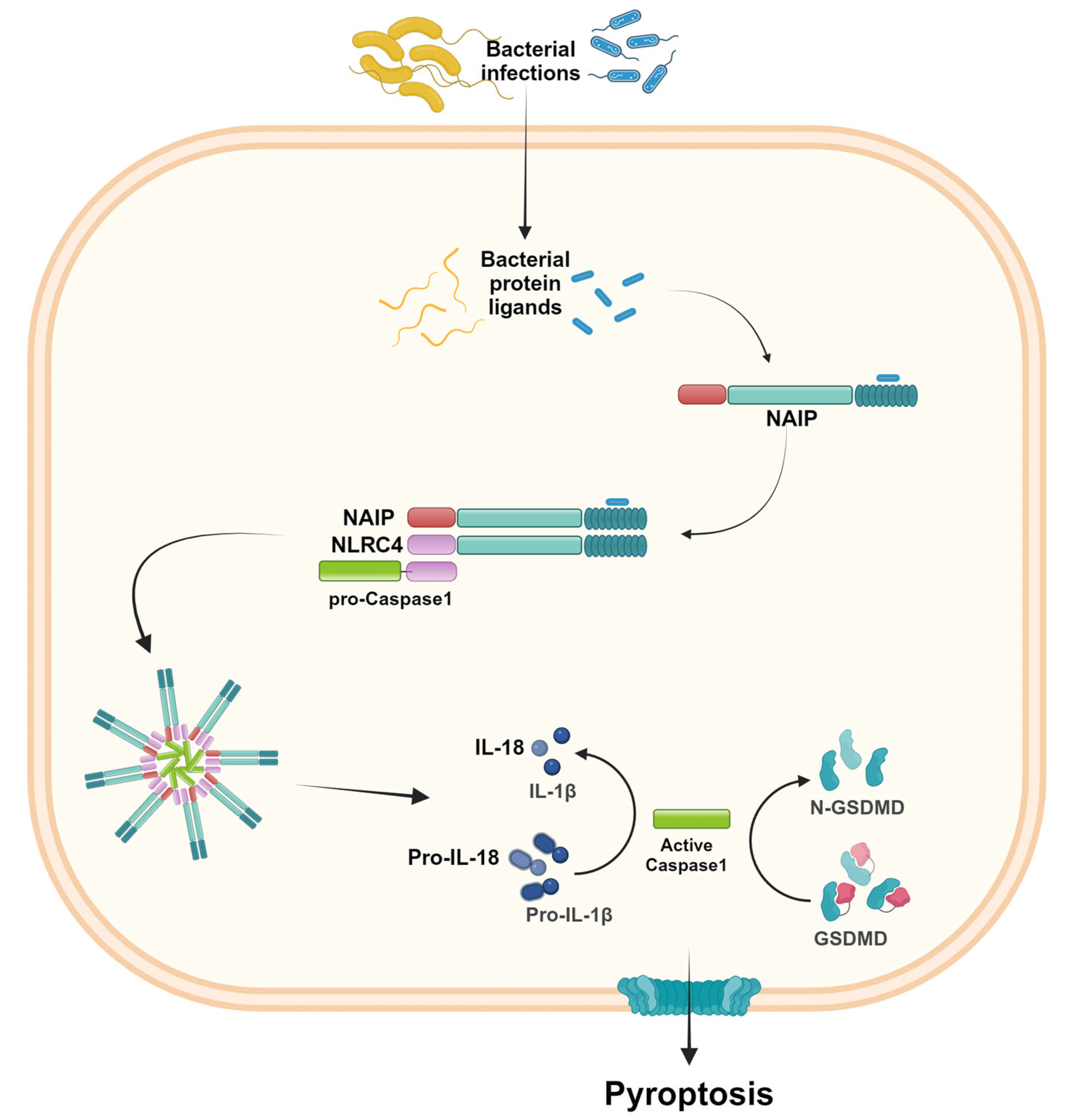
2.3. NLRC4
2.4. NLRP6
2.5. AIM2-like Receptors
2.6. Pyrin Inflammasome
3. Role of Inflammasomes in Intestinal Diseases
3.1. Irritable Bowel Disease
3.2. Celiac Disease
3.3. Inflammatory Bowel Disease
3.4. Colorectal Cancer
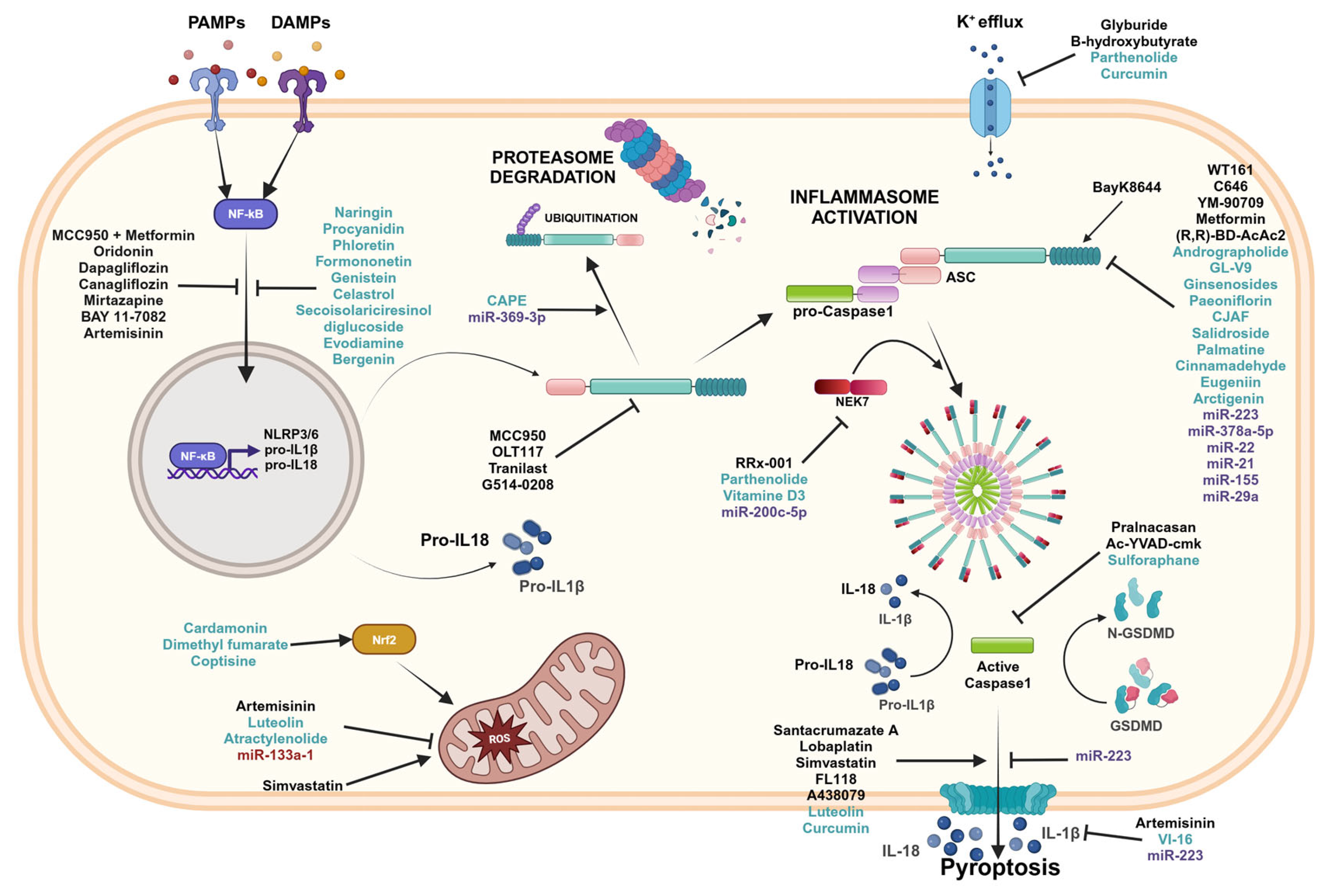
4. Strategies to Inhibit the Inflammasomes Pathways
4.1. Pharmaceutical Inhibitors
4.2. Natural Compounds
4.3. microRNA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shao, T.; Hsu, R.; Rafizadeh, D.L.; Wang, L.; Bowlus, C.L.; Kumar, N.; Mishra, J.; Timilsina, S.; Ridgway, W.M.; Gershwin, M.E.; et al. The gut ecosystem and immune tolerance. J. Autoimmun. 2023, 141, 103114. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhou, J.; Shi, C. Inflammasome: Structure, biological functions, and therapeutic targets. MedComm 2023, 4, e391. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M. Inflammasomes in the Gastrointestinal Tract: Infection, Cancer and Gut Microbiota Homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 721–737. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef]
- Platnich, J.M.; Muruve, D.A. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch. Biochem. Biophys. 2019, 30, 4–14. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and Diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An Overview of the Non-Canonical Inflammasome. Mol. Asp. Med. 2020, 76, 100924. [Google Scholar] [CrossRef]
- Babamale, A.O.; Chen, S.-T. Nod-like Receptors: Critical Intracellular Sensors for Host Protection and Cell Death in Microbial and Parasitic Infections. Int. J. Mol. Sci. 2021, 22, 11398. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, B.; Tweedell, R.E.; Prasanth Kumar, S.; Kanneganti, T.D. The NLR family of innate immune and cell death sensors. Immunity 2024, 57, 674–699. [Google Scholar] [CrossRef] [PubMed]
- Ohto, U. Activation and regulation mechanisms of NOD-like receptors based on structural biology. Front. Immunol. 2022, 13, 953530. [Google Scholar] [CrossRef] [PubMed]
- Bauernfried, S.; Hornung, V. Human NLRP1: From the shadows to center stage. J. Exp. Med. 2022, 219, e20211405. [Google Scholar] [CrossRef]
- Sandstrom, A.; Mitchell, P.S.; Goers, L.; Mu, E.W.; Lesser, C.F.; Vance, R.E. Functional Degradation: A Mechanism of NLRP1 Inflammasome Activation by Diverse Pathogen Enzymes. Science 2019, 364, eaau1330. [Google Scholar] [CrossRef]
- Barry, K.; Murphy, C.; Mansell, A. NLRP1—A Cinderella Story: A Perspective of Recent Advances in NLRP1 and the Questions They Raise. Commun. Biol. 2023, 6, 1274. [Google Scholar] [CrossRef]
- Burian, M.; Schmidt, M.F.; Yazdi, A.S. The NLRP1 Inflammasome in Skin Diseases. Front. Immunol. 2023, 14, 1111611. [Google Scholar] [CrossRef]
- Mi, L.; Min, X.; Chai, Y.; Zhang, J.; Chen, X. NLRP1 Inflammasomes: A Potential Target for the Treatment of Several Types of Brain Injury. Front. Immunol. 2022, 13, 863774. [Google Scholar] [CrossRef]
- Williams, T.M.; Leeth, R.A.; Rothschild, D.E.; Coutermarsh-Ott, S.L.; McDaniel, D.K.; Simmons, A.E.; Heid, B.; Cecere, T.E.; Allen, I.C. The NLRP1 Inflammasome Attenuates Colitis and Colitis-Associated Tumorigenesis. J. Immunol. 2015, 194, 3369–3380. [Google Scholar] [CrossRef]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Zhao, N.; Li, C.; Di, B.; Xu, L. Recent Advances in the NEK7-Licensed NLRP3 Inflammasome Activation: Mechanisms, Role in Diseases and Related Inhibitors. J. Autoimmun. 2020, 113, 102515. [Google Scholar] [CrossRef]
- Xia, J.; Jiang, S.; Dong, S.; Liao, Y.; Zhou, Y. The Role of Post-Translational Modifications in Regulation of NLRP3 Inflammasome Activation. Int. J. Mol. Sci. 2023, 24, 6126. [Google Scholar] [CrossRef]
- Scalavino, V.; Piccinno, E.; Valentini, A.M.; Schena, N.; Armentano, R.; Giannelli, G.; Serino, G. miR-369-3p Modulates Intestinal Inflammatory Response via BRCC3/NLRP3 Inflammasome Axis. Cells 2023, 12, 2184. [Google Scholar] [CrossRef]
- Li, Z.; Guo, J.; Bi, L. Role of the NLRP3 Inflammasome in Autoimmune Diseases. Biomed. Pharmacother. 2020, 130, 110542. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Zeiser, R. NLRP3 Inflammasome Activation in Cancer: A Double-Edged Sword. Front. Immunol. 2020, 11, 1444. [Google Scholar] [CrossRef]
- Qiang, R.; Li, Y.; Dai, X.; Lv, W. NLRP3 inflammasome in digestive diseases: From mechanism to therapy. Front. Immunol. 2022, 13, 978190. [Google Scholar] [CrossRef]
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H.A.; Fitzgerald, K.A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis Induced in Mice with Dextran Sulfate Sodium (DSS) Is Mediated by the NLRP3 Inflammasome. Gut 2010, 59, 1192–1199. [Google Scholar] [CrossRef]
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Vogel, P.; Body-Malapel, M.; Lamkanfi, M.; Kanneganti, T.-D. IL-18 Production Downstream of the Nlrp3 Inflammasome Confers Protection against Colorectal Tumor Formation. J. Immunol. 2010, 185, 4912–4920. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Y.; Du, Q.; Lu, P.; Fan, H.; Lu, J.; Hu, R. Inflammasome-Independent NLRP3 Is Required for Epithelial-Mesenchymal Transition in Colon Cancer Cells. Exp. Cell Res. 2016, 342, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Poyet, J.-L.; Srinivasula, S.M.; Tnani, M.; Razmara, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of Ipaf, a Human Caspase-1-Activating Protein Related to Apaf-1. J. Biol. Chem. 2001, 276, 28309–28313. [Google Scholar] [CrossRef]
- Duncan, J.A.; Canna, S.W. The NLRC4 Inflammasome. Immunol. Rev. 2018, 281, 115–123. [Google Scholar] [CrossRef]
- Wen, J.; Xuan, B.; Liu, Y.; Wang, L.; He, L.; Meng, X.; Zhou, T.; Wang, Y. Updating the NLRC4 Inflammasome: From Bacterial Infections to Autoimmunity and Cancer. Front. Immunol. 2021, 12, 702527. [Google Scholar] [CrossRef]
- Lei-Leston, A.C.; Murphy, A.G.; Maloy, K.J. Epithelial Cell Inflammasomes in Intestinal Immunity and Inflammation. Front. Immunol. 2017, 8, 1168. [Google Scholar] [CrossRef]
- An, Y.; Zhai, Z.; Wang, X.; Ding, Y.; He, L.; Li, L.; Mo, Q.; Mu, C.; Xie, R.; Liu, T.; et al. Targeting Desulfovibrio vulgaris Flagellin-Induced NAIP/NLRC4 Inflammasome Activation in Macrophages Attenuates Ulcerative Colitis. J. Adv. Res. 2023, 52, 219–232. [Google Scholar] [CrossRef]
- Tong, G.; Shen, Y.; Li, H.; Qian, H.; Tan, Z. NLRC4, inflammation and colorectal cancer (Review). Int. J. Oncol. 2024, 65, 99. [Google Scholar] [CrossRef]
- Hu, B.; Elinav, E.; Huber, S.; Booth, C.J.; Strowig, T.; Jin, C.; Eisenbarth, S.C.; Flavell, R.A. Inflammation-Induced Tumorigenesis in the Colon Is Regulated by Caspase-1 and NLRC4. Proc. Natl. Acad. Sci. USA 2010, 107, 21635–21640. [Google Scholar] [CrossRef]
- Liu, R.; Truax, A.D.; Chen, L.; Hu, P.; Li, Z.; Chen, J.; Song, C.; Chen, L.; Ting, J.P.-Y. Expression Profile of Innate Immune Receptors, NLRs and AIM2, in Human Colorectal Cancer: Correlation with Cancer Stages and Inflammasome Components. Oncotarget 2015, 6, 33456–33469. [Google Scholar] [CrossRef] [PubMed]
