
The Presence of Periodontitis Exacerbates Non-Alcoholic Fatty Liver Disease via Sphingolipid Metabolism-Associated Insulin Resistance and Hepatic Inflammation in Mice with Metabolic Syndrome


 , , and
, , and 
Abstract
1. Introduction
2. Results
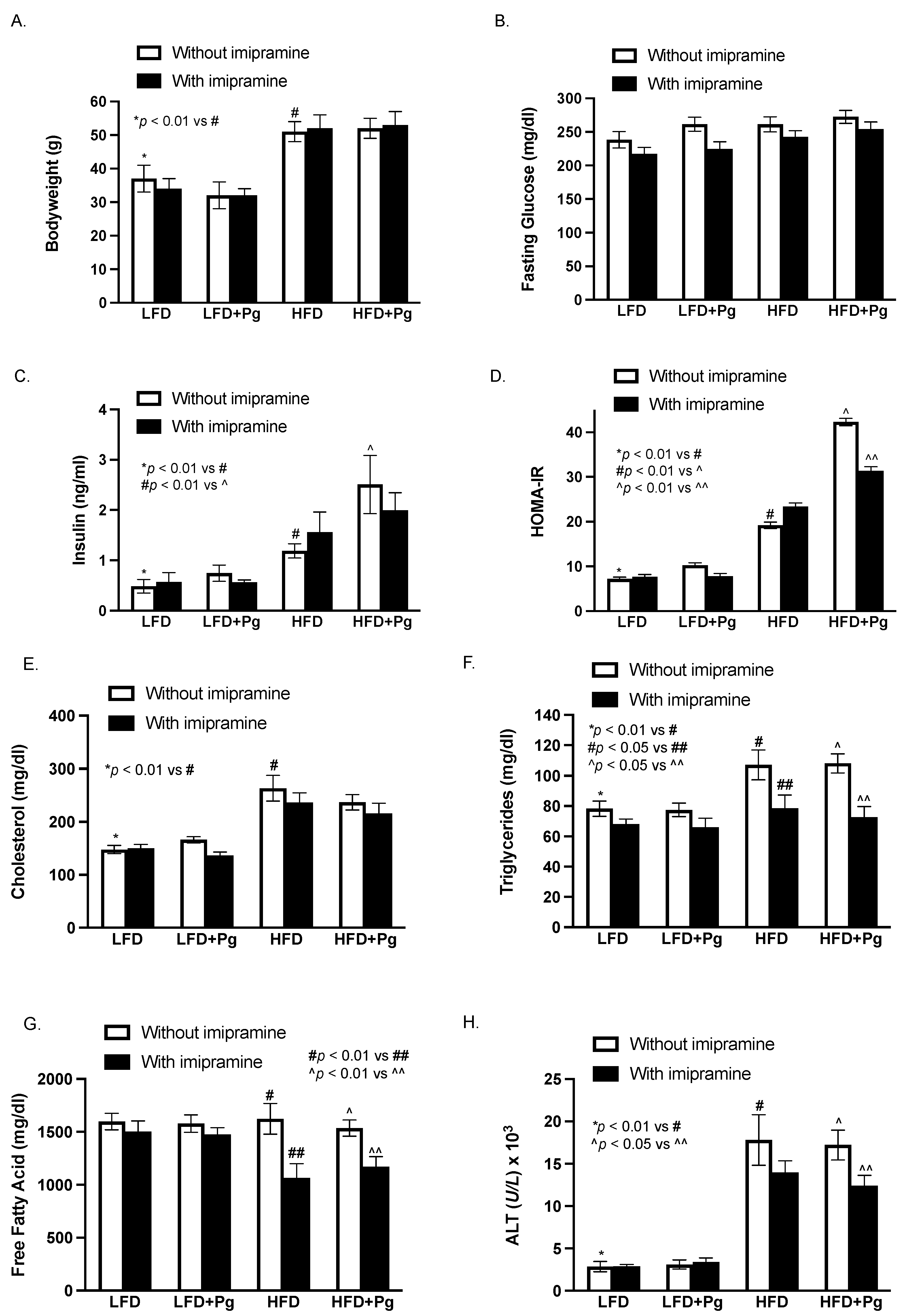
2.1. The Effect of HFD Feeding, P. gingivalis Inoculation and Imipramine on Blood Levels of Insulin, Glucose and Lipids and Hepatic Injury
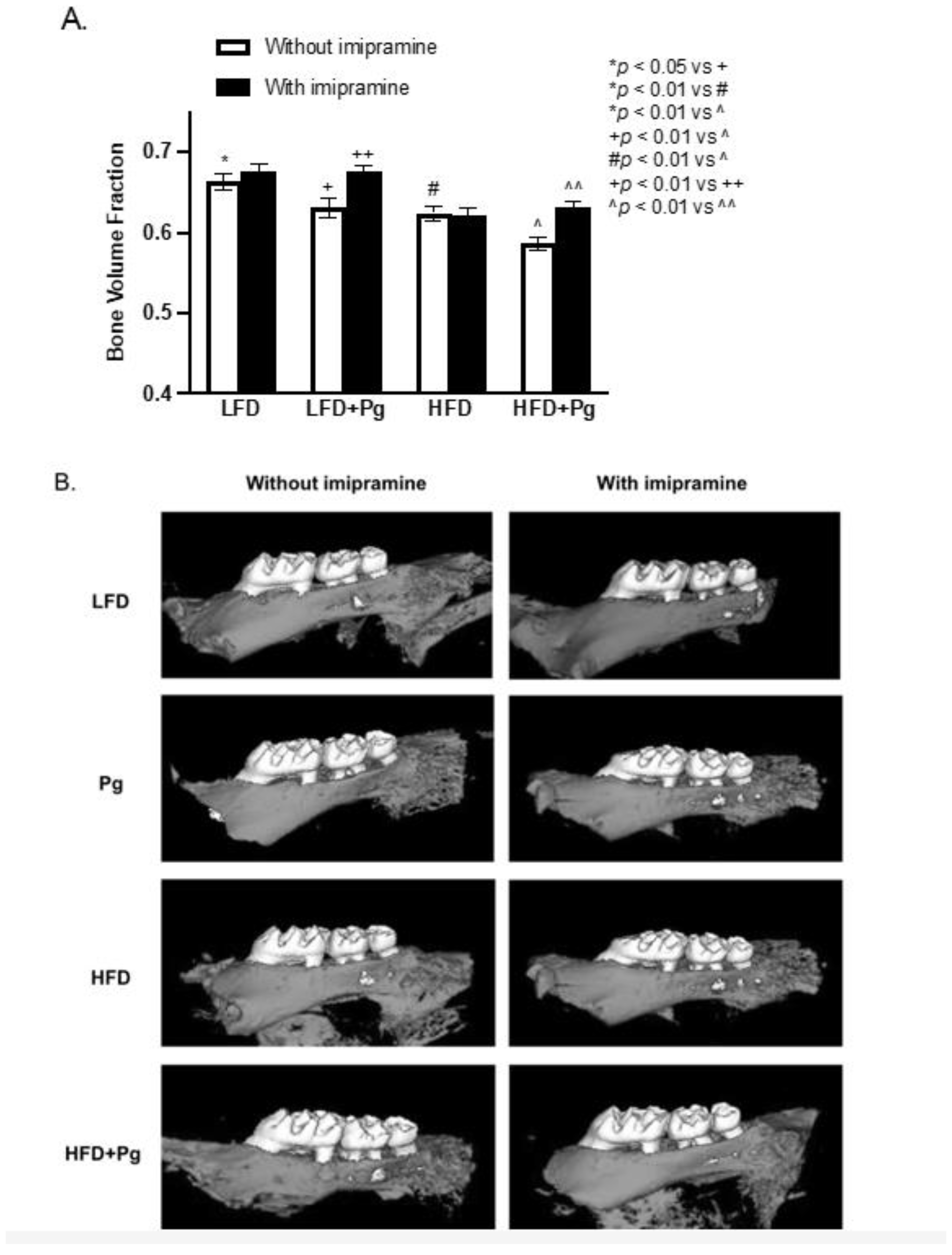
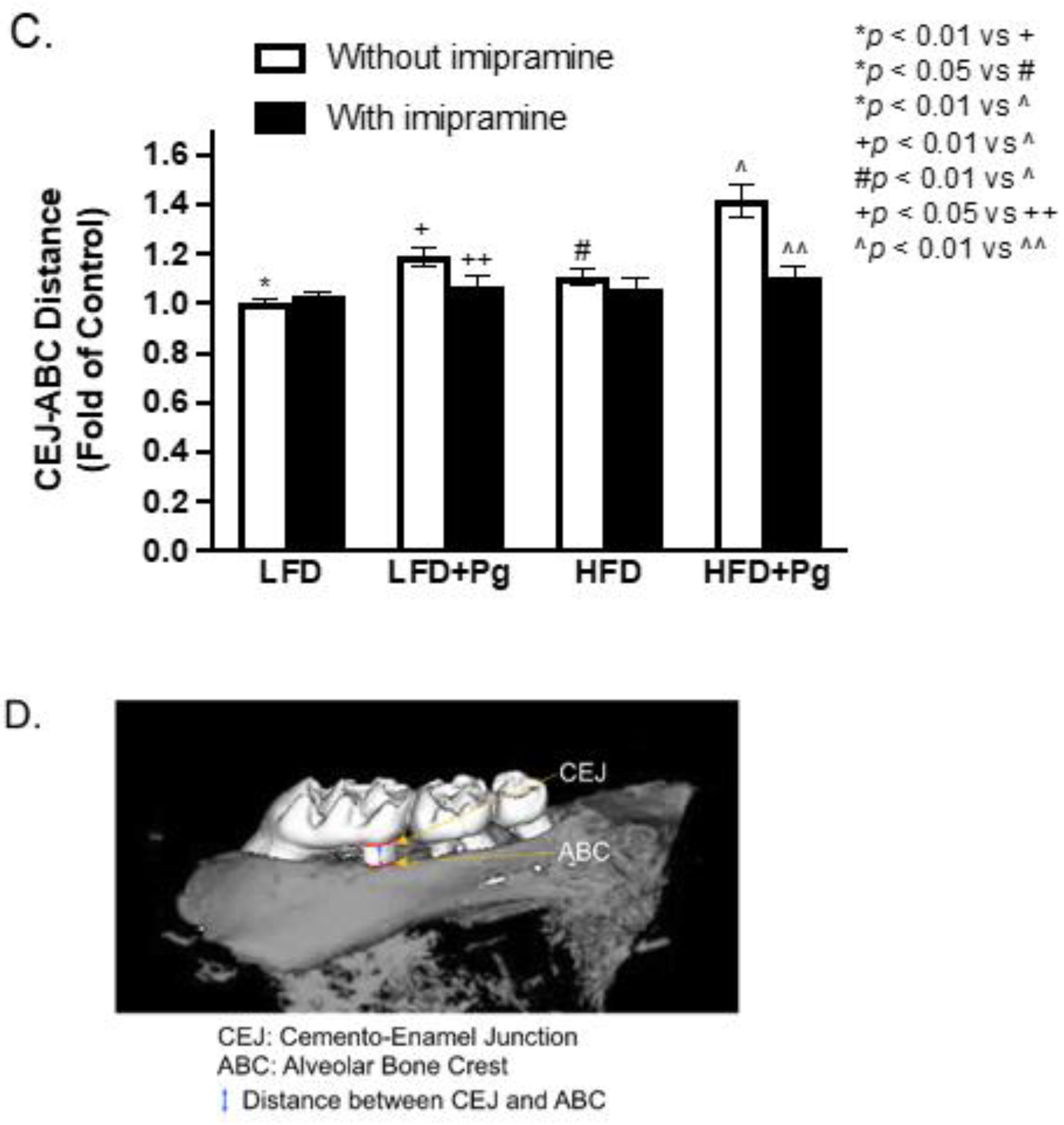
2.2. P. gingivalis Inoculation and HFD Feeding Promote, but Imipramine Alleviates Alveolar Bone Loss
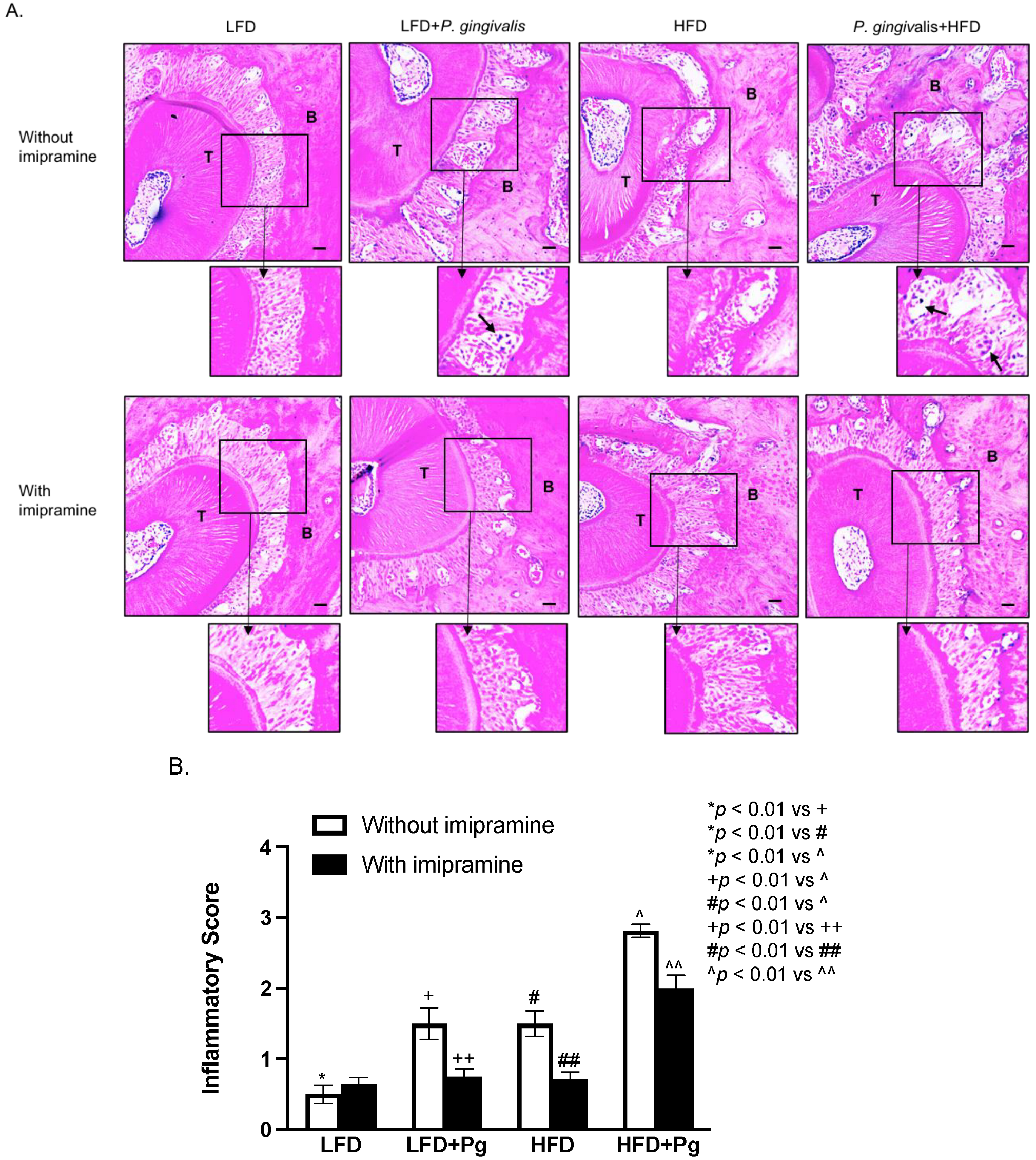
2.3. P. gingivalis Inoculation and HFD Feeding Stimulate, but Imipramine Inhibits Periodontal Inflammation and Bone Resorption
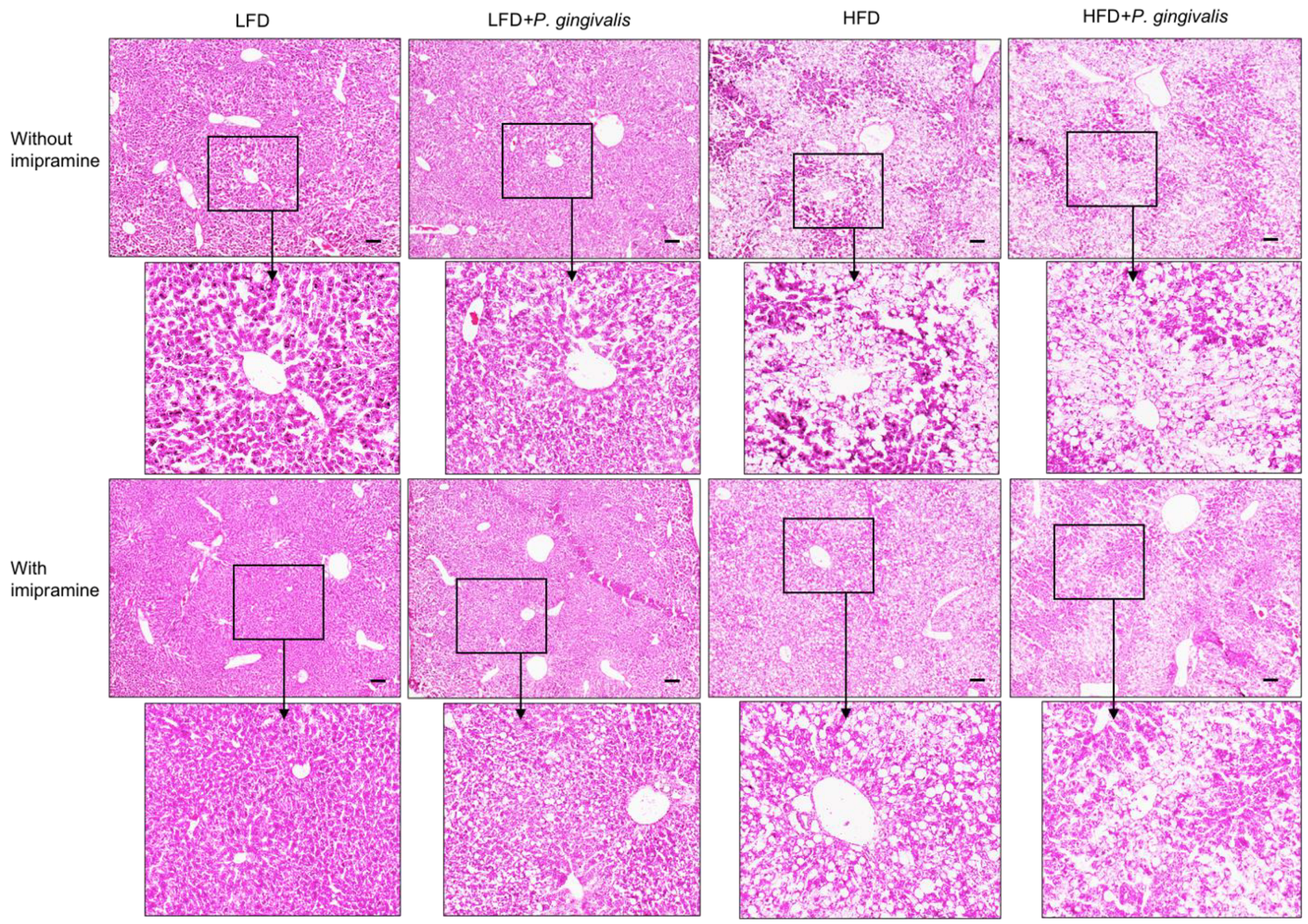
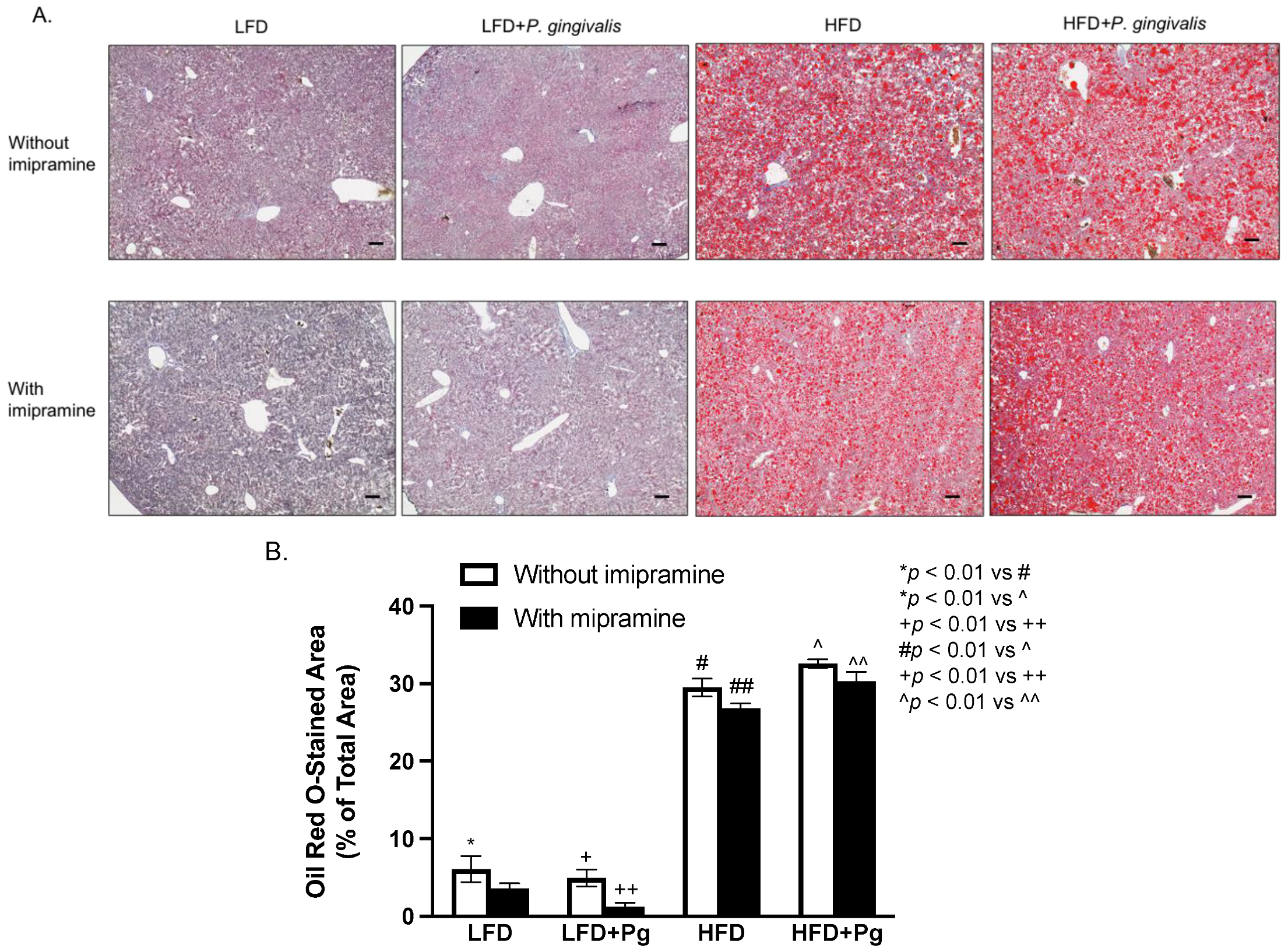
2.4. HFD Feeding or HFD Feeding plus P. gingivalis Inoculation Promotes, but Imipramine Ameliorates Hepatic Steatosis
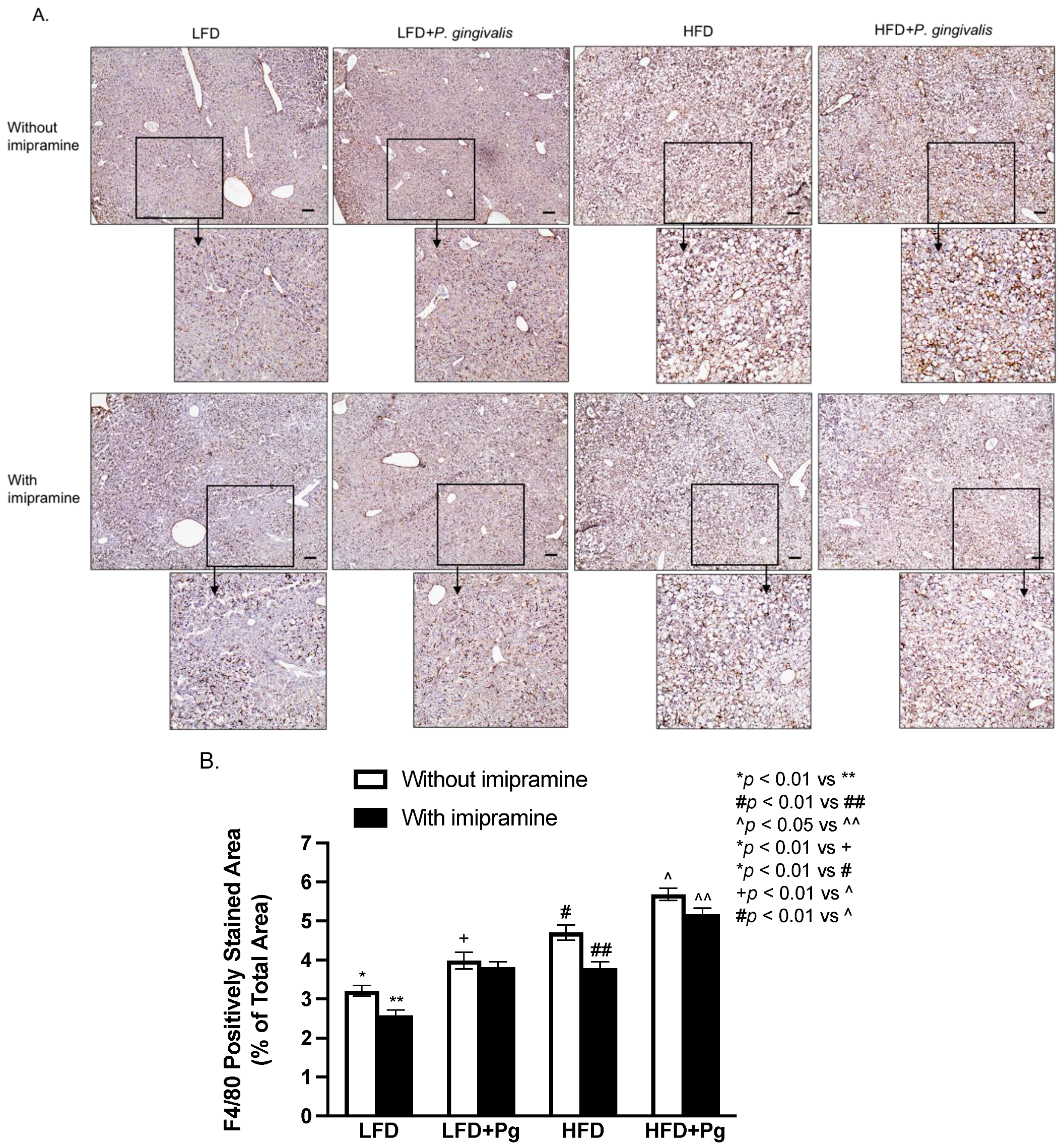
2.5. P. gingivalis Inoculation and HFD Feeding Stimulate, but Imipramine Inhibits Hepatic Inflammation
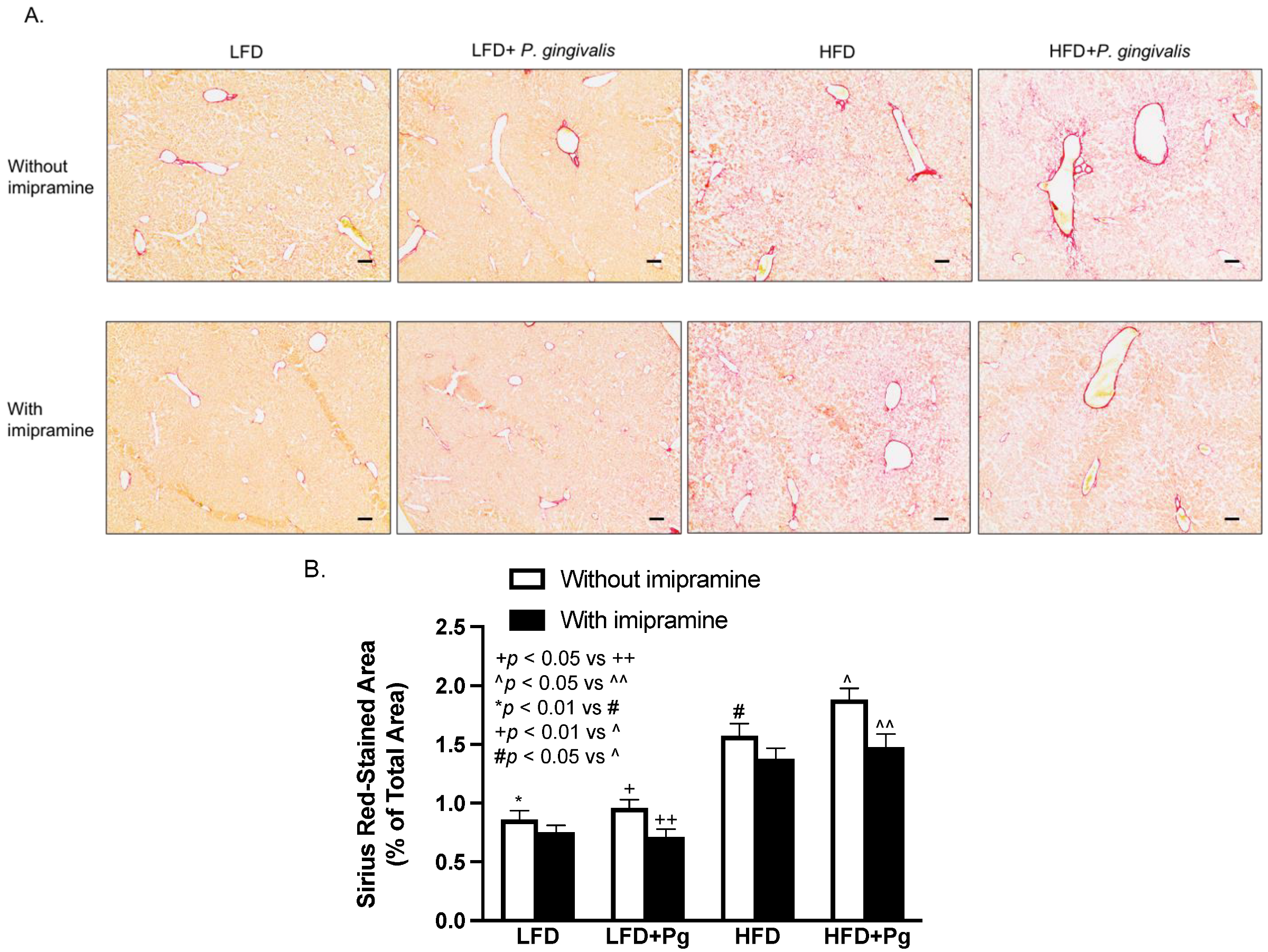
2.6. P. gingivalis Inoculation with HFD Feeding Increases, but Imipramine Attenuates Hepatic Fibrosis
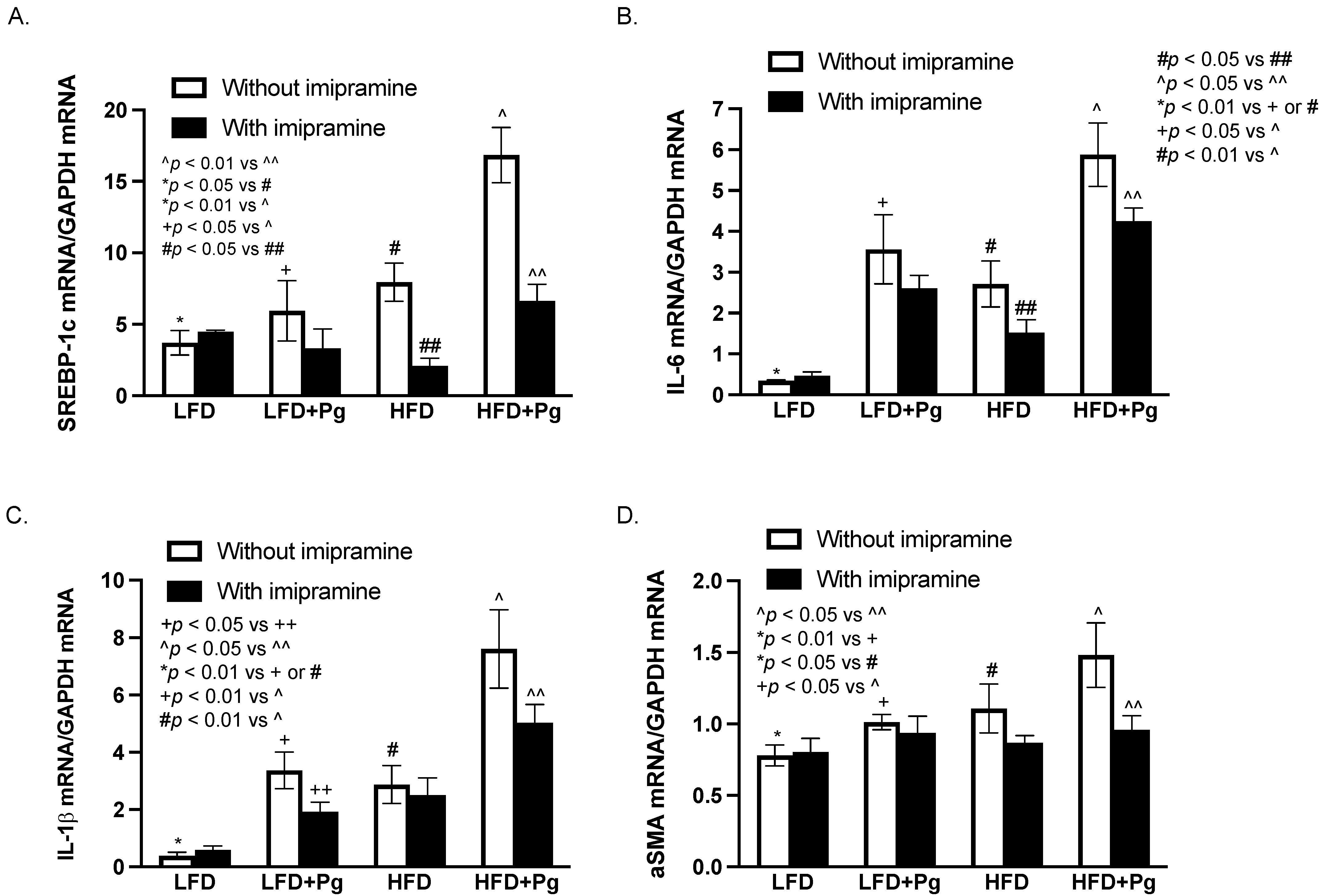
2.7. The Effects of P. gingivalis Inoculation, HFD Feeding, and Imipramine on the Liver Expression of Genes Involved in Lipogenesis, Inflammation, and Fibrosis
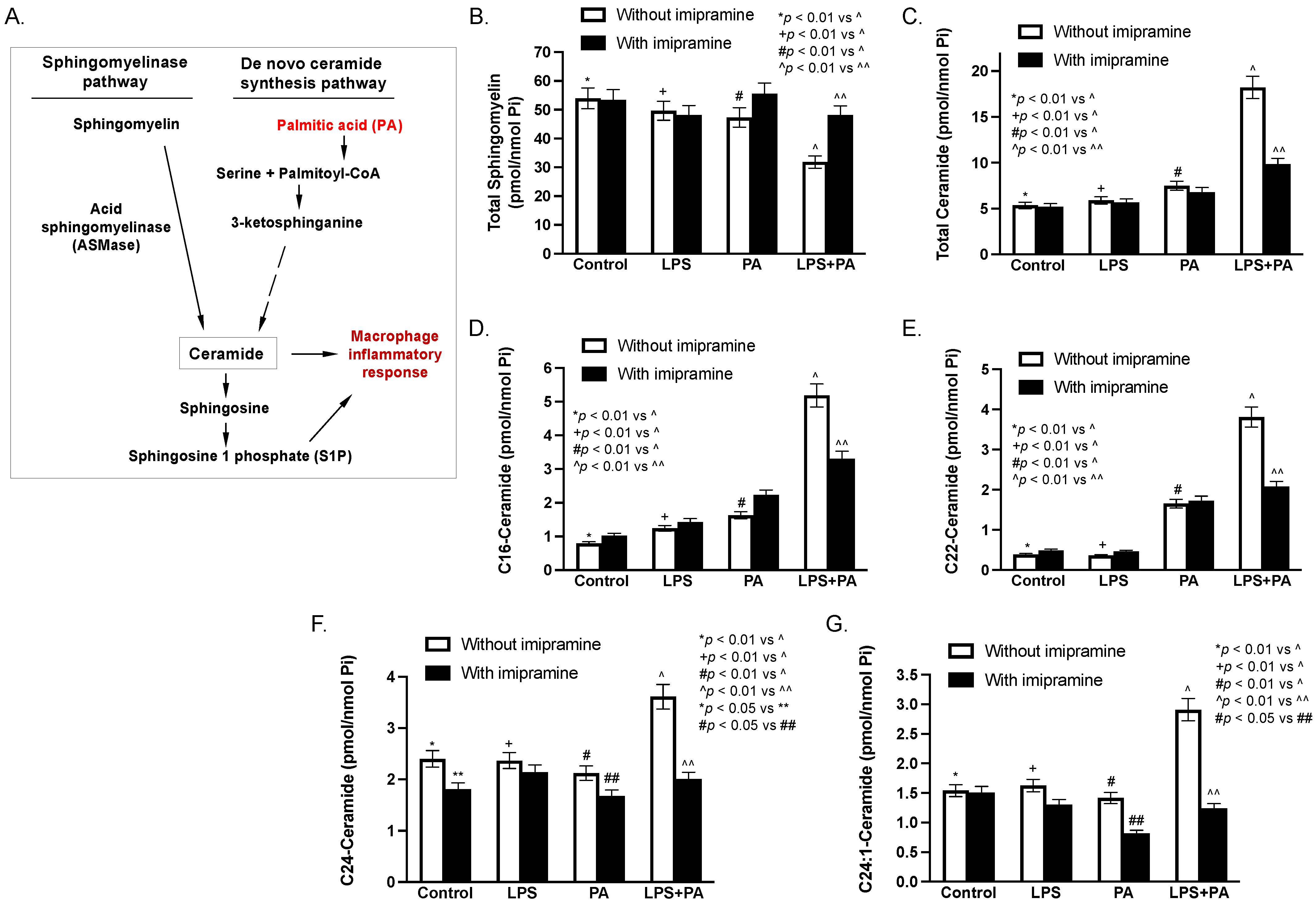
2.8. LPS and Palmitic Acid (PA) Synergistically Increase Ceramide in Macrophages In Vitro
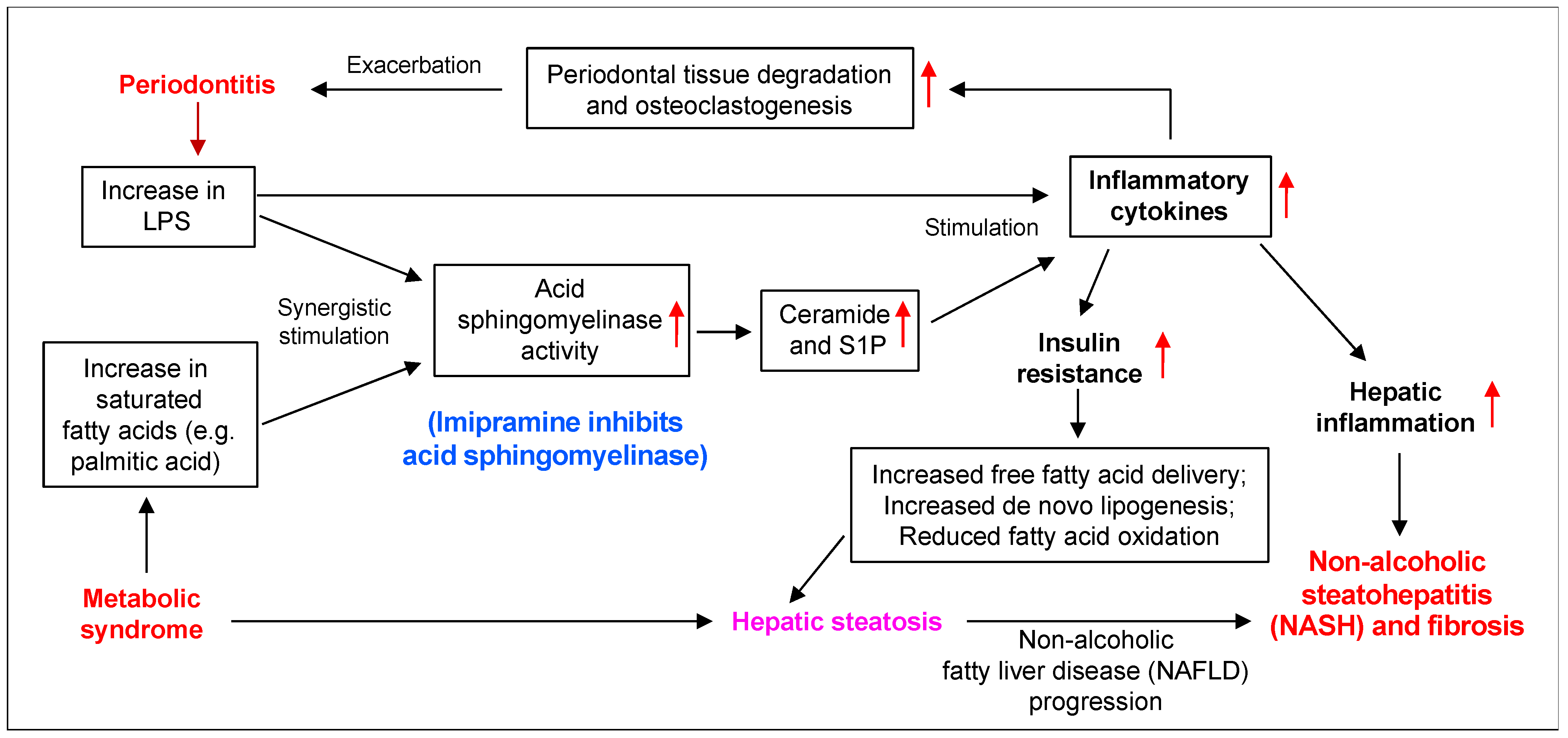
3. Discussion
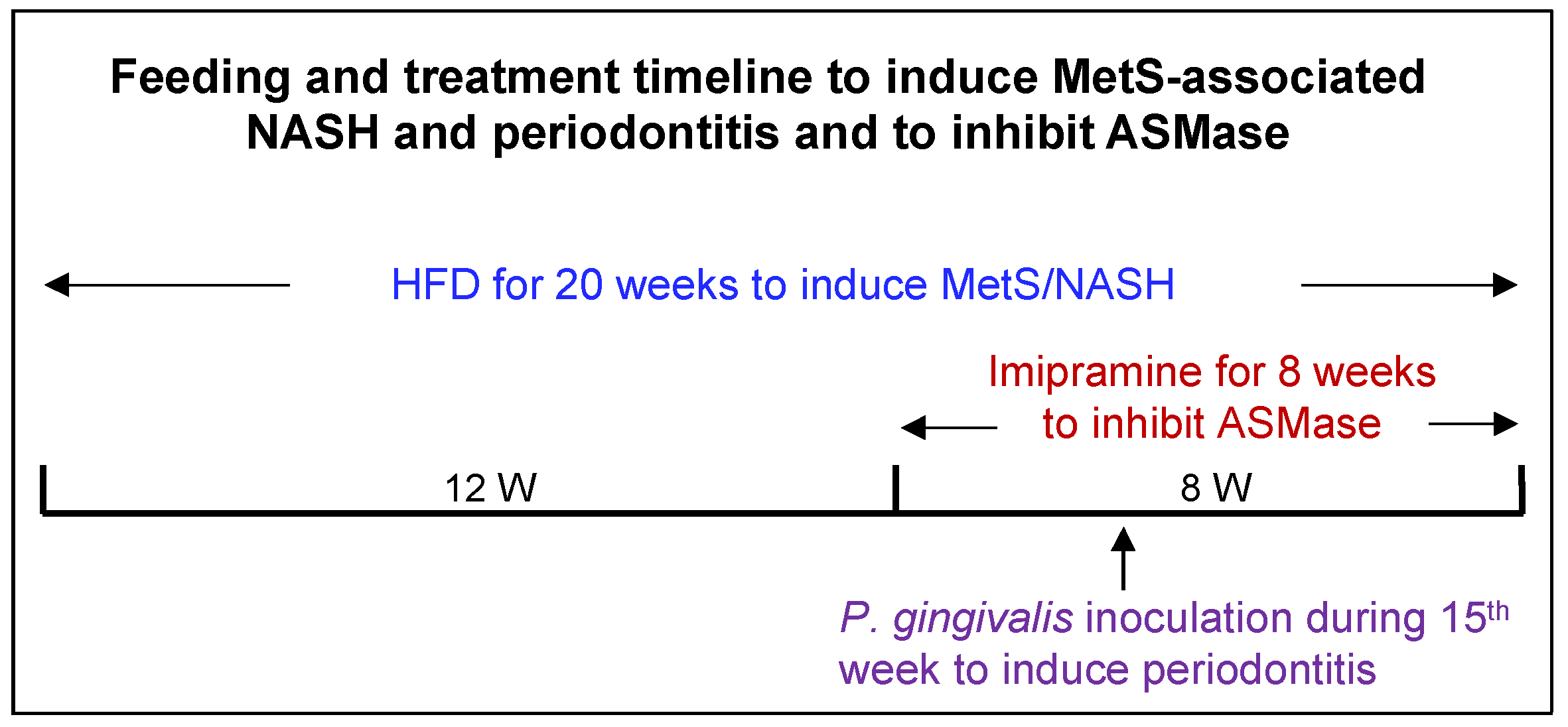
4. Materials and Methods
4.1. Animal Feeding and Treatments
4.2. Bacterial Culture
4.3. Induction of Periodontitis by Oral P. gingivalis Inoculation
4.4. Measurements of Metabolic Parameters
4.5. Micro-CT (μCT),BVF Analysis, and CEJ-ABC Distance Measurement
4.6. Histological Tissue Processing and Pathological Evaluation
4.7. Histological Examination of Liver Tissue
4.8. Oil Red O Staining
4.9. F4/80 Immunostaining
4.10. Sirius Red Staining
4.11. RNA Isolation from Liver Tissues
4.12. Real-Time Polymerase Chain Reaction (PCR)
4.13. Cell Culture
4.14. PA Preparation
4.15. Lipidomics
4.16. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beck, J.; Garcia, R.; Heiss, G.; Vokonas, P.S.; Offenbacher, S. Periodontal disease and cardiovascular disease. J. Periodontol. 1996, 67, 1123–1137. [Google Scholar] [CrossRef] [PubMed]
- Offenbacher, S. Periodontal diseases: Pathogenesis. Ann. Periodontol. 1996, 1, 821–878. [Google Scholar] [CrossRef] [PubMed]
