The Translatome Map: RNC-Seq vs. Ribo-Seq for Profiling of HBE, A549, and MCF-7 Cell Lines

Abstract
1. Introduction
2. Results
2.1. Trends in the Application of Translatome Profiling Methods
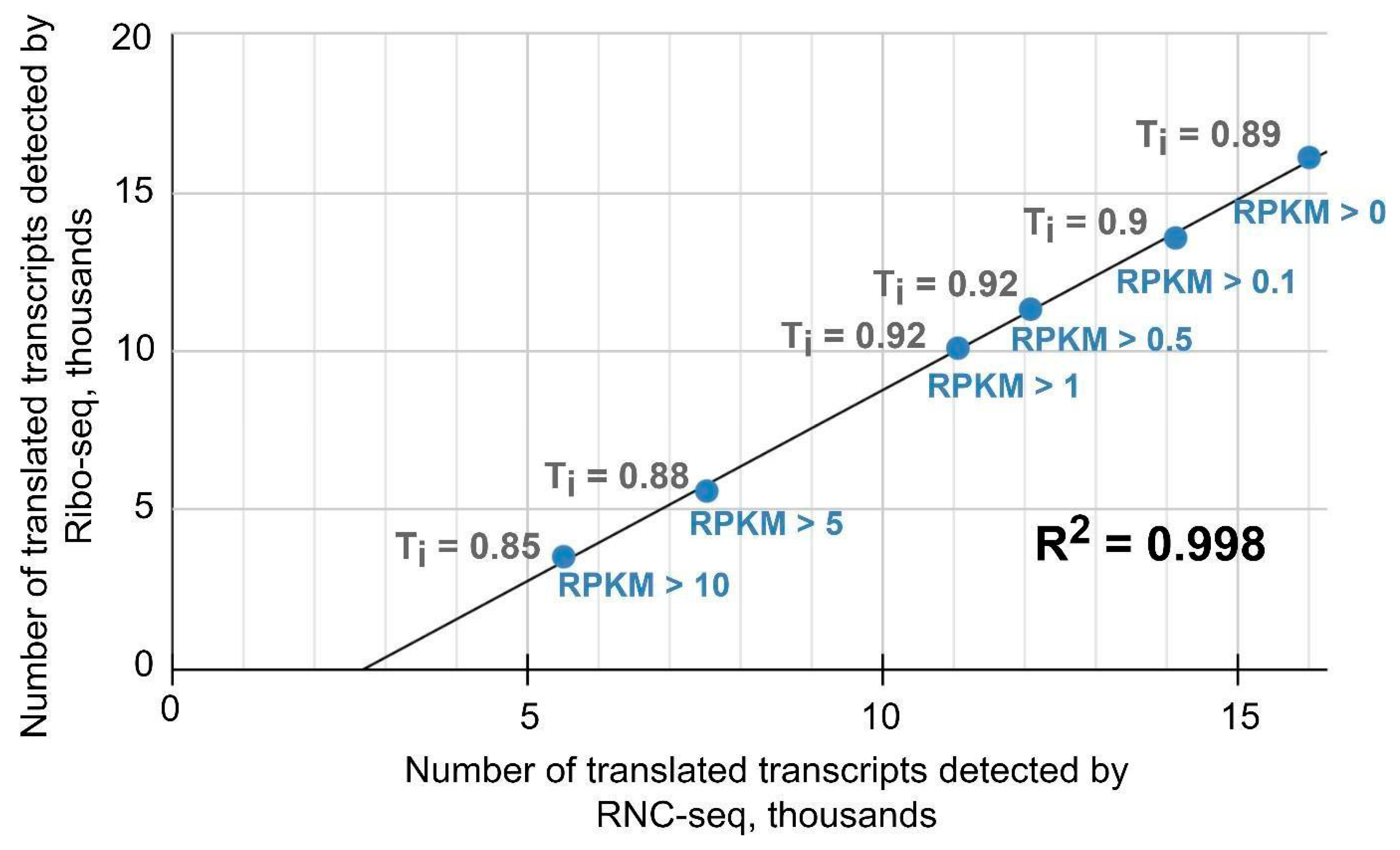
2.2. Ribo-Seq and RNC-Seq Provide Comparable Gene Quantification
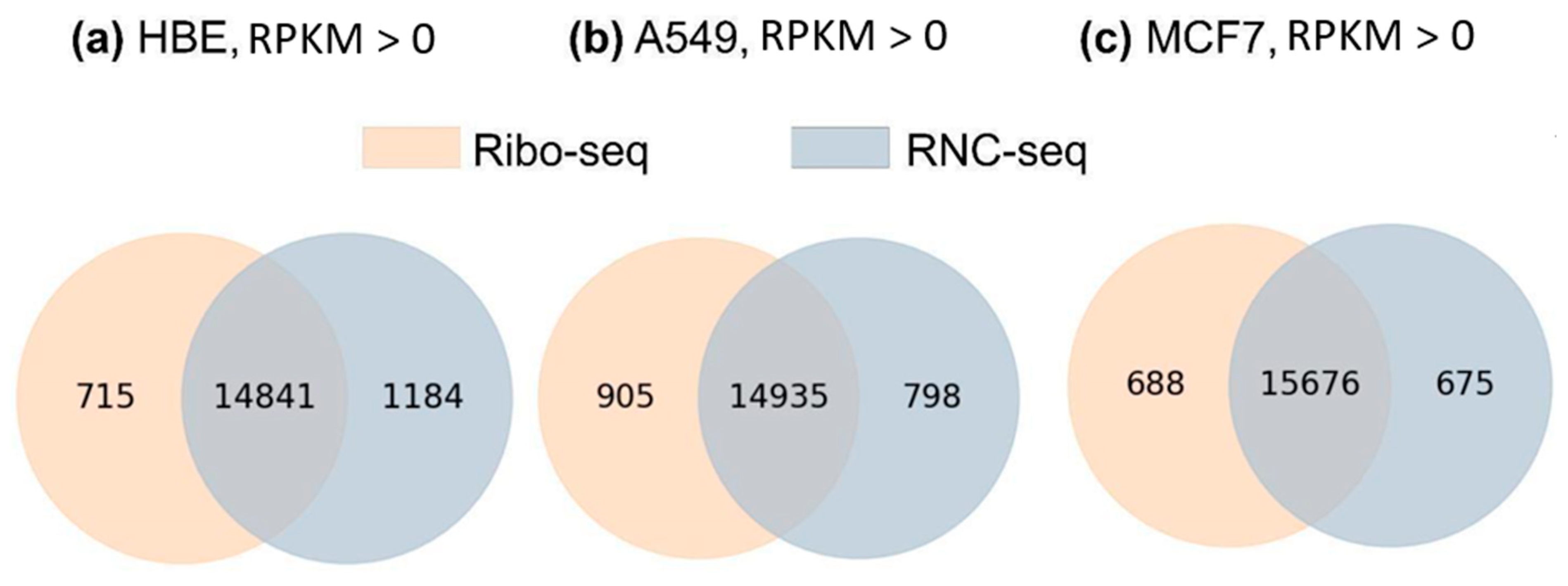
2.3. Translatome Profiling Methods Reveals Highly Comparable Sets of Genes
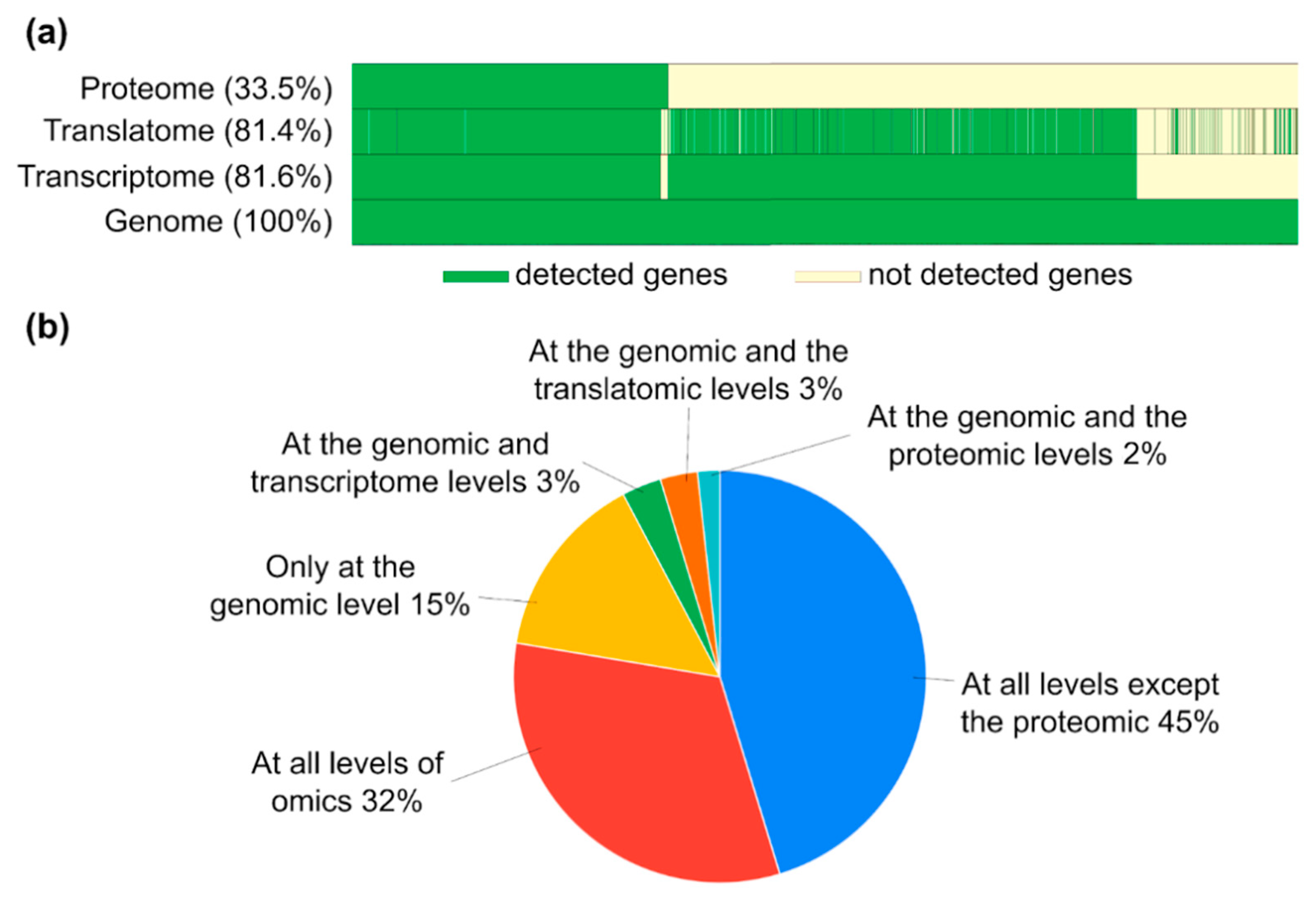
2.4. Gene-Centric Molecular “Portrait” Shows High Concordance between the Transcriptome and the Translatome
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chang, C.; Li, L.; Zhang, C.; Wu, S.; Guo, K.; Zi, J.; Chen, Z.; Jiang, J.; Ma, J.; Yu, Q.; et al. Systematic Analyses of the Transcriptome, Translatome, and Proteome Provide a Global View and Potential Strategy for the C-HPP. J. Proteome Res. 2014, 13, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Gawron, D.; Gevaert, K.; Van Damme, P. The Proteome under Translational Control. Proteomics 2014, 14, 2647–2662. [Google Scholar] [CrossRef] [PubMed]
- Qanmber, G.; You, Q.; Yang, Z.; Fan, L.; Zhang, Z.; Chai, M.; Gao, B.; Li, F.; Yang, Z. Transcriptional and Translational Landscape Fine-Tune Genome Annotation and Explores Translation Control in Cotton. J. Adv. Res. 2023, 58, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, K.; Landstorfer, R.; Fellner, L.; Simon, S.; Schafferhans, A.; Goldberg, T.; Marx, H.; Ozoline, O.N.; Rost, B.; Kuster, B.; et al. Translatomics Combined with Transcriptomics and Proteomics Reveals Novel Functional, Recently Evolved Orphan Genes in Escherichia coli O157:H7 (EHEC). BMC Genom. 2016, 17, 133. [Google Scholar] [CrossRef]
- Rendleman, J.; Cheng, Z.; Maity, S.; Kastelic, N.; Munschauer, M.; Allgoewer, K.; Teo, G.; Zhang, Y.B.M.; Lei, A.; Parker, B.; et al. New Insights into the Cellular Temporal Response to Proteostatic Stress. eLife 2018, 7, e39054. [Google Scholar] [CrossRef]