- Allam, R.; Maillard, M.H.; Tardivel, A.; Chennupati, V.; Bega, H.; Yu, C.W.; Velin, D.; Schneider, P.; Maslowski, K.M. Epithelial NAIPs Protect against Colonic Tumorigenesis. J. Exp. Med. 2015, 212, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Kern, L.; Elinav, E. The NLRP6 inflammasome. Immunology 2021, 162, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Núñez, G. A Functional Role for Nlrp6 in Intestinal Inflammation and Tumorigenesis. J. Immunol. 2011, 186, 7187–7194. [Google Scholar] [CrossRef]
- Levy, M.; Shapiro, H.; Thaiss, C.A.; Elinav, E. NLRP6: A Multifaceted Innate Immune Sensor. Trends Immunol. 2017, 38, 248–260. [Google Scholar] [CrossRef]
- Angosto-Bazarra, D.; Molina-López, C.; Pelegrín, P. Physiological and pathophysiological functions of NLRP6: Pro- and anti-inflammatory roles. Commun. Biol. 2022, 5, 524. [Google Scholar] [CrossRef]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.-P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 Inflammasome Orchestrates the Colonic Host-Microbial Interface by Regulating Goblet Cell Mucus Secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef]
- Hu, B.; Elinav, E.; Huber, S.; Strowig, T.; Hao, L.; Hafemann, A.; Jin, C.; Wunderlich, C.; Wunderlich, T.; Eisenbarth, S.C.; et al. Microbiota-Induced Activation of Epithelial IL-6 Signaling Links Inflammasome-Driven Inflammation with Transmissible Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 9862–9867. [Google Scholar] [CrossRef]
- Fan, X.; Jiao, L.; Jin, T. Activation and Immune Regulation Mechanisms of PYHIN Family During Microbial Infection. Front. Microbiol. 2022, 12, 809412. [Google Scholar] [CrossRef]
- DeYoung, K.L.; Ray, M.E.; Su, Y.A.; Anzick, S.L.; Johnstone, R.W.; Trapani, J.A.; Meltzer, P.S.; Trent, J.M. Cloning a Novel Member of the Human Interferon-Inducible Gene Family Associated with Control of Tumorigenicity in a Model of Human Melanoma. Oncogene 1997, 15, 453–457. [Google Scholar] [CrossRef]
- Lugrin, J.; Martinon, F. The AIM 2 Inflammasome: Sensor of Pathogens and Cellular Perturbations. Immunol. Rev. 2018, 281, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Russo, A.J.; Shivcharan, S.; Rathinam, V.A. AIM2 in health and disease: Inflammasome and beyond. Immunol. Rev. 2020, 297, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Perry, A.; Jiang, J.; Smith, P.; Curry, J.A.; Unterholzner, L.; Jiang, Z.; Horvath, G.; Rathinam, V.A.; Johnstone, R.W.; et al. Structures of the HIN Domain: DNA Complexes Reveal Ligand Binding and Activation Mechanisms of the AIM2 Inflammasome and IFI16 Receptor. Immunity 2012, 36, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Karki, R.; Wang, Y.; Nguyen, L.N.; Kalathur, R.C.; Kanneganti, T.-D. AIM2 Forms a Complex with Pyrin and ZBP1 to Drive PANoptosis and Host Defence. Nature 2021, 597, 415–419. [Google Scholar] [CrossRef]
- Man, S.M.; Zhu, Q.; Zhu, L.; Liu, Z.; Karki, R.; Malik, A.; Sharma, D.; Li, L.; Malireddi, R.K.S.; Gurung, P.; et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 2015, 162, 45–58. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Z.; Yu, S. AIM2 Regulates Viability and Apoptosis in Human Colorectal Cancer Cells via the PI3K/Akt Pathway. OncoTargets Ther. 2017, 10, 811–817. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, X.; Zhang, Y.; Zhu, H.; Qiao, Z.; Lu, Y.; Mi, X.; Cao, H.; Shen, G.; He, S. Absent in Melanoma 2 Attenuates Proliferation and Migration and Promotes Apoptosis of Human Colorectal Cancer Cells by Activating P38MAPK Signaling Pathway. Oncol. Res. 2024, 32, 353–360. [Google Scholar] [CrossRef]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. IFI16 DNA Sensor Is Required for Death of Lymphoid CD4 T Cells Abortively Infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef]
- Heilig, R.; Broz, P. Function and Mechanism of the Pyrin Inflammasome. Eur. J. Immunol. 2018, 48, 230–238. [Google Scholar] [CrossRef]
- Papadopoulos, V.P.; Antoniadou, C.; Ritis, K.; Skendros, P. MEFV Mutations in IBD Patients: A Systematic Review and Meta- analysis. J. Gastrointest. Liver Dis. 2022, 31, 85–97. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.D. Inflammasome signaling in colorectal cancer. Transl. Res. 2023, 252, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M. Diagnosis and Treatment of Irritable Bowel Syndrome: A Review. JAMA 2021, 325, 865–877, Erratum in JAMA 2021, 325, 1568. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.D.; Sun, N.; Canakis, A.; Park, W.Y.; Weber, H.C. Irritable Bowel Syndrome and the Gut Microbiome: A Comprehensive Review. J. Clin. Med. 2023, 12, 2558. [Google Scholar] [CrossRef] [PubMed]
- Berumen, A.; Edwinson, A.L.; Grover, M. Post-infection Irritable Bowel Syndrome. Gastroenterol. Clin. N. Am. 2021, 50, 445–461. [Google Scholar] [CrossRef]
- Gu, Q.-Y.; Zhang, J.; Feng, Y.-C. Role of NLRP3 Inflammasome in Bifidobacterium longum-Regulated Visceral Hypersensitivity of Postinfectious Irritable Bowel Syndrome. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1933–1937. [Google Scholar] [CrossRef]
- Scuderi, S.A.; Casili, G.; Lanza, M.; Filippone, A.; Paterniti, I.; Esposito, E.; Campolo, M. Modulation of NLRP3 Inflammasome Attenuated Inflammatory Response Associated to Diarrhea-Predominant Irritable Bowel Syndrome. Biomedicines 2020, 8, 519. [Google Scholar] [CrossRef]
- Yu, L.M.; Zhao, K.J.; Wang, S.S.; Wang, X.; Lu, B. Corticotropin-releasing Factor Induces Inflammatory Cytokines via the NLRP6-inflammatory Cytokine Axis in a Murine Model of Irritable Bowel Syndrome. J. Dig. Dis. 2019, 20, 143–151. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, M.; Chen, C.; Gillilland, M.; Sun, X.; El–Zaatari, M.; Huffnagle, G.B.; Young, V.B.; Zhang, J.; Hong, S.; et al. Stress-Induced Corticotropin-Releasing Hormone-Mediated NLRP6 Inflammasome Inhibition and Transmissible Enteritis in Mice. Gastroenterology 2013, 144, 1478–1487.e8. [Google Scholar] [CrossRef]
- Caio, G.; Volta, U.; Sapone, A.; Leffler, D.A.; De Giorgio, R.; Catassi, C.; Fasano, A. Celiac Disease: A Comprehensive Current Review. BMC Med. 2019, 17, 142. [Google Scholar] [CrossRef]
- Iversen, R.; Sollid, L.M. The Immunobiology and Pathogenesis of Celiac Disease. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 47–70. [Google Scholar] [CrossRef]
- Garrote, J.A.; Gómez-González, E.; Bernardo, D.; Arranz, E.; Chirdo, F. Celiac Disease Pathogenesis: The Proinflammatory Cytokine Network. J. Pediatr. Gastroenterol. Nutr. 2008, 47, S27–S32. [Google Scholar] [CrossRef] [PubMed]
- Palová-Jelínková, L.; Dáňová, K.; Drašarová, H.; Dvořák, M.; Funda, D.P.; Fundová, P.; Kotrbová-Kozak, A.; Černá, M.; Kamanová, J.; Martin, S.F.; et al. Pepsin Digest of Wheat Gliadin Fraction Increases Production of IL-1β via TLR4/MyD88/TRIF/MAPK/NF-ΚB Signaling Pathway and an NLRP3 Inflammasome Activation. PLoS ONE 2013, 8, e62426. [Google Scholar] [CrossRef] [PubMed]