- Pirih, F.Q.; Monajemzadeh, S.; Singh, N.; Sinacola, R.S.; Shin, J.M.; Chen, T.; Fenno, J.C.; Kamarajan, P.; Rickard, A.H.; Travan, S.; et al. Association between metabolic syndrome and periodontitis: The role of lipids, inflammatory cytokines, altered host response, and the microbiome. Periodontol. 2000 2021, 87, 50–75. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Spooner, R.; Chowdhury, N.; Pandey, V.; Wellslager, B.; Atanasova, K.R.; Evans, Z.; Yilmaz, Ö. In Situ Intraepithelial Localizations of Opportunistic Pathogens, Porphyromonas gingivalis and Filifactor alocis, in Human Gingiva. Curr. Res. Microb. Sci. 2020, 1, 7–17. [Google Scholar] [CrossRef]
- Olsen, I.; Yilmaz, Ö. Modulation of inflammasome activity by Porphyromonas gingivalis in periodontitis and associated systemic diseases. J. Oral Microbiol. 2016, 8, 30385. [Google Scholar] [CrossRef]
- Yilmaz, Ö.; Lee, K.L. The inflammasome and danger molecule signaling: At the crossroads of inflammation and pathogen persistence in the oral cavity. Periodontol. 2000 2015, 69, 83–95. [Google Scholar] [CrossRef]
- Martínez-García, M.; Hernández-Lemus, E. Periodontal Inflammation and Systemic Diseases: An Overview. Front. Physiol. 2021, 12, 709438. [Google Scholar] [CrossRef]
- Davidson, M.B. Metabolic syndrome/insulin resistance syndrome/pre-diabetes: New section in diabetes care. Diabetes Care 2003, 26, 3179. [Google Scholar] [CrossRef]
- DeBoer, M.D. Obesity, systemic inflammation, and increased risk for cardiovascular disease and diabetes among adolescents: A need for screening tools to target interventions. Nutrition 2013, 29, 379–386. [Google Scholar] [CrossRef]
- McPhee, J.B.; Schertzer, J.D. Immunometabolism of obesity and diabetes: Microbiota link compartmentalized immunity in the gut to metabolic tissue inflammation. Clin. Sci. 2015, 129, 1083–1096. [Google Scholar] [CrossRef]
- Lalla, E.; Lamster, I.B.; Drury, S.; Fu, C.; Schmidt, A.M. Hyperglycemia, glycoxidation and receptor for advanced glycation endproducts: Potential mechanisms underlying diabetic complications, including diabetes-associated periodontitis. Periodontology 2000 2000, 23, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Nibali, L.; Tatarakis, N.; Needleman, I.; Tu, Y.K.; D’Aiuto, F.; Rizzo, M.; Donos, N. Clinical review: Association between metabolic syndrome and periodontitis: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2013, 98, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Preshaw, P.M.; Alba, A.L.; Herrera, D.; Jepsen, S.; Konstantinidis, A.; Makrilakis, K.; Taylor, R. Periodontitis and diabetes: A two-way relationship. Diabetologia 2012, 55, 21–31. [Google Scholar] [CrossRef]
- Tonetti, M.S. Periodontitis and risk for atherosclerosis: An update on intervention trials. J. Clin. Periodontol. 2009, 36, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Leira, Y.; Seoane, J.; Blanco, M.; Rodríguez-Yáñez, M.; Takkouche, B.; Blanco, J.; Castillo, J. Association between periodontitis and ischemic stroke: A systematic review and meta-analysis. Eur. J. Epidemiol. 2017, 32, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, G.; Guo, J.F.; Tan, Y.H. Associations between osteoporosis and risk of periodontitis: A pooled analysis of observational studies. Oral Dis. 2021, 27, 357–369. [Google Scholar] [CrossRef]
