

Uncovering the Underworld of Axial Spondyloarthritis


Abstract
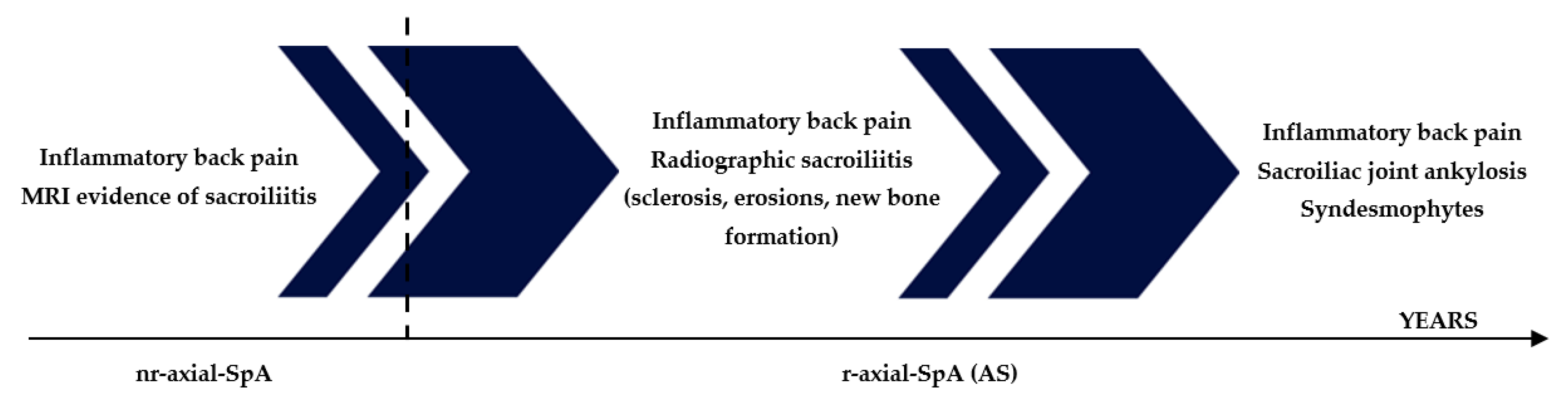
1. Introduction
2. HLA-B27: The Main Genetic Player in Axial-SpA Pathogenesis
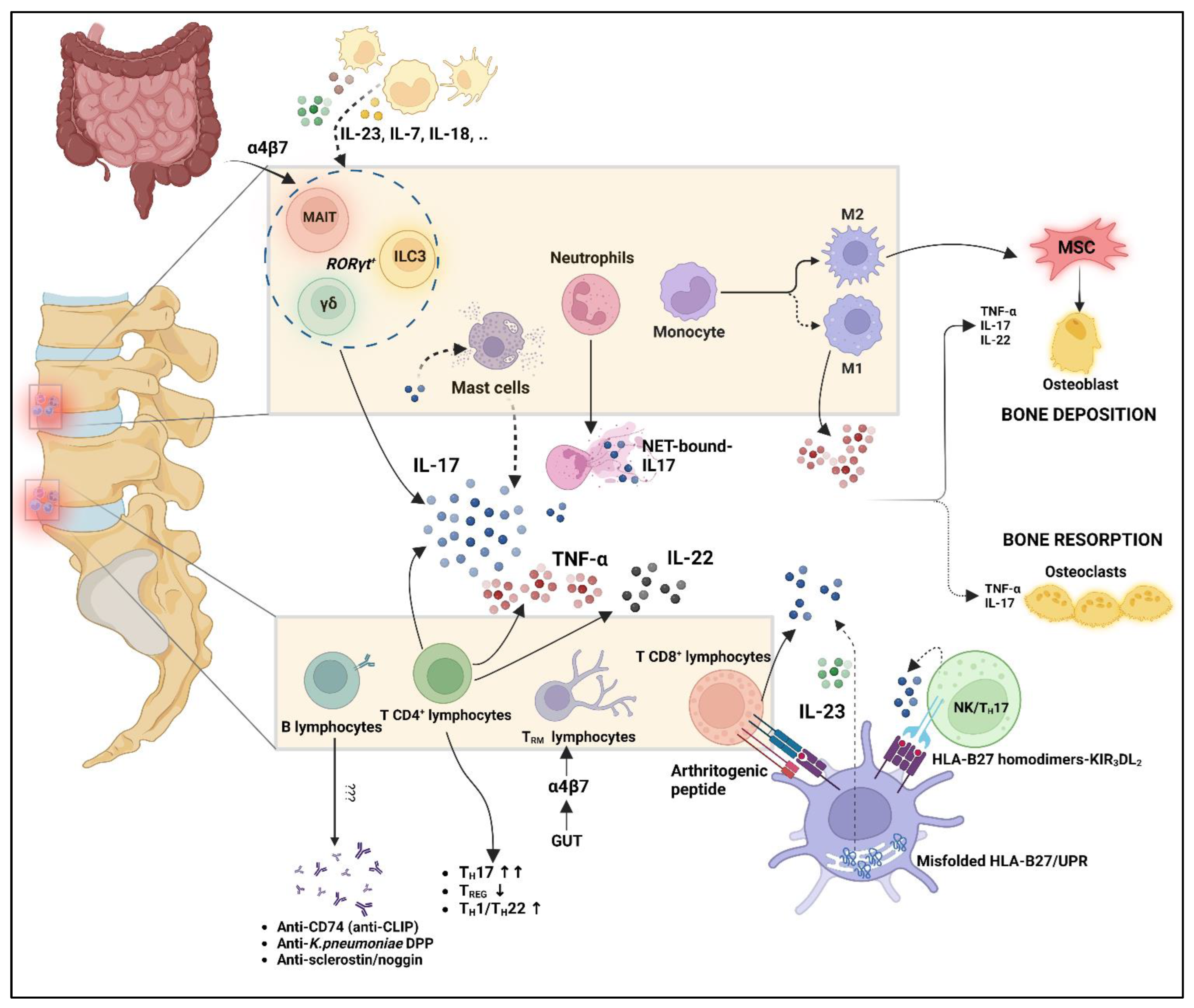
3. Role of Innate Immunity
4. Role of Adaptive Immunity
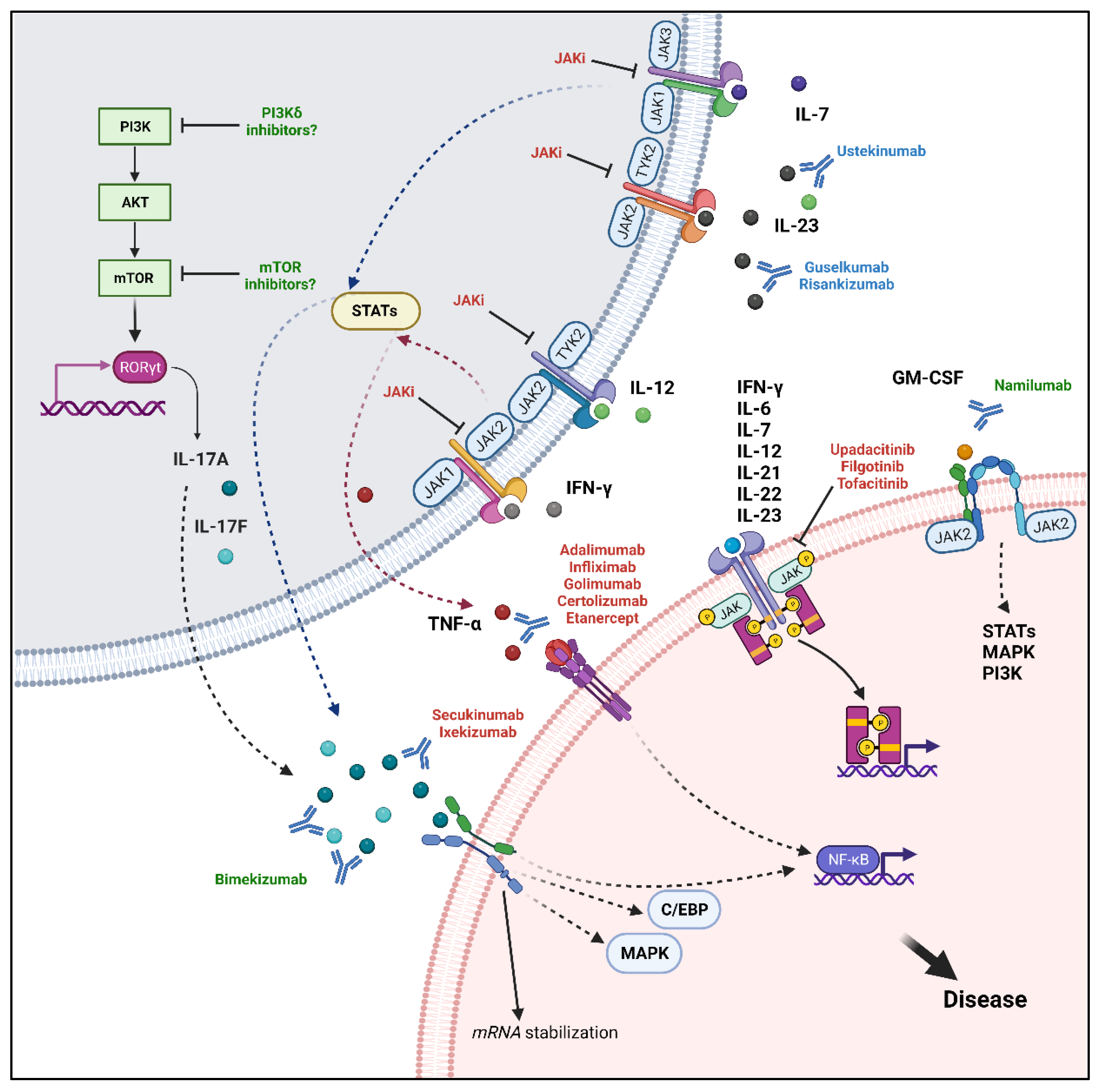
5. Therapeutic Targets in Axial-SpA
5.1. TNF-α
5.2. IL-17
5.3. IL-23: A Debated Role in Axial-SpA
5.4. JAK–STAT Pathway
5.5. Future Perspectives: Potential Therapeutic Targets
6. Closing Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robinson, P.C.; van der Linden, S.; Khan, M.A.; Taylor, W.J. Axial Spondyloarthritis: Concept, Construct, Classification and Implications for Therapy. Nat. Rev. Rheumatol. 2021, 17, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Sieper, J.; Braun, J.; Dougados, M.; Baeten, D. Axial Spondyloarthritis. Nat. Rev. Dis. Prim. 2015, 1, 15013. [Google Scholar] [CrossRef] [PubMed]
- Sieper, J.; van der Heijde, D.; Landewé, R.; Brandt, J.; Burgos-Vagas, R.; Collantes-Estevez, E.; Dijkmans, B.; Dougados, M.; Khan, M.A.; Leirisalo-Repo, M.; et al. New Criteria for Inflammatory Back Pain in Patients with Chronic Back Pain: A Real Patient Exercise by Experts from the Assessment of SpondyloArthritis International Society (ASAS). Ann. Rheum. Dis. 2009, 68, 784–788. [Google Scholar] [CrossRef] [PubMed]
- De Koning, A.; Schoones, J.W.; van der Heijde, D.; van Gaalen, F.A. Pathophysiology of Axial Spondyloarthritis: Consensus and Controversies. Eur. J. Clin. Investig. 2018, 48, e12913. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Goronzy, J.J. The Immunology of Rheumatoid Arthritis. Nat. Immunol. 2021, 22, 10–18. [Google Scholar] [CrossRef]
- Fang, W.; Zhang, Y.; Chen, Z. Innate Lymphoid Cells in Inflammatory Arthritis. Arthritis Res. Ther. 2020, 22, 25. [Google Scholar] [CrossRef]
- Wu, X. Innate Lymphocytes in Inflammatory Arthritis. Front. Immunol. 2020, 11, 565275. [Google Scholar] [CrossRef]
- Rizzo, A.; Guggino, G.; Ferrante, A.; Ciccia, F. Role of Subclinical Gut Inflammation in the Pathogenesis of Spondyloarthritis. Front. Med. 2018, 5, 63. [Google Scholar] [CrossRef]
- Danve, A.; Deodhar, A. Treatment of Axial Spondyloarthritis: An Update. Nat. Rev. Rheumatol. 2022, 18, 205–216. [Google Scholar] [CrossRef]
- Stolwijk, C.; Boonen, A.; van Tubergen, A.; Reveille, J.D. Epidemiology of Spondyloarthritis. Rheum. Dis. Clin. N. Am. 2012, 38, 441–476. [Google Scholar] [CrossRef]
- Navid, F.; Holt, V.; Colbert, R.A. The Enigmatic Role of HLA-B*27 in Spondyloarthritis Pathogenesis. Semin. Immunopathol. 2021, 43, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A. An Update on the Genetic Polymorphism of HLA-B*27 With 213 Alleles Encompassing 160 Subtypes (and Still Counting). Curr. Rheumatol. Rep. 2017, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Guiliano, D.B.; North, H.; Panayoitou, E.; Campbell, E.C.; McHugh, K.; Cooke, F.G.M.; Silvestre, M.; Bowness, P.; Powis, S.J.; Antoniou, A.N. Polymorphisms in the F Pocket of HLA–B27 Subtypes Strongly Affect Assembly, Chaperone Interactions, and Heavy-Chain Misfolding. Arthritis Rheumatol. 2017, 69, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.K.; Luthra-Guptasarma, M. Differences in Conformational Stability of the Two Alpha Domains of the Disease-Associated and Non-Disease-Associated Subtypes of HLA-B27. Int. J. Biol. Macromol. 2017, 94, 233–245. [Google Scholar] [CrossRef]
- Cauli, A.; Shaw, J.; Giles, J.; Hatano, H.; Rysnik, O.; Payeli, S.; McHugh, K.; Dessole, G.; Porru, G.; Desogus, E.; et al. The Arthritis-Associated HLA-B*27:05 Allele Forms More Cell Surface B27 Dimer and Free Heavy Chain Ligands for KIR3DL2 than HLA-B*27:09. Rheumatology 2013, 52, 1952–1962. [Google Scholar] [CrossRef]
- Lim Kam Sian, T.C.C.; Indumathy, S.; Halim, H.; Greule, A.; Cryle, M.J.; Bowness, P.; Rossjohn, J.; Gras, S.; Purcell, A.W.; Schittenhelm, R.B. Allelic Association with Ankylosing Spondylitis Fails to Correlate with Human Leukocyte Antigen B27 Homodimer Formation. J. Biol. Chem. 2019, 294, 20185–20195. [Google Scholar] [CrossRef]
- Loll, B.; Fabian, H.; Huser, H.; Hee, C.-S.; Ziegler, A.; Uchanska-Ziegler, B.; Ziegler, A. Increased Conformational Flexibility of HLA–B*27 Subtypes Associated With Ankylosing Spondylitis. Arthritis Rheumatol. 2016, 68, 1172–1182. [Google Scholar] [CrossRef]
- Loll, B.; Rückert, C.; Uchanska-Ziegler, B.; Ziegler, A. Conformational Plasticity of HLA-B27 Molecules Correlates Inversely With Efficiency of Negative T Cell Selection. Front. Immunol. 2020, 11, 179. [Google Scholar] [CrossRef]
- Yin, L.; Dai, S.; Clayton, G.; Gao, W.; Wang, Y.; Kappler, J.; Marrack, P. Recognition of Self and Altered Self by T Cells in Autoimmunity and Allergy. Protein Cell 2013, 4, 8–16. [Google Scholar] [CrossRef]
- Bodmer, W.F. The HLA System: Structure and Function. J. Clin. Pathol. 1987, 40, 948–958. [Google Scholar] [CrossRef]
- Laloux, L.; Voisin, M.-C.; Allain, J.; Martin, N.; Kerboull, L.; Chevalier, X.; Claudepierre, P. Immunohistological Study of Entheses in Spondyloarthropathies: Comparison in Rheumatoid Arthritis and Osteoarthritis. Ann. Rheum. Dis. 2001, 60, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Gracey, E.; Yao, Y.; Qaiyum, Z.; Lim, M.; Tang, M.; Inman, R.D. Altered Cytotoxicity Profile of CD8+ T Cells in Ankylosing Spondylitis. Arthritis Rheumatol. 2020, 72, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Atagunduz, P.; Appel, H.; Kuon, W.; Wu, P.; Thiel, A.; Kloetzel, P.-M.; Sieper, J. HLA–B27–Restricted CD8+ T Cell Response to Cartilage-Derived Self Peptides in Ankylosing Spondylitis. Arthritis Rheum. 2005, 52, 892–901. [Google Scholar] [CrossRef]
- Lau, M.C.; Keith, P.; Costello, M.-E.; Bradbury, L.A.; Hollis, K.A.; Thomas, R.; Thomas, G.P.; Brown, M.A.; Kenna, T.J. Genetic Association of Ankylosing Spondylitis with TBX21 Influences T-Bet and pro-Inflammatory Cytokine Expression in Humans and SKG Mice as a Model of Spondyloarthritis. Ann. Rheum. Dis. 2017, 76, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Faham, M.; Carlton, V.; Moorhead, M.; Zheng, J.; Klinger, M.; Pepin, F.; Asbury, T.; Vignali, M.; Emerson, R.O.; Robins, H.S.; et al. Discovery of T Cell Receptor β Motifs Specific to HLA-B27-Positive Ankylosing Spondylitis by Deep Repertoire Sequence Analysis. Arthritis Rheumatol. 2017, 69, 774–784. [Google Scholar] [CrossRef]
- Hanson, A.L.; Nel, H.J.; Bradbury, L.; Phipps, J.; Thomas, R.; Lê Cao, K.-A.; Kenna, T.J.; Brown, M.A. Altered Repertoire Diversity and Disease-Associated Clonal Expansions Revealed by T Cell Receptor Immunosequencing in Ankylosing Spondylitis Patients. Arthritis Rheumatol. 2020, 72, 1289–1302. [Google Scholar] [CrossRef]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The Long Road to Destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef]
- Guiliano, D.B.; Fussell, H.; Lenart, I.; Tsao, E.; Nesbeth, D.; Fletcher, A.J.; Campbell, E.C.; Yousaf, N.; Williams, S.; Santos, S.; et al. Endoplasmic Reticulum Degradation–Enhancing α-Mannosidase–like Protein 1 Targets Misfolded HLA–B27 Dimers for Endoplasmic Reticulum–Associated Degradation. Arthritis Rheumatol. 2014, 66, 2976–2988. [Google Scholar] [CrossRef] [PubMed]
- Navid, F.; Colbert, R.A. Causes and Consequences of Endoplasmic Reticulum Stress in Rheumatic Disease. Nat. Rev. Rheumatol. 2017, 13, 25–40. [Google Scholar] [CrossRef]
- Goodall, J.C.; Wu, C.; Zhang, Y.; McNeill, L.; Ellis, L.; Saudek, V.; Gaston, J.S.H. Endoplasmic Reticulum Stress-Induced Transcription Factor, CHOP, Is Crucial for Dendritic Cell IL-23 Expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17698–17703. [Google Scholar] [CrossRef]
- Turner, M.J.; Sowders, D.P.; DeLay, M.L.; Mohapatra, R.; Bai, S.; Smith, J.A.; Brandewie, J.R.; Taurog, J.D.; Colbert, R.A. HLA-B27 Misfolding in Transgenic Rats Is Associated with Activation of the Unfolded Protein Response. J. Immunol. 2005, 175, 2438–2448. [Google Scholar] [CrossRef] [PubMed]