- Gómez Castro, M.F.; Miculán, E.; Herrera, M.G.; Ruera, C.; Perez, F.; Prieto, E.D.; Barrera, E.; Pantano, S.; Carasi, P.; Chirdo, F.G. P31-43 Gliadin Peptide Forms Oligomers and Induces NLRP3 Inflammasome/Caspase 1-Dependent Mucosal Damage in Small Intestine. Front. Immunol. 2019, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Ruera, C.N.; Perez, F.; Iribarren, M.L.; Guzman, L.; Menendez, L.; Garbi, L.; Chirdo, F.G. Coexistence of Apoptosis, Pyroptosis, and Necroptosis Pathways in Celiac Disease. Clin. Exp. Immunol. 2023, 214, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Pontillo, A.; Vendramin, A.; Catamo, E.; Fabris, A.; Crovella, S. The Missense Variation Q705K in CIAS1/NALP3/NLRP3 Gene and an NLRP1 Haplotype Are Associated with Celiac Disease. Am. J. Gastroenterol. 2011, 106, 539–544. [Google Scholar] [CrossRef]
- Diez-Martin, E.; Hernandez-Suarez, L.; Muñoz-Villafranca, C.; Martin-Souto, L.; Astigarraga, E.; Ramirez-Garcia, A.; Barreda-Gómez, G. Inflammatory Bowel Disease: A Comprehensive Analysis of Molecular Bases, Predictive Biomarkers, Diagnostic Methods, and Therapeutic Options. Int. J. Mol. Sci. 2024, 25, 7062. [Google Scholar] [CrossRef]
- Liu, L.; Dong, Y.; Ye, M.; Jin, S.; Yang, J.; Joosse, M.E.; Sun, Y.; Zhang, J.; Lazarev, M.; Brant, S.R.; et al. The Pathogenic Role of NLRP3 Inflammasome Activation in Inflammatory Bowel Diseases of Both Mice and Humans. J. Crohn’s Colitis 2016, 11, 737–750. [Google Scholar] [CrossRef]
- Tourkochristou, E.; Aggeletopoulou, I.; Konstantakis, C.; Triantos, C. Role of NLRP3 inflammasome in inflammatory bowel diseases. World J. Gastroenterol. 2019, 25, 4796–4804. [Google Scholar] [CrossRef]
- Shao, B.Z.; Wang, S.L.; Pan, P.; Yao, J.; Wu, K.; Li, Z.S.; Bai, Y.; Linghu, E.Q. Targeting NLRP3 Inflammasome in Inflammatory Bowel Disease: Putting out the Fire of Inflammation. Inflammation 2019, 42, 1147–1159. [Google Scholar] [CrossRef]
- Gao, S.-J. Interleukin-18 Genetic Polymorphisms Contribute Differentially to the Susceptibility to Crohn’s Disease. World J. Gastroenterol. 2015, 21, 8711–8722. [Google Scholar] [CrossRef]
- Siegmund, B.; Lehr, H.-A.; Fantuzzi, G.; Dinarello, C.A. IL-1β-Converting Enzyme (Caspase-1) in Intestinal Inflammation. Proc. Natl. Acad. Sci. USA 2001, 98, 13249–13254. [Google Scholar] [CrossRef] [PubMed]
- Ishikura, T.; Kanai, T.; Uraushihara, K.; Iiyama, R.; Makita, S.; Totsuka, T.; Yamazaki, M.; Sawada, T.; Nakamura, T.; Miyata, T.; et al. Interleukin-18 Overproduction Exacerbates the Development of Colitis with Markedly Infiltrated Macrophages in Interleukin-18 Transgenic Mice. J. Gastroenterol. Hepatol. 2003, 18, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Steiner, A.; Reygaerts, T.; Pontillo, A.; Ceccherini, I.; Moecking, J.; Moghaddas, F.; Davidson, S.; Caroli, F.; Grossi, A.; Castro, F.F.M.; et al. Recessive NLRC4-Autoinflammatory Disease Reveals an Ulcerative Colitis Locus. J. Clin. Immunol. 2022, 42, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Ringel-Scaia, V.M.; Qin, Y.; Thomas, C.A.; Huie, K.E.; McDaniel, D.K.; Eden, K.; Wade, P.A.; Allen, I.C. Maternal Influence and Murine Housing Confound Impact of NLRP1 Inflammasome on Microbiome Composition. J. Innate Immun. 2019, 11, 416–431. [Google Scholar] [CrossRef]
- Cummings, J.R.F.; Cooney, R.M.; Clarke, G.; Beckly, J.; Geremia, A.; Pathan, S.; Hancock, L.; Guo, C.; Cardon, L.R.; Jewell, D.P. The Genetics of NOD-like Receptors in Crohn’s Disease. Tissue Antigens 2010, 76, 48–56. [Google Scholar] [CrossRef]
- Chang, L.; Tian, Y.; Xu, L.; Hao, Q.; Song, L.; Lu, Y.; Zhen, Y. Spotlight on NLRP6 and Tumor Research Situation: A Potential Cancer Participant. J. Immunol. Res. 2023, 2023, 6613064. [Google Scholar] [CrossRef]
- Lemire, P.; Robertson, S.J.; Maughan, H.; Tattoli, I.; Streutker, C.J.; Platnich, J.M.; Muruve, D.A.; Philpott, D.J.; Girardin, S.E. The NLR Protein NLRP6 Does Not Impact Gut Microbiota Composition. Cell Rep. 2017, 21, 3653–3661. [Google Scholar] [CrossRef]
- Mamantopoulos, M.; Ronchi, F.; Van Hauwermeiren, F.; Vieira-Silva, S.; Yilmaz, B.; Martens, L.; Saeys, Y.; Drexler, S.K.; Yazdi, A.S.; Raes, J.; et al. Nlrp6- and ASC-Dependent Inflammasomes Do Not Shape the Commensal Gut Microbiota Composition. Immunity 2017, 47, 339–348.e4. [Google Scholar] [CrossRef]
- Hu, S.; Peng, L.; Kwak, Y.-T.; Tekippe, E.M.; Pasare, C.; Malter, J.S.; Hooper, L.V.; Zaki, H. The DNA Sensor AIM2 Maintains Intestinal Homeostasis via Regulation of Epithelial Antimicrobial Host Defense. Cell Rep. 2015, 13, 1922–1936. [Google Scholar] [CrossRef]
- Ratsimandresy, R.A.; Indramohan, M.; Dorfleutner, A.; Stehlik, C. The AIM2 Inflammasome Is a Central Regulator of Intestinal Homeostasis through the IL-18/IL-22/STAT3 Pathway. Cell. Mol. Immunol. 2017, 14, 127–142. [Google Scholar] [CrossRef]
- Postwala, H.; Shah, Y.; Parekh, P.S.; Chorawala, M.R. Unveiling the genetic and epigenetic landscape of colorectal cancer: New insights into pathogenic pathways. Med. Oncol. 2023, 40, 334. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Nisar, S.; Singh, M.; Ashraf, B.; Masoodi, T.; Prasad, C.P.; Sharma, A.; Maacha, S.; Karedath, T.; Hashem, S.; et al. Cytokine- and Chemokine-induced Inflammatory Colorectal Tumor Microenvironment: Emerging Avenue for Targeted Therapy. Cancer Commun. 2022, 42, 689–715. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Lei, Z.; Zhou, C. NLRP3 Promotes Colorectal Cancer Cell Proliferation and Metastasis via Regulating Epithelial Mesenchymal Transformation. Anticancer Agents Med. Chem. 2020, 20, 820–827. [Google Scholar] [CrossRef]
- Hong, S.; Hwang, I.; Lee, Y.-S.; Park, S.; Lee, W.-K.; Fernandes-Alnemri, T.; Alnemri, E.S.; Kim, Y.-S.; Yu, J.-W. Restoration of ASC Expression Sensitizes Colorectal Cancer Cells to Genotoxic Stress-Induced Caspase-Independent Cell Death. Cancer Lett. 2013, 331, 183–191. [Google Scholar] [CrossRef]
- Salcedo, R.; Worschech, A.; Cardone, M.; Jones, Y.; Gyulai, Z.; Dai, R.-M.; Wang, E.; Ma, W.; Haines, D.; O’hUigin, C.; et al. MyD88-Mediated Signaling Prevents Development of Adenocarcinomas of the Colon: Role of Interleukin 18. J. Exp. Med. 2010, 207, 1625–1636. [Google Scholar] [CrossRef]
- Sharma, D.; Malik, A.; Guy, C.S.; Karki, R.; Vogel, P.; Kanneganti, T.-D. Pyrin Inflammasome Regulates Tight Junction Integrity to Restrict Colitis and Tumorigenesis. Gastroenterology 2018, 154, 948–964.e8. [Google Scholar] [CrossRef]
- Dihlmann, S.; Tao, S.; Echterdiek, F.; Herpel, E.; Jansen, L.; Chang-Claude, J.; Brenner, H.; Hoffmeister, M.; Kloor, M. Lack of Absent in Melanoma 2 (AIM2) Expression in Tumor Cells Is Closely Associated with Poor Survival in Colorectal Cancer Patients. Int. J. Cancer 2014, 135, 2387–2396. [Google Scholar] [CrossRef]
- Patsos, G.; Germann, A.; Gebert, J.; Dihlmann, S. Restoration of Absent in Melanoma 2 (AIM2) Induces G2/M Cell Cycle Arrest and Promotes Invasion of Colorectal Cancer Cells. Int. J. Cancer 2010, 126, 1838–1849. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, M.; Jin, C.; Ding, Y.; Yang, M.; Wang, R.; Zhou, Y.; Zhou, Y.; Li, T.; Wang, K.; et al. Absent in Melanoma 2 Suppresses Epithelial-mesenchymal Transition via Akt and Inflammasome Pathways in Human Colorectal Cancer Cells. J. Cell. Biochem. 2019, 120, 17744–17756. [Google Scholar] [CrossRef]