- Mao, S.; Huang, C.P.; Lan, H.; Lau, H.G.; Chiang, C.P.; Chen, Y.W. Association of periodontitis and oral microbiomes with Alzheimer’s disease: A narrative systematic review. J. Dent. Sci. 2022, 17, 1762–1779. [Google Scholar] [CrossRef]
- Patel, S.; Howard, D.; Chowdhury, N.; Derieux, C.; Wellslager, B.; Yilmaz, Ö.; French, L. Characterization of Human Genes Modulated by Porphyromonas gingivalis Highlights the Ribosome, Hypothalamus, and Cholinergic Neurons. Front. Immunol. 2021, 12, 646259. [Google Scholar] [CrossRef]
- Atanasova, K.R.; Yilmaz, Ö. Prelude to oral microbes and chronic diseases: Past, present and future. Microbes. Infect. 2015, 17, 473–483. [Google Scholar] [CrossRef]
- Atanasova, K.R.; Yilmaz, O. Looking in the Porphyromonas gingivalis cabinet of curiosities: The microbium, the host and cancer association. Mol. Oral Microbiol. 2014, 29, 55–66. [Google Scholar] [CrossRef]
- Tomeno, W.; Imajo, K.; Takayanagi, T.; Ebisawa, Y.; Seita, K.; Takimoto, T.; Honda, K.; Kobayashi, T.; Nogami, A.; Kato, T.; et al. Complications of Non-Alcoholic Fatty Liver Disease in Extrahepatic Organs. Diagnostics 2020, 10, 912. [Google Scholar] [CrossRef]
- Alakhali, M.S.; Al-Maweri, S.A.; Al-Shamiri, H.M.; Al-Haddad, K.; Halboub, E. The potential association between periodontitis and non-alcoholic fatty liver disease: A systematic review. Clin. Oral Investig. 2018, 22, 2965–2974. [Google Scholar] [CrossRef] [PubMed]
- Rosato, V.; Masarone, M.; Dallio, M.; Federico, A.; Aglitti, A.; Persico, M. NAFLD and Extra-Hepatic Comorbidities: Current Evidence on a Multi-Organ Metabolic Syndrome. Int. J. Environ. Res. Public Health 2019, 16, 3415. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef]
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef] [PubMed]
- Albhaisi, S.; Noureddin, M. Current and Potential Therapies Targeting Inflammation in NASH. Front Endocrinol. 2021, 12, 767314. [Google Scholar] [CrossRef]
- Shin, H.S.; Hong, M.H.; Moon, J.Y.; Sim, S.J. Periodontal disease could be a potential risk factor for non-alcoholic fatty liver disease: An 11-year retrospective follow-up study. Clin. Oral Investig. 2022, 26, 5503–5514. [Google Scholar] [CrossRef]
- Ram, D.; Wilensky, A.; Zur, D.; Almoznino, G. The Triangle of Nonalcoholic Fatty Liver Disease, Metabolic Dysfunction, and Periodontitis: Analysis of the Dental, Oral, Medical and Epidemiological (DOME) Records-Based Nationwide Research. Metabolites 2022, 12, 1212. [Google Scholar] [CrossRef]
- Weintraub, J.A.; Lopez Mitnik, G.; Dye, B.A. Oral Diseases Associated with Nonalcoholic Fatty Liver Disease in the United States. J. Dent. Res. 2019, 98, 1219–1226. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, G.N.; Song, H.C.; Park, Y.M.; Ahn, Y.B.; Han, K.; Ko, S.H. Association between Fatty Liver Index and Periodontitis: The Korea National Health and Nutrition Examination Survey. Sci. Rep. 2020, 10, 3805. [Google Scholar] [PubMed]