- DeLay, M.L.; Turner, M.J.; Klenk, E.I.; Smith, J.A.; Sowders, D.P.; Colbert, R.A. HLA–B27 Misfolding and the Unfolded Protein Response Augment Interleukin-23 Production and Are Associated with Th17 Activation in Transgenic Rats. Arthritis Rheum. 2009, 60, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Rezaiemanesh, A.; Mahmoudi, M.; Amirzargar, A.A.; Vojdanian, M.; Babaie, F.; Mahdavi, J.; Rajabinejad, M.; Jamshidi, A.R.; Nicknam, M.H. Upregulation of Unfolded Protein Response and ER Stress-Related IL-23 Production in M1 Macrophages from Ankylosing Spondylitis Patients. Inflammation 2022, 45, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Ambarus, C.A.; Yeremenko, N.; Baeten, D.L. Altered Cytokine Expression by Macrophages from HLA-B27-Positive Spondyloarthritis Patients without Evidence of Endoplasmic Reticulum Stress. Rheumatol. Adv. Pract. 2018, 2, rky014. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Accardo-Palumbo, A.; Rizzo, A.; Guggino, G.; Raimondo, S.; Giardina, A.; Cannizzaro, A.; Colbert, R.A.; Alessandro, R.; Triolo, G. Evidence That Autophagy, but Not the Unfolded Protein Response, Regulates the Expression of IL-23 in the Gut of Patients with Ankylosing Spondylitis and Subclinical Gut Inflammation. Ann. Rheum. Dis. 2014, 73, 1566–1574. [Google Scholar] [CrossRef]
- Navid, F.; Layh-Schmitt, G.; Sikora, K.A.; Cougnoux, A.; Colbert, R.A. The Role of Autophagy in the Degradation of Misfolded HLA-B27 Heavy Chains. Arthritis Rheumatol. 2018, 70, 746–755. [Google Scholar] [CrossRef]
- Thakur, A.K.; Luthra-Guptasarma, M. Differences in Cellular Clearing Mechanisms of Aggregates of Two Subtypes of HLA-B27. Front. Immunol. 2021, 12, 795053. [Google Scholar] [CrossRef]
- Bird, L.A.; Peh, C.A.; Kollnberger, S.; Elliott, T.; McMichael, A.J.; Bowness, P. Lymphoblastoid Cells Express HLA-B27 Homodimers Both Intracellularly and at the Cell Surface Following Endosomal Recycling. Eur. J. Immunol. 2003, 33, 748–759. [Google Scholar] [CrossRef]
- Zhang, Z.; Hatano, H.; Shaw, J.; Olde Nordkamp, M.; Jiang, G.; Li, D.; Kollnberger, S. The Leukocyte Immunoglobulin-Like Receptor Family Member LILRB5 Binds to HLA-Class I Heavy Chains. PLoS ONE 2015, 10, e0129063. [Google Scholar] [CrossRef]
- Kollnberger, S.; Chan, A.; Sun, M.-Y.; Ye Chen, L.; Wright, C.; di Gleria, K.; McMichael, A.; Bowness, P. Interaction of HLA-B27 Homodimers with KIR3DL1 and KIR3DL2, Unlike HLA-B27 Heterotrimers, Is Independent of the Sequence of Bound Peptide. Eur. J. Immunol. 2007, 37, 1313–1322. [Google Scholar] [CrossRef]
- Wong-Baeza, I.; Ridley, A.; Shaw, J.; Hatano, H.; Rysnik, O.; McHugh, K.; Piper, C.; Brackenbridge, S.; Fernandes, R.; Chan, A.; et al. KIR3DL2 Binds to HLA-B27 Dimers and Free H Chains More Strongly than Other HLA Class I and Promotes the Expansion of T Cells in Ankylosing Spondylitis. J. Immunol. 2013, 190, 3216–3224. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.T.; Kollnberger, S.D.; Wedderburn, L.R.; Bowness, P. Expansion and Enhanced Survival of Natural Killer Cells Expressing the Killer Immunoglobulin-like Receptor KIR3DL2 in Spondylarthritis. Arthritis Rheum. 2005, 52, 3586–3595. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-C.; Huang, K.-Y.; Lu, M.-C.; Huang Tseng, H.-Y.; Liu, S.-Q.; Lai, N.-S.; Huang, H.-B. HLA-B*27 Heavy Chain Homo-Oligomers Promote the Cytotoxicity of NK Cells via Activation of PI3K/AKT Signaling. Medicina 2022, 58, 1411. [Google Scholar] [CrossRef] [PubMed]
- Bowness, P.; Ridley, A.; Shaw, J.; Chan, A.T.; Wong-Baeza, I.; Fleming, M.; Cummings, F.; McMichael, A.; Kollnberger, S. Th17 Cells Expressing KIR3DL2+ and Responsive to HLA-B27 Homodimers Are Increased in Ankylosing Spondylitis. J. Immunol. 2011, 186, 2672–2680. [Google Scholar] [CrossRef]
- Neerinckx, B.; Kollnberger, S.; Shaw, J.; Lories, R. No Evidence for a Direct Role of HLA-B27 in Pathological Bone Formation in Axial SpA. RMD Open 2017, 3, e000451. [Google Scholar] [CrossRef]
- Aschermann, S.; Englbrecht, M.; Bergua, A.; Spriewald, B.M.; Said-Nahal, R.; Breban, M.; Schett, G.; Rech, J. Presence of HLA-B27 Is Associated with Changes of Serum Levels of Mediators of the Wnt and Hedgehog Pathway. Jt. Bone Spine 2016, 83, 43–46. [Google Scholar] [CrossRef]
- Grandon, B.; Rincheval-Arnold, A.; Jah, N.; Corsi, J.-M.; Araujo, L.M.; Glatigny, S.; Prevost, E.; Roche, D.; Chiocchia, G.; Guénal, I.; et al. HLA-B27 Alters BMP/TGFβ Signalling in Drosophila, Revealing Putative Pathogenic Mechanism for Spondyloarthritis. Ann. Rheum. Dis. 2019, 78, 1653–1662. [Google Scholar] [CrossRef]
- Liu, C.-H.; Raj, S.; Chen, C.-H.; Hung, K.-H.; Chou, C.-T.; Chen, I.-H.; Chien, J.-T.; Lin, I.-Y.; Yang, S.-Y.; Angata, T.; et al. HLA-B27–Mediated Activation of TNAP Phosphatase Promotes Pathogenic Syndesmophyte Formation in Ankylosing Spondylitis. J. Clin. Investig. 2019, 129, 5357–5373. [Google Scholar] [CrossRef]
- Martín-Esteban, A.; Sanz-Bravo, A.; Guasp, P.; Barnea, E.; Admon, A.; López de Castro, J.A. Separate Effects of the Ankylosing Spondylitis Associated ERAP1 and ERAP2 Aminopeptidases Determine the Influence of Their Combined Phenotype on the HLA-B*27 Peptidome. J. Autoimmun. 2017, 79, 28–38. [Google Scholar] [CrossRef]
- De Castro, J.A. How ERAP1 and ERAP2 Shape the Peptidomes of Disease-Associated MHC-I Proteins. Front. Immunol. 2018, 9, 2463. [Google Scholar] [CrossRef]
- Reeves, E.; James, E. The Role of Polymorphic ERAP1 in Autoinflammatory Disease. Biosci. Rep. 2018, 38, BSR20171503. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.; Pulit, S.L.; Leo, P.J.; Pointon, J.J.; Robinson, P.C.; Weisman, M.H.; Ward, M.; Gensler, L.S.; Zhou, X.; Garchon, H.-J.; et al. Major Histocompatibility Complex Associations of Ankylosing Spondylitis Are Complex and Involve Further Epistasis with ERAP1. Nat. Commun. 2015, 6, 7146. [Google Scholar] [CrossRef] [PubMed]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; Ouwehand, W.H.; Samani, N.J.; et al. Association Scan of 14,500 Nonsynonymous SNPs in Four Diseases Identifies Autoimmunity Variants. Nat. Genet. 2007, 39, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Barnea, E.; Melamed Kadosh, D.; Haimovich, Y.; Satumtira, N.; Dorris, M.L.; Nguyen, M.T.; Hammer, R.E.; Tran, T.M.; Colbert, R.A.; Taurog, J.D.; et al. The Human Leukocyte Antigen (HLA)-B27 Peptidome in Vivo, in Spondyloarthritis-Susceptible HLA-B27 Transgenic Rats and the Effect of Erap1 Deletion. Mol. Cell. Proteom. 2017, 16, 642–662. [Google Scholar] [CrossRef]
- Reeves, E.; Colebatch-Bourn, A.; Elliott, T.; Edwards, C.J.; James, E. Functionally Distinct ERAP1 Allotype Combinations Distinguish Individuals with Ankylosing Spondylitis. Proc. Natl. Acad. Sci. USA 2014, 111, 17594–17599. [Google Scholar] [CrossRef] [PubMed]
- Haroon, N.; Tsui, F.W.; Uchanska-Ziegler, B.; Ziegler, A.; Inman, R.D. Endoplasmic Reticulum Aminopeptidase 1 (ERAP1) Exhibits Functionally Significant Interaction with HLA-B27 and Relates to Subtype Specificity in Ankylosing Spondylitis. Ann. Rheum. Dis. 2012, 71, 589–595. [Google Scholar] [CrossRef]
- Chen, L.; Ridley, A.; Hammitzsch, A.; Al-Mossawi, M.H.; Bunting, H.; Georgiadis, D.; Chan, A.; Kollnberger, S.; Bowness, P. Silencing or Inhibition of Endoplasmic Reticulum Aminopeptidase 1 (ERAP1) Suppresses Free Heavy Chain Expression and Th17 Responses in Ankylosing Spondylitis. Ann. Rheum. Dis. 2016, 75, 916–923. [Google Scholar] [CrossRef]
- Tran, T.M.; Hong, S.; Edwan, J.H.; Colbert, R.A. ERAP1 Reduces Accumulation of Aberrant and Disulfide-Linked Forms of HLA-B27 on the Cell Surface. Mol. Immunol. 2016, 74, 10–17. [Google Scholar] [CrossRef]
- Kenna, T.J.; Lau, M.C.; Keith, P.; Ciccia, F.; Costello, M.-E.; Bradbury, L.; Low, P.-L.; Agrawal, N.; Triolo, G.; Alessandro, R.; et al. Disease-Associated Polymorphisms in ERAP1 Do Not Alter Endoplasmic Reticulum Stress in Patients with Ankylosing Spondylitis. Genes Immun. 2015, 16, 35–42. [Google Scholar] [CrossRef]
- Noordenbos, T.; Yeremenko, N.; Gofita, I.; van de Sande, M.; Tak, P.P.; Caňete, J.D.; Baeten, D. Interleukin-17-Positive Mast Cells Contribute to Synovial Inflammation in Spondylarthritis. Arthritis Rheum. 2012, 64, 99–109. [Google Scholar] [CrossRef]
- Noordenbos, T.; Blijdorp, I.; Chen, S.; Stap, J.; Mul, E.; Cañete, J.D.; Lubberts, E.; Yeremenko, N.; Baeten, D. Human Mast Cells Capture, Store, and Release Bioactive, Exogenous IL-17A. J. Leukoc. Biol. 2016, 100, 453–462. [Google Scholar] [CrossRef]
- Paramarta, J.E.; Turina, M.C.; Noordenbos, T.; Heijda, T.F.; Blijdorp, I.C.; Yeremenko, N.; Baeten, D. A Proof-of-Concept Study with the Tyrosine Kinase Inhibitor Nilotinib in Spondyloarthritis. J. Transl. Med. 2016, 14, 308. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhan, X.; Qu, H.; Liang, T.; Li, H.; Chen, L.; Huang, S.; Sun, X.; Jiang, W.; Chen, J.; et al. Upregulated of ANXA3, SORL1, and Neutrophils May Be Key Factors in the Progressionof Ankylosing Spondylitis. Front. Immunol. 2022, 13, 861459. [Google Scholar] [CrossRef] [PubMed]
- Rosine, N.; Rowe, H.; Koturan, S.; Yahia-Cherbal, H.; Leloup, C.; Watad, A.; Berenbaum, F.; Sellam, J.; Dougados, M.; Aimanianda, V.; et al. Characterization of Blood Mucosal-Associated Invariant T Cells in Patients with Axial Spondyloarthritis and of Resident Mucosal-Associated Invariant T Cells From the Axial Entheses of Non-Axial Spondyloarthritis Control Patients. Arthritis Rheumatol. 2022, 74, 1786–1795. [Google Scholar] [CrossRef]