- Wilson, J.E.; Petrucelli, A.S.; Chen, L.; Koblansky, A.A.; Truax, A.D.; Oyama, Y.; Rogers, A.B.; Brickey, W.J.; Wang, Y.; Schneider, M.; et al. Inflammasome-Independent Role of AIM2 in Suppressing Colon Tumorigenesis via DNA-PK and Akt. Nat. Med. 2015, 21, 906–913. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 Directly Targets the NLRP3 ATP-Hydrolysis Motif for Inflammasome Inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Perera, A.P.; Fernando, R.; Shinde, T.; Gundamaraju, R.; Southam, B.; Sohal, S.S.; Robertson, A.A.B.; Schroder, K.; Kunde, D.; Eri, R. MCC950, a Specific Small Molecule Inhibitor of NLRP3 Inflammasome Attenuates Colonic Inflammation in Spontaneous Colitis Mice. Sci. Rep. 2018, 8, 8618. [Google Scholar] [CrossRef] [PubMed]
- Umiker, B.; Lee, H.-H.; Cope, J.; Ajami, N.J.; Laine, J.-P.; Fregeau, C.; Ferguson, H.; Alves, S.E.; Sciammetta, N.; Kleinschek, M.; et al. The NLRP3 Inflammasome Mediates DSS-Induced Intestinal Inflammation in Nod2 Knockout Mice. Innate Immun. 2019, 25, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Mehto, S.; Jena, K.K.; Nath, P.; Chauhan, S.; Kolapalli, S.P.; Das, S.K.; Sahoo, P.K.; Jain, A.; Taylor, G.A.; Chauhan, S. The Crohn’s Disease Risk Factor IRGM Limits NLRP3 Inflammasome Activation by Impeding Its Assembly and by Mediating Its Selective Autophagy. Mol. Cell 2019, 73, 429–445.e7. [Google Scholar] [CrossRef] [PubMed]
- Oizumi, T.; Mayanagi, T.; Toya, Y.; Sugai, T.; Matsumoto, T.; Sobue, K. NLRP3 Inflammasome Inhibitor OLT1177 Suppresses Onset of Inflammation in Mice with Dextran Sulfate Sodium-Induced Colitis. Dig. Dis. Sci. 2022, 67, 2912–2921. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast Directly Targets NLRP 3 to Treat Inflammasome-driven Diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- Seto, Y.; Kato, K.; Tsukada, R.; Suzuki, H.; Kaneko, Y.; Kojo, Y.; Sato, H.; Onoue, S. Protective Effects of Tranilast on Experimental Colitis in Rats. Biomed. Pharmacother. 2017, 90, 842–849. [Google Scholar] [CrossRef]
- Sun, X.; Suzuki, K.; Nagata, M.; Kawauchi, Y.; Yano, M.; Ohkoshi, S.; Matsuda, Y.; Kawachi, H.; Watanabe, K.; Asakura, H.; et al. Rectal Administration of Tranilast Ameliorated Acute Colitis in Mice through Increased Expression of Heme Oxygenase-1. Pathol. Int. 2010, 60, 93–101. [Google Scholar] [CrossRef]
- Nozu, T.; Arie, H.; Miyagishi, S.; Ishioh, M.; Takakusaki, K.; Okumura, T. Tranilast Alleviates Visceral Hypersensitivity and Colonic Hyperpermeability by Suppressing NLRP3 Inflammasome Activation in Irritable Bowel Syndrome Rat Models. Int. Immunopharmacol. 2024, 133, 112099. [Google Scholar] [CrossRef]
- Dai, Z.; Chen, X.; An, L.; Li, C.; Zhao, N.; Yang, F.; You, S.; Hou, C.; Li, K.; Jiang, C.; et al. Development of Novel Tetrahydroquinoline Inhibitors of NLRP3 Inflammasome for Potential Treatment of DSS-Induced Mouse Colitis. J. Med. Chem. 2021, 64, 871–889. [Google Scholar] [CrossRef]
- Saber, S.; El-Kader, E.M.A. Novel Complementary Coloprotective Effects of Metformin and MCC950 by Modulating HSP90/NLRP3 Interaction and Inducing Autophagy in Rats. Inflammopharmacology 2021, 29, 237–251. [Google Scholar] [CrossRef] [PubMed]
- El-Rous, M.A.; Saber, S.; Raafat, E.M.; Ahmed, A.A.E. Dapagliflozin, an SGLT2 Inhibitor, Ameliorates Acetic Acid-Induced Colitis in Rats by Targeting NFκB/AMPK/NLRP3 Axis. Inflammopharmacology 2021, 29, 1169–1185. [Google Scholar] [CrossRef] [PubMed]
- Nasr, M.; Cavalu, S.; Saber, S.; Youssef, M.E.; Abdelhamid, A.M.; Elagamy, H.I.; Kamal, I.; Gaafar, A.G.A.; El-Ahwany, E.; Amin, N.A.; et al. Canagliflozin-Loaded Chitosan-Hyaluronic Acid Microspheres Modulate AMPK/NF-ΚB/NLRP3 Axis: A New Paradigm in the Rectal Therapy of Ulcerative Colitis. Biomed. Pharmacother. 2022, 153, 113409. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.Q.; Wang, H.L.; Chen, K.; Wang, S.B.; Xu, Y.; Ye, Q.; Sun, Y.W. Oridonin Derivative Ameliorates Experimental Colitis by Inhibiting Activated T-cells and Translocation of Nuclear Factor-kappa B. J. Dig. Dis. 2016, 17, 104–112. [Google Scholar] [CrossRef]
- Hafez, H.M.; Ibrahim, M.A.; Yehia Abdelzaher, W.; Gad, A.A.; Mohammed Naguib Abdel Hafez, S.; Abdel-Gaber, S.A. Protective Effect of Mirtazapine against Acetic Acid-Induced Ulcerative Colitis in Rats: Role of NLRP3 Inflammasome Pathway. Int. Immunopharmacol. 2021, 101, 108174. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.-W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-Inflammatory Compounds Parthenolide and Bay 11-7082 Are Direct Inhibitors of the Inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef]
- Hua, L.; Liang, S.; Zhou, Y.; Wu, X.; Cai, H.; Liu, Z.; Ou, Y.; Chen, Y.; Chen, X.; Yan, Y.; et al. Artemisinin-Derived Artemisitene Blocks ROS-Mediated NLRP3 Inflammasome and Alleviates Ulcerative Colitis. Int. Immunopharmacol. 2022, 113, 109431. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide Inhibits the Cryopyrin/Nalp3 Inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The Ketone Metabolite β-Hydroxybutyrate Blocks NLRP3 Inflammasome–Mediated Inflammatory Disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Abdelhady, R.; Saber, S.; Ahmed Abdel-Reheim, M.; Alamri, M.M.S.; Alfaifi, J.; Adam, M.I.E.; Saleh, L.A.; Farag, A.I.; Elmorsy, E.A.; El-Wakeel, H.S.; et al. Unveiling the Therapeutic Potential of Exogenous β-Hydroxybutyrate for Chronic Colitis in Rats: Novel Insights on Autophagy, Apoptosis, and Pyroptosis. Front. Pharmacol. 2023, 14, 1239025. [Google Scholar] [CrossRef]
- Saber, S.; Alamri, M.M.S.; Alfaifi, J.; Saleh, L.A.; Abdel-Ghany, S.; Aboregela, A.M.; Farrag, A.A.; Almaeen, A.H.; Adam, M.I.E.; AlQahtani, A.A.J.; et al. (R,R)-BD-AcAc2 Mitigates Chronic Colitis in Rats: A Promising Multi-Pronged Approach Modulating Inflammasome Activity, Autophagy, and Pyroptosis. Pharmaceuticals 2023, 16, 953. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; He, H.; Lin, B.; Chen, Y.; Deng, X.; Jiang, W.; Zhou, R. RRx-001 Ameliorates Inflammatory Diseases by Acting as a Potent Covalent NLRP3 Inhibitor. Cell. Mol. Immunol. 2021, 18, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Yu, X.; Gong, P.; Wang, X.; Tian, L. Identification of WT161 as a Potent Agent for the Treatment of Colitis by Targeting the Nucleotide-Binding Domain-Like Receptor Family Pyrin Domain Containing 3 Inflammasome. Front. Pharmacol. 2022, 13, 780179. [Google Scholar] [CrossRef]
- Xu, X.; Li, J.; Long, X.; Tao, S.; Yu, X.; Ruan, X.; Zhao, K.; Tian, L. C646 Protects Against DSS-Induced Colitis Model by Targeting NLRP3 Inflammasome. Front. Pharmacol. 2021, 12, 707610. [Google Scholar] [CrossRef]
- Liu, G.; Wang, F.; Feng, Y.; Tang, H. Metformin Inhibits NLRP3 Inflammasome Expression and Regulates Inflammatory Microenvironment to Delay the Progression of Colorectal Cancer. Recent Pat. Anti-Cancer Drug Discov. 2024, in press. [Google Scholar] [CrossRef]
- Segovia, M.; Russo, S.; Jeldres, M.; Mahmoud, Y.D.; Perez, V.; Duhalde, M.; Charnet, P.; Rousset, M.; Victoria, S.; Veigas, F.; et al. Targeting TMEM176B Enhances Antitumor Immunity and Augments the Efficacy of Immune Checkpoint Blockers by Unleashing Inflammasome Activation. Cancer Cell 2019, 35, 767–781.e6. [Google Scholar] [CrossRef]