- Wijarnpreecha, K.; Panjawatanan, P.; Cheungpasitporn, W.; Lukens, F.J.; Harnois, D.M.; Pungpapong, S.; Ungprasert, P. The Association between Periodontitis and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis. J. Gastrointestin. Liver Dis. 2020, 29, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Tang, J. Is There An Association Between Periodontitis And Non-Alcoholic Fatty Liver Disease? A Systematic Review and Meta-Analysis. Community Dent. Health 2023, 40, 47–52. [Google Scholar] [PubMed]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef]
- Hanayama, M.; Yamamoto, Y.; Utsunomiya, H.; Yoshida, O.; Liu, S.; Mogi, M.; Matsuura, B.; Takeshita, E.; Ikeda, Y.; Hiasa, Y. The mechanism of increased intestinal palmitic acid absorption and its impact on hepatic stellate cell activation in nonalcoholic steatohepatitis. Sci. Rep. 2021, 11, 13380. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhang, X.; Lu, Z.; Perry, D.M.; Li, Y.; Russo, S.B.; Cowart, L.A.; Hannun, Y.A.; Huang, Y. Acid sphingomyelinase plays a key role in palmitic acid-amplified inflammatory signaling triggered by lipopolysaccharide at low concentrations in macrophages. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E853–E867. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Lu, Z.; Li, Y.; Ru, J.H.; Lopes-Virella, M.F.; Huang, Y. LPS and palmitate synergistically stimulate sphingosine kinase 1 and increase sphingosine 1 phosphate in RAW264.7 macrophages. J. Leukoc. Biol. 2018, 104, 843–853. [Google Scholar]
- Lu, Z.; Li, Y.; Syn, W.K.; Wang, Z.; Lopes-Virella, M.F.; Lyons, T.J.; Huang, Y. Amitriptyline inhibits nonalcoholic steatohepatitis and atherosclerosis induced by high-fat diet and LPS through modulation of sphingolipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E131–E144. [Google Scholar] [CrossRef]
- Li, Y.; Lu, Z.; Zhang, L.; Kirkwood, C.L.; Kirkwood, K.L.; Lopes-Virella, M.F.; Huang, Y. Inhibition of acid sphingomyelinase by imipramine abolishes the synergy between metabolic syndrome and periodontitis on alveolar bone loss. J. Periodontal. Res. 2022, 57, 173–185. [Google Scholar] [CrossRef]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Kang, B.S.; Lee, S.H.; Suh, S.W. Administration of an Acidic Sphingomyelinase (ASMase) Inhibitor, Imipramine, Reduces Hypoglycemia-Induced Hippocampal Neuronal Death. Cells 2022, 11, 667. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Abramson, Z.R.; Taba, M., Jr.; Jin, Q.; Chang, J.; Kreider, J.M.; Goldstein, S.A.; Giannobile, W.V. Three-dimensional micro-computed tomographic imaging of alveolar bone in experimental bone loss or repair. J. Periodontol. 2007, 78, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Machado, E.R.; Yu, H.; Zhang, X.; Lu, Z.; Li, Y.; Lopes-Virella, M.F.; Kirkwood, K.L.; Huang, Y. Simvastatin inhibits LPS-induced alveolar bone loss during metabolic syndrome. J. Dent. Res. 2014, 93, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Austyn, J.M.; Gordon, S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur. J. Immunol. 1981, 11, 805–815. [Google Scholar] [CrossRef] [PubMed]