- Appel, H.; Maier, R.; Wu, P.; Scheer, R.; Hempfing, A.; Kayser, R.; Thiel, A.; Radbruch, A.; Loddenkemper, C.; Sieper, J. Analysis of IL-17(+) Cells in Facet Joints of Patients with Spondyloarthritis Suggests That the Innate Immune Pathway Might Be of Greater Relevance than the Th17-Mediated Adaptive Immune Response. Arthritis Res. Ther. 2011, 13, R95. [Google Scholar] [CrossRef]
- Papagoras, C.; Chrysanthopoulou, A.; Mitsios, A.; Ntinopoulou, M.; Tsironidou, V.; Batsali, A.K.; Papadaki, H.A.; Skendros, P.; Ritis, K. IL-17A Expressed on Neutrophil Extracellular Traps Promotes Mesenchymal Stem Cell Differentiation toward Bone-Forming Cells in Ankylosing Spondylitis. Eur. J. Immunol. 2021, 51, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Zambrano-Zaragoza, J.F.; Gutiérrez-Franco, J.; de Jesús Durán-Avelar, M.; Vibanco-Pérez, N.; Ortiz-Martínez, L.; Ayón-Pérez, M.F.; Vázquez-Reyes, A.; Agraz-Cibrián, J.M. Neutrophil Extracellular Traps and Inflammatory Response: Implications for the Immunopathogenesis of Ankylosing Spondylitis. Int. J. Rheum. Dis. 2021, 24, 426–433. [Google Scholar] [CrossRef]
- Ruiz-Limon, P.; Ladehesa-Pineda, M.L.; Castro-Villegas, M.D.C.; Abalos-Aguilera, M.D.C.; Lopez-Medina, C.; Lopez-Pedrera, C.; Barbarroja, N.; Espejo-Peralbo, D.; Gonzalez-Reyes, J.A.; Villalba, J.M.; et al. Enhanced NETosis Generation in Radiographic Axial Spondyloarthritis: Utility as Biomarker for Disease Activity and Anti-TNF-α Therapy Effectiveness. J. Biomed. Sci. 2020, 27, 54. [Google Scholar] [CrossRef] [PubMed]
- Salemme, R.; Peralta, L.N.; Meka, S.H.; Pushpanathan, N.; Alexander, J.J. The Role of NETosis in Systemic Lupus Erythematosus. J. Cell. Immunol. 2019, 1, 33–42. [Google Scholar] [CrossRef]
- Bollow, M.; Fischer, T.; Reisshauer, H.; Backhaus, M.; Sieper, J.; Hamm, B.; Braun, J. Quantitative Analyses of Sacroiliac Biopsies in Spondyloarthropathies: T Cells and Macrophages Predominate in Early and Active Sacroiliitis- Cellularity Correlates with the Degree of Enhancement Detected by Magnetic Resonance Imaging. Ann. Rheum. Dis. 2000, 59, 135–140. [Google Scholar] [CrossRef]
- Akhtari, M.; Zargar, S.J.; Vojdanian, M.; Jamshidi, A.; Mahmoudi, M. Monocyte-Derived and M1 Macrophages from Ankylosing Spondylitis Patients Released Higher TNF-α and Expressed More IL1B in Response to BzATP than Macrophages from Healthy Subjects. Sci. Rep. 2021, 11, 17842. [Google Scholar] [CrossRef] [PubMed]
- Bridgewood, C.; Watad, A.; Russell, T.; Palmer, T.M.; Marzo-Ortega, H.; Khan, A.; Millner, P.A.; Dunsmuir, R.; Rao, A.; Loughenbury, P.; et al. Identification of Myeloid Cells in the Human Enthesis as the Main Source of Local IL-23 Production. Ann. Rheum. Dis. 2019, 78, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Guggino, G.; Zeng, M.; Thomas, R.; Ranganathan, V.; Rahman, A.; Alessandro, R.; Rizzo, A.; Saieva, L.; Macaluso, F.; et al. Proinflammatory CX3CR1+CD59+Tumor Necrosis Factor-Like Molecule 1A+Interleukin-23+ Monocytes Are Expanded in Patients With Ankylosing Spondylitis and Modulate Innate Lymphoid Cell 3 Immune Functions. Arthritis Rheumatol. 2018, 70, 2003–2013. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yuan, W.; Tao, C.; Sun, P.; Yang, Z.; Xu, W. M2 Polarization of Monocytes in Ankylosing Spondylitis and Relationship with Inflammation and Structural Damage. APMIS 2017, 125, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liang, T.; Zhang, Z.; Chen, J.; Xue, J.; Zhan, X.; Ren, L. Transfer of MicroRNA-22-3p by M2 Macrophage-Derived Extracellular Vesicles Facilitates the Development of Ankylosing Spondylitis through the PER2-Mediated Wnt/β-Catenin Axis. Cell Death Discov. 2022, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, V.; Ciccia, F.; Zeng, F.; Sari, I.; Guggino, G.; Muralitharan, J.; Gracey, E.; Haroon, N. Macrophage Migration Inhibitory Factor Induces Inflammation and Predicts Spinal Progression in Ankylosing Spondylitis. Arthritis Rheumatol. 2017, 69, 1796–1806. [Google Scholar] [CrossRef]
- Kok, T.; Wasiel, A.A.; Cool, R.H.; Melgert, B.N.; Poelarends, G.J.; Dekker, F.J. Small-Molecule Inhibitors of Macrophage Migration Inhibitory Factor (MIF) as an Emerging Class of Therapeutics for Immune Disorders. Drug Discov. Today 2018, 23, 1910–1918. [Google Scholar] [CrossRef]
- Serafini, N.; Jarade, A.; Surace, L.; Goncalves, P.; Sismeiro, O.; Varet, H.; Legendre, R.; Coppee, J.-Y.; Disson, O.; Durum, S.K.; et al. Trained ILC3 Responses Promote Intestinal Defense. Science 2022, 375, 859–863. [Google Scholar] [CrossRef]
- Clottu, A.S.; Humbel, M.; Fluder, N.; Karampetsou, M.P.; Comte, D. Innate Lymphoid Cells in Autoimmune Diseases. Front. Immunol. 2021, 12, 789788. [Google Scholar] [CrossRef]
- Hoorweg, K.; Peters, C.P.; Cornelissen, F.; Aparicio-Domingo, P.; Papazian, N.; Kazemier, G.; Mjösberg, J.M.; Spits, H.; Cupedo, T. Functional Differences between Human NKp44(-) and NKp44(+) RORC(+) Innate Lymphoid Cells. Front. Immunol. 2012, 3, 72. [Google Scholar] [CrossRef]
- Longman, R.S.; Diehl, G.E.; Victorio, D.A.; Huh, J.R.; Galan, C.; Miraldi, E.R.; Swaminath, A.; Bonneau, R.; Scherl, E.J.; Littman, D.R. CX3CR1+ Mononuclear Phagocytes Support Colitis-Associated Innate Lymphoid Cell Production of IL-22. J. Exp. Med. 2014, 211, 1571–1583. [Google Scholar] [CrossRef]
- Cuthbert, R.J.; Fragkakis, E.M.; Dunsmuir, R.; Li, Z.; Coles, M.; Marzo-Ortega, H.; Giannoudis, P.V.; Jones, E.; El-Sherbiny, Y.M.; McGonagle, D. Brief Report: Group 3 Innate Lymphoid Cells in Human Enthesis. Arthritis Rheumatol. 2017, 69, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Saieva, L.; Peralta, S.; Giardina, A.; Cannizzaro, A.; Sireci, G.; De Leo, G.; Alessandro, R.; et al. Type 3 Innate Lymphoid Cells Producing IL-17 and IL-22 Are Expanded in the Gut, in the Peripheral Blood, Synovial Fluid and Bone Marrow of Patients with Ankylosing Spondylitis. Ann. Rheum. Dis. 2015, 74, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Rosine, N.; Miceli-Richard, C. Innate Cells: The Alternative Source of IL-17 in Axial and Peripheral Spondyloarthritis? Front. Immunol. 2020, 11, 553742. [Google Scholar] [CrossRef]
- Debusschere, K.; Lories, R.J.; Elewaut, D. MAIT Cells: Not Just Another Brick in the Wall. Ann. Rheum. Dis. 2016, 75, 2057–2059. [Google Scholar] [CrossRef] [PubMed]
- Toussirot, E.; Saas, P. MAIT Cells: Potent Major Cellular Players in the IL-17 Pathway of Spondyloarthritis? RMD Open 2018, 4, e000821. [Google Scholar] [CrossRef] [PubMed]
- Gracey, E.; Qaiyum, Z.; Almaghlouth, I.; Lawson, D.; Karki, S.; Avvaru, N.; Zhang, Z.; Yao, Y.; Ranganathan, V.; Baglaenko, Y.; et al. IL-7 Primes IL-17 in Mucosal-Associated Invariant T (MAIT) Cells, Which Contribute to the Th17-Axis in Ankylosing Spondylitis. Ann. Rheum. Dis. 2016, 75, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, E.; Chiba, A.; Tada, K.; Haga, K.; Kitagaichi, M.; Nakajima, S.; Kusaoi, M.; Sekiya, F.; Ogasawara, M.; Yamaji, K.; et al. Involvement of Mucosal-Associated Invariant T Cells in Ankylosing Spondylitis. J. Rheumatol. 2016, 43, 1695–1703. [Google Scholar] [CrossRef]
- Toussirot, É.; Laheurte, C.; Gaugler, B.; Gabriel, D.; Saas, P. Increased IL-22- and IL-17A-Producing Mucosal-Associated Invariant T Cells in the Peripheral Blood of Patients With Ankylosing Spondylitis. Front. Immunol. 2018, 9, 1610. [Google Scholar] [CrossRef]
- Hogquist, K.; Georgiev, H. Recent Advances in INKT Cell Development. F1000Research 2020, 9, 127. [Google Scholar] [CrossRef]
- Jacques, P.; Venken, K.; Van Beneden, K.; Hammad, H.; Seeuws, S.; Drennan, M.B.; Deforce, D.; Verbruggen, G.; Apostolaki, M.; Kollias, G.; et al. Invariant Natural Killer T Cells Are Natural Regulators of Murine Spondylarthritis. Arthritis Rheum. 2010, 62, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Noto Llana, M.; Sarnacki, S.H.; Morales, A.L.; Aya Castañeda, M.D.R.; Giacomodonato, M.N.; Blanco, G.; Cerquetti, M.C. Activation of INKT Cells Prevents Salmonella-Enterocolitis and Salmonella-Induced Reactive Arthritis by Downregulating IL-17-Producing ΓδT Cells. Front. Cell. Infect. Microbiol. 2017, 7, 398. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.; Jacques, P.; Mortier, C.; Labadia, M.E.; Decruy, T.; Coudenys, J.; Hoyt, K.; Wayne, A.L.; Hughes, R.; Turner, M.; et al. RORγt Inhibition Selectively Targets IL-17 Producing INKT and Γδ-T Cells Enriched in Spondyloarthritis Patients. Nat. Commun. 2019, 10, 9. [Google Scholar] [CrossRef]