- Zhang, J.; Fu, S.; Sun, S.; Li, Z.; Guo, B. Inflammasome Activation Has an Important Role in the Development of Spontaneous Colitis. Mucosal Immunol. 2014, 7, 1139–1150. [Google Scholar] [CrossRef]
- Guan, X.; Liu, R.; Wang, B.; Xiong, R.; Cui, L.; Liao, Y.; Ruan, Y.; Fang, L.; Lu, X.; Yu, X.; et al. Inhibition of HDAC2 Sensitises Antitumour Therapy by Promoting NLRP3/GSDMD-mediated Pyroptosis in Colorectal Cancer. Clin. Transl. Med. 2024, 14, e1692. [Google Scholar] [CrossRef]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of GSDME by Caspase-3 Determines Lobaplatin-Induced Pyroptosis in Colon Cancer Cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef]
- Xie, W.; Peng, M.; Liu, Y.; Zhang, B.; Yi, L.; Long, Y. Simvastatin Induces Pyroptosis via ROS/Caspase-1/GSDMD Pathway in Colon Cancer. Cell Commun. Signal. 2023, 21, 329. [Google Scholar] [CrossRef]
- Tang, Z.; Ji, L.; Han, M.; Xie, J.; Zhong, F.; Zhang, X.; Su, Q.; Yang, Z.; Liu, Z.; Gao, H.; et al. Pyroptosis Is Involved in the Inhibitory Effect of FL118 on Growth and Metastasis in Colorectal Cancer. Life Sci. 2020, 257, 118065. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, F.; Wang, L.; Lou, Y. A438079 Affects Colorectal Cancer Cell Proliferation, Migration, Apoptosis, and Pyroptosis by Inhibiting the P2X7 Receptor. Biochem. Biophys. Res. Commun. 2021, 558, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Hashemzehi, M.; Yavari, N.; Rahmani, F.; Asgharzadeh, F.; Soleimani, A.; Shakour, N.; Avan, A.; Hadizadeh, F.; Fakhraie, M.; Marjaneh, R.M.; et al. Inhibition of Transforming Growth Factor-Beta by Tranilast Reduces Tumor Growth and Ameliorates Fibrosis in Colorectal Cancer. EXCLI J. 2021, 20, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Chen, W.; Fan, H.; Chen, X.; Zhang, J.; Zhu, J.-S. Dihydroartemisinin Prevents Dextran Sodium Sulphate-Induced Colitis through Inhibition of the Activation of NLRP3 Inflammasome and P38 MAPK Signaling. Int. Immunopharmacol. 2020, 88, 106949. [Google Scholar] [CrossRef]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 Is an Essential Mediator of NLRP3 Activation Downstream of Potassium Efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef]
- Magupalli, V.G.; Negro, R.; Tian, Y.; Hauenstein, A.V.; Di Caprio, G.; Skillern, W.; Deng, Q.; Orning, P.; Alam, H.B.; Maliga, Z.; et al. HDAC6 Mediates an Aggresome-like Mechanism for NLRP3 and Pyrin Inflammasome Activation. Science 2020, 369, eaas8995. [Google Scholar] [CrossRef]
- Fang, Q.; Xu, Y.; Tan, X.; Wu, X.; Li, S.; Yuan, J.; Chen, X.; Huang, Q.; Fu, K.; Xiao, S. The Role and Therapeutic Potential of Pyroptosis in Colorectal Cancer: A Review. Biomolecules 2024, 14, 874. [Google Scholar] [CrossRef]
- Duarte, J.A.; de Barros, A.L.B.; Leite, E.A. The Potential Use of Simvastatin for Cancer Treatment: A Review. Biomed. Pharmacother. 2021, 141, 111858. [Google Scholar] [CrossRef]
- Li, M.; Wang, L.; Wei, Y.; Wang, W.; Liu, Z.; Zuo, A.; Liu, W.; Tian, J.; Wang, H. Anti-Colorectal Cancer Effects of a Novel Camptothecin Derivative PCC0208037 In Vitro and In Vivo. Pharmaceuticals 2022, 16, 53. [Google Scholar] [CrossRef]
- Cao, H.; Liu, J.; Shen, P.; Cai, J.; Han, Y.; Zhu, K.; Fu, Y.; Zhang, N.; Zhang, Z.; Cao, Y. Protective Effect of Naringin on DSS-Induced Ulcerative Colitis in Mice. J. Agric. Food Chem. 2018, 66, 13133–13140. [Google Scholar] [CrossRef]
- Chen, L.; You, Q.; Hu, L.; Gao, J.; Meng, Q.; Liu, W.; Wu, X.; Xu, Q. The Antioxidant Procyanidin Reduces Reactive Oxygen Species Signaling in Macrophages and Ameliorates Experimental Colitis in Mice. Front. Immunol. 2018, 8, 1910. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, P.; An, Y.; Ren, J.; Yan, D.; Cui, J.; Li, D.; Li, M.; Wang, M.; Zhong, G. Phloretin Ameliorates Dextran Sulfate Sodium-Induced Ulcerative Colitis in Mice by Regulating the Gut Microbiota. Pharmacol. Res. 2019, 150, 104489. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wu, K.; Zhu, Q.; Xiao, W.; Shan, Q.; Yan, Z.; Wu, J.; Deng, B.; Xue, Y.; Gong, W.; et al. Formononetin Administration Ameliorates Dextran Sulfate Sodium-Induced Acute Colitis by Inhibiting NLRP3 Inflammasome Signaling Pathway. Mediators Inflamm. 2018, 2018, 3048532. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Le, T.H.; Du, Q.; Zhao, Z.; Liu, Y.; Zou, J.; Hua, W.; Liu, C.; Zhu, Y. Genistein Protects against DSS-Induced Colitis by Inhibiting NLRP3 Inflammasome via TGR5-CAMP Signaling. Int. Immunopharmacol. 2019, 71, 144–154. [Google Scholar] [CrossRef]
- Shaker, M.E.; Ashamallah, S.A.; Houssen, M.E. Celastrol Ameliorates Murine Colitis via Modulating Oxidative Stress, Inflammatory Cytokines and Intestinal Homeostasis. Chem. Biol. Interact. 2014, 210, 26–33. [Google Scholar] [CrossRef]
- Shen, P.; Zhang, Z.; Zhu, K.; Cao, H.; Liu, J.; Lu, X.; Li, Y.; Jing, Y.; Yuan, X.; Fu, Y.; et al. Evodiamine Prevents Dextran Sulfate Sodium-Induced Murine Experimental Colitis via the Regulation of NF-ΚB and NLRP3 Inflammasome. Biomed. Pharmacother. 2019, 110, 786–795. [Google Scholar] [CrossRef]
- Lopes de Oliveira, G.A.; Alarcón de la Lastra, C.; Rosillo, M.Á.; Castejon Martinez, M.L.; Sánchez-Hidalgo, M.; Rolim Medeiros, J.V.; Villegas, I. Preventive Effect of Bergenin against the Development of TNBS-Induced Acute Colitis in Rats Is Associated with Inflammatory Mediators Inhibition and NLRP3/ASC Inflammasome Signaling Pathways. Chem. Biol. Interact. 2019, 297, 25–33. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, T.; Yang, C.; Bao, T.; Yang, X.; He, F.; Zhang, Y.; Zhu, L.; Chen, H.; Rong, S.; et al. Secoisolariciresinol Diglucoside Suppresses Dextran Sulfate Sodium Salt-Induced Colitis through Inhibiting NLRP1 Inflammasome. Int. Immunopharmacol. 2020, 78, 105931. [Google Scholar] [CrossRef]
- Wang, K.; Lv, Q.; Miao, Y.; Qiao, S.; Dai, Y.; Wei, Z. Cardamonin, a Natural Flavone, Alleviates Inflammatory Bowel Disease by the Inhibition of NLRP3 Inflammasome Activation via an AhR/Nrf2/NQO1 Pathway. Biochem. Pharmacol. 2018, 155, 494–509. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, W.; Zhang, X.; Lu, P.; Du, Q.; Tao, L.; Ding, Y.; Wang, Y.; Hu, R. Dimethyl Fumarate Ameliorates Dextran Sulfate Sodium-Induced Murine Experimental Colitis by Activating Nrf2 and Suppressing NLRP3 Inflammasome Activation. Biochem. Pharmacol. 2016, 112, 37–49. [Google Scholar] [CrossRef]
- Xiong, Y.; Wei, H.; Chen, C.; Jiao, L.; Zhang, J.; Tan, Y.; Zeng, L. Coptisine Attenuates Post-infectious IBS via Nrf2-dependent Inhibition of the NLPR3 Inflammasome. Mol. Med. Rep. 2022, 26, 362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.-C.; Li, Z.; Xu, W.; Xiang, C.-H.; Ma, Y.-F. Luteolin Alleviates NLRP3 Inflammasome Activation and Directs Macrophage Polarization in Lipopolysaccharide-Stimulated RAW264.7 Cells. Am. J. Transl. Res. 2018, 10, 265–273. [Google Scholar] [PubMed]