- On, S.; Kim, H.Y.; Kim, H.S.; Park, J.; Kang, K.W. Involvement of G-Protein-Coupled Receptor 40 in the Inhibitory Effects of Docosahexaenoic Acid on SREBP1-Mediated Lipogenic Enzyme Expression in Primary Hepatocytes. Int. J. Mol. Sci. 2019, 20, 2625. [Google Scholar] [CrossRef]
- Fu, J.; Wu, B.; Zhong, S.; Deng, W.; Lin, F. miR-29a-3p suppresses hepatic fibrosis pathogenesis by modulating hepatic stellate cell proliferation via targeting PIK3R3 gene expression. Biochem. Biophys. Res. Commun. 2020, 529, 922–929. [Google Scholar] [CrossRef]
- Fang, H.; Yu, L.; You, D.; Peng, N.; Guo, W.; Wang, J.; Zhang, X. In vivo Therapeutic Effects and Mechanisms of Hydroxyasiaticoside Combined With Praziquantel in the Treatment of Schistosomiasis Induced Hepatic Fibrosis. Front. Bioeng. Biotechnol. 2020, 8, 613784. [Google Scholar] [CrossRef]
- Simon, J.; Ouro, A.; Ala-Ibanibo, L.; Presa, N.; Delgado, T.C.; Martínez-Chantar, M.L. Sphingolipids in Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma: Ceramide Turnover. Int. J. Mol. Sci. 2019, 21, 40. [Google Scholar] [CrossRef]
- Yu, X.D.; Wang, J.W. Ceramide de novo synthesis in non-alcoholic fatty liver disease: Pathogenic mechanisms and therapeutic perspectives. Biochem. Pharmacol. 2022, 202, 115157. [Google Scholar] [CrossRef]
- Nagahashi, M.; Abe, M.; Sakimura, K.; Takabe, K.; Wakai, T. The role of sphingosine-1-phosphate in inflammation and cancer progression. Cancer Sci. 2018, 109, 3671–3678. [Google Scholar] [CrossRef]
- Gao, D.; Pararasa, C.; Dunston, C.R.; Bailey, C.J.; Griffiths, H.R. Palmitate promotes monocyte atherogenicity via de novo ceramide synthesis. Free Radic. Biol. Med. 2012, 53, 796–806. [Google Scholar] [CrossRef]
- Sun, X.; Gao, J.; Meng, X.; Lu, X.; Zhang, L.; Chen, R. Polarized Macrophages in Periodontitis: Characteristics, Function, and Molecular Signaling. Front. Immunol. 2021, 12, 763334. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zheng, C.; Yang, J.; Li, B. Intersection between macrophages and periodontal pathogens in periodontitis. J. Leukoc. Biol. 2021, 110, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhou, Y.; Wang, H.; Zhang, M.; Qiu, P.; Zhang, M.; Zhang, R.; Zhao, Q.; Liu, J. Crosstalk Between Liver Macrophages and Surrounding Cells in Nonalcoholic Steatohepatitis. Front. Immunol. 2020, 11, 1169. [Google Scholar] [CrossRef]
- George, A.K.; Narayan, V.; Kurian, N.; Joseph, A.E.; Anil, S. A pilot study on glycemia and insulin resistance in patients with severe periodontitis. J. Indian Soc. Periodontol. 2021, 25, 393–398. [Google Scholar] [CrossRef]