- Kitchen, R.R.; Rozowsky, J.S.; Gerstein, M.B.; Nairn, A.C. Decoding Neuroproteomics: Integrating the Genome, Translatome and Functional Anatomy. Nat. Neurosci. 2014, 17, 1491–1499. [Google Scholar] [CrossRef]
- Anderson, L.; Seilhamer, J. A comparison of selected mRNA and protein abundances in human liver. Electrophoresis 1997, 18, 533–537. [Google Scholar] [CrossRef]
- Gygi, S.P.; Rochon, Y.; Franza, B.R.; Aebersold, R. Correlation between protein and mRNA abundance in yeast. Mol. Cell Biol. 1999, 19, 1720–1730. [Google Scholar] [CrossRef]
- Ghazalpour, A.; Bennett, B.; Petyuk, V.A.; Orozco, L.; Hagopian, R.; Mungrue, I.N.; Farber, C.R.; Sinsheimer, J.; Kang, H.M.; Furlotte, N.; et al. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 2011, 7, e1001393. [Google Scholar] [CrossRef]
- Yan, X.; Hoek, T.A.; Vale, R.D.; Tanenbaum, M.E. Dynamics of Translation of Single mRNA Molecules In Vivo. Cell 2016, 165, 976–989. [Google Scholar] [CrossRef]
- Wunner, W.H.; Bell, J.; Munro, H.N. The Effect of Feeding with a Tryptophan-Free Amino Acid Mixture on Rat-Liver Polysomes and Ribosomal Ribonucleic Acid. Biochem. J. 1966, 101, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Mašek, T.; Valášek, L.; Pospíšek, M. Polysome Analysis and RNA Purification from Sucrose Gradients. In RNA; Nielsen, H., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; Volume 703, pp. 293–309. ISBN 978-1-58829-913-0. [Google Scholar]
- Spangenberg, L.; Shigunov, P.; Abud, A.P.R.; Cofré, A.R.; Stimamiglio, M.A.; Kuligovski, C.; Zych, J.; Schittini, A.V.; Costa, A.D.T.; Rebelatto, C.K.; et al. Polysome Profiling Shows Extensive Posttranscriptional Regulation during Human Adipocyte Stem Cell Differentiation into Adipocytes. Stem Cell Res. 2013, 11, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cui, Y.; Jin, J.; Guo, J.; Wang, G.; Yin, X.; He, Q.