- Qin, Y.; Yu, Y.; Yang, C.; Wang, Z.; Yang, Y.; Wang, C.; Zheng, Q.; Li, D.; Xu, W. Atractylenolide I Inhibits NLRP3 Inflammasome Activation in Colitis-Associated Colorectal Cancer via Suppressing Drp1-Mediated Mitochondrial Fission. Front. Pharmacol. 2021, 12, 674340. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, L.; Gao, J.; Hu, J.; Li, A.; Zhu, Y.; Zhang, C.; Qiu, B.; Shen, Z. Parthenolide Targets NLRP3 to Treat Inflammasome-Related Diseases. Int. Immunopharmacol. 2023, 119, 110229. [Google Scholar] [CrossRef]
- Gong, Z.; Zhao, S.; Zhou, J.; Yan, J.; Wang, L.; Du, X.; Li, H.; Chen, Y.; Cai, W.; Wu, J. Curcumin Alleviates DSS-Induced Colitis via Inhibiting NLRP3 Inflammsome Activation and IL-1β Production. Mol. Immunol. 2018, 104, 11–19. [Google Scholar] [CrossRef]
- Cao, R.; Ma, Y.; Li, S.; Shen, D.; Yang, S.; Wang, X.; Cao, Y.; Wang, Z.; Wei, Y.; Li, S.; et al. 1,25(OH)2D3 Alleviates DSS-Induced Ulcerative Colitis via Inhibiting NLRP3 Inflammasome Activation. J. Leukoc. Biol. 2020, 108, 283–295. [Google Scholar] [CrossRef]
- Liu, C.; Wang, J.; Yang, Y.; Liu, X.; Zhu, Y.; Zou, J.; Peng, S.; Le, T.H.; Chen, Y.; Zhao, S.; et al. Ginsenoside Rd Ameliorates Colitis by Inducing P62-Driven Mitophagy-Mediated NLRP3 Inflammasome Inactivation in Mice. Biochem. Pharmacol. 2018, 155, 366–379. [Google Scholar] [CrossRef]
- Tian, M.; Ma, P.; Zhang, Y.; Mi, Y.; Fan, D. Ginsenoside Rk3 Alleviated DSS-Induced Ulcerative Colitis by Protecting Colon Barrier and Inhibiting NLRP3 Inflammasome Pathway. Int. Immunopharmacol. 2020, 85, 106645. [Google Scholar] [CrossRef]
- Liu, J.; Cai, J.; Fan, P.; Zhang, N.; Cao, Y. The Abilities of Salidroside on Ameliorating Inflammation, Skewing the Imbalanced Nucleotide Oligomerization Domain-Like Receptor Family Pyrin Domain Containing 3/Autophagy, and Maintaining Intestinal Barrier Are Profitable in Colitis. Front. Pharmacol. 2019, 10, 1385. [Google Scholar] [CrossRef]
- Mai, C.-T.; Wu, M.-M.; Wang, C.-L.; Su, Z.-R.; Cheng, Y.-Y.; Zhang, X.-J. Palmatine Attenuated Dextran Sulfate Sodium (DSS)-Induced Colitis via Promoting Mitophagy-Mediated NLRP3 Inflammasome Inactivation. Mol. Immunol. 2019, 105, 76–85. [Google Scholar] [CrossRef]
- Qu, S.; Shen, Y.; Wang, M.; Wang, X.; Yang, Y. Suppression of miR-21 and miR-155 of Macrophage by Cinnamaldehyde Ameliorates Ulcerative Colitis. Int. Immunopharmacol. 2019, 67, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-L.; Wong, W.-T.; Weng, Y.-M.; Ho, C.-L.; Hsu, H.-T.; Hua, K.-F.; Wu, C.-H.; Li, L.-H. Cinnamaldehyde, a Bioactive Compound from the Leaves of Cinnamomum osmophloeum Kaneh, Ameliorates Dextran Sulfate Sodium-Induced Colitis in Mice by Inhibiting the NLRP3 Inflammasome. J. Physiol. Investig. 2024, 67, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Ke, W.; Wang, Y.; Huang, S.; Liu, S.; Zhu, H.; Xie, X.; Yang, H.; Lu, Q.; Gan, J.; He, G.; et al. Paeoniflorin Alleviates Inflammatory Response in IBS-D Mouse Model via Downregulation of the NLRP3 Inflammasome Pathway with Involvement of miR-29a. Heliyon 2022, 8, e12312. [Google Scholar] [CrossRef]
- Ke, W.; Wu, J.; Li, H.; Huang, S.; Li, H.; Wang, Y.; Wu, Y.; Yuan, J.; Zhang, S.; Tang, H.; et al. Network Pharmacology and Experimental Validation to Explore the Mechanism of Changji’an Formula against Irritable Bowel Syndrome with Predominant Diarrhea. Heliyon 2024, 10, e33102. [Google Scholar] [CrossRef]
- Dai, G.; Jiang, Z.; Sun, B.; Liu, C.; Meng, Q.; Ding, K.; Jing, W.; Ju, W. Caffeic Acid Phenethyl Ester Prevents Colitis-Associated Cancer by Inhibiting NLRP3 Inflammasome. Front. Oncol. 2020, 10, 721. [Google Scholar] [CrossRef]
- Guo, W.; Sun, Y.; Liu, W.; Wu, X.; Guo, L.; Cai, P.; Wu, X.; Wu, X.; Shen, Y.; Shu, Y.; et al. Small Molecule-Driven Mitophagy-Mediated NLRP3 Inflammasome Inhibition Is Responsible for the Prevention of Colitis-Associated Cancer. Autophagy 2014, 10, 972–985. [Google Scholar] [CrossRef]
- Xu, L.; Cai, P.; Li, X.; Wu, X.; Gao, J.; Liu, W.; Yang, J.; Xu, Q.; Guo, W.; Gu, Y. Inhibition of NLRP3 Inflammasome Activation in Myeloid-Derived Suppressor Cells by Andrographolide Sulfonate Contributes to 5-FU Sensitization in Mice. Toxicol. Appl. Pharmacol. 2021, 428, 115672. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, Q.; Zhao, K.; Zhou, Y.; Li, W.; Pan, C.; Qiang, L.; Li, Z.; Lu, N. Small Molecule GL-V9 Protects against Colitis-Associated Colorectal Cancer by Limiting NLRP3 Inflammasome through Autophagy. Oncoimmunology 2018, 7, e1375640. [Google Scholar] [CrossRef]
- Chen, Y.; Ma, S.; Pi, D.; Wu, Y.; Zuo, Q.; Li, C.; Ouyang, M. Luteolin Induces Pyroptosis in HT-29 Cells by Activating the Caspase1/Gasdermin D Signalling Pathway. Front. Pharmacol. 2022, 13, 952587. [Google Scholar] [CrossRef]
- Dal, Z.; Aru, B. The Role of Curcumin on Apoptosis and NLRP3 Inflammasome-Dependent Pyroptosis on Colorectal Cancer in Vitro. Turk. J. Med. Sci. 2023, 53, 883–893. [Google Scholar] [CrossRef]
- Greaney, A.J.; Maier, N.K.; Leppla, S.H.; Moayeri, M. Sulforaphane Inhibits Multiple Inflammasomes through an Nrf2-Independent Mechanism. J. Leukoc. Biol. 2016, 99, 189–199. [Google Scholar] [CrossRef]
- Zhou, Z.; Dong, J.; Qiu, Y.; Zhang, G.; Wei, K.; He, L.; Sun, Y.; Jiang, H.; Zhang, S.; Guo, X.; et al. Sulforaphane Decreases Oxidative Stress and Inhibits NLRP3 Inflammasome Activation in a Mouse Model of Ulcerative Colitis. Biomed. Pharmacother. 2024, 175, 116706. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, Q.; Zhu, Q.; Tan, R.; Bai, D.; Bu, X.; Lin, B.; Zhao, K.; Pan, C.; Chen, H.; et al. Flavonoid VI-16 Protects against DSS-Induced Colitis by Inhibiting Txnip-Dependent NLRP3 Inflammasome Activation in Macrophages via Reducing Oxidative Stress. Mucosal Immunol. 2019, 12, 1150–1163. [Google Scholar] [CrossRef]
- Cascão, R.; Fonseca, J.E.; Moita, L.F. Celastrol: A Spectrum of Treatment Opportunities in Chronic Diseases. Front. Med. 2017, 4, 69. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.; Lv, C.; Tao, Y.; Miao, Y.; Zhu, Y.; Zhang, W.; Sun, D.; Yun, X.; Xia, Y.; Wei, Z.; et al. Arctigenin Disrupts NLRP3 Inflammasome Assembly in Colonic Macrophages via Downregulating Fatty Acid Oxidation to Prevent Colitis-Associated Cancer. Cancer Lett. 2020, 491, 162–179. [Google Scholar] [CrossRef]
- Deng, Z.; Rong, Y.; Teng, Y.; Mu, J.; Zhuang, X.; Tseng, M.; Samykutty, A.; Zhang, L.; Yan, J.; Miller, D.; et al. Broccoli-Derived Nanoparticle Inhibits Mouse Colitis by Activating Dendritic Cell AMP-Activated Protein Kinase. Mol. Ther. 2017, 25, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Rieger, A.; Schildberg, F.A.; Knolle, P.A.; Schmid-Burgk, J.L.; Hornung, V. NLRP3 Inflammasome Activity Is Negatively Controlled by miR-223. J. Immunol. 2012, 189, 4175–4181. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Lane, T.; Venugopal, R.; Parthasarathy, P.T.; Cho, Y.; Galam, L.; Lockey, R.; Kolliputi, N. MicroRNA-133a-1 Regulates Inflammasome Activation through Uncoupling Protein-2. Biochem. Biophys. Res. Commun. 2013, 439, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zhang, D.; Yang, L.; Wu, Q.; Yuan, L. MicroRNA-200c-5p Targets NIMA Related Kinase 7 (NEK7) to Inhibit NOD-like Receptor 3 (NLRP3) Inflammasome Activation, MODE-K Cell Pyroptosis, and Inflammatory Bowel Disease in Mice. Mol. Immunol. 2022, 146, 57–68. [Google Scholar] [CrossRef]