- Pulido-Moran, M.; Bullon, P.; Morillo, J.M.; Battino, M.; Quiles, J.L.; Ramirez-Tortosa, M. The relationship between insulin resistance and periodontitis is not affected by Mediterranean diet in a Spanish population. Arch. Oral Biol. 2017, 77, 62–67. [Google Scholar] [CrossRef]
- Mammen, J.; Vadakkekuttical, R.J.; George, J.M.; Kaziyarakath, J.A.; Radhakrishnan, C. Effect of non-surgical periodontal therapy on insulin resistance in patients with type II diabetes mellitus and chronic periodontitis, as assessed by C-peptide and the Homeostasis Assessment Index. J. Investig. Clin. Dent. 2017, 8, e12221. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Kahn, S.E. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef]
- Athyros, V.G.; Alexandrides, T.K.; Bilianou, H.; Cholongitas, E.; Doumas, M.; Ganotakis, E.S.; Goudevenos, J.; Elisaf, M.S.; Germanidis, G.; Giouleme, O.; et al. The use of statins alone, or in combination with pioglitazone and other drugs, for the treatment of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and related cardiovascular risk. An Expert Panel Statement. Metabolism 2017, 71, 17–32. [Google Scholar] [CrossRef]
- Carter-Kent, C.; Zein, N.N.; Feldstein, A.E. Cytokines in the pathogenesis of fatty liver and disease progression to steatohepatitis: Implications for treatment. Am. J. Gastroenterol. 2008, 103, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, Z.; Zhang, L.; Kirkwood, K.L.; Lopes-Virella, M.F.; Huang, Y. Acid sphingomyelinase deficiency exacerbates LPS-induced experimental periodontitis. Oral Dis. 2020, 26, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Al-Mansoori, L.; Al-Jaber, H.; Prince, M.S.; Elrayess, M.A. Role of Inflammatory Cytokines, Growth Factors and Adipokines in Adipogenesis and Insulin Resistance. Inflammation 2022, 45, 31–44. [Google Scholar] [CrossRef]
- Copaci, I.; Micu, L.; Voiculescu, M. The role of cytokines in non-alcoholic steatohepatitis. A review. J. Gastrointest. Liver Dis. 2006, 15, 363–373. [Google Scholar]
- Tan, J.; Dai, A.; Pan, L.; Zhang, L.; Wang, Z.; Ke, T.; Sun, W.; Wu, Y.; Ding, P.H.; Chen, L. Inflamm-Aging-Related Cytokines of IL-17 and IFN-γ Accelerate Osteoclastogenesis and Periodontal Destruction. J. Immunol. Res. 2021, 2021, 9919024. [Google Scholar] [CrossRef]
- Thibaut, R.; Gage, M.C.; Pineda-Torra, I.; Chabrier, G.; Venteclef, N.; Alzaid, F. Liver macrophages and inflammation in physiology and physiopathology of non-alcoholic fatty liver disease. FEBS J. 2021, 289, 3024–3057. [Google Scholar] [CrossRef]
- Becker, K.A.; Riethmüller, J.; Lüth, A.; Döring, G.; Kleuser, B.; Gulbins, E. Acid sphingomyelinase inhibitors normalize pulmonary ceramide and inflammation in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 42, 716–724. [Google Scholar] [CrossRef]
- Colombini, M. Ceramide channels and their role in mitochondria-mediated apoptosis. Biochim. Biophys. Acta 2010, 1797, 1239–1244. [Google Scholar] [CrossRef]