- Akitsu, A.; Iwakura, Y. Interleukin-17-Producing Γδ T (Γδ17) Cells in Inflammatory Diseases. Immunology 2018, 155, 418–426. [Google Scholar] [CrossRef]
- Kenna, T.J.; Davidson, S.I.; Duan, R.; Bradbury, L.A.; McFarlane, J.; Smith, M.; Weedon, H.; Street, S.; Thomas, R.; Thomas, G.P.; et al. Enrichment of Circulating Interleukin-17-Secreting Interleukin-23 Receptor-Positive γ/δ T Cells in Patients with Active Ankylosing Spondylitis. Arthritis Rheum. 2012, 64, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, R.J.; Watad, A.; Fragkakis, E.M.; Dunsmuir, R.; Loughenbury, P.; Khan, A.; Millner, P.A.; Davison, A.; Marzo-Ortega, H.; Newton, D.; et al. Evidence That Tissue Resident Human Enthesis ΓδT-Cells Can Produce IL-17A Independently of IL-23R Transcript Expression. Ann. Rheum. Dis. 2019, 78, 1559–1565. [Google Scholar] [CrossRef]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.-C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 Induces Spondyloarthropathy by Acting on ROR-Γt+ CD3+CD4-CD8- Entheseal Resident T Cells. Nat. Med. 2012, 18, 1069–1076. [Google Scholar] [CrossRef]
- Reinhardt, A.; Yevsa, T.; Worbs, T.; Lienenklaus, S.; Sandrock, I.; Oberdörfer, L.; Korn, T.; Weiss, S.; Förster, R.; Prinz, I. Interleukin-23-Dependent γ/δ T Cells Produce Interleukin-17 and Accumulate in the Enthesis, Aortic Valve, and Ciliary Body in Mice. Arthritis Rheumatol. 2016, 68, 2476–2486. [Google Scholar] [CrossRef]
- Romero-López, J.P.; Gómez-Martínez, D.; Domínguez-López, M.L.; Jiménez-Zamudio, L.; Casasola-Vargas, J.C.; Burgos-Vargas, R.; García-Latorre, E. Differential Expression of TLR2 and TLR4 in A4β7-Positive Leukocytes of Patients with Axial Spondyloarthritis. Rheumatology 2020, 59, 879–888. [Google Scholar] [CrossRef]
- Taurog, J.D.; Dorris, M.L.; Satumtira, N.; Tran, T.M.; Sharma, R.; Dressel, R.; van den Brandt, J.; Reichardt, H.M. Spondylarthritis in HLA-B27/Human Beta2-Microglobulin-Transgenic Rats Is Not Prevented by Lack of CD8. Arthritis Rheum. 2009, 60, 1977–1984. [Google Scholar] [CrossRef]
- Ruutu, M.; Thomas, G.; Steck, R.; Degli-Esposti, M.A.; Zinkernagel, M.S.; Alexander, K.; Velasco, J.; Strutton, G.; Tran, A.; Benham, H.; et al. β-Glucan Triggers Spondylarthritis and Crohn’s Disease–like Ileitis in SKG Mice. Arthritis Rheum. 2012, 64, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Yasutomo, K.; Doyle, C.; Miele, L.; Fuchs, C.; Germain, R.N. The Duration of Antigen Receptor Signalling Determines CD4+ versus CD8+ T-Cell Lineage Fate. Nature 2000, 404, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Hombauer, M.; Bilic, I.; Naoe, Y.; Schebesta, A.; Taniuchi, I.; Ellmeier, W. The Zinc-Finger Protein MAZR Is Part of the Transcription Factor Network That Controls the CD4 versus CD8 Lineage Fate of Double-Positive Thymocytes. Nat. Immunol. 2010, 11, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Zhang, X.; Zhou, Y.; Tang, J.; Han, Q.; Zhang, Y.; Ni, Q.; Chen, G.; Jia, Q.; Yu, H.; et al. TCR Repertoire and CDR3 Motif Analyses Depict the Role of Aβ T Cells in Ankylosing Spondylitis. EBioMedicine 2019, 47, 414–426. [Google Scholar] [CrossRef]
- Komech, E.A.; Koltakova, A.D.; Barinova, A.A.; Minervina, A.A.; Salnikova, M.A.; Shmidt, E.I.; Korotaeva, T.V.; Loginova, E.Y.; Erdes, S.F.; Bogdanova, E.A.; et al. TCR Repertoire Profiling Revealed Antigen-Driven CD8+ T Cell Clonal Groups Shared in Synovial Fluid of Patients with Spondyloarthritis. Front. Immunol. 2022, 13, 973243. [Google Scholar] [CrossRef]
- Yang, M.; Lv, Q.; Wei, Q.; Jiang, Y.; Qi, J.; Xiao, M.; Fang, L.; Xie, Y.; Cao, S.; Lin, Z.; et al. TNF-α Inhibitor Therapy Can Improve the Immune Imbalance of CD4+ T Cells and Negative Regulatory Cells but Not CD8+ T Cells in Ankylosing Spondylitis. Arthritis Res. Ther. 2020, 22, 149. [Google Scholar] [CrossRef]
- Liu, D.; Liu, B.; Lin, C.; Gu, J. Imbalance of Peripheral Lymphocyte Subsets in Patients with Ankylosing Spondylitis: A Meta-Analysis. Front. Immunol. 2021, 12, 696973. [Google Scholar] [CrossRef]
- Lejon, K.; Hellman, U.; Do, L.; Kumar, A.; Forsblad-d’Elia, H. Increased Proportions of Inflammatory T Cells and Their Correlations with Cytokines and Clinical Parameters in Patients with Ankylosing Spondylitis from Northern Sweden. Scand. J. Immunol. 2022, 96, e13190. [Google Scholar] [CrossRef]
- Li, M.; Zhou, X.; Zhou, L.; Yu, Z.; Fu, L.; Yang, P. Meta-Analysis of Changes in the Number and Proportion of Regulatory T Cells in Patients with Ankylosing Spondylitis. Biomed Res. Int. 2020, 2020, 8709804. [Google Scholar] [CrossRef]
- Gracey, E.; Yao, Y.; Green, B.; Qaiyum, Z.; Baglaenko, Y.; Lin, A.; Anton, A.; Ayearst, R.; Yip, P.; Inman, R.D. Sexual Dimorphism in the Th17 Signature of Ankylosing Spondylitis. Arthritis Rheumatol. 2016, 68, 679–689. [Google Scholar] [CrossRef]
- Bautista-Caro, M.-B.; Arroyo-Villa, I.; Castillo-Gallego, C.; de Miguel, E.; Peiteado, D.; Puig-Kröger, A.; Martín-Mola, E.; Miranda-Carús, M.-E. Decreased Th17 and Th1 Cells in the Peripheral Blood of Patients with Early Non-Radiographic Axial Spondyloarthritis: A Marker of Disease Activity in HLA-B27+ Patients. Rheumatology 2013, 52, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Takeuchi, Y.; Hirota, K. The Pathogenicity of Th17 Cells in Autoimmune Diseases. Semin. Immunopathol. 2019, 41, 283–297. [Google Scholar] [CrossRef]
- Fragoulis, G.E.; Siebert, S. Treatment Strategies in Axial Spondyloarthritis: What, When and How? Rheumatology 2020, 59, iv79–iv89. [Google Scholar] [CrossRef] [PubMed]
- Klasen, C.; Meyer, A.; Wittekind, P.S.; Waqué, I.; Nabhani, S.; Kofler, D.M. Prostaglandin Receptor EP4 Expression by Th17 Cells Is Associated with High Disease Activity in Ankylosing Spondylitis. Arthritis Res. Ther. 2019, 21, 159. [Google Scholar] [CrossRef] [PubMed]
- Guggino, G.; Rizzo, A.; Mauro, D.; Macaluso, F.; Ciccia, F. Gut-Derived CD8(+) Tissue-Resident Memory T Cells Are Expanded in the Peripheral Blood and Synovia of SpA Patients. Ann. Rheum. Dis. 2021, 80, e174. [Google Scholar] [CrossRef] [PubMed]
- Qaiyum, Z.; Gracey, E.; Yao, Y.; Inman, R.D. Integrin and Transcriptomic Profiles Identify a Distinctive Synovial CD8+ T Cell Subpopulation in Spondyloarthritis. Ann. Rheum. Dis. 2019, 78, 1566–1575. [Google Scholar] [CrossRef]
- Zhu, W.; He, X.; Cheng, K.; Zhang, L.; Chen, D.; Wang, X.; Qiu, G.; Cao, X.; Weng, X. Ankylosing Spondylitis: Etiology, Pathogenesis, and Treatments. Bone Res. 2019, 7, 22. [Google Scholar] [CrossRef]
- Wang, M.; Liu, C.; Bond, A.; Yang, J.; Zhou, X.; Wang, J.; Ji, B. Dysfunction of Regulatory T Cells in Patients with Ankylosing Spondylitis Is Associated with a Loss of Tim-3. Int. Immunopharmacol. 2018, 59, 53–60. [Google Scholar] [CrossRef]
- Miao, J.; Zhu, P. Functional Defects of Treg Cells: New Targets in Rheumatic Diseases, Including Ankylosing Spondylitis. Curr. Rheumatol. Rep. 2018, 20, 30. [Google Scholar] [CrossRef]
- Xie, J.; Wang, Z.; Wang, W. Semaphorin 4D Induces an Imbalance of Th17/Treg Cells by Activating the Aryl Hydrocarbon Receptor in Ankylosing Spondylitis. Front. Immunol. 2020, 11, 2151. [Google Scholar] [CrossRef]
- Peng, J.; Gong, Y.; Zhang, Y.; Wang, D.; Xiao, Z. Immunohistological Analysis of Active Sacroiliitis in Patients with Axial Spondyloarthritis. Medicine 2017, 96, e6605. [Google Scholar] [CrossRef]
- Lin, Q.; Gu, J.; Li, T.; Zhang, F.; Lin, Z.; Liao, Z.; Wei, Q.; Cao, S.; Li, L. Value of the Peripheral Blood B-Cells Subsets in Patients with Ankylosing Spondylitis. Chin. Med. J. 2009, 122, 1784–1789. [Google Scholar] [PubMed]
- Bautista-Caro, M.-B.; de Miguel, E.; Peiteado, D.; Plasencia-Rodríguez, C.; Villalba, A.; Monjo-Henry, I.; Puig-Kröger, A.; Sánchez-Mateos, P.; Martín-Mola, E.; Miranda-Carús, M.-E. Increased Frequency of Circulating CD19+CD24hiCD38hi B Cells with Regulatory Capacity in Patients with Ankylosing Spondylitis (AS) Naïve for Biological Agents. PLoS ONE 2017, 12, e0180726. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, L.; Ren, Y.; Zhang, K.; Yang, Y.; Fang, Y.; Yan, X.; Peng, D.; Gao, C.; Li, S. Defective Function of CD24(+)CD38(+) Regulatory B Cells in Ankylosing Spondylitis. DNA Cell Biol. 2016, 35, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Wilbrink, R.; Spoorenberg, A.; Verstappen, G.M.P.J.; Kroese, F.G.M. B Cell Involvement in the Pathogenesis of Ankylosing Spondylitis. Int. J. Mol. Sci. 2021, 22, 13325. [Google Scholar] [CrossRef]
- Wendling, D.; Augé, B.; Streit, G.; Toussirot, E.; Mathieu, S. Lack of Short-Term Efficacy of Rituximab upon Symptoms of Ankylosing Spondylitis Treated for an Associated Vasculitis. Jt. Bone Spine 2008, 75, 510–511. [Google Scholar] [CrossRef] [PubMed]