- Cong, J.; Gong, J.; Yang, C.; Xia, Z.; Zhang, H. miR-22 Suppresses Tumor Invasion and Metastasis in Colorectal Cancer by Targeting NLRP3. Cancer Manag. Res. 2020, 12, 5419–5429. [Google Scholar] [CrossRef] [PubMed]
- Neudecker, V.; Haneklaus, M.; Jensen, O.; Khailova, L.; Masterson, J.C.; Tye, H.; Biette, K.; Jedlicka, P.; Brodsky, K.S.; Gerich, M.E.; et al. Myeloid-Derived miR-223 Regulates Intestinal Inflammation via Repression of the NLRP3 Inflammasome. J. Exp. Med. 2017, 214, 1737–1752. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Pan, S.; Luo, W.; Shen, Z.; Meng, X.; Xiao, M.; Tan, B.; Nie, K.; Tong, T.; Wang, X. Roseburiaintestinalis-derived Flagellin Ameliorates Colitis by Targeting miR-223-3p-mediated Activation of NLRP3 Inflammasome and Pyroptosis. Mol. Med. Rep. 2020, 22, 2695–2704. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhang, Z.; Yuan, J.; Ocansey, D.K.W.; Tu, Q.; Zhang, X.; Qian, H.; Xu, W.; Qiu, W.; Mao, F. hucMSC-Derived Exosomes Attenuate Colitis by Regulating Macrophage Pyroptosis via the miR-378a-5p/NLRP3 Axis. Stem Cell Res. Ther. 2021, 12, 416. [Google Scholar] [CrossRef]
- Polytarchou, C.; Oikonomopoulos, A.; Mahurkar, S.; Touroutoglou, A.; Koukos, G.; Hommes, D.W.; Iliopoulos, D. Assessment of Circulating MicroRNAs for the Diagnosis and Disease Activity Evaluation in Patients with Ulcerative Colitis by Using the Nanostring Technology. Inflamm. Bowel Dis. 2015, 21, 2533–2539. [Google Scholar] [CrossRef]
- Onisor, D.; Brusnic, O.; Banescu, C.; Carstea, C.; Sasaran, M.; Stoian, M.; Avram, C.; Boicean, A.; Boeriu, A.; Dobru, D. miR-155 and miR-21 as Diagnostic and Therapeutic Biomarkers for Ulcerative Colitis: There Is Still a Long Way to Go. Biomedicines 2024, 12, 1315. [Google Scholar] [CrossRef]
- Quaglio, A.E.V.; Santaella, F.J.; Rodrigues, M.A.M.; Sassaki, L.Y.; Di Stasi, L.C. MicroRNAs Expression Influence in Ulcerative Colitis and Crohn’s Disease: A Pilot Study for the Identification of Diagnostic Biomarkers. World J. Gastroenterol. 2021, 27, 7801–7812. [Google Scholar] [CrossRef]
- Xia, S.-S.; Zhang, G.-J.; Liu, Z.-L.; Tian, H.-P.; He, Y.; Meng, C.-Y.; Li, L.-F.; Wang, Z.-W.; Zhou, T. MicroRNA-22 Suppresses the Growth, Migration and Invasion of Colorectal Cancer Cells through a Sp1 Negative Feedback Loop. Oncotarget 2017, 8, 36266–36278. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Inhibitor | Target | Mechanism of Action | Experimental Models | References |
|---|---|---|---|---|---|
| Inflammasome Sensors | MCC950 | NLRP3 | Blocks canonical and non-canonical inflammasome activation binding NACHT domain and reduces IL-1β secretion | Spontaneous colitis mice | [101,102,103,104] |
| DSS-induced colitis using Nod2 knockout mice | |||||
| DSS-induced colitis mice | |||||
| OLT117 | NLRP3 | Inhibits canonical and non-canonical inflammasome activation binding NACHT domain | DSS-induced colitis mice | [105] | |
| Tranilast | NLRP3 | Influences the activation of inflammasome by binding NACHT domain Inhibits visceral hypersensitivity and colon permeability | DSS-induced colitis mice | [106,107,108,109] | |
| DSS-induced colitis rats | |||||
| Mouse BMDMs and THP1 | |||||
| IBS rat models | |||||
| G514-0208 | NLRP3 | Selectively inhibitor via NACHT domain binding | DSS-induced colitis mice | [110] | |
| NF-κB Pathway | MCC950 | NF-κB | In combination with metformin, inhibits the priming and activation of inflammasomes via TLR4/NF-κB and induces autophagy | DSS-induced colitis rats | [111] |
| Dapagliflozin | NLRP3 | Inhibits inflammasome activation via the NF-κB/AMPK/NLRP3 axis | Acetic acid-induced colitis in rats | [112] | |
| Canagliflozin | NLRP3 | Inhibits inflammasome activation via the NF-κB/AMPK/NLRP3 axis | Acetic acid-induced colitis in rats. | [113] | |
| Oridonin | NF-κB | Inhibits inflammasome activation via NF-κB and reduced cytokines expression | TNBS-induced colitis mice | [114] | |
| Mirtazapine | NLRP3 | Blocks inflammasome activation, modulating NF-κB activation and the NLRP3/caspase-1 signaling pathway | Acetic acid-induced colitis in rats | [115] | |
| BAY 11-7082 | NLRP3 | Inhibits inflammasome activation by suppressing both the NF-κB and NLRP3 pathways | Mouse BMDMs | [66,116] | |
| IBS rat models | |||||
| Artemisinin | NLRP3 | Blocks NLRP3 inflammasome assembly by inhibiting ROS production and NF-κB and MAPK signaling | Mouse BMDMs | [117] | |
| DSS-induced colitis mice | |||||
| Inflammasome Activation | Glyburide | K+ efflux | Prevents inflammasome activation through the inhibition of K+ efflux | Mouse BMDMs | [77,118] |
| IL10−/− mouse model | |||||
| BHB | K+ efflux | Prevents inflammasome activation through the inhibition of K+ efflux and ASC oligomerization Reduces cytokine inflammasome-related release and mitigates pyroptosis | Mouse BMDMs | [119,120] | |
| DSS-induced colitis rats | |||||
| (R,R)-BD-AcAc2 | NLRP3 | Prevents inflammasome activation and pyroptosis | DSS-induced colitis rats | [121] | |
| RRx-001 | NEK7 | Covalent binding to the cysteine residue, preventing NLRP3-NEK7 interaction; attenuates experimental colitis | Mouse BMDMs; | [122] | |
| DSS-induced colitis mice | |||||
| WT161 | NLRP3 | Inhibits NLRP3 activation by blocking HDAC6 | DSS-induced colitis mice | [123] | |
| Mouse BMDMs | |||||
| C646 | NLRP3 | Inhibits NLRP3 activation by blocking histone acetyltransferase p300 | DSS-induced colitis mice | [124] | |
| Mouse BMDMs | |||||
| Metformin | NLRP3 | Suppresses inflammatory processes and regulates the inflammatory microenvironment | LS1034 | [125] | |
| BayK8644 | TMEM176B | Triggers NLRP3 inflammasomes activation and limits the tumor growth | CRC mouse model | [126] | |
| THP-1 | |||||
| Mouse BMDMs | |||||
| Caspase-1 Activation and Pyroptosis | Artemisinin | NLRC4 AIM2 | Inhibits Il-1β secretion | Mouse BMDMs | [117] |
| DSS-induced colitis mice | |||||
| Pralnacasan | Caspase-1 | Inhibits caspase-1 activation | DSS-induced colitis mice | [30] | |
| Mouse BMDMs | |||||
| THP-1 | |||||
| Ac-YVAD-cmk | Caspase-1 | Inhibits caspase-1 activation | DSS-induced colitis mice | [127] | |
| Mouse BMDMs | |||||
| Santacrumazate A | NLRP3 | Inhibits HDAC2 expression and, in combination with antitumor agents, activates NLRP3/GSDMD-mediated pyroptosis | SW620 | [128] | |
| LS174T | |||||
| Lobaplatin | GSDME | Induces pyroptosis via GSDME activation | HT29 | [129] | |
| HCT116 | |||||
| Simvastatin | NLRP3 | Induces intracellular ROS production and triggers the NLRP3/caspase-1 pathway and pyroptosis | HCT116 | [130] | |
| SW620 | |||||
| FL118 | NLRP3 | Induces NLRP3-ASC-caspase-1 mediated pyroptosis | SW480 | [131] | |
| HT29 | |||||
| A438079 | P2X7 | Induces Bcl2/caspase-9/caspase-3 apoptosis and NLRP3/caspase-1 pyroptosis | HCT116 | [132] | |
| SW620 |
| Phase | Natural Compounds | Target | Mechanism of Action | Experimental Models | References |
|---|---|---|---|---|---|