- Li, C.; Wang, A.; Wu, Y.; Gulbins, E.; Grassmé, H.; Zhao, Z. Acid Sphingomyelinase-Ceramide System in Bacterial Infections. Cell Physiol. Biochem. 2019, 52, 280–301. [Google Scholar]
- Hammerschmidt, P.; Brüning, J.C. Contribution of specific ceramides to obesity-associated metabolic diseases. Cell Mol. Life Sci. 2022, 79, 395. [Google Scholar] [CrossRef]
- Gupta, A.; Muralidharan, S.; Torta, F.; Wenk, M.R.; Wohland, T. Long acyl chain ceramides govern cholesterol and cytoskeleton dependence of membrane outer leaflet dynamics. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183153. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, Ö. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell Microbiol. 2013, 15, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Roberts, J.S.; Choi, C.H.; Atanasova, K.R.; Yilmaz, Ö. Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence 2018, 9, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Kang, J.; Andriankaja, O.; Wada, K.; Rossa, C., Jr. Animal models to study host-bacteria interactions involved in periodontitis. Front. Oral Biol. 2012, 15, 117–132. [Google Scholar]
- Hajishengallis, G.; Shakhatreh, M.A.; Wang, M.; Liang, S. Complement receptor 3 blockade promotes IL-12-mediated clearance of Porphyromonas gingivalis and negates its virulence in vivo. J. Immunol. 2007, 179, 2359–2367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ju, J.; Rigney, T.; Tribble, G. Porphyromonas gingivalis infection increases osteoclastic bone resorption and osteoblastic bone formation in a periodontitis mouse model. BMC Oral Health 2014, 14, 89. [Google Scholar] [CrossRef]
- Steinkamp, H.M.; Hathaway-Schrader, J.D.; Chavez, M.B.; Aartun, J.D.; Zhang, L.; Jensen, T.; Shojaee Bakhtiari, A.; Helke, K.L.; Stumpo, D.J.; Alekseyenko, A.V.; et al. Tristetraprolin Is Required for Alveolar Bone Homeostasis. J. Dent. Res. 2018, 97, 946–953. [Google Scholar] [CrossRef]
- Bouxsein, M.L.; Boyd, S.K.; Christiansen, B.A.; Guldberg, R.E.; Jepsen, K.J.; Muller, R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Min. Res. 2010, 25, 1468–1486. [Google Scholar] [CrossRef]
- Van Veldhoven, P.P.; Bell, R.M. Effect of harvesting methods, growth conditions and growth phase on diacylglycerol levels in cultured human adherent cells. Biochim. Biophys. Acta 1988, 959, 185–196. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mice Without Imipramine Treatment | Mice with Imipramine Treatment | |||
|---|---|---|---|---|
| BVF | % BVF Change Compared to Control Mice | BVF | % BVF Change Compared to Mice without Imipramine Treatment | |
| Control (LFD-fed) | 0.663 ± 0.010 | − | 0.676 ± 0.009 | +1.96% |
| P. gingivalis inoculation | 0.626 ± 0.009 | −5.58% * | 0.679 ± 0.007 | +8.47% ** |
| MetS (HFD-fed) | 0.624 ± 0.009 | −5.88% ** | 0.621 ± 0.010 | −0.48% |
| MetS + P. gingivalis inoculation | 0.587 ± 0.008 | −11.46% ** | 0.631 ± 0.008 | +7.50% ** |
| C16-SM | C18-SM | C20-SM | C22-SM | C24-SM | C24:1-SM | Total-SM | |
|---|---|---|---|---|---|---|---|