-Y.; Zhang, G. Translating mRNAs Strongly Correlate to Proteins in a Multivariate Manner and Their Translation Ratios Are Phenotype Specific. Nucleic Acids Res. 2013, 41, 4743–4754. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Brar, G.A.; Rouskin, S.; McGeachy, A.M.; Weissman, J.S. The Ribosome Profiling Strategy for Monitoring Translation in Vivo by Deep Sequencing of Ribosome-Protected mRNA Fragments. Nat. Protoc. 2012, 7, 1534–1550. [Google Scholar] [CrossRef]
- McGlincy, N.J.; Ingolia, N.T. Transcriptome-Wide Measurement of Translation by Ribosome Profiling. Methods 2017, 126, 112–129. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Hussmann, J.A.; Weissman, J.S. Ribosome Profiling: Global Views of Translation. Cold Spring Harb. Perspect. Biol. 2019, 11, a032698. [Google Scholar] [CrossRef]
- Shirokikh, N.E.; Archer, S.K.; Beilharz, T.H.; Powell, D.; Preiss, T. Translation Complex Profile Sequencing to Study the in Vivo Dynamics of mRNA–Ribosome Interactions during Translation Initiation, Elongation and Termination. Nat. Protoc. 2017, 12, 697–731. [Google Scholar] [CrossRef]
- Heiman, M.; Kulicke, R.; Fenster, R.J.; Greengard, P.; Heintz, N. Cell Type-Specific mRNA Purification by Translating Ribosome Affinity Purification (TRAP). Nat. Protoc. 2014, 9, 1282–1291. [Google Scholar] [CrossRef]
- Hafner, M.; Katsantoni, M.; Köster, T.; Marks, J.; Mukherjee, J.; Staiger, D.; Ule, J.; Zavolan, M. CLIP and Complementary Methods. Nat. Rev. Methods Primers 2021, 1, 1–23. [Google Scholar] [CrossRef]
- Tarbeeva, S.; Lyamtseva, E.; Lisitsa, A.; Kozlova, A.; Ponomarenko, E.; Ilgisonis, E. ScanBious: Survey for Obesity Genes Using PubMed Abstracts and DisGeNET. J. Pers. Med. 2021, 11, 246. [Google Scholar] [CrossRef]