- Song, I.-H.; Heldmann, F.; Rudwaleit, M.; Listing, J.; Appel, H.; Haug-Rost, I.; Braun, J.; Sieper, J. One-Year Follow-up of Ankylosing Spondylitis Patients Responding to Rituximab Treatment and Re-Treated in Case of a Flare. Ann. Rheum. Dis. 2013, 72, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Song, I.-H.; Heldmann, F.; Rudwaleit, M.; Listing, J.; Appel, H.; Braun, J.; Sieper, J. Different Response to Rituximab in Tumor Necrosis Factor Blocker-Naive Patients with Active Ankylosing Spondylitis and in Patients in Whom Tumor Necrosis Factor Blockers Have Failed: A Twenty-Four-Week Clinical Trial. Arthritis Rheum. 2010, 62, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Na, N.; Zhang, X.; Zhao, Y. The Biological Function and Significance of CD74 in Immune Diseases. Inflamm. Res. 2017, 66, 209–216. [Google Scholar] [CrossRef]
- Van Kempen, T.S.; Leijten, E.F.A.; Lindenbergh, M.F.S.; Nordkamp, M.O.; Driessen, C.; Lebbink, R.-J.; Baerlecken, N.; Witte, T.; Radstake, T.R.D.J.; Boes, M. Impaired Proteolysis by SPPL2a Causes CD74 Fragment Accumulation That Can Be Recognized by Anti-CD74 Autoantibodies in Human Ankylosing Spondylitis. Eur. J. Immunol. 2020, 50, 1209–1219. [Google Scholar] [CrossRef]
- Baraliakos, X.; Baerlecken, N.; Witte, T.; Heldmann, F.; Braun, J. High Prevalence of Anti-CD74 Antibodies Specific for the HLA Class II-Associated Invariant Chain Peptide (CLIP) in Patients with Axial Spondyloarthritis. Ann. Rheum. Dis. 2014, 73, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, A.; Dolcino, M.; Tinazzi, E.; Moretta, F.; D’Angelo, S.; Olivieri, I.; Lunardi, C. Antibodies Directed against a Peptide Epitope of a Klebsiella Pneumoniae-Derived Protein Are Present in Ankylosing Spondylitis. PLoS ONE 2017, 12, e0171073. [Google Scholar] [CrossRef] [PubMed]
- Tsui, F.W.L.; Tsui, H.W.; Las Heras, F.; Pritzker, K.P.H.; Inman, R.D. Serum Levels of Novel Noggin and Sclerostin-Immune Complexes Are Elevated in Ankylosing Spondylitis. Ann. Rheum. Dis. 2014, 73, 1873–1879. [Google Scholar] [CrossRef]
- Tanaka, S.; Matsumoto, T. Sclerostin: From Bench to Bedside. J. Bone Miner. Metab. 2021, 39, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Krause, C.; Guzman, A.; Knaus, P. Noggin. Int. J. Biochem. Cell Biol. 2011, 43, 478–481. [Google Scholar] [CrossRef]
- Appel, H.; Ruiz-Heiland, G.; Listing, J.; Zwerina, J.; Herrmann, M.; Mueller, R.; Haibel, H.; Baraliakos, X.; Hempfing, A.; Rudwaleit, M.; et al. Altered Skeletal Expression of Sclerostin and Its Link to Radiographic Progression in Ankylosing Spondylitis. Arthritis Rheum. 2009, 60, 3257–3262. [Google Scholar] [CrossRef]
- Jang, D.-I.; Lee, A.-H.; Shin, H.-Y.; Song, H.-R.; Park, J.-H.; Kang, T.-B.; Lee, S.-R.; Yang, S.-H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Bradley, J.R. TNF-Mediated Inflammatory Disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Bollow, M.; Neure, L.; Seipelt, E.; Seyrekbasan, F.; Herbst, H.; Eggens, U.; Distler, A.; Sieper, J. Use of Immunohistologic and in Situ Hybridization Techniques in the Examination of Sacroiliac Joint Biopsy Specimens from Patients with Ankylosing Spondylitis. Arthritis Rheum. 1995, 38, 499–505. [Google Scholar] [CrossRef]
- Asadbeik, M.; Farazmand, A.; Vanaki, N.; Mostafaei, S.; Jamshidi, A.R.; Ahmadzadeh, N.; Vojdanian, M.; Mohammad–Amoli, M.; Mahmoudi, M. Gene Expression Profile of Proinflammatory Cytokines in Iranian Patients with Ankylosing Spondylitis. Rheumatol. Res. 2017, 2, 31–38. [Google Scholar] [CrossRef]
- Christodoulou-Vafeiadou, E.; Geka, C.; Ntari, L.; Kranidioti, K.; Argyropoulou, E.; Meier, F.; Armaka, M.; Mourouzis, I.; Pantos, C.; Rouchota, M.; et al. Ectopic Bone Formation and Systemic Bone Loss in a Transmembrane TNF-Driven Model of Human Spondyloarthritis. Arthritis Res. Ther. 2020, 22, 232. [Google Scholar] [CrossRef] [PubMed]
- Landewé, R.; Sieper, J.; Mease, P.; Inman, R.D.; Lambert, R.G.; Deodhar, A.; Marzo-Ortega, H.; Magrey, M.; Kiltz, U.; Wang, X.; et al. Efficacy and Safety of Continuing versus Withdrawing Adalimumab Therapy in Maintaining Remission in Patients with Non-Radiographic Axial Spondyloarthritis (ABILITY-3): A Multicentre, Randomised, Double-Blind Study. Lancet 2018, 392, 134–144. [Google Scholar] [CrossRef]
- Sieper, J.; van der Heijde, D.; Dougados, M.; Mease, P.J.; Maksymowych, W.P.; Brown, M.A.; Arora, V.; Pangan, A.L. Efficacy and Safety of Adalimumab in Patients with Non-Radiographic Axial Spondyloarthritis: Results of a Randomised Placebo-Controlled Trial (ABILITY-1). Ann. Rheum. Dis. 2013, 72, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Guillot, X.; Prati, C.; Sondag, M.; Wendling, D. Etanercept for Treating Axial Spondyloarthritis. Expert Opin. Biol. Ther. 2017, 17, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Proft, F.; Weiß, A.; Torgutalp, M.; Protopopov, M.; Rodriguez, V.R.; Haibel, H.; Behmer, O.; Sieper, J.; Poddubnyy, D. Sustained Clinical Response and Safety of Etanercept in Patients with Early Axial Spondyloarthritis: 10-Year Results of the ESTHER Trial. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720X20987700. [Google Scholar] [CrossRef]
- Inman, R.D.; Davis, J.C.J.; van der Heijde, D.; Diekman, L.; Sieper, J.; Kim, S., Il; Mack, M.; Han, J.; Visvanathan, S.; Xu, Z.; et al. Efficacy and Safety of Golimumab in Patients with Ankylosing Spondylitis: Results of a Randomized, Double-Blind, Placebo-Controlled, Phase III Trial. Arthritis Rheum. 2008, 58, 3402–3412. [Google Scholar] [CrossRef]
- Mitoma, H.; Horiuchi, T.; Tsukamoto, H.; Ueda, N. Molecular Mechanisms of Action of Anti-TNF-α Agents—Comparison among Therapeutic TNF-α Antagonists. Cytokine 2018, 101, 56–63. [Google Scholar] [CrossRef]
- Van der Heijde, D.; Dijkmans, B.; Geusens, P.; Sieper, J.; DeWoody, K.; Williamson, P.; Braun, J. Efficacy and Safety of Infliximab in Patients with Ankylosing Spondylitis: Results of a Randomized, Placebo-Controlled Trial (ASSERT). Arthritis Rheum. 2005, 52, 582–591. [Google Scholar] [CrossRef]
- Van der Heijde, D.; Baraliakos, X.; Hermann, K.-G.A.; Landewé, R.B.M.; Machado, P.M.; Maksymowych, W.P.; Davies, O.R.; de Peyrecave, N.; Hoepken, B.; Bauer, L.; et al. Limited Radiographic Progression and Sustained Reductions in MRI Inflammation in Patients with Axial Spondyloarthritis: 4-Year Imaging Outcomes from the RAPID-AxSpA Phase III Randomised Trial. Ann. Rheum. Dis. 2018, 77, 699–705. [Google Scholar] [CrossRef]
- Sepriano, A.; Ramiro, S.; Wichuk, S.; Chiowchanwisawakit, P.; Paschke, J.; van der Heijde, D.; Landewé, R.; Maksymowych, W.P. Tumor Necrosis Factor Inhibitors Reduce Spinal Radiographic Progression in Patients With Radiographic Axial Spondyloarthritis: A Longitudinal Analysis From the Alberta Prospective Cohort. Arthritis Rheumatol. 2021, 73, 1211–1219. [Google Scholar] [CrossRef]
- Karmacharya, P.; Duarte-Garcia, A.; Dubreuil, M.; Murad, M.H.; Shahukhal, R.; Shrestha, P.; Myasoedova, E.; Crowson, C.S.; Wright, K.; Davis, J.M. 3rd Effect of Therapy on Radiographic Progression in Axial Spondyloarthritis: A Systematic Review and Meta-Analysis. Arthritis Rheumatol. 2020, 72, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Monin, L.; Gaffen, S.L. Interleukin 17 Family Cytokines: Signaling Mechanisms, Biological Activities, and Therapeutic Implications. Cold Spring Harb. Perspect. Biol. 2018, 10, a028522. [Google Scholar] [CrossRef] [PubMed]
- Burkett, P.R.; zu Horste, G.M.; Kuchroo, V.K. Pouring Fuel on the Fire: Th17 Cells, the Environment, and Autoimmunity. J. Clin. Investig. 2015, 125, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Park, C.O.; Kupper, T.S. The Emerging Role of Resident Memory T Cells in Protective Immunity and Inflammatory Disease. Nat. Med. 2015, 21, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, M.B.M.; Yeremenko, N.G.; Baeten, D.L.P.; Chielie, S.; Spuls, P.I.; de Rie, M.A.; Lantz, O.; Res, P.C.M. The IL-17A-Producing CD8+ T-Cell Population in Psoriatic Lesional Skin Comprises Mucosa-Associated Invariant T Cells and Conventional T Cells. J. Investig. Dermatol. 2014, 134, 2898–2907. [Google Scholar] [CrossRef]
- Pickens, S.R.; Volin, M.V.; Mandelin, A.M., II; Kolls, J.K.; Pope, R.M.; Shahrara, S. IL-17 Contributes to Angiogenesis in Rheumatoid Arthritis. J. Immunol. 2010, 184, 3233–3241. [Google Scholar] [CrossRef]
- Tsukazaki, H.; Kaito, T. The Role of the IL-23/IL-17 Pathway in the Pathogenesis of Spondyloarthritis. Int. J. Mol. Sci. 2020, 21, 6401. [Google Scholar] [CrossRef]