| NF-κB Pathway | Naringin | NF-κB | Suppresses the activation of NF-κB, preventing the NLRP3 inflammasome signaling pathway | DSS-induced colitis mice | [140] |
| Procyanidin | NF-κB | Suppresses the activation of NF-κB, preventing NLRP3 inflammasome activation | DSS-induced colitis mice | [141] | |
| THP-1 | |||||
| Phloretin | NF-κB | Suppresses the activation of NF-κB, preventing NLRP3 inflammasome activation | DSS-induced colitis mice | [142] | |
| Formononetin | NLRP3 | Inhibits the NLRP3 signaling pathway | DSS-induced colitis mice | [143] | |
| Genistein | NLRP3 | Inhibits the NLRP3 signaling pathway | DSS-induced colitis mice | [144] | |
| THP-1 | |||||
| Celastrol | NF-κB | Inhibits NLRP3 inflammasome activation and contrasts oxidative stress | DSS-induced colitis mice | [145] | |
| Evodiamine | NF-κB | Modulates NLRP3 inflammasome activation | DSS-induced colitis mice | [146] | |
| Bergenin | NF-κB | Blocks the canonical and non-canonical NLRP3 inflammasome pathways | DSS-induced colitis rats | [147] | |
| Secoisolariciresinol diglucoside | NF-κB | Inhibits NLRP1 inflammasome activation | DSS-induced colitis mice Raw264.7 | [148] | |
| Inflammasome Activation | Cardamonin | Nrf2 | Inhibits NLRP3 inflammasome activation by inducing activation of the AhR/Nrf2/NQO1 pathway | DSS-induced colitis mice | [149] |
| TNBS-induced colitis mice | |||||
| BMDMs | |||||
| THP-1 | |||||
| Dimethyl fumarate | Nrf2 | Reduces NLRP3 inflammasome activation by inducing Nrf2 pathway activation | DSS-induced colitis mice | [150] | |
| THP-1 | |||||
| Coptisine | Nrf2 | Modulates the expression levels of NLRP3 inflammasome components, increasing the expression of Nrf2 | PI-IBS rats | [151] | |
| Luteolin | ROS | Inhibits NLRP3 inflammasomes activation and promotes macrophage polarization | Raw264.7 | [152] | |
| Atractylenolide | DRP1 | Inhibits NLRP3 inflammasome activation via mitochondrial damage suppression | AOM/DSS mice | [153] | |
| Parthenolide | NEK7 K+ efflux | Inhibits NLRP3 inflammasome assembly, affecting NLRP3/NEK7 interaction Inhibits NLRP3 inflammasome activation by suppressing K+ efflux | Murine BMDMs | [116,154] | |
| THP-1 | |||||
| Curcumin | K+ efflux | Inhibits activation of the NLRP3 inflammasome, reducing ROS and Cathepsin B release | DSS-induced colitis mice | [155] | |
| Vitamine D3 | NEK7 | Impairs NLRP3/NEK7 interaction, suppressing NLRP3 inflammasome assembly and activation | DSS-induced colitis mice | [156] | |
| Ginsenosides | NLRP3 | Reduces NLRP3 inflammasome activation | DSS-induced colitis mice | [157,158] | |
| Salidroside | NLRP3 | Modulates NLRP3 inflammasome activation and downstream pro-inflammatory mediators | DSS-induced colitis mice | [159] | |
| Palmatine | NLRP3 | Modulates NLRP3 inflammasome activation and increases mitophagy | DSS-induced colitis mice | [160] | |
| Cinnamadehyde | NLRP3 | Inhibits activation of the NLRP3 inflammasome and restores the NLRP3-related inflammatory mediators | DSS-induced colitis mice | [161,162] | |
| Paeoniflorin | miR-29a | Modulates NLRP3 inflammasome activation | IBS mice | [163] | |
| CJAF | NLRP3 | Modulates NLRP3 inflammasome activation and downstream pro-inflammatory mediators | IBS mice | [164] | |
| Eugeniin | NLRC4 | Reduces NAIP/NLRC4 inflammasome activation by bacterial pathogenic components | DSS-induced colitis mice | [38] | |
| Raw264.7 | |||||
| THP-1 | |||||
| Arctigenin | NLRP3 | Inhibits NLRP3 inflammasome assembly and activation | AOM/DSS mice | [165] | |
| Andrographolide | NLRP3 | Inhibits NLRP3 inflammasome activation In combination with antitumor agents, enhances the effects of cancer therapies | DSS-induced colitis mice | [166,167] | |
| AOM/DSS mice | |||||
| Murine BMDMs | |||||
| GL-V9 | NLRP3 | Triggers autophagy, inducing NLRP3 inflammasome degradation | DSS-induced colitis mice | [168] | |
| AOM/DSS mice | |||||
| THP-1 | |||||
| Murine BMDMs | |||||
| Caspase-1 Activation and Pyroptosis | Luteolin | Caspase-1 GSDMD IL-1β | Inhibits cell proliferation and induces pyroptosis | HT29 | [169] |
| Curcumin | NLRP3 | Promotes NLRP3-mediated pyroptosis | SW480 | [170] | |
| HCT116 | |||||
| LoVo | |||||
| HT29 | |||||
| Sulforaphane | Caspase-1 | Inhibits caspase-1 cleavage and IL-1β maturation Reduces ROS levels | Raw264.7 | [171,172] | |
| Murine BMDMs | |||||
| DSS-induced colitis mice | |||||
| VI-16 | IL-1β | Inhibits NLRP3 inflammasome activation and IL-1β release | DSS-induced colitis mice | [173] | |
| THP-1 | |||||
| Post-Transcriptional Modification | CAPE | NLRP3 | Promotes NLRP3 ubiquitination and degradation | AOM/DSS mice | [165] |
| Phase | miRNAs | Target | Mechanism of Action | Experimental Models | References |
|---|---|---|---|---|---|
| Inflammasome Activation | miR-223 | NLRP3 | Suppresses NLRP3 expression and activation | DSS-induced colitis mice | [178] |
| Murine BMDMs | |||||
| THP-1 | |||||
| miR-21 miR-155 | NLRP3 | Cinnamadehyde inhibits activation of the NLRP3 inflammasome by reducing the expression levels of miR-21 and miR-155 | DSS-induced colitis mice | [161] | |
| miR-29a | NLRP3 | Peoniflorin modulates NLRP3 inflammasome signaling by inhibiting miR-29a | IBS mice | [163] | |
| miR-133a-1 | UCP2 | Reduces the expression of NLRP3 inflammasome components and downstream cytokines | THP-1 | [179] | |
| miR-200c-5p | NEK7 | Reduces NLRP3 inflammasome activation and pyroptosis | DSS-induced colitis mice | [180] | |
| miR-22 | NLRP3 | Reduces NLRP3 inflammasome expression, cell proliferation, migration, and invasion | HCT116 | [181] | |
| mouse xenograft model | |||||
| Pyroptosis | miR-223 | NLRP3 | Reduces IL-1β production and inhibits pyroptosis | DSS-induced colitis mice | [182,183] |
| Murine BMDMs | |||||
| THP-1 | |||||
| miR-378a-5p | NLRP3 | Inhibits NLRP3 activation and pyroptosis | DSS-induced colitis mice | [184] | |
| Murine BMDMs | |||||
| THP-1 | |||||
| Post-Transcriptional Modification | miR-369-3p | BRCC3 | Inhibits deubiquitination of NLRP3, modulating inflammasome assembly and activation | Murine BMDMs | [26] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scalavino, V.; Piccinno, E.; Giannelli, G.; Serino, G. Inflammasomes in Intestinal Disease: Mechanisms of Activation and Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 13058. https://doi.org/10.3390/ijms252313058
Scalavino V, Piccinno E, Giannelli G, Serino G. Inflammasomes in Intestinal Disease: Mechanisms of Activation and Therapeutic Strategies. International Journal of Molecular Sciences. 2024; 25(23):13058. https://doi.org/10.3390/ijms252313058
Chicago/Turabian StyleScalavino, Viviana, Emanuele Piccinno, Gianluigi Giannelli, and Grazia Serino. 2024. "Inflammasomes in Intestinal Disease: Mechanisms of Activation and Therapeutic Strategies" International Journal of Molecular Sciences 25, no. 23: 13058. https://doi.org/10.3390/ijms252313058
APA StyleScalavino, V., Piccinno, E., Giannelli, G., & Serino, G. (2024). Inflammasomes in Intestinal Disease: Mechanisms of Activation and Therapeutic Strategies. International Journal of Molecular Sciences, 25(23), 13058. https://doi.org/10.3390/ijms252313058