- Liu, W.; Xiang, L.; Zheng, T.; Jin, J.; Zhang, G. TranslatomeDB: A Comprehensive Database and Cloud-Based Analysis Platform for Translatome Sequencing Data. Nucleic Acids Res. 2018, 46, D206–D212. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.; Amstislavskiy, V.; Risch, T.; Schuette, M.; Dökel, S.; Ralser, M.; Balzereit, D.; Lehrach, H.; Yaspo, M.-L. Influence of RNA Extraction Methods and Library Selection Schemes on RNA-Seq Data. BMC Genom. 2014, 15, 675. [Google Scholar] [CrossRef] [PubMed]
- Ilgisonis, E.V.; Ponomarenko, E.A.; Tarbeeva, S.N.; Lisitsa, A.V.; Zgoda, V.G.; Radko, S.P.; Archakov, A.I. Gene-Centric Coverage of the Human Liver Transcriptome: QPCR, Illumina, and Oxford Nanopore RNA-Seq. Front. Mol. Biosci. 2022, 9, 944639. [Google Scholar] [CrossRef] [PubMed]
- Ilgisonis, E.; Vavilov, N.; Ponomarenko, E.; Lisitsa, A.; Poverennaya, E.; Zgoda, V.; Radko, S.; Archakov, A. Genome of the Single Human Chromosome 18 as a “Gold Standard” for Its Transcriptome. Front. Genet. 2021, 12, 674534. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, Y.; Wang, Y.; Zhang, P.; Chen, G.; Zhou, Y. Insights Into Translatomics in the Nervous System. Front. Genet. 2020, 11, 599548. [Google Scholar] [CrossRef]
- Crick, F. Central Dogma of Molecular Biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Leushkin, E.; Liechti, A.; Ovchinnikova, S.; Mößinger, K.; Brüning, T.; Rummel, C.; Grützner, F.; Cardoso-Moreira, M.; Janich, P.; et al. Transcriptome and Translatome Co-Evolution in Mammals. Nature 2020, 588, 642–647. [Google Scholar] [CrossRef]
- Lane, A.N.; Fan, T.W.-M. Regulation of Mammalian Nucleotide Metabolism and Biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef]
- Riba, A.; Di Nanni, N.; Mittal, N.; Arhné, E.; Schmidt, A.; Zavolan, M. Protein Synthesis Rates and Ribosome Occupancies Reveal Determinants of Translation Elongation Rates. Proc. Natl. Acad. Sci. USA 2019, 116, 15023–15032. [Google Scholar] [CrossRef]
- Sørensen, M.A.; Pedersen, S. Absolute in Vivo Translation Rates of Individual Codons in Escherichia coli. J. Mol. Biol. 1991, 222, 265–280. [Google Scholar] [CrossRef]