- Schinocca, C.; Rizzo, C.; Fasano, S.; Grasso, G.; La Barbera, L.; Ciccia, F.; Guggino, G. Role of the IL-23/IL-17 Pathway in Rheumatic Diseases: An Overview. Front. Immunol. 2021, 12, 637829. [Google Scholar] [CrossRef]
- Sieper, J.; Poddubnyy, D.; Miossec, P. The IL-23-IL-17 Pathway as a Therapeutic Target in Axial Spondyloarthritis. Nat. Rev. Rheumatol. 2019, 15, 747–757. [Google Scholar] [CrossRef]
- Danoy, P.; Pryce, K.; Hadler, J.; Bradbury, L.A.; Farrar, C.; Pointon, J.; Ward, M.; Weisman, M.; Reveille, J.D.; Wordsworth, B.P.; et al. Association of Variants at 1q32 and STAT3 with Ankylosing Spondylitis Suggests Genetic Overlap with Crohn’s Disease. PLoS Genet. 2010, 6, e1001195. [Google Scholar] [CrossRef]
- Yang, B.; Xu, Y.; Liu, X.; Huang, Z.; Wang, L. IL-23R and IL-17A Polymorphisms Correlate with Susceptibility of Ankylosing Spondylitis in a Southwest Chinese Population. Oncotarget 2017, 8, 70310–70316. [Google Scholar] [CrossRef]
- Wielińska, J.; Świerkot, J.; Kolossa, K.; Bugaj, B.; Chaszczewska-Markowska, M.; Jeka, S.; Bogunia-Kubik, K. Polymorphisms within Genes Coding for IL-17A and F and Their Receptor as Clinical Hallmarks in Ankylosing Spondylitis. Mediators Inflamm. 2021, 2021, 3125922. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.; Hadler, J.; Pointon, J.P.; Robinson, P.C.; Karaderi, T.; Leo, P.; Cremin, K.; Pryce, K.; Harris, J.; Lee, S.; et al. Identification of Multiple Risk Variants for Ankylosing Spondylitis through High-Density Genotyping of Immune-Related Loci. Nat. Genet. 2013, 45, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.-Y.; Zheng, J.-W.; Yang, J.-Y.; Zhang, T.-Y.; Song, S.; Zhao, R.; Di, J.-K.; Zhang, S.-X.; Wang, C.-H.; Gao, H.-Y. Levels of Peripheral Th17 Cells and Th17-Related Cytokines in Patients with Ankylosing Spondylitis: A Meta-Analysis. Adv. Ther. 2022, 39, 4423–4439. [Google Scholar] [CrossRef]
- Devogelaer, J.P.; Maldague, B.; Malghem, J.; Nagant de Deuxchaisnes, C. Appendicular and Vertebral Bone Mass in Ankylosing Spondylitis. A Comparison of Plain Radiographs with Single- and Dual-Photon Absorptiometry and with Quantitative Computed Tomography. Arthritis Rheum. 1992, 35, 1062–1067. [Google Scholar] [CrossRef]
- Okamoto, K.; Nakashima, T.; Shinohara, M.; Negishi-Koga, T.; Komatsu, N.; Terashima, A.; Sawa, S.; Nitta, T.; Takayanagi, H. Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems. Physiol. Rev. 2017, 97, 1295–1349. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, Y.; Lei, L.; Zhong, J.; Shen, Y.; Tan, J.; Xia, M.; Wu, Y.; Sun, W.; Chen, L. RANKL Expression of Primary Osteoblasts Is Enhanced by an IL-17-Mediated JAK2/STAT3 Pathway through Autophagy Suppression. Connect. Tissue Res. 2021, 62, 411–426. [Google Scholar] [CrossRef]
- Ono, T.; Okamoto, K.; Nakashima, T.; Nitta, T.; Hori, S.; Iwakura, Y.; Takayanagi, H. IL-17-Producing Γδ T Cells Enhance Bone Regeneration. Nat. Commun. 2016, 7, 10928. [Google Scholar] [CrossRef]
- Liao, C.; Zhang, C.; Jin, L.; Yang, Y. IL-17 Alters the Mesenchymal Stem Cell Niche towards Osteogenesis in Cooperation with Osteocytes. J. Cell. Physiol. 2020, 235, 4466–4480. [Google Scholar] [CrossRef]
- Osta, B.; Lavocat, F.; Eljaafari, A.; Miossec, P. Effects of Interleukin-17A on Osteogenic Differentiation of Isolated Human Mesenchymal Stem Cells. Front. Immunol. 2014, 5, 425. [Google Scholar] [CrossRef]
- Gravallese, E.M.; Schett, G. Effects of the IL-23-IL-17 Pathway on Bone in Spondyloarthritis. Nat. Rev. Rheumatol. 2018, 14, 631–640. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Huang, Y.; Zhang, C.; Liu, D.; Cheng, C.; Xu, W.; Zhang, X. Interleukin-17A-Promoted MSC2 Polarization Related with New Bone Formation of Ankylosing Spondylitis. Oncotarget 2017, 8, 96993–97008. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Wang, S.E.; Lee, Y.L.; Kang, S.; Lee, B.; Han, J.; Sung, I.-H.; Park, Y.-S.; Bae, S.-C.; Kim, T.-H. IL-17A Induces Osteoblast Differentiation by Activating JAK2/STAT3 in Ankylosing Spondylitis. Arthritis Res. Ther. 2018, 20, 115. [Google Scholar] [CrossRef] [PubMed]
- Daoussis, D.; Kanellou, A.; Panagiotopoulos, E.; Papachristou, D. DKK-1 Is Underexpressed in Mesenchymal Stem Cells from Patients with Ankylosing Spondylitis and Further Downregulated by IL-17. Int. J. Mol. Sci. 2022, 23, 6660. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.A.A.; Hueber, A.J.; Wilson, S.; Galm, M.; Baum, W.; Kitson, C.; Auer, J.; Lorenz, S.H.; Moelleken, J.; Bader, M.; et al. Combined Inhibition of Tumor Necrosis Factor α and Interleukin-17 as a Therapeutic Opportunity in Rheumatoid Arthritis: Development and Characterization of a Novel Bispecific Antibody. Arthritis Rheumatol. 2015, 67, 51–62. [Google Scholar] [CrossRef]
- Taams, L.S.; Steel, K.J.A.; Srenathan, U.; Burns, L.A.; Kirkham, B.W. IL-17 in the Immunopathogenesis of Spondyloarthritis. Nat. Rev. Rheumatol. 2018, 14, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Henness, S.; van Thoor, E.; Ge, Q.; Armour, C.L.; Hughes, J.M.; Ammit, A.J. IL-17A Acts via P38 MAPK to Increase Stability of TNF-Alpha-Induced IL-8 MRNA in Human ASM. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1283–L1290. [Google Scholar] [CrossRef][Green Version]
- Guler, M.A.; Celik, O.F.; Ayhan, F.F. The Important Role of Central Sensitization in Chronic Musculoskeletal Pain Seen in Different Rheumatic Diseases. Clin. Rheumatol. 2020, 39, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Kieskamp, S.C.; Paap, D.; Carbo, M.J.G.; Wink, F.; Bos, R.; Bootsma, H.; Arends, S.; Spoorenberg, A. Central Sensitization Has Major Impact on Quality of Life in Patients with Axial Spondyloarthritis. Semin. Arthritis Rheum. 2022, 52, 151933. [Google Scholar] [CrossRef]
- Aykurt Karlıbel, I.; Kasapoğlu Aksoy, M. The Relationship between Central Sensitization and Disease Activity, Quality of Life, and Sleep Quality among Patients with Axial Spondyloarthritis. Irish J. Med. Sci. 2022, 192, 481–489. [Google Scholar] [CrossRef]
- Zhou, L.; Li, T.; Wu, X.; Lu, H.; Lin, L.; Ye, L.; Yin, J.; Zhao, J.; Wang, X.; Bian, J.; et al. Assessment of Neuropathic Pain in Ankylosing Spondylitis: Prevalence and Characteristics. Pain Ther. 2021, 10, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Borman, P.; Kaygisiz, F.; Yaman, A. Neuropathic Component of Low Back Pain in Patients with Ankylosing Spondylitis. Mod. Rheumatol. 2021, 31, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Gok, K.; Cengiz, G.; Erol, K.; Ozgocmen, S. Neuropathic Pain Component in Axial Spondyloarthritis and the Influence on Disease Burden. J. Clin. Rheumatol. Pract. Rep. Rheum. Musculoskelet. Dis. 2018, 24, 324–327. [Google Scholar] [CrossRef]
- Kim, T.W.; Son, S.M.; Lee, J.S. Neuropathic Pain in Ankylosing Spondylitis: A Meta-Analysis. Z. Rheumatol. 2020, 79, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.G.; Cunha, T.M.; Vieira, S.M.; Lemos, H.P.; Verri, W.A.; Cunha, F.Q.; Ferreira, S.H. IL-17 Mediates Articular Hypernociception in Antigen-Induced Arthritis in Mice. Pain 2010, 148, 247–256. [Google Scholar] [CrossRef]
- Pinho-Ribeiro, F.A.; Verri, W.A.; Chiu, I.M. Nociceptor Sensory Neuron–Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef]
- Richter, F.; Natura, G.; Ebbinghaus, M.; von Banchet, G.S.; Hensellek, S.; König, C.; Bräuer, R.; Schaible, H.-G. Interleukin-17 Sensitizes Joint Nociceptors to Mechanical Stimuli and Contributes to Arthritic Pain through Neuronal Interleukin-17 Receptors in Rodents. Arthritis Rheum. 2012, 64, 4125–4134. [Google Scholar] [CrossRef]
- Ebbinghaus, M.; Natura, G.; Segond von Banchet, G.; Hensellek, S.; Böttcher, M.; Hoffmann, B.; Salah, F.S.; Gajda, M.; Kamradt, T.; Schaible, H.-G. Interleukin-17A Is Involved in Mechanical Hyperalgesia but Not in the Severity of Murine Antigen-Induced Arthritis. Sci. Rep. 2017, 7, 10334. [Google Scholar] [CrossRef]
- Segond von Banchet, G.; Boettger, M.K.; König, C.; Iwakura, Y.; Bräuer, R.; Schaible, H.-G. Neuronal IL-17 Receptor Upregulates TRPV4 but Not TRPV1 Receptors in DRG Neurons and Mediates Mechanical but Not Thermal Hyperalgesia. Mol. Cell. Neurosci. 2013, 52, 152–160. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, Y.; Lao, L.; Saito, R.; Li, A.; Bäckman, C.M.; Berman, B.M.; Ren, K.; Wei, P.-K.; Zhang, R.-X. Spinal Interleukin-17 Promotes Thermal Hyperalgesia and NMDA NR1 Phosphorylation in an Inflammatory Pain Rat Model. Pain 2013, 154, 294–305. [Google Scholar] [CrossRef]