| Control | 34.09 | 2.13 | 0.71 | 3.65 | 2.46 | 6.64 | 53.70 |
| Control + IMP | 34.13 | 2.30 | 0.77 | 3.82 | 2.40 | 6.21 | 53.46 |
| LPS | 33.40 | 1.55 | 0.55 | 2.74 | 2.02 | 5.16 | 49.59 |
| LPS + IMP | 32.24 | 1.89 | 0.61 | 3.04 | 2.39 | 4.62 | 48.22 |
| PA | 26.55 | 4.23 | 1.33 | 5.98 | 2.54 | 6.87 | 50.67 |
| PA + IMP | 33.29 | 3.89 | 1.08 | 4.99 | 2.25 | 6.76 | 55.54 |
| LPS + PA | 18.84 | 2.20 | 0.64 | 2.75 | 1.15 | 4.16 | 31.80 |
| LPS + P + IMP | 30.63 | 2.76 | 0.81 | 3.52 | 1.58 | 5.84 | 48.14 |
| C14-Cer | C16-Cer | C18-Cer | C20-Cer | C22-Cer | C24-Cer | C24:1-Cer | |
|---|---|---|---|---|---|---|---|
| Control | 0.79 | 0.05 | 0.03 | 0.39 | 2.40 | 1.54 | 5.36 |
| Control + IMP | 1.03 | 0.13 | 0.04 | 0.49 | 1.81 | 1.51 | 5.20 |
| LPS | 1.24 | 0.06 | 0.03 | 0.36 | 2.37 | 1.62 | 5.90 |
| LPS + IMP | 1.42 | 0.11 | 0.05 | 0.46 | 2.14 | 1.30 | 5.69 |
| PA | 1.62 | 0.30 | 0.18 | 1.65 | 2.12 | 1.42 | 7.50 |
| PA + IMP | 2.23 | 0.49 | 0.24 | 1.73 | 1.68 | 0.81 | 7.32 |
| LPS + PA | 5.18 | 1.31 | 0.70 | 3.81 | 3.61 | 2.91 | 18.21 |
| LPS + PA + IMP | 3.30 | 0.66 | 0.30 | 2.08 | 2.01 | 1.24 | 9.85 |
| Total Fat | Fatty Acid | Saturated Fatty Acid | Palmitic Acid | |
|---|---|---|---|---|
| D12492 | 60% kcal fat | 254.5 g/kg | 81.5 g/kg | 51.0 g/kg |
| D12450B | 10% kcal fat | 43.7 g/kg | 9.9 g/kg | 1.1 g/kg |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Z.; Li, Y.; Chowdhury, N.; Yu, H.; Syn, W.-K.; Lopes-Virella, M.; Yilmaz, Ö.; Huang, Y. The Presence of Periodontitis Exacerbates Non-Alcoholic Fatty Liver Disease via Sphingolipid Metabolism-Associated Insulin Resistance and Hepatic Inflammation in Mice with Metabolic Syndrome. Int. J. Mol. Sci. 2023, 24, 8322. https://doi.org/10.3390/ijms24098322
Lu Z, Li Y, Chowdhury N, Yu H, Syn W-K, Lopes-Virella M, Yilmaz Ö, Huang Y. The Presence of Periodontitis Exacerbates Non-Alcoholic Fatty Liver Disease via Sphingolipid Metabolism-Associated Insulin Resistance and Hepatic Inflammation in Mice with Metabolic Syndrome. International Journal of Molecular Sciences. 2023; 24(9):8322. https://doi.org/10.3390/ijms24098322
Chicago/Turabian StyleLu, Zhongyang, Yanchun Li, Nityananda Chowdhury, Hong Yu, Wing-Kin Syn, Maria Lopes-Virella, Özlem Yilmaz, and Yan Huang. 2023. "The Presence of Periodontitis Exacerbates Non-Alcoholic Fatty Liver Disease via Sphingolipid Metabolism-Associated Insulin Resistance and Hepatic Inflammation in Mice with Metabolic Syndrome" International Journal of Molecular Sciences 24, no. 9: 8322. https://doi.org/10.3390/ijms24098322
APA StyleLu, Z., Li, Y., Chowdhury, N., Yu, H., Syn, W.-K., Lopes-Virella, M., Yilmaz, Ö., & Huang, Y. (2023). The Presence of Periodontitis Exacerbates Non-Alcoholic Fatty Liver Disease via Sphingolipid Metabolism-Associated Insulin Resistance and Hepatic Inflammation in Mice with Metabolic Syndrome. International Journal of Molecular Sciences, 24(9), 8322. https://doi.org/10.3390/ijms24098322