- Zhu, M.; Dai, X.; Wang, Y.-P. Real Time Determination of Bacterial in Vivo Ribosome Translation Elongation Speed Based on LacZα Complementation System. Nucleic Acids Res. 2016, 44, e155. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, A.; Choi, J.; Wang, J.; Petrov, A.; Puglisi, J.D. Dynamic Basis of Fidelity and Speed in Translation: Coordinated Multistep Mechanisms of Elongation and Termination. Protein Sci. 2017, 26, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Gerashchenko, M.V.; Peterfi, Z.; Yim, S.H.; Gladyshev, V.N. Translation Elongation Rate Varies among Organs and Decreases with Age. Nucleic Acids Res. 2021, 49, e9. [Google Scholar] [CrossRef] [PubMed]
- Metelev, M.; Lundin, E.; Volkov, I.L.; Gynnå, A.H.; Elf, J.; Johansson, M. Direct Measurements of mRNA Translation Kinetics in Living Cells. Nat. Commun. 2022, 13, 1852. [Google Scholar] [CrossRef] [PubMed]
- Gholami, A.M.; Hahne, H.; Wu, Z.; Auer, F.J.; Meng, C.; Wilhelm, M.; Kuster, B. Global Proteome Analysis of the NCI-60 Cell Line Panel. Cell Rep. 2013, 4, 609–620. [Google Scholar] [CrossRef]
- Sinitcyn, P.; Richards, A.L.; Weatheritt, R.J.; Brademan, D.R.; Marx, H.; Shishkova, E.; Meyer, J.G.; Hebert, A.S.; Westphall, M.S.; Blencowe, B.J.; et al. Global detection of human variants and isoforms by deep proteome sequencing. Nat. Biotechnol. 2023, 41, 1776–1786. [Google Scholar] [CrossRef]
- Lu, J.; Hua, Z.; Kobertz, W.R.; Deutsch, C. Nascent Peptide Side Chains Induce Rearrangements in Distinct Locations of the Ribosomal Tunnel. J. Mol. Biol. 2011, 411, 499–510. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, J.; Lian, X.; Sun, L.; Meng, K.; Chen, Y.; Sun, Z.; Yin, X.; Li, Y.; Zhao, J.; et al. A Hidden Human Proteome Encoded by ‘Non-Coding’ Genes. Nucleic Acids Res. 2019, 47, 8111–8125. [Google Scholar] [CrossRef]