- Sieper, J.; Deodhar, A.; Marzo-Ortega, H.; Aelion, J.A.; Blanco, R.; Jui-Cheng, T.; Andersson, M.; Porter, B.; Richards, H.B. Secukinumab Efficacy in Anti-TNF-Naive and Anti-TNF-Experienced Subjects with Active Ankylosing Spondylitis: Results from the MEASURE 2 Study. Ann. Rheum. Dis. 2017, 76, 571–592. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, M.; Taniguchi, A.; Fujishige, A.; Kaneko, S.; Haemmerle, S.; Porter, B.O.; Kobayashi, S. Efficacy and Safety of Secukinumab in Japanese Patients with Active Ankylosing Spondylitis: 24-Week Results from an Open-Label Phase 3 Study (MEASURE 2-J). Mod. Rheumatol. 2020, 30, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Kivitz, A.J.; Wagner, U.; Dokoupilova, E.; Supronik, J.; Martin, R.; Talloczy, Z.; Richards, H.B.; Porter, B. Efficacy and Safety of Secukinumab 150 Mg with and Without Loading Regimen in Ankylosing Spondylitis: 104-Week Results from MEASURE 4 Study. Rheumatol. Ther. 2018, 5, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Baraliakos, X.; Braun, J.; Deodhar, A.; Poddubnyy, D.; Kivitz, A.; Tahir, H.; Van den Bosch, F.; Delicha, E.-M.; Talloczy, Z.; Fierlinger, A. Long-Term Efficacy and Safety of Secukinumab 150 Mg in Ankylosing Spondylitis: 5-Year Results from the Phase III MEASURE 1 Extension Study. RMD Open 2019, 5, e001005. [Google Scholar] [CrossRef]
- Baeten, D.; Sieper, J.; Braun, J.; Baraliakos, X.; Dougados, M.; Emery, P.; Deodhar, A.; Porter, B.; Martin, R.; Andersson, M.; et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N. Engl. J. Med. 2015, 373, 2534–2548. [Google Scholar] [CrossRef]
- Van der Heijde, D.; Cheng-Chung Wei, J.; Dougados, M.; Mease, P.; Deodhar, A.; Maksymowych, W.P.; Van den Bosch, F.; Sieper, J.; Tomita, T.; Landewé, R.; et al. Ixekizumab, an Interleukin-17A Antagonist in the Treatment of Ankylosing Spondylitis or Radiographic Axial Spondyloarthritis in Patients Previously Untreated with Biological Disease-Modifying Anti-Rheumatic Drugs (COAST-V): 16 Week Results of a Phase 3 R. Lancet 2018, 392, 2441–2451. [Google Scholar] [CrossRef]
- Dougados, M.; Wei, J.C.-C.; Landewé, R.; Sieper, J.; Baraliakos, X.; den Bosch, F.; Maksymowych, W.P.; Ermann, J.; Walsh, J.A.; Tomita, T.; et al. Efficacy and Safety of Ixekizumab through 52 Weeks in Two Phase 3, Randomised, Controlled Clinical Trials in Patients with Active Radiographic Axial Spondyloarthritis (COAST-V and COAST-W). Ann. Rheum. Dis. 2020, 79, 176–185. [Google Scholar] [CrossRef]
- Ramiro, S.; Nikiphorou, E.; Sepriano, A.; Ortolan, A.; Webers, C.; Baraliakos, X.; Landewé, R.B.M.; Van den Bosch, F.E.; Boteva, B.; Bremander, A.; et al. ASAS-EULAR Recommendations for the Management of Axial Spondyloarthritis: 2022 Update. Ann. Rheum. Dis. 2022, 82, 19–34. [Google Scholar] [CrossRef]
- Paul, C. Ixekizumab or Secukinumab in Psoriasis: What Difference Does It Make? Br. J. Dermatol. 2018, 178, 1003–1005. [Google Scholar] [CrossRef]
- Braun, J.; Baraliakos, X.; Deodhar, A.; Poddubnyy, D.; Emery, P.; Delicha, E.M.; Talloczy, Z.; Porter, B. Secukinumab Shows Sustained Efficacy and Low Structural Progression in Ankylosing Spondylitis: 4-Year Results from the MEASURE 1 Study. Rheumatology 2019, 58, 859–868. [Google Scholar] [CrossRef]
- Shah, M.; Maroof, A.; Al-Hosni, R.N.; Gikas, P.; Gozzard, N.; Shaw, S.; Roberts, S. Bimekizumab Blocks T Cell-Mediated Osteogenic Differentiation of Periosteal Stem Cells: Coupling Pathological Bone Formation to IL17A and IL-17F Signaling. Arthritis Rheumatol. 2017, 69 (Suppl. 10), 1936. [Google Scholar]
- Adams, R.; Maroof, A.; Baker, T.; Lawson, A.D.G.; Oliver, R.; Paveley, R.; Rapecki, S.; Shaw, S.; Vajjah, P.; West, S.; et al. Bimekizumab, a Novel Humanized IgG1 Antibody That Neutralizes Both IL-17A and IL-17F. Front. Immunol. 2020, 11, 1894. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijde, D.; Gensler, L.S.; Deodhar, A.; Baraliakos, X.; Poddubnyy, D.; Kivitz, A.; Farmer, M.K.; Baeten, D.; Goldammer, N.; Coarse, J.; et al. Dual Neutralisation of Interleukin-17A and Interleukin-17F with Bimekizumab in Patients with Active Ankylosing Spondylitis: Results from a 48-Week Phase IIb, Randomised, Double-Blind, Placebo-Controlled, Dose-Ranging Study. Ann. Rheum. Dis. 2020, 79, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijde, D.; Deodhar, A.; Baraliakos, X.; Brown, M.A.; Dobashi, H.; Dougados, M.; Elewaut, D.; Ellis, A.M.; Fleurinck, C.; Gaffney, K.; et al. Efficacy and Safety of Bimekizumab in Axial Spondyloarthritis: Results of Two Parallel Phase 3 Randomised Controlled Trials. Ann. Rheum. Dis. 2023, 82, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Deodhar, A.; Gensler, L.S.; Sieper, J.; Clark, M.; Calderon, C.; Wang, Y.; Zhou, Y.; Leu, J.H.; Campbell, K.; Sweet, K.; et al. Three Multicenter, Randomized, Double-Blind, Placebo-Controlled Studies Evaluating the Efficacy and Safety of Ustekinumab in Axial Spondyloarthritis. Arthritis Rheumatol. 2019, 71, 258–270. [Google Scholar] [CrossRef]
- Baeten, D.; Østergaard, M.; Wei, J.C.-C.; Sieper, J.; Järvinen, P.; Tam, L.-S.; Salvarani, C.; Kim, T.-H.; Solinger, A.; Datsenko, Y.; et al. Risankizumab, an IL-23 Inhibitor, for Ankylosing Spondylitis: Results of a Randomised, Double-Blind, Placebo-Controlled, Proof-of-Concept, Dose-Finding Phase 2 Study. Ann. Rheum. Dis. 2018, 77, 1295–1302. [Google Scholar] [CrossRef]
- McInnes, I.B.; Kavanaugh, A.; Gottlieb, A.B.; Puig, L.; Rahman, P.; Ritchlin, C.; Brodmerkel, C.; Li, S.; Wang, Y.; Mendelsohn, A.M.; et al. Efficacy and Safety of Ustekinumab in Patients with Active Psoriatic Arthritis: 1 Year Results of the Phase 3, Multicentre, Double-Blind, Placebo-Controlled PSUMMIT 1 Trial. Lancet 2013, 382, 780–789. [Google Scholar] [CrossRef]
- Östör, A.; Van den Bosch, F.; Papp, K.; Asnal, C.; Blanco, R.; Aelion, J.; Alperovich, G.; Lu, W.; Wang, Z.; Soliman, A.M.; et al. Efficacy and Safety of Risankizumab for Active Psoriatic Arthritis: 24-Week Results from the Randomised, Double-Blind, Phase 3 KEEPsAKE 2 Trial. Ann. Rheum. Dis. 2022, 81, 351–358. [Google Scholar] [CrossRef]
- Kristensen, L.E.; Keiserman, M.; Papp, K.; McCasland, L.; White, D.; Lu, W.; Wang, Z.; Soliman, A.M.; Eldred, A.; Barcomb, L.; et al. Efficacy and Safety of Risankizumab for Active Psoriatic Arthritis: 24-Week Results from the Randomised, Double-Blind, Phase 3 KEEPsAKE 1 Trial. Ann. Rheum. Dis. 2022, 81, 225–231. [Google Scholar] [CrossRef]
- Araujo, E.G.; Englbrecht, M.; Hoepken, S.; Finzel, S.; Kampylafka, E.; Kleyer, A.; Bayat, S.; Schoenau, V.; Hueber, A.; Rech, J.; et al. Effects of Ustekinumab versus Tumor Necrosis Factor Inhibition on Enthesitis: Results from the Enthesial Clearance in Psoriatic Arthritis (ECLIPSA) Study. Semin. Arthritis Rheum. 2019, 48, 632–637. [Google Scholar] [CrossRef]
- McGonagle, D.; Lories, R.J.U.; Tan, A.L.; Benjamin, M. The Concept of a “Synovio-Entheseal Complex” and Its Implications for Understanding Joint Inflammation and Damage in Psoriatic Arthritis and Beyond. Arthritis Rheum. 2007, 56, 2482–2491. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, A.; Puig, L.; Gottlieb, A.B.; Ritchlin, C.; You, Y.; Li, S.; Song, M.; Randazzo, B.; Rahman, P.; McInnes, I.B. Efficacy and Safety of Ustekinumab in Psoriatic Arthritis Patients with Peripheral Arthritis and Physician-Reported Spondylitis: Post-Hoc Analyses from Two Phase III, Multicentre, Double-Blind, Placebo-Controlled Studies (PSUMMIT-1/PSUMMIT-2). Ann. Rheum. Dis. 2016, 75, 1984–1988. [Google Scholar] [CrossRef]
- Mease, P.J.; Helliwell, P.S.; Gladman, D.D.; Poddubnyy, D.; Baraliakos, X.; Chakravarty, S.D.; Kollmeier, A.P.; Hsia, E.C.; Xu, X.L.; Sheng, S.; et al. Efficacy of Guselkumab on Axial Involvement in Patients with Active Psoriatic Arthritis and Sacroiliitis: A Post-Hoc Analysis of the Phase 3 DISCOVER-1 and DISCOVER-2 Studies. Lancet Rheumatol. 2021, 3, e715–e723. [Google Scholar] [CrossRef]
- Cole, S.; Murray, J.; Simpson, C.; Okoye, R.; Tyson, K.; Griffiths, M.; Baeten, D.; Shaw, S.; Maroof, A. Interleukin (IL)-12 and IL-18 Synergize to Promote MAIT Cell IL-17A and IL-17F Production Independently of IL-23 Signaling. Front. Immunol. 2020, 11, 585134. [Google Scholar] [CrossRef] [PubMed]
- Benham, H.; Rehaume, L.M.; Hasnain, S.Z.; Velasco, J.; Baillet, A.C.; Ruutu, M.; Kikly, K.; Wang, R.; Tseng, H.-W.; Thomas, G.P.; et al. Interleukin-23 Mediates the Intestinal Response to Microbial β-1,3-Glucan and the Development of Spondyloarthritis Pathology in SKG Mice. Arthritis Rheumatol. 2014, 66, 1755–1767. [Google Scholar] [CrossRef]