- Clamer, M.; Tebaldi, T.; Lauria, F.; Bernabò, P.; Gómez-Biagi, R.F.; Marchioretto, M.; Kandala, D.T.; Minati, L.; Perenthaler, E.; Gubert, D.; et al. Active Ribosome Profiling with RiboLace. Cell Rep. 2018, 25, 1097–1108.e5. [Google Scholar] [CrossRef]
- Pyatnitskiy, M.A.; Arzumanian, V.A.; Radko, S.P.; Ptitsyn, K.G.; Vakhrushev, I.V.; Poverennaya, E.V.; Ponomarenko, E.A. Oxford Nanopore MinION Direct RNA-Seq for Systems Biology. Biology 2021, 10, 1131. [Google Scholar] [CrossRef]
- Zhao, S.; Ye, Z.; Stanton, R. Misuse of RPKM or TPM Normalization When Comparing across Samples and Sequencing Protocols. RNA 2020, 26, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Luber, C.A.; Cox, J.; Lauterbach, H.; Fancke, B.; Selbach, M.; Tschopp, J.; Akira, S.; Wiegand, M.; Hochrein, H.; O’Keeffe, M.; et al. Quantitative Proteomics Reveals Subset-Specific Viral Recognition in Dendritic Cells. Immunity 2010, 32, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.M.; Chiu, S.F.; Akbarpour, M.; Bharat, A.; Ridge, K.M.; Bartom, E.T.; Winter, D.R. A Beginner’s Guide to Analysis of RNA Sequencing Data. Am. J. Respir. Cell Mol. Biol. 2018, 59, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Dall’Agnol, H.P.; Baraúna, R.A.; De Sá, P.H.; Ramos, R.T.; Nóbrega, F.; Nunes, C.I.; Das Graças, D.A.; Carneiro, A.R.; Santos, D.M.; Pimenta, A.M.; et al. Omics Profiles Used to Evaluate the Gene Expression of Exiguobacterium Antarcticum B7 during Cold Adaptation. BMC Genom. 2014, 15, 986. [Google Scholar] [CrossRef]
- Abdullah, H.M.; Akbari, P.; Paulose, B.; Schnell, D.; Qi, W.; Park, Y.; Pareek, A.; Dhankher, O.P. Transcriptome Profiling of Camelina Sativa to Identify Genes Involved in Triacylglycerol Biosynthesis and Accumulation in the Developing Seeds. Biotechnol. Biofuels 2016, 9, 136. [Google Scholar] [CrossRef]
- Łabaj, P.P.; Kreil, D.P. Sensitivity, Specificity, and Reproducibility of RNA-Seq Differential Expression Calls. Biol. Direct 2016, 11, 66. [Google Scholar] [CrossRef]
- Yang, J.-R.; Chen, X. Dosage Sensitivity of X-Linked Genes in Human Embryonic Single Cells. BMC Genom. 2019, 20, 42. [Google Scholar] [CrossRef]
- Wright, H.L.; Thomas, H.B.; Moots, R.J.; Edwards, S.W. RNA-Seq Reveals Activation of Both Common and Cytokine-Specific Pathways Following Neutrophil Priming. PLoS ONE 2013, 8, e58598. [Google Scholar] [CrossRef]
- Rogers, D.J.; Tanimoto, T.T. A Computer Program for Classifying Plants: The Computer Is Programmed to Simulate the Taxonomic Process of Comparing Each Case with Every Other Case. Science 1960, 132, 1115–1118. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Ribo-Seq 1 | RNC-Seq 2 |
|---|---|---|
| Number of entries | ||
| Pubmed | 1454 | 210 |
| PMC Full-Text | 8265 | 1278 |
| BioProject | 1339 | 27 |
| GEO DataSets | 8168 | 36 |
| DB Translatome | 4054 | 216 |
| Level | Method | Number of PCGs (% of the Total PCGs) | ||
|---|---|---|---|---|
| HBE (Normal Human Bronchial Epithelial Cells) | A549 (Lung Adenocarcinoma Epithelial Cells) | MCF-7 (Hormone- Responsive Breast Cancer Cell Line) | ||
| Transcriptome | RNA-seq | 16,375 (80.2%) | 16,807 (82.3%) | 16,655 (81.6%) |
| Translatome | RNC-seq | 16,300 (79.8%) | 15,999 (78.3%) | 16,622 (81.4%) |
| Ribo-seq | 15,815 (77.4%) | 16,112 (78.9%) | 16,641 (81.5%) | |
| Proteome | LC-MS/MS | – | 5850 (28.6%) | 6843 (33.5%) |
| (a) HBE | |||
|---|---|---|---|
| RPKM > 0 | RPKM > 1 | RPKM > 10 | |
| RNA-seq vs. RNC-seq | 0.91 | 0.95 | 0.87 |
| RNA-seq vs. Ribo-seq | 0.89 | 0.83 | 0.53 |
| (b) A549 | |||
| RPKM > 0 | RPKM > 1 | RPKM > 10 | |
| RNA-seq vs. RNC-seq | 0.91 | 0.93 | 0.81 |
| RNA-seq vs. Ribo-seq | 0.9 | 0.86 | 0.57 |
| Proteome vs. RNC-seq | 0.36 | 0.32 | 0.17 |
| Proteome vs. Ribo-seq | 0.35 | 0.34 | 0.19 |
| (c) MCF-7 | |||
| RPKM > 0 | RPKM > 1 | RPKM > 10 | |
| RNA-seq vs. RNC-seq | 0.92 | 0.97 | 0.93 |
| RNA-seq vs. Ribo-seq | 0.92 | 0.95 | 0.86 |