- Van Tok, M.N.; Na, S.; Lao, C.R.; Alvi, M.; Pots, D.; van de Sande, M.G.H.; Taurog, J.D.; Sedgwick, J.D.; Baeten, D.L.; van Duivenvoorde, L.M. The Initiation, but Not the Persistence, of Experimental Spondyloarthritis Is Dependent on Interleukin-23 Signaling. Front. Immunol. 2018, 9, 1550. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Holland, S.M.; Staudt, L.M. JAKs and STATs in Immunity, Immunodeficiency, and Cancer. N. Engl. J. Med. 2013, 368, 161–170. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The Molecular Details of Cytokine Signaling via the JAK/STAT Pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Tanaka, Y.; Luo, Y.; O’Shea, J.J.; Nakayamada, S. Janus Kinase-Targeting Therapies in Rheumatology: A Mechanisms-Based Approach. Nat. Rev. Rheumatol. 2022, 18, 133–145. [Google Scholar] [CrossRef]
- McInnes, I.B.; Szekanecz, Z.; McGonagle, D.; Maksymowych, W.P.; Pfeil, A.; Lippe, R.; Song, I.-H.; Lertratanakul, A.; Sornasse, T.; Biljan, A.; et al. A Review of JAK–STAT Signalling in the Pathogenesis of Spondyloarthritis and the Role of JAK Inhibition. Rheumatology 2022, 61, 1783–1794. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, X.; Wang, Y. Analysis of JAK2 and STAT3 Polymorphisms in Patients with Ankylosing Spondylitis in Chinese Han Population. Clin. Immunol. 2010, 136, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Cortes, A.; Shipman, L.; Evans, H.G.; Attfield, K.E.; Jostins, L.; Barber, T.; Kaur, G.; Kuttikkatte, S.B.; Leach, O.A.; et al. Resolving TYK2 Locus Genotype-to-Phenotype Differences in Autoimmunity. Sci. Transl. Med. 2016, 8, 363ra149. [Google Scholar] [CrossRef]
- Maeda, Y.; Stavre, Z.; Huang, T.; Manning, C.; Shaughn, B.; Macoritto, M.; Hyland, D.; Waegell, W.; Gravallese, E.M. Blockade of the JAK/STAT Pathway Inhibits Inflammation and Bone Formation in Two Murine Models of Spondyloarthritis. In Proceedings of the 2018 ACR/ARHP Annual Meeting, Chicago, IL, USA, 19–24 October 2018; Volume 7. [Google Scholar]
- Hammitzsch, A.; Chen, L.; de Wit, J.; Al-Mossawi, M.H.; Ridley, A.; Sekine, T.; Simone, D.; Doig, K.; Skapenko, A.; Bowness, P. Inhibiting Ex-Vivo Th17 Responses in Ankylosing Spondylitis by Targeting Janus Kinases. Sci. Rep. 2018, 8, 15645. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK Inhibition as a Therapeutic Strategy for Immune and Inflammatory Diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijde, D.; Deodhar, A.; Wei, J.C.; Drescher, E.; Fleishaker, D.; Hendrikx, T.; Li, D.; Menon, S.; Kanik, K.S. Tofacitinib in Patients with Ankylosing Spondylitis: A Phase II, 16-Week, Randomised, Placebo-Controlled, Dose-Ranging Study. Ann. Rheum. Dis. 2017, 76, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Deodhar, A.; Sliwinska-Stanczyk, P.; Xu, H.; Baraliakos, X.; Gensler, L.S.; Fleishaker, D.; Wang, L.; Wu, J.; Menon, S.; Wang, C.; et al. Tofacitinib for the Treatment of Ankylosing Spondylitis: A Phase III, Randomised, Double-Blind, Placebo-Controlled Study. Ann. Rheum. Dis. 2021, 80, 1004–1013. [Google Scholar] [CrossRef]
- Van der Heijde, D.; Baraliakos, X.; Gensler, L.S.; Maksymowych, W.P.; Tseluyko, V.; Nadashkevich, O.; Abi-Saab, W.; Tasset, C.; Meuleners, L.; Besuyen, R.; et al. Efficacy and Safety of Filgotinib, a Selective Janus Kinase 1 Inhibitor, in Patients with Active Ankylosing Spondylitis (TORTUGA): Results from a Randomised, Placebo-Controlled, Phase 2 Trial. Lancet 2018, 392, 2378–2387. [Google Scholar] [CrossRef]
- Van der Heijde, D.; Song, I.-H.; Pangan, A.L.; Deodhar, A.; van den Bosch, F.; Maksymowych, W.P.; Kim, T.-H.; Kishimoto, M.; Everding, A.; Sui, Y.; et al. Efficacy and Safety of Upadacitinib in Patients with Active Ankylosing Spondylitis (SELECT-AXIS 1): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 2/3 Trial. Lancet 2019, 394, 2108–2117. [Google Scholar] [CrossRef]
- Deodhar, A.; den Bosch, F.; Poddubnyy, D.; Maksymowych, W.P.; der Heijde, D.; Kim, T.H.; Kishimoto, M.; Duan, Y.; Li, Y.; Pangan, A.; et al. OP0016 efficacy and safety of upadacitinib in patients with active non-radiographic axial spondyloarthritis: A double-blind, randomized, placebo-controlled phase 3 trial. Ann. Rheum. Dis. 2022, 81, 9–10. [Google Scholar] [CrossRef]
- Rodriguez, R.M.; Suarez-Alvarez, B.; Lavín, J.L.; Ascensión, A.M.; Gonzalez, M.; Lozano, J.J.; Raneros, A.B.; Bulnes, P.D.; Vidal-Castiñeira, J.R.; Huidobro, C.; et al. Signal Integration and Transcriptional Regulation of the Inflammatory Response Mediated by the GM-/M-CSF Signaling Axis in Human Monocytes. Cell Rep. 2019, 29, 860–872.e5. [Google Scholar] [CrossRef]
- Papagoras, C.; Tsiami, S.; Chrysanthopoulou, A.; Mitroulis, I.; Baraliakos, X. Serum Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) Is Increased in Patients with Active Radiographic Axial Spondyloarthritis and Persists despite Anti-TNF Treatment. Arthritis Res. Ther. 2022, 24, 195. [Google Scholar] [CrossRef] [PubMed]
- Worth, C.; Bowness, P.; Hussein Al-Mossawi, M. Novel Therapeutic Targets in Axial Spondyloarthritis. Curr. Treat. Options Rheumatol. 2018, 4, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Nagai, S.; Kurebayashi, Y.; Koyasu, S. Role of PI3K/Akt and MTOR Complexes in Th17 Cell Differentiation. Ann. N. Y. Acad. Sci. 2013, 1280, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Long, F. MTOR Signaling in Skeletal Development and Disease. Bone Res. 2018, 6, 1. [Google Scholar] [CrossRef]
- Chen, S.; van Tok, M.N.; Knaup, V.L.; Kraal, L.; Pots, D.; Bartels, L.; Gravallese, E.M.; Taurog, J.D.; van de Sande, M.; van Duivenvoorde, L.M.; et al. MTOR Blockade by Rapamycin in Spondyloarthritis: Impact on Inflammation and New Bone Formation in Vitro and in Vivo. Front. Immunol. 2019, 10, 2344. [Google Scholar] [CrossRef]
- Chen, S.; Paveley, R.; Kraal, L.; Sritharan, L.; Stevens, E.; Dedi, N.; Shock, A.; Shaw, S.; Juarez, M.; Yeremenko, N.; et al. Selective Targeting of PI3Kδ Suppresses Human IL-17-Producing T Cells and Innate-like Lymphocytes and May Be Therapeutic for IL-17-Mediated Diseases. J. Autoimmun. 2020, 111, 102435. [Google Scholar] [CrossRef]
- Van Tok, M.N.; Mandour, M.; Wahle, J.; Labadia, M.E.; van de Sande, M.G.H.; Nabozny, G.; Baeten, D.L.; van Duivenvoorde, L.M. Paradoxical Augmentation of Experimental Spondyloarthritis by RORC Inhibition in HLA-B27 Transgenic Rats. Front. Immunol. 2021, 12, 699987. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Zhang, P.; Song, C.; Pan, F.; Li, G.; Peng, L.; Yang, Y.; Wei, Z.; Huang, F. Gut Microbiota Changes in Patients with Spondyloarthritis: A Systematic Review. Semin. Arthritis Rheum. 2022, 52, 151925. [Google Scholar] [CrossRef]
- So, J.; Tam, L.-S. Gut Microbiome and Its Interaction with Immune System in Spondyloarthritis. Microorganisms 2020, 8, 1727. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target | Drug/Inhibitor | Clinical Stage |
|---|---|---|
| TNF-α | Infliximab, adalimumab, golimumab, certolizumab etanercept | Efficacy confirmed in phase III clinical trials [142,143,144,145,146,147,148] |
| IL-17A IL-17A/F | Secukinumab, ixekizumab Bimekizumab | Efficacy confirmed in phase III clinical trials [191,192,193,194,195,196,197,198] Efficacy confirmed in phase III clinical trials [204] |
| IL-23 | IL-23/12 p40 inhibitor: ustekinumab IL-23 p19 inhibitor: risankizumab, guselkumab | Efficacy in axial-SpA was not confirmed in phase III clinical trials [205] Post hoc analysis from phase III clinical trials showed efficacy in axial-PsA [212] Efficacy in axial-SpA was not confirmed in a phase II clinical trial [206] Post hoc analysis from phase III clinical trials showed efficacy in axial-PsA [213] |
| JAK/STAT pathway | JAKs inhibitors: tofacitinib, upadacitinib, filgotinib | Efficacy confirmed in phase III clinical trials [226,227,228,229,230] |
| GM-CSF | Namilumab | Efficacy was not confirmed in a phase IIa clinical trial (NCT03622658) |
| PI3K/Akt/mTor pathway | PI3Kδ inhibitor (seletalisib) mTor inhibitor (rapamycin) | NA NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Vescovo, S.; Venerito, V.; Iannone, C.; Lopalco, G. Uncovering the Underworld of Axial Spondyloarthritis. Int. J. Mol. Sci. 2023, 24, 6463. https://doi.org/10.3390/ijms24076463
Del Vescovo S, Venerito V, Iannone C, Lopalco G. Uncovering the Underworld of Axial Spondyloarthritis. International Journal of Molecular Sciences. 2023; 24(7):6463. https://doi.org/10.3390/ijms24076463
Chicago/Turabian StyleDel Vescovo, Sergio, Vincenzo Venerito, Claudia Iannone, and Giuseppe Lopalco. 2023. "Uncovering the Underworld of Axial Spondyloarthritis" International Journal of Molecular Sciences 24, no. 7: 6463. https://doi.org/10.3390/ijms24076463
APA StyleDel Vescovo, S., Venerito, V., Iannone, C., & Lopalco, G. (2023). Uncovering the Underworld of Axial Spondyloarthritis. International Journal of Molecular Sciences, 24(7), 6463. https://doi.org/10.3390/ijms24076463