| Proteome vs. RNC-seq | 0.4 | 0.32 | 0.17 |
| Proteome vs. Ribo-seq | 0.4 | 0.31 | 0.17 |
| Cell Line | Assay 1 | Number of Reads × 106 | Sequence Read Archive (SRA) | Study Reference | Illumina Sequencer Model |
|---|---|---|---|---|---|
| HBE (normal human bronchial epithelial cells) | RNA-seq | 12 | SRR611121 | [14] | Genome Analyzer IIx |
| RNC-seq | 16 | SRR611122 | |||
| Ribo-seq | 110 | SRR3286543 | [39] | HiSeq 2500 | |
| A549 (lung adenocarcinoma epithelial cells) | RNA-seq | 19 | SRR611119 | [14] | Genome Analyzer IIx |
| RNC-seq | 13 | SRR611120 | |||
| Ribo-seq | 95 | SRR3286544 | [39] | HiSeq 2500 | |
| MCF-7 (hormone—responsive breast cancer cell line) | RNA-seq | 19 | SRR6892923 | [40] | HiSeq 2000 |
| RNC-seq | 17 | SRR6892909 | |||
| Ribo-seq | 13 | SRR6892903 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozlova, A.; Sarygina, E.; Ilgisonis, E.; Tarbeeva, S.; Ponomarenko, E. The Translatome Map: RNC-Seq vs. Ribo-Seq for Profiling of HBE, A549, and MCF-7 Cell Lines. Int. J. Mol. Sci. 2024, 25, 10970. https://doi.org/10.3390/ijms252010970
Kozlova A, Sarygina E, Ilgisonis E, Tarbeeva S, Ponomarenko E. The Translatome Map: RNC-Seq vs. Ribo-Seq for Profiling of HBE, A549, and MCF-7 Cell Lines. International Journal of Molecular Sciences. 2024; 25(20):10970. https://doi.org/10.3390/ijms252010970
Chicago/Turabian StyleKozlova, Anna, Elizaveta Sarygina, Ekaterina Ilgisonis, Svetlana Tarbeeva, and Elena Ponomarenko. 2024. "The Translatome Map: RNC-Seq vs. Ribo-Seq for Profiling of HBE, A549, and MCF-7 Cell Lines" International Journal of Molecular Sciences 25, no. 20: 10970. https://doi.org/10.3390/ijms252010970
APA StyleKozlova, A., Sarygina, E., Ilgisonis, E., Tarbeeva, S., & Ponomarenko, E. (2024). The Translatome Map: RNC-Seq vs. Ribo-Seq for Profiling of HBE, A549, and MCF-7 Cell Lines. International Journal of Molecular Sciences, 25(20), 10970. https://doi.org/10.3390/ijms252010970